 Zhenjie Li
Zhenjie Li Dehai Chen
Dehai Chen- Department of Cardiovascular Surgery, The First People’s Hospital of Zhaoqing, The First Affiliated Hospital of Zhaoqing Medical College, Zhaoqing, China
Background: Previous studies have shown that Alzheimer’s disease (AD) can cause myocardial damage. However, whether there is a causal association between AD and non-ischemic cardiomyopathy (NICM) remains unclear. Using a comprehensive two-sample Mendelian randomization (MR) method, we aimed to determine whether AD and family history of AD (FHAD) affect left ventricular (LV) structure and function and lead to NICM, including hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM).
Methods: The summary statistics for exposures [AD, paternal history of AD (PH-AD), and maternal history of AD (MH-AD)] and outcomes (NICM, HCM, DCM, and LV traits) were obtained from the large European genome-wide association studies. The causal effects were estimated using inverse variance weighted, MR-Egger, and weighted median methods. Sensitivity analyses were conducted, including Cochran’s Q test, MR-Egger intercept test, MR pleiotropy residual sum and outlier, MR Steiger test, leave-one-out analysis, and the funnel plot.
Results: Genetically predicted AD was associated with a lower risk of NICM [odds ratio (OR) 0.9306, 95% confidence interval (CI) 0.8825–0.9813, p = 0.0078], DCM (OR 0.8666, 95% CI 0.7752–0.9689, p = 0.0119), and LV remodeling index (OR 0.9969, 95% CI 0.9940–0.9998, p = 0.0337). Moreover, genetically predicted PH-AD was associated with a decreased risk of NICM (OR 0.8924, 95% CI 0.8332–0.9557, p = 0.0011). MH-AD was also strongly associated with a decreased risk of NICM (OR 0.8958, 95% CI 0.8449–0.9498, p = 0.0002). Different methods of sensitivity analysis demonstrated the robustness of the results.
Conclusion: Our study revealed that AD and FHAD were associated with a decreased risk of NICM, providing a new genetic perspective on the pathogenesis of NICM.
1 Introduction
Non-ischemic cardiomyopathy (NICM) refers to a group of myocardial diseases that are not caused by coronary artery disease or ischemic injury. NICM has numerous etiologies and leads to high mortality rates, making it the most common cause of non-ischemic systolic heart failure (HF) (Elliott et al., 2008; Pimentel et al., 2016). It was reported that about 30% of patients with NICM die from sudden cardiac death (Kusumoto et al., 2014). NICM includes several subtypes, including hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), and restrictive cardiomyopathy (RCM) (Elliott et al., 2008). HCM and DCM are the most commonly diagnosed forms of NICM in clinical practice, with an estimated prevalence of 1/200 and 1/250–2500 among adults, respectively (Wang et al., 2023). Although the exact etiology of NICM is not fully understood, a hereditary component is known to be involved. HCM, an autosomal dominant cardiomyopathy, is characterized by an asymmetric increase in the thickness of the ventricular wall. Approximately 60% of patients with HCM are caused by genetic mutations related to sarcomeric genes. Other genetic diseases, such as mitochondrial diseases, metabolic disorders, and Fabry’s disease, can also lead to HCM. In addition, cardiac amyloidosis may contribute to the development of HCM by acting as an intermediate phenotype (Wang et al., 2023). DCM, on the other hand, is characterized by left ventricular diastolic dysfunction and poor ventricular remodeling (Japp et al., 2016). It is estimated that 30%–48% of DCM cases are hereditary (Weintraub et al., 2017). Genetic factors may also play a role in the pathogenesis of DCM in patients with chemotherapy-related cardiac dysfunction or alcohol intolerance (Wang et al., 2023). Further studies are needed to better understand the underlying causes of NICM and develop effective treatments for this devastating condition.
Alzheimer’s disease (AD), a progressive neurodegenerative disorder that accounts for about 70% of all cases of dementia, has also been implicated in the development of cardiomyopathy (Tublin et al., 2019). The risk of AD is 60%–80% depending on heritable factors and is characterized by the presence of β-amyloid (Aβ) plaques and tau neural fibrillary tangles (Scheltens et al., 2021). Genetic factors commonly associated with AD, such as apolipoprotein E (apoE) 4 and presenilin mutations, were also found to be associated with HF and DCM, suggesting an underlying genetic link between AD and cardiovascular diseases (Tublin et al., 2019). Moreover, misfolded proteins play a key role not only in AD but also in cardiac hypertrophy and cardiomyopathy (Willis and Patterson, 2013). Studies on the relationship between plasma Aβ40 and cardiac function have shown that Aβ40 is associated with decreased left ventricular end-systolic volume (LVESV), LV stroke volume index, and VO2 Max, in the absence of overt cardiovascular diseases (Stamatelopoulos et al., 2018).
