
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 28 October 2024
Sec. Genetics of Common and Rare Diseases
Volume 15 - 2024 | https://doi.org/10.3389/fgene.2024.1383033
 Elena Kondratyeva*
Elena Kondratyeva* Yuliya MelyanovskayaVictoriya ShermanAnna Voronkova
Yuliya MelyanovskayaVictoriya ShermanAnna Voronkova Elena ZhekaiteStanislav Krasovsky
Elena ZhekaiteStanislav Krasovsky Elena Amelina
Elena Amelina Nataliya KashirskayaVera ShadrinaAlexander PolyakovTagui Adyan
Nataliya KashirskayaVera ShadrinaAlexander PolyakovTagui Adyan Olga SсhaginaMarina StarinovaElena Enina
Olga SсhaginaMarina StarinovaElena Enina Andrey Vasilyev
Andrey Vasilyev Andrey MarakhonovRena ZinchenkoSergey Kutsev
Andrey MarakhonovRena ZinchenkoSergey KutsevCystic fibrosis (CF) is a genetically inherited disorder characterized by a wide range of clinical manifestations and genetic variations. This study focuses on the genetic and molecular epidemiology of CF in the Russian population, utilizing data from the national CF registry. The birth prevalence of CF in Russia has been analyzed over a span of years, revealing variations in frequency. The study delves into the genetic landscape of CFTR gene variants in Russian patients, showcasing a diverse spectrum with a predominance of severe variants, some of which are rare and distinct from global populations. A total of 233 variants have been documented, exhibiting frequencies ranging from 0.01% to 51.5%, with 47 of these variants remaining uncharted within international genetic databases. As of 2021, CFTR modulator therapy has been introduced for patients under 19 years, heightening the importance of genetic diagnosis. In 2023, more than 1,850 patients under 19 received CFTR modulator therapy. Notably, the impact of complex alleles on disease progression and response to targeted therapies is gaining recognition. Comparisons with European registries highlight distinctive features of the Russian population, such as differences in age distribution among patients. Additionally, the study emphasizes the need to ascertain clinical significance and pathogenicity of newly identified genetic variants, along with exploring their suitability for targeted therapies. The integration of genetic insights into the management of CF offers potential for enhanced personalized therapeutic interventions. In conclusion, this thorough analysis provides a comprehensive understanding of the genetic nuances within the Russian CF population. By illuminating the intricate relationship between genetic variations and disease manifestation, the study underscores the essential role of genetics in shaping therapeutic strategies and improving patient outcomes. Further research and ongoing genetic exploration are crucial for optimizing the care of individuals with CF in the era of evolving therapeutic options.
Cystic fibrosis (CF; OMIM #219700) is an autosomal recessive disorder caused by pathogenic variants in the nucleotide sequence of the CFTR gene (OMIM *602421). The clinical presentation of CF varies widely, ranging from mild monosymptomatic manifestations to severe multi-organ involvement (Mall et al., 2024; Grasemann and Ratjen, 2023). In European countries, the average prevalence of CF is 1 in 7,000 newborns (Castellani et al., 2018), while in the Russian Federation, it ranges from 1 in 8,000 to 1 in 10,000 newborns, showing year-to-year fluctuations and variation among populations and federal districts (Kashirskaya et al., 2021).
The spectrum and frequency of pathogenic variants in the CFTR gene exhibit significant diversity across different populations and ethnic groups. To date, over 2000 genetic variants of the CFTR gene have been identified, of which 719 pathogenic variants are cataloged on the CFTR 2 international project website (https://cftr2.org) as of 2023. Utilizing data from the federal register, genetic and molecular epidemiology studies not only help elucidate potential variations in CF prevalence among different federal districts of the Russian Federation and distinguish prevalent genetic mutations from novel ones, but also offer insights into the prospects of CFTR modulator prescriptions for patient care.
This study aims to investigate the genetic and molecular epidemiology of cystic fibrosis within the Russian population, utilizing epidemiological data and information from the 2021 registry. By analyzing epidemiological data and leveraging the 2021 CF patent Registry (RCFPR), this research seeks to provide a comprehensive understanding of the genetic and molecular aspects of cystic fibrosis in the Russian population. This investigation will contribute to a deeper comprehension of the prevalence, distribution, and molecular variations of the disease’s genetic mutations among different regions and demographic groups within Russia. Additionally, the study intends to identify common genetic variants as well as previously unreported mutations, shedding light on the disease’s genetic landscape in this specific population. Ultimately, the findings of this research hold the potential to advance our knowledge of cystic fibrosis in the Russian population, paving the way for more accurate diagnosis, better targeted treatment approaches, and potential advancements in therapeutic interventions.
The birth prevalence rates of individuals with cystic fibrosis (pwCF) between 2007 and 2022 were meticulously investigated by analyzing data from the federal register. This analysis involved calculating the ratio of identified CF patients for a specific year to the total number of newborns during that same year. The data source for this comprehensive study was the website of the federal register, available at (https://amg-genetics.ru/) (Krasovsky et al., 2021).
The research process encompassed the collection and examination of information from various regions within the Russian Federation (RF). The focal point was the meticulous analysis of the Register of Patients with Cystic Fibrosis in Russia for the year 2021. This database is a compilation of data from all regions that are considered constituent entities of the Russian Federation, with the exception of the Nenets and Chukotka Autonomous Districts. These two districts were not included due to official records from the Ministry of Health of the Russian Federation, which indicate the presence of only one CF patient within these areas.
Furthermore, special attention was given to the prominent urban centers of Moscow and St. Petersburg. The data related to these cities were treated separately, and specific indicators were presented for each. For a more comprehensive overview, Table 1 has been provided, offering insight into the population figures and the corresponding numbers of CF patients. The information is categorized according to the relevant Federal Districts, providing a comprehensive view of the distribution of patients across the Russian regions.

Table 1. Information regarding population and the number of CF patients, categorized by Federal Districts.
The 2021 registry encompasses a comprehensive dataset consisting of clinical and laboratory information pertaining to a total of 3,969 patients. Among these, 3,563 individuals were actively living, with 46 cases sadly deceased within the same year, and an additional 360 patients not being under observation during this period. Notably, the cumulative count pwCF within the entirety of the Russian Federation, as delineated by the “Program of 14 High-Cost Nosologies” sanctioned by the Ministry of Health of the Russian Federation, is documented at 4,259 individuals.
The comprehensive summary of the 2021 registry is meticulously presented in Table 2. Within the context of this dataset, the average age of patients during the year 2021 was calculated at 14.0 years, with a standard deviation of 9.8 years. The median age stood at 11.9 years, encompassing a range from 6.7 to 19.0 years. Among the pwCF population, there was a slight male predominance, constituting 51.8% of the cohort, while women accounted for the remaining 48.2%.
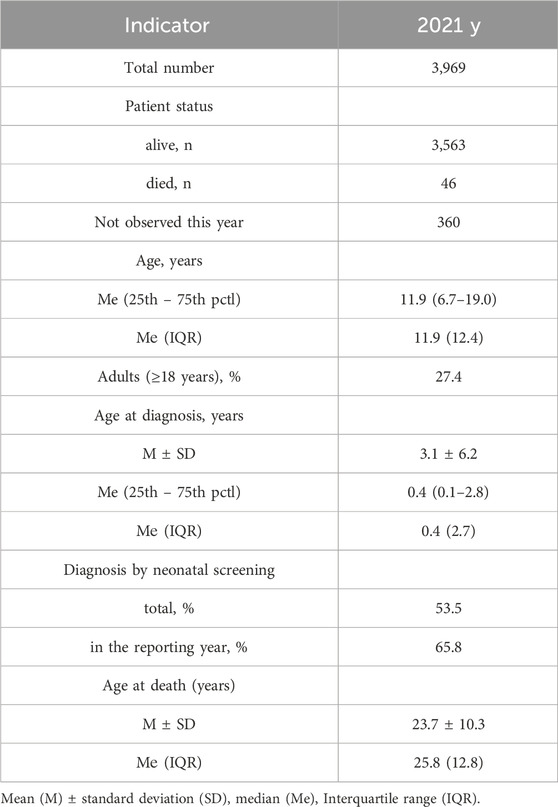
Table 2. The main indicators reflecting the organization of care for pwCF and their health status for 2021.