Although traditional observational studies have suggested a link between Alzheimer’s disease (AD) and cardiomyopathy, the causal relationship remains uncertain. Mendelian randomization (MR) analysis offers a potential solution by using genetic variations. It uses single nucleotide polymorphisms (SNPs), from genome-wide association studies (GWAS) as instrumental variables (IVs) for environmental exposure. This approach can help clarify the causal relationship between exposure and outcome (Lawlor et al., 2008). Given that genetic factors play a significant role in the pathogenesis of AD (Scheltens et al., 2021), a two-sample MR study was conducted to investigate the causal effects of AD and family history of AD (FHAD) on NICM and LV structure and function.
2 Materials and methods
2.1 Study design
A two-sample MR analysis was conducted to investigate the causal effects of AD and FHAD on NICM and LV structure and function (Figure 1). The MR design satisfied three key assumptions: 1) the instrumental variables (IVs) were strongly associated with the exposure; 2) IVs were independent of confounding factors on the exposure-outcome pathway; and 3) IVs could only be associated with the outcomes through exposure, not through other pathways (Lawlor, 2016). In this MR analysis, three AD-related traits were used as exposures: AD itself, paternal history of AD (PH-AD), and maternal history of AD (MH-AD). NICM, HCM, and DCM were the three types of cardiomyopathy considered as outcomes. Five structural and functional parameters were the LV traits measured in this study: LV end-diastolic volume (LVEDV), LVESV, ejection fraction (LVEF), mass (LVM), and left ventricular mass-to-end-diastolic volume ratio (LVMVR, also known as LV remodeling index) (Buakhamsri et al., 2009; Tadros et al., 2021).
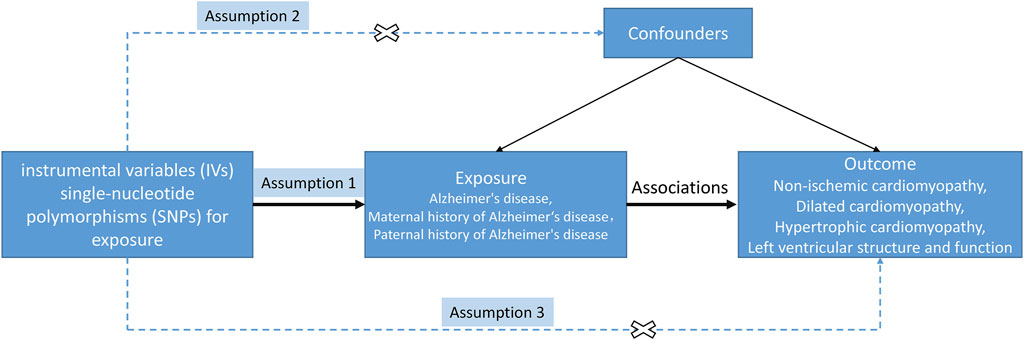
Figure 1. Study design of MR between AD (family history included) and NICM, and left ventricular structure and function. The solid/dash lines indicate relevance/irrelevance. Dash lines with a cross means that instrumental variants (IVs) were independent of confounders (assumption 2) and IVs only affected the outcome via exposure but not via other vicegerent routes (assumption 3).
2.2 Data resource
GWAS summary statistics for the outcomes of non-ischemic cardiomyopathy (NICM) and hypertrophic cardiomyopathy (HCM) were obtained from the FinnGen research project. The FinnGen project provided GWAS-level data on 11,400 cases of NICM and 175,752 control subjects, and 556 cases of HCM and 218,236 control subjects (Kurki et al., 2023). For the genetic predictors of dilated cardiomyopathy (DCM), data were extracted from a GWAS conducted on European ancestry. This study included 1,444 cases and 353,937 controls (Sakaue et al., 2021). Additionally, GWAS data on five LV traits (LVEDV, LVESV, LVEF, LVM, and LVMVR) were derived from cardiac magnetic resonance (CMR) imaging of 19,260 UK Biobank participants. The mean age of these participants was 63 ± 8 years, with 46% being male. Exclusion criteria were uncontrolled hypertension and extreme body mass indexes. None of the participants had heart failure, cardiomyopathy, myocardial infarction, or structural heart disease at the time of CMR imaging (Tadros et al., 2021).
Genetic instruments for AD were derived from the UK Biobank, including 39,106 cases and 46,828 controls (Bellenguez et al., 2022). In addition, genetic instruments for FHAD, including paternal history of AD (PH-AD) and maternal history of AD (MH-AD), were derived from a UK Biobank dataset. This dataset included 314,278 participants, with 27,696 maternal cases and 14,338 paternal cases (Bellenguez et al., 2022). To ensure the reliability of the results, a different GWAS dataset (AD-DD) published in 2019, was used to repeat the MR analysis. The AD-DD dataset was extracted from the Alzheimer Disease Genetics Consortium and included 21,982 cases and 41,944 controls (Kunkle et al., 2019). In addition to these datasets, genetic instruments for FHAD, including PH-AD and MH-AD, were extracted from the same UK Biobank dataset. This dataset included 314,278 participants, with 27,696 maternal cases and 14,338 paternal cases (Marioni et al., 2018). All participants in these datasets were of European origin. A summary description of the GWAS datasets is provided in Table 1.
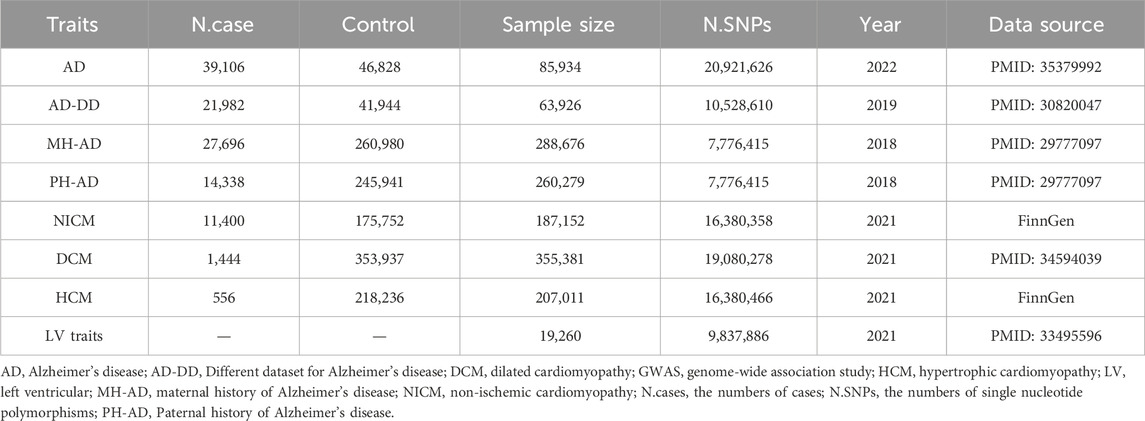
Table 1. Summary of the GWAS included in the current MR study.
2.3 Instrumental variables selection
SNPs genetically associated with exposures at the genome-wide significance level of p < 5 × 10−8 were selected as IVs, and linkage disequilibrium (LD) was measured using r2 < 0.001 and LD distance >10,000 kb (Clarke et al., 2012). Then, we calculated the F statistic to distinguish weak instrumental bias (F < 10) (Burgess and Thompson, 2011). The correlation hypothesis was evaluated using the F statistic: F = R2(N-1-K)/(1-R2)K, where R2 reflects the proportion of SNPs in explaining the exposure variable, N is the sample size, and K is the number of SNPs (Davies et al., 2018). Following the MR assumptions mentioned above, we extracted and screened SNPs associated with corresponding exposures and confounders from the Open GWAS catalog (https://www.ebi.ac.uk/gwas/) and PhenoScanner V2 database (Kamat et al., 2019).
2.4 Mendelian randomization analyses
2.4.1 Estimation of causal association
Three MR methods were used to investigate the causal effects of exposures on outcomes. The inverse variance weighted (IVW) method was used as the main MR method to determine the MR estimate for each exposure. The IVW method can be used for meta-analyses to combine the causal effects of SNPs on the outcome with the Wald ratio in the absence of horizontal pleiotropy (Burgess et al., 2016). A random-effects IVW model was adopted in the presence of heterogeneity. The other two methods, including the MR-Egger method (Bowden et al., 2015) and the weighted median (WM) method (Bowden et al., 2016), helped provide the consistency of the direction of MR results. The MR-Egger method and weighted median (WM) method were used to obtain reliable estimates. The latter can provide a robust estimate when more than half of the SNPs satisfy the IV assumptions (Bowden et al., 2016). The consistency of the direction of MR results suggests the solidness of the MR results. The causal estimates are presented as odds ratios (ORs) and 95% confidence intervals (CIs). The Bonferroni correction was executed to correct p-values in the presence of multiple tests (Armstrong, 2014). A p-value less than 7.82 × 10−4 (0.05/64) was considered strong evidence of a causal association. Correlation with the original p-value less than 0.05 was considered a nominal causal association.