Molecular diagnostics were conducted following the clinical guidelines of 2021 (Ministry of Health of the Russian Federation, 2021; Castellani et al., 2018). The approach encompassed a concise three-stage algorithm: (i) analysis of 33 frequent CFTR variants; (ii) Sanger sequencing of the coding regions, intron–exon junctions, 5′- and 3′-UTRs of the CFTR gene; and (iii) MLPA analysis for CNV—in DNA samples of CF patients according to the protocol described previously (Petrova et al., 2020). This succinct algorithmic framework ensured a systematic and comprehensive approach to molecular diagnostics, enabling the identification of genetic anomalies associated with cystic fibrosis in patients’ DNA samples.
The variants were analyzed in several databases, namely,: the “RCMG” database on genetics (http://seqdb.med-gen.ru), the CFTR1 database (http://www.genet.sickkids.on.ca/cftr), CFTR2 Base (https://cftr2.org), CFTR-France Base (https://cftr.iurc.montp.inserm.fr/cgi-bin/affiche_var2.cgi), Exome Aggregation Consortium (http://exac.broadinstitute.org), Genome Aggregation Database (http://gnomad.broadinstitute.org), dbSNP (http://www.ncbi.nlm.nih.gov/snp), Exome Variant Server (http://evs.gs.washington.edu/EVS), 1000 Genomes Project (http://browser.1000geno-mes.org/index.html), OMIM (http://www.omim.org), dbVar (http://www. ncbi.nlm.nih.gov/dbvar), Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/in-dex.php), Clin Var (http://www.ncbi. nlm.nih.gov/clin-var), Human Genome Variation Society (http://www.hgvs.org/dblist/dblist.html), DECIPHER (https://decipher).
Statistical data processing was performed using the SPSS software package. Depending on the type of distribution, the mean (M) ± standard deviation (SD) or median (Me) (interquartile range) served as measures of central tendency and dispersion. Statistical processing was performed using the Mann-Whitney test, Pearson chi-square test, Fisher’s exact test, and Kruskal–Wallis test. Linear correlation analysis was used. Differences were considered statistically significant at p < 0.05.
The study and the form of informed voluntary consent were approved by the Ethics Committee of the “RCMG” of the Ministry of Education and Science of the Russian Federation on 10 February 2021 (the chairman of the Ethics Committee is Prof. L. F. Kurilo).
The fluctuation of the birth prevalence index of patients with cystic fibrosis (pwCF) between the years 2007 and 2022 was assessed using data extracted from the RCFPR. The data, as presented in Table 3, details the count of identified patients in each respective reporting year, along with the birth prevalence values calculated in relation to the number of newborns. Notably, the analysis of Table 3 reveals a statistically significant variance in birth prevalence values across the examined years (χ2 = 26.48; p ≤ 0.05; Degrees of Freedom = 14).
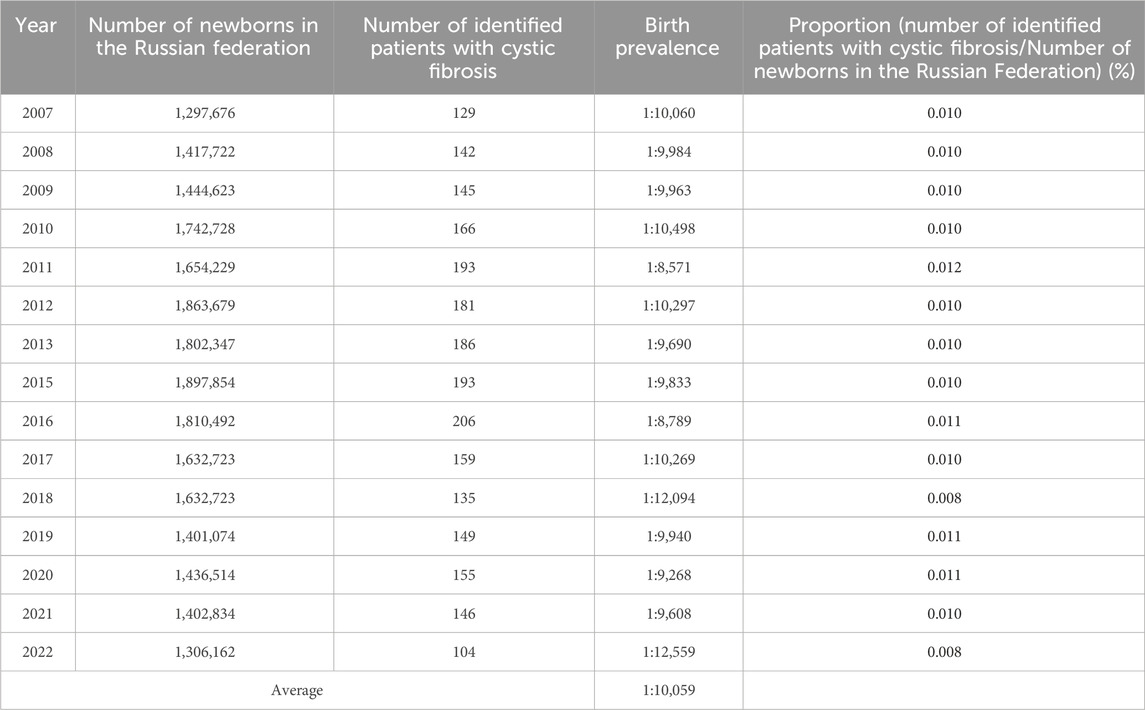
Table 3. Number of identified patients with CF and birth prevalence values (2007–2022).
The calculated average birth prevalence values were determined to be 1 per 10,059 births. This observation signifies the dynamic nature of birth prevalence rates for pwCF throughout the investigated timeframe, indicating the importance of continued monitoring and exploration in this field.
Given the reduced life expectancy of pwCF, we conducted an assessment of CF prevalence within the child population across different federal districts, providing a more realistic representation of this metric (refer to Table 4). Notably, the average prevalence value estimated across the federal districts was found to be 1 per 10,168 children, a figure that aligns closely with the birth prevalence rate of 1 per 10,059. These findings underscore the limited impact of childhood mortality on the overall prevalence of CF.
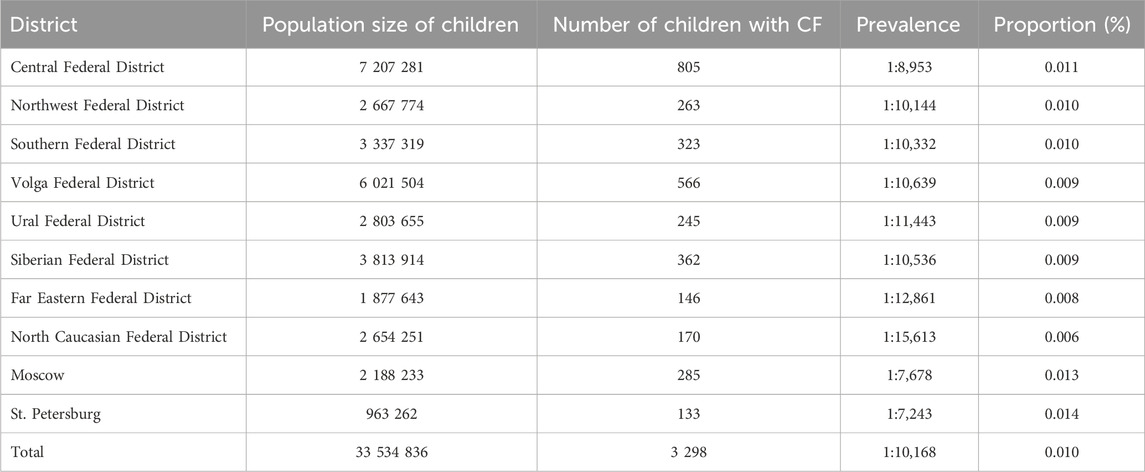
Table 4. Prevalence of CF in federal districts among child population.
Our analysis indicates notably low mortality rates for pwCF during childhood. Quantitative evaluation of CF prevalence further underscores these observations, revealing a statistically significant range of prevalence rates across federal districts, spanning from 1 per 7,243 in St. Petersburg to 1 per 15,613 in the North Caucasus Federal District (χ2 = 96.10; p ≤ 0.05; Degrees of Freedom = 9).
In the subsequent phase, an analysis of molecular genetic data pertaining to the identification of genetic variants in the CFTR gene was conducted. The scope of genetic investigation coverage in the year 2021 encompassed a substantial 93.6% of CF patients (see Table 5). Notably, this coverage consisted of 94.4% of children and 91.3% of adults among the cases examined.