2.4.2 Sensitivity analysis
For associations with a p-value less than 0.05 in the IVW method, sensitivity analyses were performed to assess the robustness of the primary causal estimate. The MR-Egger method was used to detect directional pleiotropy indicated by the intercept. A p-value of more than 0.05 indicated the absence of directional pleiotropy; however, it was not sensitive to outliers (Bowden et al., 2016). The MR-multi-directional residual sum and outliers (MR-PRESSO) method was employed to efficiently identify and address outlier SNPs and potential horizontal pleiotropy (Verbanck et al., 2018). The leave-one-out test was used to assess the stability of MR results by excluding SNPs one by one. The Cochran’s Q statistic was adopted to evaluate the heterogeneity for the IVW model (Bowden et al., 2019). Moreover, the MR Steiger directionality test was used to assess the accuracy of the direction of the causal association between exposures and outcomes (Hemani et al., 2017). All analyses were conducted using the TwoSampleMR (version 0.5.6) and MR-PRESSO (version 1.0) packages of R Studio version 4.3.0.
3 Results
3.1 Selection of instrumental variables
The MR analysis of the causal effects of AD/AD-DD/PH-AD/MH-AD on NICM/DCM/HCM indicated 56/59/56 IVs (AD:NICM/DCM/HCM), 18/18/18 IVs (AD-DD:NICM/DCM/HCM), 3/3/3 IVs (PH-AD:NICM/DCM/HCM), and 4/4/4 IVs (MH-AD:NICM/DCM/HCM). For the causal effects of AD on five LV traits, MR analysis found 58/58/58/57 IVs (AD:LVEDV/LVESV/LVEF/LVM/LVMVR). SNPs explained 1.0151% (AD:NICM), 1.0259% (AD:DCM), 0.7044% (AD-DD:DCM), 0.4645% (PH-AD:NICM), 1.0412% (MH-AD:NICM), and 5.8094% (AD:LVMVR) of the variance. Specific information about IVs with significant associations is provided in Supplementary Tables S1–S5. All the F statistics for genetic instruments were >10, indicating no evidence of weak instrument bias.
3.2 Mendelian randomization analyses
3.2.1 The causal effect of genetically predicted AD on NICM/DCM/HCM
According to the standard IVW method, we estimated the causal effects of AD on NICM/DCM/HCM. Based on the IVW estimates, the effects of SNPs (ORs) genetically associated with AD on NICM, DCM, and HCM were 0.9306 (95% CI 0.8825–0.9813, p = 0.0078), 0.8666 (95% CI 0.7752–0.9689, p = 0.0119), and 1.0469 (95% CI 0.8324–1.3167, p = 0.6953), respectively (Figure 2). In addition, other MR methods, including MR-Egger method and WM method, yielded consistent direction of the results except for the causal estimate between AD and HCM. Using another dataset of AD (AD-DD) on DCM, the IVW model showed similar results (OR 0.9002, 95% CI 0.8170–0.9920, p = 0.0338). The results provided suggestive evidence for the effect of AD on NICM/DCM when referring to our preset statistically significant threshold (p < 7.82 × 10−4). The scatter plots depicting the effect of AD on NICM/DCM are shown in Figure 5. The details can be obtained in Supplementary Table S6.
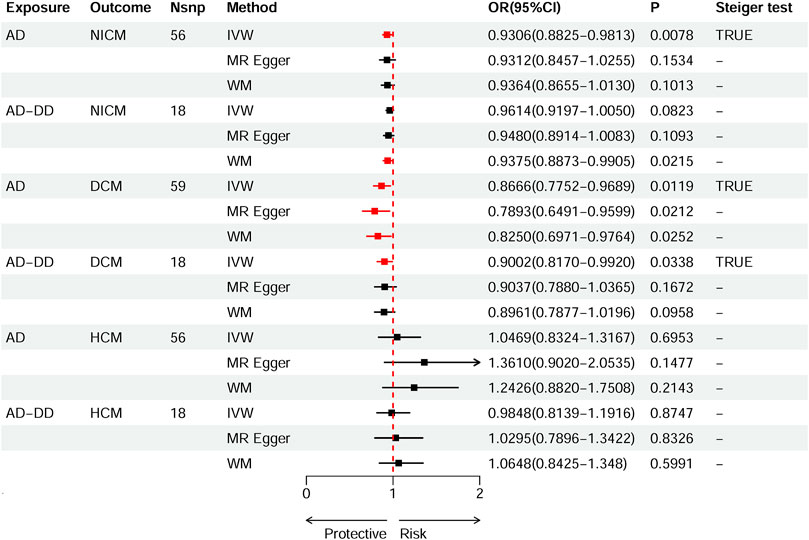
Figure 2. Causal effects of AD on NICM, DCM, and HCM. Summary of the Mendelian randomization (MR) estimates derived from the inverse variance weighted (IVW), weighted median (WM), and MR-Egger methods. AD, Alzheimer’s disease; AD-DD, different dataset for Alzheimer’s disease; NICM, non-ischemic cardiomyopathy; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy.