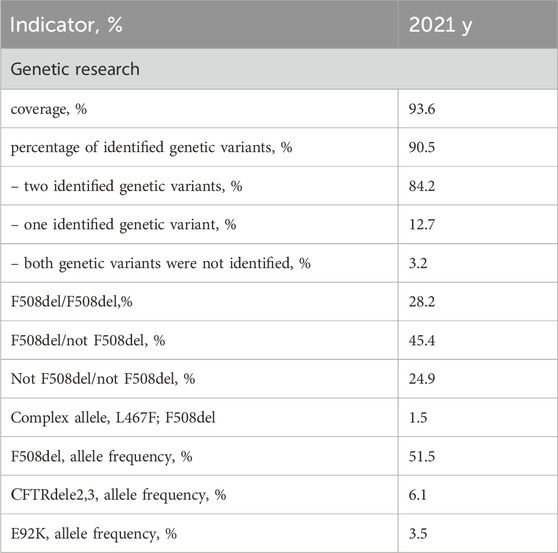
Table 5. Coverage and genetic variant distribution in CFTR gene analysis according to the RCFPR 2021.
The cumulative frequency of identified alleles was determined to be 90.5%, with a breakdown of 91.5% for children and 90.2% for adults. Among patients subjected to genetic examination, two distinct variants of the CFTR gene nucleotide sequence were discerned in 84.2% of cases, while a single variant was detected in 12.7%. In 3.2% of patients, a genetic variant could not be identified.
Within the subset of patients who underwent genetic analysis, a comparable pattern was observed for the detection of genetic variants. Specifically, two genetic variants of the CFTR gene nucleotide sequence were identified in 84.2% of children and 84.3% of adults. Conversely, a single genetic variant was ascertained in 13.6% of children and 10.0% of adults, while a minute subset – 2.2% of children and 5.7% of adults–displayed an absence of detected genetic variants.
Supplementary Table S1 provides an overview of the 233 identified pathogenic variants within the CFTR gene, organized in descending order of frequency. Among these variants, 132 have been repeatedly observed. Remarkably, 83 of these variants are not documented within the CFTR2 database; however, they are comprehensively described in publications authored by both Russian and international researchers (Krasovsky et al., 2021).
47 genetic variants are not presented in international CFTR databases—W1282R, 3272-16T>A, A96E, D579Y, G509R, E403D, G1047S, G480S, I175V, D993A, G509V, P205T, T277X, Q493R, R153I, Y569H, −461A->G, −741T->G, c.1584+18672A>G, c.1761del, c.2312delA, c.264_268del, c.353delC, c.3615_3625del, c.3717+1219C>A, c.37dupT, c.3873+4485A>T, c.3983T>A, c.546T>A, c.743+2T>A, C590Y, D572N, F1286S, G1249E, G314R, G509D; E217G,I506T, L1093P, L159S, L233F, L568F, N505H, E1433G, K1468N, F1078I, T604I, V392G. Of the 47 identified rare genetic variants: 18 are severe, 16 are mild variants, 13 are of unclear clinical significance: G480S, I175V, G509V, Q493R, R153I, −461A->G, −741T->G, c.3983T>A, c.546T>A, C590Y, G1249E, L233F, F1078I. In addition to the newly identified variants, clinical significance or pathogenicity has not been established for some of the previously described variants. Previously undescribed variants that are missing in the literature and databases are highlighted in Supplementary Table S2 in grey.
The first 44 genetic variants are major for the Russian Federation and occur with a frequency more often than 0.10%. According to the European Register of pwCF in 2020, compiled by analyzing 49,111 genotypes of pwCF from 39 countries, the allelic frequencies of 18 frequent CF-causing variants in the CFTR gene that differ from the Russian Federation were determined: F508del – 60.41% (in the RF – 51.55%), G542%X – 2.75% (in the RF– 1.49%), N1303K – 2.18% (in the RF – 1.52%), G551D – 1.26% (in the RF – 0.04%), W1282%X – 1.07% (in the RF – 1.72%), 2,789+5G->A – 1.07% (in the RF - 0.39%), 3,849+10kbC->T – 1.00% (in the RF – 2.22%), CFTRdele2,3%–0.96% (in the RF – 6.11%), R117H – 0.95% (in the RF – 0.03%), 1717-1G->A – 0.88% (in the RF - 0.05%), R553%X– 0.85% (in the RF – 0.18%), 2183AA->G – 0.71% (in the RF – 0.11%), D1152H – 0.63% (in the RF – 0.12%), 621+1G->T – 0.62% (in the RF – 0.19%), R347P – 0.60% (in the RF – 0.12%), G85E – 0.53% (in the RF – 0.11%), 3272-26A->G – 0.52% (in the RF – 0.05%), R1162%X – 0.51% (in the RF – 0.16%) (ECFSPR Annual report 2020., 2022). The roster of prevalent genetic variants within the Russian Federation is distinctive due to its unique spectrum of pathogenic variants and their frequencies, setting it apart from Europe and other global regions. As follows from the information presented in Table 6, it is evident that there is an observable variation in the frequencies of these significant genetic variants when considering the geographical distribution in different federal districts. The genetic variants F508del and CFTRdele2.3 are less common (p < 0.05) in the North Caucasian Federal District, with a higher frequency (p < 0.05) of two genetic variants W1282X (13.5) and 1677delTA (28.4). The E92K variant with a higher frequency (p < 0.05) was detected in the North Caucasian Federal District (7.3) and Volga Federal District (9.2). This variation highlights the influence of regional factors on the prevalence of these genetic variants.
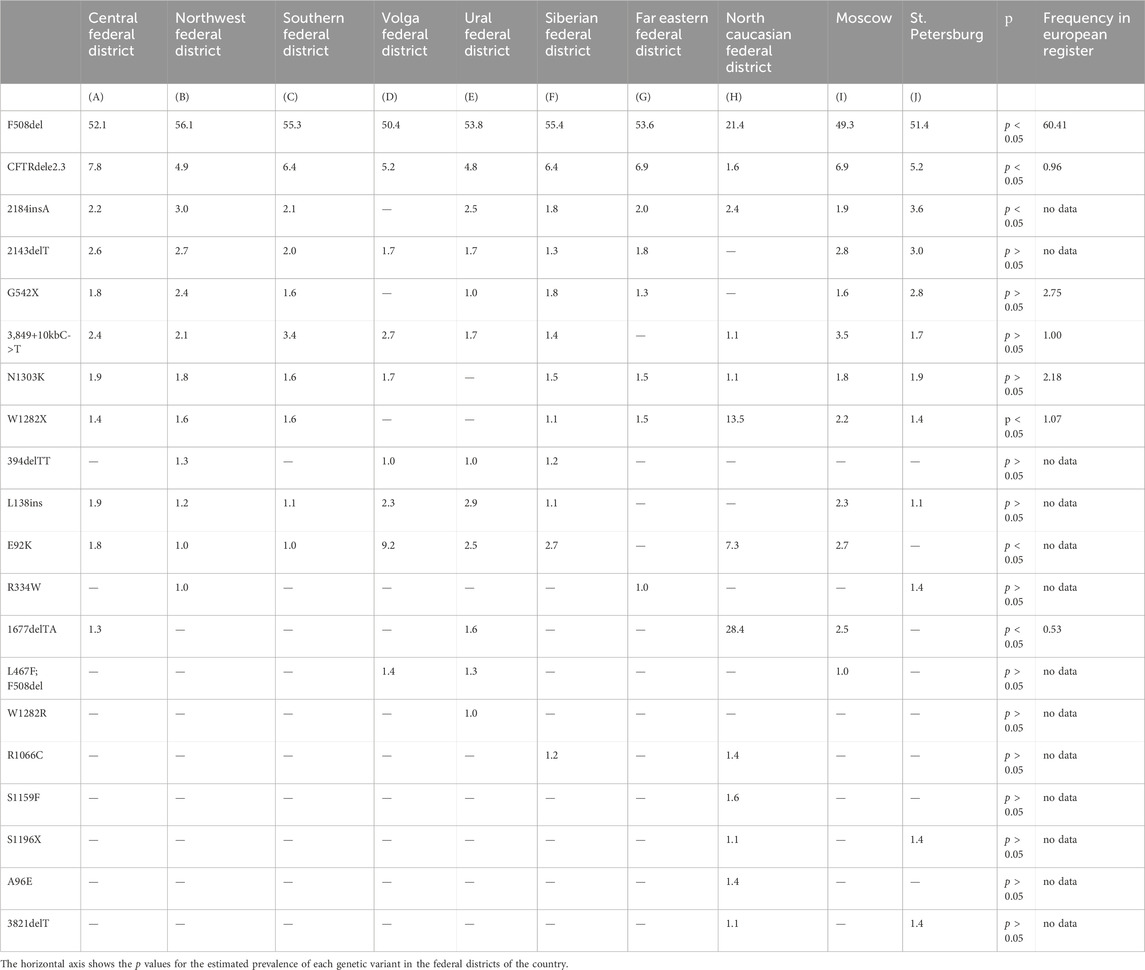
Table 6. Frequency of major genetic variants (frequency > 1.0%) in CFTR gene nucleotide sequence across federal districts of RF.
Guided by these disparities identified across European countries, the subsequent phase involved an in-depth analysis of the spectrum and frequencies of major genetic variants, defined as those occurring with a frequency surpassing 1.0%, across the Federal Districts of the Russian Federation. The outcomes of this analysis are succinctly summarized in Table 6 and Figure 1.
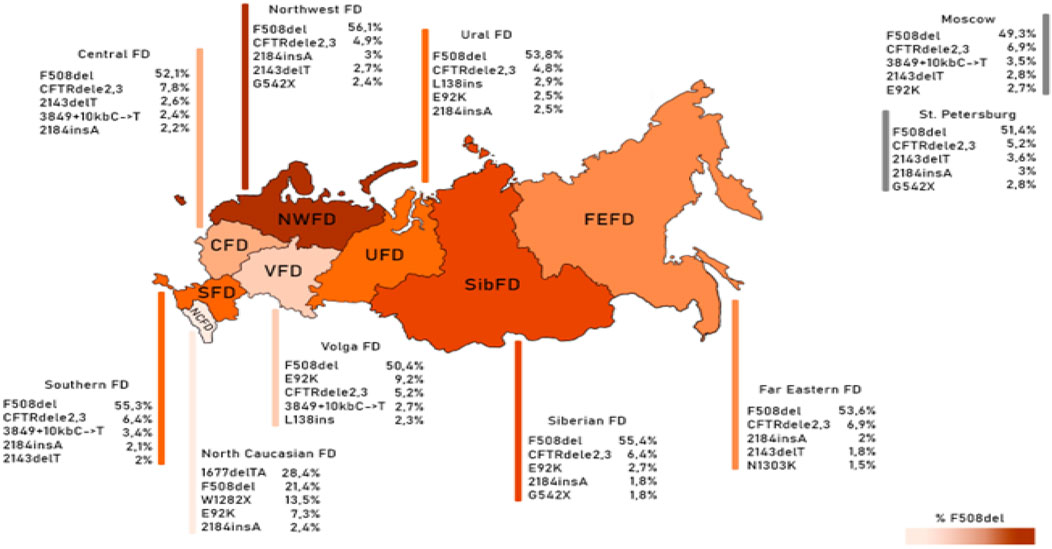
Figure 1. Frequency of the first five variants of the CFTR gene nucleotide sequence in different federal districts.
As deduced from the information presented in Table 6, it is evident that there exists an observable variation in the frequencies of these significant genetic variants when considering the geographical distribution across the various Federal Districts. This variation underscores the impact of regional factors on the prevalence of these genetic variants.
The genetic landscape of Europe highlights the prevalence of F508del as the most frequent genetic variant within the CFTR gene. This variation exhibits distinct frequencies across different countries, with its prevalence ranging from under 5% in Georgia and Armenia to over 82% in Albania (Festini et al., 2008). Figure 2A illustrates the frequency distribution of the pathogenic F508del variant within the Federal Districts of the Russian Federation. Notably, the average frequency in Russia rests at 51.55%, showcasing a range that spans from 56.1% in the Northwestern Federal District—dominantly inhabited by the Slavic population—to 28.4% in the North Caucasian Federal District.
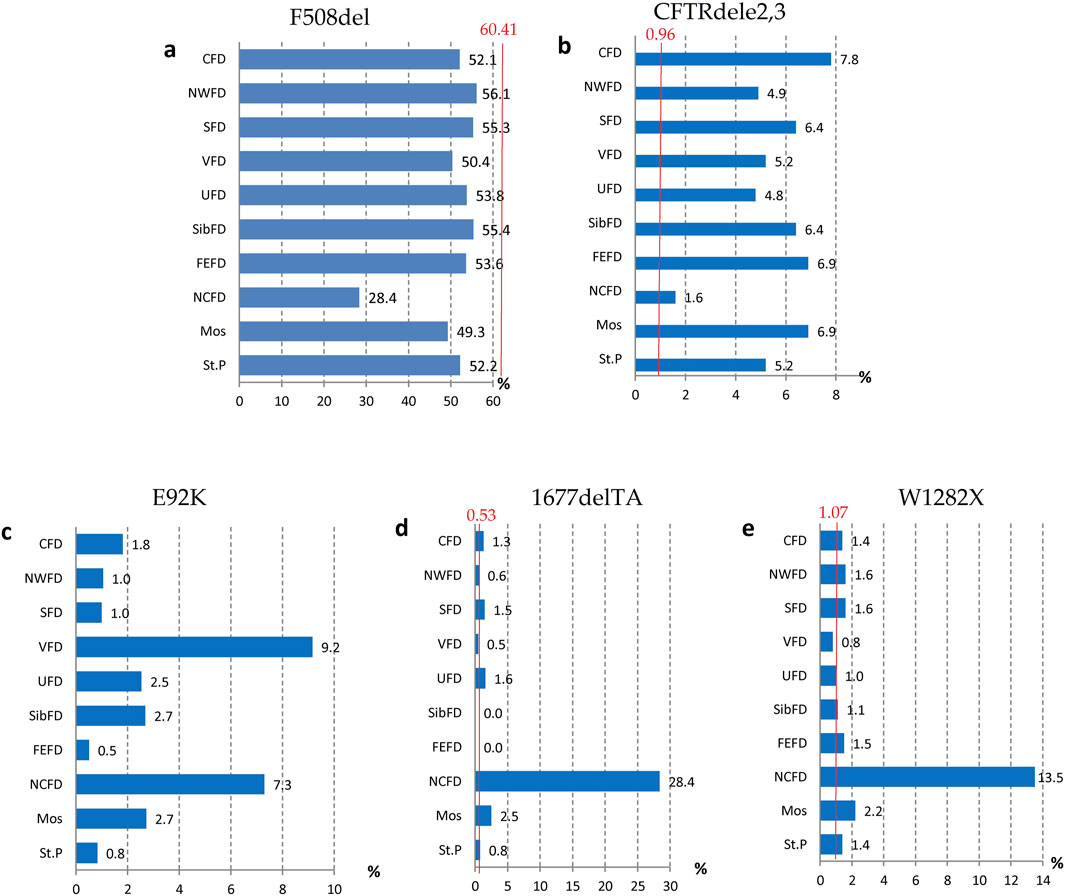
Figure 2. Variation of frequencies of genetic variants in different federal districts of the Russian Federation. (A) Frequency of the genetic variant of the F508del CFTR gene; (B) Frequency of the genetic variant of the CFTRdele2,3 CFTR gene; (C) Frequency of the genetic variant of the E92K CFTR gene; (D) Frequency of the genetic variant of the 1677delTA CFTR gene; (E) Frequency of the genetic variant of the W1282X CFTR gene. Note: CFD - Central Federal District; NWFD - Northwest Federal District; SFD - Southern Federal District; VFD - Volga Federal District; UFD - Ural Federal District; SibFD - Siberian Federal District; FEFD - Far Eastern Federal District; NCFD - North Caucasian Federal District; Mos–Moscow; St.P - St. Petersburg The red line indicates the frequency in the European registry.
Another prevalent variation in the Russian Federation is the CFTRdele2,3 deletion, commonly referred to as the “Slavic” deletion. The average frequency of this variation is 6.11%, with fluctuation from 1.6% in the North Caucasian Federal District to 7.8% in the Central Federal District, as depicted in Figure 2B. In European countries, the average frequency of this genetic variant stands at 0.96%, with its peak prevalence observed in Belarus at 10.9%.
The most significant variation among the prevalent genetic variants was identified in pathogenic variants E92K, 1677delTA, and W1282X, particularly pronounced in the North Caucasian and Volga Federal Districts—polyethnic regions renowned for their diverse populations (Petrova et al., 2021). Figure 2C illustrates the frequency distribution of the E92K nucleotide sequence variant within the Federal Districts. Nationally, the average allele frequency for E92K was determined as 3.46%. The E92K mutation emerged as the third most frequent pathogenic variant in the Russian Federation. Notably, it exhibited a higher prevalence of 9.2% in the Volga region and 7.3% in the North Caucasus Federal District, while being comparatively rarer in other Federal Districts (Kondratyeva et al., 2023; Petrova et al., 2019).
Concerning the genetic variant 1677delTA, it exhibited high allelic frequency solely in the North Caucasian Federal District at 28.4%. However, variants with frequencies exceeding 1.0% were found in the Central, Southern, and Ural Federal Districts as illustrated in Figure 2D. Notably, the variant 1677delTA was most prevalent among Chechen individuals at 67.3%, and it was also detected in other small ethnic groups in the North Caucasus region (Petrova et al., 2019). This variant, originally identified in Georgian individuals with a frequency of around 25%, is common in Mediterranean countries, albeit at significantly lower frequencies (e.g., Bulgaria (2.1%), Romania (0.8%), Greece (0.7%), Turkey (4.1%), Cyprus (approximately 6%)) (World Health Organization, 2004).
The genetic variant W1282X (Figure 2E and Figure 3), originating in theMiddle East, dominates in Israel with an allelic frequency of 22.6%, especially prominent among Ashkenazi Jews. It’s also notably present in Georgia at 10.2% (Kerem et al., 1995). Within the North Caucasus Federal District, the W1282X variant exhibited an average frequency of 13.5%, with particular prominence in certain populations like the Karachay (88.9%) and Ossetians (37.5%) (Petrova et al., 2016; Petrova et al., 2020).
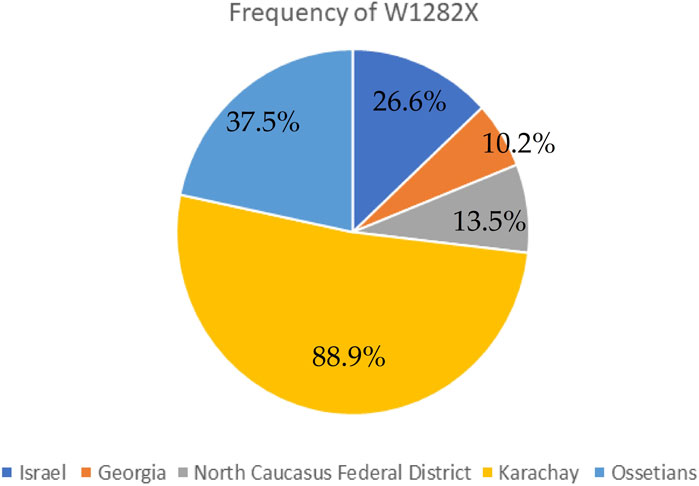
Figure 3. Frequency of W1282X in different countries and certain populations.
The identification of these genetic variants holds pivotal implications for the potential of pathogenetic therapy for pwCF. Among the 233 genetic variants identified, the majority (116) belong to class I disorders, with 6 in class II, 5 in class III, 11 in class IV, 11 in class V, and 1 in class VI. The class remains undefined in 84 variants.
Determining the mutation class and genotype type (severe or mild) based on age is also essential for enabling pathogenetic therapy. The first eleven variants within the nucleotide sequence of the CFTR gene in both children and adults are provided in Table 7.
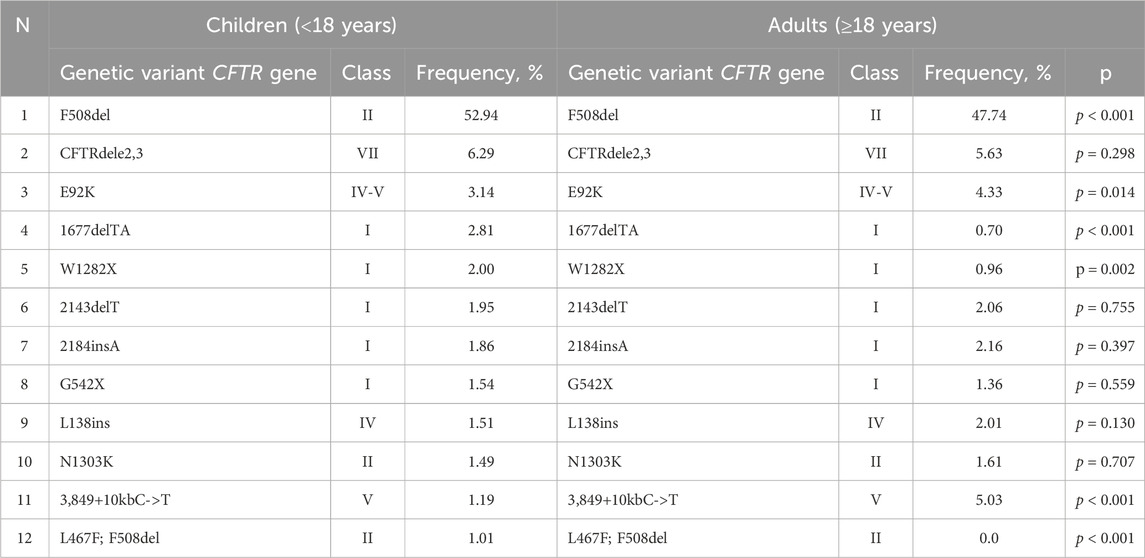
Table 7. The allelic frequencies of the most common CFTR genetic variants among children and adults (in descending order).
The frequency of homozygotes, heterozygotes according to F508del and genotypes without F508del among children and adults is shown in Table 8.

Table 8. The frequencies of homozygotes and heterozygotes for the F508del variant, as well as genotypes without F508del among children and adults.
The occurrence of the “mild” genotype was identified in 24.1% of patients. The distribution of these “mild” genotypes in relation to age is graphically illustrated in Figures 4, 5. We found that ‘severe’ genotypes are predominant among both children and adults - amounting to 80.6% before the age of 18, and slightly diminishing to 63.3% after the age of 18.
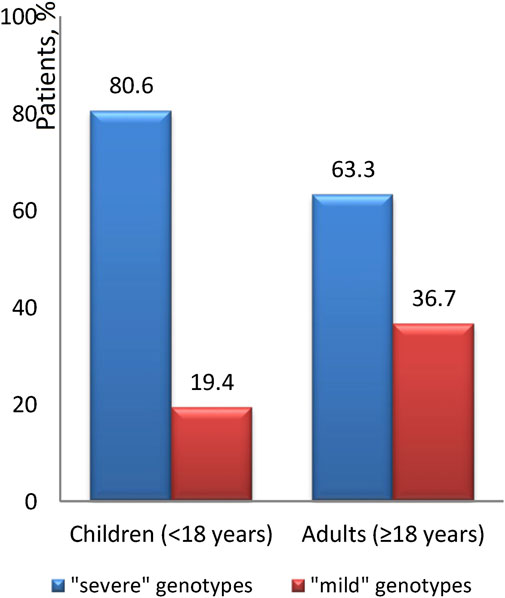
Figure 4. The ratio of the “severity” of genotypes depending on age.

Figure 5. Distribution of “mild” genotypes in different age groups.
Upon closer examination of age-related distinctions, it's revealed that the “mild” genotype was detected in 19.6% of patients under 12 years of age and a notable 66.7% of patients over 36 years of age. Interestingly, the average age of patients with the “mild” genotype is 17.7 ± 12.4, with a median of 14.2 (18.5). Conversely, the average age of patients with the “severe” genotype is 12.9 ± 8.6, with a median of 11.2 (10.7).
Of the 3,714 patients who underwent DNA diagnosis, one or both variants were found only in 3,598 patients (97%) (Table 9). Of the 3,598 patients, F508del/F508del was found in 30.5%, F508del/not F508del in 47.0%, and not F508del/not F508del in 22.5% (Table 9). In 7.5% of patients, 1 variant in the genotype is among 177 FDA-approved variants for ETI, 6.9% of patients can receive ETI because at least one variant is expected to respond to ETI, in 9.8% both variants are not suitable for treatment with targeted drugs due to the absence of CFTR protein (Table 9).
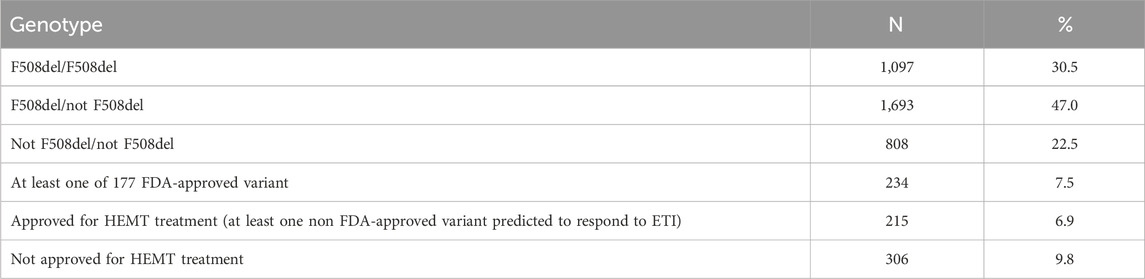
Table 9. Distribution of genetic variants with possible efficacy of targeted therapy according to the data of the Registry 2021.
In the Russian Federation, the drug Lumacaftor/Ivacaftor is registered in 2020, and Elexacaftor/Tezacaftor/Ivacaftor + Ivacaftor in 2023. The frequency of modulators use was 6.47% in 2021 (Table 10).
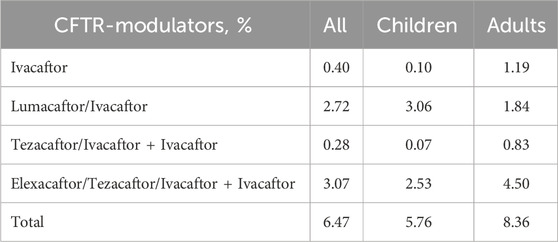
Table 10. Frequency of use of different types of CFTR modulators in children and adults in 2021.
In 2023 (as of 01.09.2023), 1,504 children (52.2% of children with CF) are provided with targeted therapy, of which 1,046 (69.55%) receive ETI therapy, taking into account the severity of the children’s condition. Lumacaftor/Ivacaftor is received by 458 children (30.45%). The number of adults on targeted therapy is 350 patients (32,2%).
The efficacy of targeting drugs not specified in the instructions for use in RF was proved by Forskolin-induced swelling (FIS) test on intestinal organoids in patients with variants: E92K, N1303K, L138ins, G1047S, 3272-16T>A in the genotype (Table 11).

Table 11. Pathogenic variants in the patient’s genotype that are not included in the instructions for the drugs, but there is data from a study of effectiveness on Forskolin-induced swelling (FIS) test using intestinal organoids.
In patients with variants S466X; R1070Q, L467F; F508del, c.1083G>A, c.831G>A, c.1513A>C, c.1329_1350del in the genotype, ineffective treatment with targeted drugs was proved by FIS (Table 12).
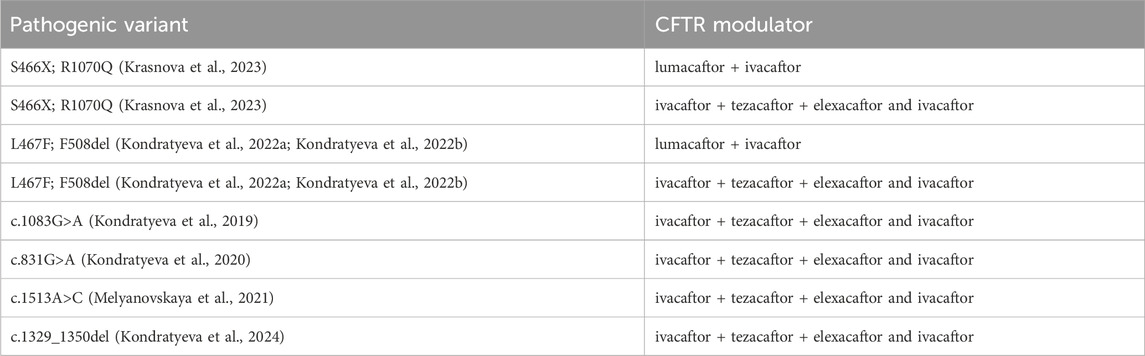
Table 12. Pathogenic variants in the patient’s genotype that did not respond to targeting drugs in the Forskolin-induced swelling (FIS) test using intestinal organoids.
Since 2011, a meticulous RCFPR has been methodically cultivated within the Russian Federation, adhering to the stringent standards set forth by the European register. This comprehensive database encompasses a range of clinical indicators, therapeutic interventions, and, notably, an in-depth section dedicated to genetic epidemiology and molecular genetic diagnostics. This segment provides valuable insights into disease prevalence, the gamut of genetic variations within Russian patients, the availability of DNA diagnostics (including CFTR gene sequencing), and the potential for pathogenetic therapies.
The primary objective of this publication is threefold: to present the birth prevalence of pwCF, exhibiting variations over the years; to outline the spectrum and frequency of CFTR gene disorders within the Russian patient population; and to underscore the distinct aspects characterizing the Russian cohort in comparison to the European registry, as well as the role of the registry in planning the availability of CFTR modulators and identifying a group of patients with rare pathogenic variants who may receive ETI if at least one variant is expected to respond to ETI after forskolin testing on intestinal organoids (Forskolin-induced swelling (FIS) assay using intestinal organoids).
These differences encompass noteworthy disparities, such as the proportion of patients aged over 18, which constitutes a mere 27.4% in contrast to the 53% observed in the European Cystic Fibrosis Society Patient Registry (ECFSPR) for the year 2020 (Orenti et al., 2022). An imminent increase in this statistic is anticipated post-2025, attributed to the transition of children identified through neonatal screening to the adult category, and the implementation of CFTR modulators.
Although the Russian cohort boasts a commendable genetic diagnostic coverage of 93%, as compared to Europe’s aggregate of 99%, the share of identified alleles is noted at 90.5%. It's noteworthy that 12.7% of Russian patients possess an incomplete genetic diagnosis—a figure higher than the 5.48% documented among European patients in 2020. Furthermore, a subset of 3.2% of Russian patients remain in the dark about both alleles. This discrepancy primarily stems from the disparate accessibility to comprehensive DNA diagnostics across different regions of the vast country, with limited availability of extended genetic examination. Another distinctive hallmark of the Russian national pwCF register lies in the extensive diversity of genetic variants identified. A total of 233 variants have been documented, exhibiting frequencies ranging from 0.01% to 51.5%, with 47 of these variants remaining uncharted within international genetic databases. Notably, the clinical significance of certain variants remains undetermined, contributing to the intricate tapestry of genetic diversity within the Russian population. Substantial disparities exist in the allelic frequency of the most prevalent variants within the CFTR gene. For instance, the widely recognized F508del variant, which is identified on average in 60.4% of European cases and can reach as high as 80% in certain countries, demonstrates a frequency of 51% of alleles within the Russian Federation. Additionally, the proportion of homozygotes for F508del in Russia (28%) significantly trails behind the European average of 40%. This variation highlights distinct genetic characteristics within the Russian population.
Another noteworthy pathogenic variant, G542X, which holds second place in the European register at 2.75%, occupies the eleventh position in the Russian register at 1.49%. Conversely, the E92K variant, prevalent in Russia at 3.46%, is notably absent from the European list of 18 variants with an allelic frequency of up to 0.5%. This divergence underscores the unique genetic landscape within Russia, contributing to variations in the prevalence of specific pathogenic variants.
The spectrum of frequent genetic variants further diverges based on Federal Districts, as it was presented in Table 6 and Figure 1. The Russian patient population showcases an extensive array of genetic variants within the CFTR gene, with a notable 116 variants belonging to class I variants—a distinctive hallmark of the genetic diversity in the region.
Starting from 2021, CFTR modulator therapy has become accessible in the Russian Federation for patients, significantly elevating the importance of genetic diagnosis. In 2023, more than 1,850 patients received CFTR modulator therapy. Rare genetic variants have been studied using the ICM method and FIS assay to improve patient coverage of targeted therapy as in other countries (Dreano et al., 2023).
As insights accumulate regarding the potential impact of complex alleles—comprising two or more variants in the cis position—there’s an emerging recognition of their influence not only on the progression of cystic fibrosis but also on the efficacy of targeted therapies tailored to this condition (Baatallah et al., 2018).
This genetic variant exploration among Russian patients specifically highlights the prevalence of the complex allele [L467F; F508del], a combination that has demonstrated a tangible impact on the effectiveness of targeted ivacaftor/lumacaftor therapy in F508del homozygotes. Russian research into the prevalence of this specific cis combination of pathogenic variants and a number of rare variants (E92K, N1303K, L138ins, G1047S) has prompted a reevaluation of clinical recommendations concerning the administration of this particular CFTR modulator (Table 12) (Petrova et al., 2022; Kondratyeva et al., 2022b; Sondo et al., 2022).
In the context of the modern capabilities for disease-modifying therapy, it's imperative to extend efforts towards comprehending the clinical significance and pathogenicity of newly identified variants. Furthermore, exploring the feasibility of implementing targeted therapies for individuals carrying these variants is a pivotal undertaking.
In conclusion, the Russian population of pwCF is characterized by a rich diversity of variants within the CFTR gene. This diversity is marked by a preponderance of severe variants, some of which are rare and distinct from those observed in other populations. In light of contemporary advancements in disease-modifying therapies, it is imperative to focus on the comprehensive evaluation of the clinical significance and pathogenicity of these new genetic variants. Equally vital is exploring the feasibility of employing targeted therapies for individuals who carry these specific variants.
As the medical landscape continues to evolve, understanding the genetic intricacies that contribute to disease progression and the efficacy of therapeutic interventions is of paramount importance. The dynamic interaction between genetic variations and modern treatment modalities underscores the necessity of ongoing research efforts aimed at enhancing the quality of care for individuals with cystic fibrosis.
This comprehensive analysis sheds light on the intricate genetic mosaic that defines the cystic fibrosis landscape in Russia, emphasizing the critical role of genetic research in guiding therapeutic strategies. For a more comprehensive grasp of the nuances within the Russian population’s genetic makeup, their clinical implications, and the ongoing advancements in cystic fibrosis research, referring to the original source or document is highly recommended.
The data analyzed in this study is subject to the following licenses/restrictions: All patient data should be anonymized. Requests to access these datasets should be directed to Elena Kondratyeva, ZWxlbmFmcGtAbWFpbC5ydQ==.
The studies involving humans were approved by The local ethics committee of the Research Centre for Medical Genetics (approval number 2012–2/1). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
EK: Conceptualization, Formal Analysis, Supervision, Writing–original draft, Writing–review and editing. YM: Writing–review and editing, Data curation. VS: Methodology, Resources, Writing–review and editing. AVo: Investigation, Writing–review and editing. EZ: Resources, Validation, Writing–review and editing. SKr: Resources, Writing–review and editing. EA: Writing–review and editing, Resources. NK: Formal Analysis, Writing–review and editing. VS: Resources, Writing–review and editing. AP: Validation, Writing–review and editing. TA: Validation, Writing–review and editing. OS: Validation, Writing–review and editing. MS: Data curation, Software, Writing–review and editing. EE: Resources, Writing–review and editing. AVa: Investigation, Writing–review and editing. AM: Visualization, Writing–review and editing. RZ: Conceptualization, Writing–original draft, Writing–review and editing. SKu: Project administration, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by The Ministry of Science and Higher Education of the Russian Federation (the Federal Scien-tific-technical programme for genetic technologies development for 2019-2030, agreement No 075-15-2021-1061, RF 193021X0029).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1383033/full#supplementary-material
Amelina, E. L., Efremova, A. S., Melyanovskaya, Yu.L., Bulatenko, N. V., Bukharova, T. B., Kashirskaya, N.Yu., et al. (2021). Functional tests for assessment of residual CFTR channel activity and personalized selection of efficacious CFTR-modulators for cystic fibrosis patients with ‘mild’ and ‘severe’ genetic variants. PULMONOLOGIYA 31 (2), 167–177. doi:10.18093/0869-0189-2021-31-2-167-177
Baatallah, N., Bitam, S., Martin, N., Servel, N., Costes, B., Mekki, C., et al. (2018). Cis variants identified in F508del complex alleles modulate CFTR channel rescue by small molecules. Hum. Mutat. 39 (4), 506–514. doi:10.1002/humu.23389
Burgel, P. R., Sermet-Gaudelus, I., Durieu, I., Kanaan, R., Macey, J., and Grenet, D.Reference Network study group (2023). The French Compassionate Program of elexacaftor-tezacaftor-ivacaftor in people with cystic fibrosis with advanced lung disease and no F508del CFTR variant. Eur. Respir. J., 2202437. doi:10.1183/13993003.02437-2022
Castellani, C., Duff, A. J. A., Bell, S. C., Heijerman, H. G. M., Munck, A., Ratjen, F., et al. (2018). ECFS best practice guidelines: the 2018 revision. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 17 (2), 153–178. doi:10.1016/j.jcf.2018.02.006
Cohen-Cymberknoh, M., Sadras, I., Kerem, E., Livnat, G., Sarouk, I., Breuer, O., et al. (2023). P077 Clinical and functional efficacy of elexacaftor/tezacaftor/ivacaftor in people with cystic fibrosis carrying the N1303K mutation. J. Cyst. Fibros. 22S2, S87. doi:10.1016/s1569-1993(23)00452-6
DeStefano, S., Gees, M., and Hwang, T. C. (2018). Physiological and pharmacological characterization of the N1303K mutant CFTR. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 17 (5), 573–581. doi:10.1016/j.jcf.2018.05.011
Dreano, E., Burgel, P. R., Hatton, A., Bouazza, N., Chevalier, B., and Macey, J.French CF Reference Network study group (2023). Theratyping cystic fibrosis patients to guide elexacaftor/tezacaftor/ivacaftor out-of-label prescription. Eur. Respir. J. 62 (4), 2300110. doi:10.1183/13993003.00110-2023
Ensinck, M. M., De Keersmaecker, L., Ramalho, A. S., Cuyx, S., Van Biervliet, S., Dupont, L., et al. (2022). Novel CFTR modulator combinations maximise rescue of G85E and N1303K in rectal organoids. ERJ open Res. 8 (2), 00716-2021–02021. doi:10.1183/23120541.00716-2021
Festini, F., Taccetti, G., Repetto, T., Mannini, C., Neri, S., Bisogni, S., et al. (2008). Incidence of cystic fibrosis in the Albanian population. Pediatr. Pulmonol. 43 (11), 1124–1129. doi:10.1002/ppul.20920
Gona-Hoepler, L. M., Dehlink, E., Strasser, N., Nachbaur, E., and Gruber, S. (2023). P078 Case report: elexacaftor/tezacaftor/ivacaftor as a game changer in an individual with CFTR class II mutation N1303k. J. Cyst. Fibros. 22S2. S. 87. doi:10.1016/S1569-1993(23)00453-8
Grasemann, H., and Ratjen, F. (2023). Cystic fibrosis. N. Engl. J. Med. 389 (18), 1693–1707. doi:10.1056/NEJMra2216474
Kashirskaya, N.Yu., Kapranov, N. I., and Kondratieva, E. I. (2021). Monograph cystic fibrosis: 2nd edition revised and supplemented. Moscow: Medpraсtika-M.
Kerem, E., Kalman, Y. M., Yahav, Y., Shoshani, T., Abeliovich, D., Szeinberg, A., et al. (1995). Highly variable incidence of cystic fibrosis and different mutation distribution among different Jewish ethnic groups in Israel. Hum. Genet. 96 (2), 193–197. doi:10.1007/BF00207378
Kondratyeva, E., Bukharova, T., Efremova, A., Melyanovskaya, Y., Bulatenko, N., Davydenko, K., et al. (2021). Health characteristics of patients with cystic fibrosis whose genotype includes a variant of the nucleotide sequence c.3140-16t>A and functional analysis of this variant. Genes 12 (6), 837. doi:10.3390/genes12060837
Kondratyeva, E., Bulatenko, N., Melyanovskaya, Y., Efremova, A., Zhekaite, E., Sherman, V., et al. (2022a). Personalized Selection of a CFTR Modulator for a Patient with a Complex Allele [L467F;F508del]. Curr. issues Mol. Biol. 44 (10), 5126–5138. doi:10.3390/cimb44100349
Kondratyeva, E., Efremova, A., Melyanovskaya, Y., Petrova, N., Satsuk, N., Bulatenko, N., et al. (2020). Clinical and genetic characterization of patients with cystic fibrosis and functional assessment of the chloride channel with the pathogenic variant c.831G>A (p.Trp277*), described for the first time. Gene 761, 145023. doi:10.1016/j.gene.2020.145023
Kondratyeva, E., Efremova, A., Melyanovskaya, Y., Voronkova, A., Polyakov, A., Bulatenko, N., et al. (2022b). Evaluation of the Complex p.[Leu467Phe;Phe508del] CFTR Allele in the Intestinal Organoids Model: Implications for Therapy. Int. J. Mol. Sci. 23 (18), 10377. doi:10.3390/ijms231810377
Kondratyeva, E., Melyanovskaya, Y., Bulatenko, N., Davydenko, K., Filatova, A., Efremova, A., et al. (2023). Clinical and functional characteristics of the E92K CFTR gene variant in the Russian and Turkish population of people with cystic fibrosis. Int. J. Mol. Sci. 24 (7), 6351. doi:10.3390/ijms24076351
Kondratyeva, E. I., Krasnova, M. G., Melyanovskaya, Yu.L., Sherman, V. D., Mokrousova, D. O., Efremova, A. S., et al. (2024). Study of a rare variant of the CFTR c.1329_1350del gene in a homozygous state in a child with cystic fibrosis using functional tests. Med. Genet. 23 (1), 60–68. doi:10.25557/2073-7998.2024.01.60-68
Kondratyeva, E. I., Melyanovskaya, Y. L., Efremova, A. S., Bulatenko, N. V., Bukharova, T. B., Petrova, N. V., et al. (2019). Clinical and genetic features of cystic fibrosis patients with novel pathogenic variant CFTR c.1083G> A (p.Trp361*) and functional assessment of the activity of the chloride channel. Med. Genet. 18 (9), 9–18. doi:10.25557/2073-7998.2019.09.9-18
Krasnova, M. G., Melianovskaya, Y. L., Krasovskiy, S. A., Bulatenko, N. V., Efremova, A. S., Bukharova, T. B., et al. (2023). Description of the clinical picture and assessment of functional activity of the CFTR channel in a patient with a complex allele [S466X; R1070Q]. PULMONOLOGIYA 33 (2), 233–242. doi:10.18093/0869-0189-2023-33-2-233-242
Krasovsky, S. A., Starinova, M. A., Voronkova, A.Yu., Amelina, E. L., Kashirskaya, N.Yu., Kondratyeva, E. I., et al. (2021). Russian federation cystic fibrosis patient registry. MEDPRACTICA-M, 95.
Mall, M. A., Burgel, P. R., Castellani, C., Davies, J. C., Salathe, M., and Taylor-Cousar, J. L. (2024). Cystic fibrosis. Nat. Rev. Dis. Prim. 10 (1), 53. doi:10.1038/s41572-024-00538-6
Melyanovskaya, Y., Kondratyeva, E., Zhekaite, E., Voronkova, A., Petrova, N., and Kutsev, S. (2021). 650: clinical and genetic characteristics of a patient with a newly described pathogenic variant CFTR p.Asn505His c.1513A >C p.(Asn505His) de novo and functional assessment of the chloride channel. J. Cyst. Fibros. 20, S308. doi:10.1016/s1569-1993(21)02073-7
Melyanovskaya, Y. L., Krasovsky, S. A., Efremova, A. S., Bulatenko, N. V., and Makarova, M. A. (2020). Course of the disease with evaluation of the chloride channel function and selection of target therapy in vitro in an adult with cystic fibrosis with 2184insa/l138ins genotype. Med. News North Cauc. 15 (2), 170–174. doi:10.14300/mnnc.2020.15041
Ministry of Health of the Russian Federation (2021). Clinical guidelines for cystic fibrosis. Available at: https://cr.minzdrav.gov.ru/recomend/372_2.
Orenti, A., Zolin, A., Jung, A., van Rens, J., Fox, A., Krasnyk, M., et al. (2022). ECFSPR annual report 2020. Available at: https://www.ecfs.eu/sites/default/files/ECFSPR_Report_2020_v1.0%20%2807Jun2022%29_website.pdf.
Petrova, N. V., Balinova, N. V., Marakhonov, A. V., Vasilyeva, T. A., Kashirskaya, N., Galkina, V. A., et al. (2021). Ethnic differences in the frequency of CFTR gene mutations in populations of the European and North Caucasian part of the Russian federation. Front. Genet. 12, 678374. doi:10.3389/fgene.2021.678374
Petrova, N. V., Kashirskaya, N. Y., Saydaeva, D. K., Polyakov, A. V., Adyan, T. A., Simonova, O. I., et al. (2019). Spectrum of CFTR mutations in Chechen cystic fibrosis patients: high frequency of c.1545_1546delTA (p.Tyr515X; 1677delTA) and c.274G>A (p.Glu92Lys, E92K) mutations in North Caucasus. BMC Med. Genet. 20, 44. doi:10.1186/s12881-019-0785-z
Petrova, N. V., Kashirskaya, N. Y., Vasilyeva, T. A., Balinova, N. V., Marakhonov, A. V., Kondratyeva, E. I., et al. (2022). High frequency of complex CFTR alleles associated with c.1521_1523delCTT (F508del) in Russian cystic fibrosis patients. BMC genomics 23 (1), 252. doi:10.1186/s12864-022-08466-z
Petrova, N. V., Kashirskaya, N. Y., Vasilyeva, T. A., Kondratyeva, E. I., Zhekaite, E. K., Voronkova, A. Y., et al. (2020). Analysis of CFTR mutation spectrum in ethnic Russian cystic fibrosis patients. Genes 11 (5), 554. doi:10.3390/genes11050554
Petrova, N. V., Kashirskaya, N. Y., Vasilyeva, T. A., Timkovskaya, E. E., Voronkova, A. Y., Shabalova, L. A., et al. (2016). High prevalence of W1282x mutation in cystic fibrosis patients from Karachay-Cherkessia. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 15 (3), e28–e32. doi:10.1016/j.jcf.2016.02.003
Sadras, I., Kerem, E., Livnat, G., Sarouk, I., Breuer, O., Reiter, J., et al. (2023). Clinical and functional efficacy of elexacaftor/tezacaftor/ivacaftor in people with cystic fibrosis carrying the N1303K mutation. J. Cyst. Fibros. official J. Eur. Cyst. Fibros. Soc. 22 (6), 1062–1069. doi:10.1016/j.jcf.2023.06.001
Sondo, E., Cresta, F., Pastorino, C., Tomati, V., Capurro, V., Pesce, E., et al. (2022). The L467F-F508del Complex Allele Hampers Pharmacological Rescue of Mutant CFTR by Elexacaftor/Tezacaftor/Ivacaftor in Cystic Fibrosis Patients: The Value of the ex vivo Nasal Epithelial Model to Address Non-Responders to CFTR-Modulating Drugs. Int. J. Mol. Sci. 23 (6), 3175. doi:10.3390/ijms23063175
World Health Organization (WHO) (2004). The molecular genetic epidemiology of cystic fibrosis. Report of a Joint Meeting of WHO/ECFTN/ICF(M)/ECFS. Genoa, Italy, 19 June 2002. Hum. Genet. Program–24. Available at: https://iris.who.int/bitstream/handle/10665/68702/WHO_HGN_CF_WG_04.02.pdf;jsessionid=501E48A03FC4243C194E1981F7584D5E?sequence=1 (Accessed February 02, 2024).
Keywords: cystic fibrosis, genetic epidemiology, CFTR gene variants, targeted therapy, disease prevalence
Citation: Kondratyeva E, Melyanovskaya Y, Sherman V, Voronkova A, Zhekaite E, Krasovsky S, Amelina E, Kashirskaya N, Shadrina V, Polyakov A, Adyan T, Sсhagina O, Starinova M, Enina E, Vasilyev A, Marakhonov A, Zinchenko R and Kutsev S (2024) Study of the genetic and molecular epidemiology of cystic fibrosis based on the patient registry for planning targeted therapy in Russian Federation. Front. Genet. 15:1383033. doi: 10.3389/fgene.2024.1383033
Received: 11 February 2024; Accepted: 08 October 2024;
Published: 28 October 2024.
Edited by:
Ahmed Rebai, Centre of Biotechnology of Sfax, TunisiaReviewed by:
Milan Macek, Charles University, CzechiaCopyright © 2024 Kondratyeva, Melyanovskaya, Sherman, Voronkova, Zhekaite, Krasovsky, Amelina, Kashirskaya, Shadrina, Polyakov, Adyan, Sсhagina, Starinova, Enina, Vasilyev, Marakhonov, Zinchenko and Kutsev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena Kondratyeva, ZWxlbmFmcGtAbWFpbC5ydQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.