3.2.2 The causal effect of genetically predicted family history of AD on NICM/DCM/HCM
Based on IVW estimates, genetically predicted PH-AD was nominally associated with a decreased risk of NICM (OR 0.8924, 95% CI 0.8332–0.9557, p = 0.0011), and genetically predicted MH-AD was strongly associated with a decreased risk of NICM (OR 0.8958, 95% CI 0.8449–0.9498, p = 0.0002) (Figure 3). We found no causal effect of genetically predicted family history of AD on DCM (PH-AD: OR 0.9730, 95% CI 0.7333–1.2910, p-IVW = 0.8495; MH-AD: OR 0.9223, 95% CI 0.6929–1.2277, p-IVW = 0.5795) or HCM (PH-AD: OR 0.9480, 95% CI 0.6244–1.4393, p-IVW = 0.8022; MH-AD: OR 1.0521, 95% CI 0.7598–1.2234, p-IVW = 0.7636). The scatter plots for the effect of PH-AD/MH-AD on NICM are shown in Figure 5. The details are provided in Supplementary Table S7.
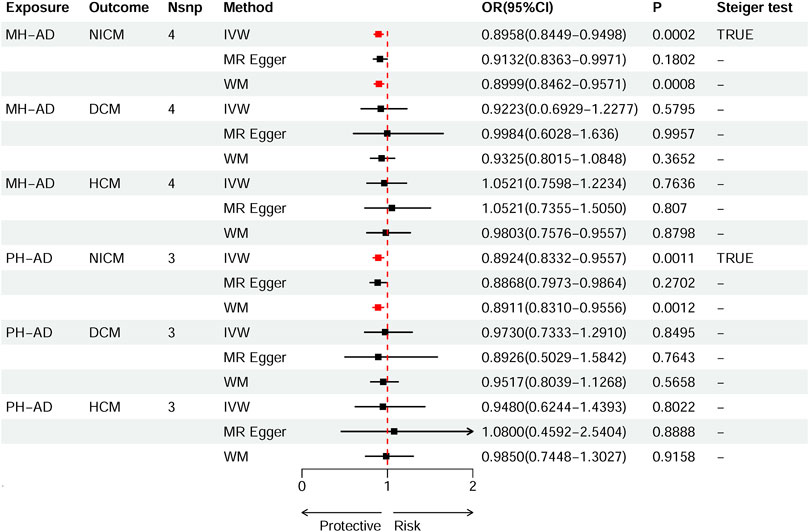
Figure 3. Causal effects of MH-AD on NICM, DCM, and HCM. Summary of the Mendelian randomization (MR) estimates derived from the inverse variance weighted (IVW), weighted median (WM), and MR-Egger methods. AD, Alzheimer’s disease; AD-DD, different dataset for Alzheimer’s disease; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; MH-AD, maternal history of Alzheimer’s disease; NICM, non-ischemic cardiomyopathy; PH-AD, paternal history of Alzheimer’s disease.
3.2.3 The causal effect of genetically predicted AD on LV traits
Based on IVW estimates, genetically predicted AD was nominally associated with decreased LVMVR (OR 0.9969, 95% CI 0.9940–0.9998, p = 0.0337). MR-Egger method (OR 0.9978, 95% CI 0.9927–1.0029, p = 0.3921) and WM method (OR 0.9962, 95% CI 0.9925–0.9999, p = 0.0448) showed consistent direction of the results (Figure 4). No causal effect of genetically predicted AD was found on the remaining four LV traits: LVEDV (OR 1.2806, 95% CI 0.5421–3.0251, p-IVW = 0.5729), LVESV (OR 0.8787, 95% CI 0.5438–1.4198, p-IVW = 0.5973), LVM (OR 0.7264, 95% CI 0.4548–1.1602, p-IVW = 0.1809), and LVEF (OR 1.1783, 95% CI 0.9746–1.4246, p-IVW = 0.0902). The scatter plots for the effect of AD on LVMVR are shown in Figure 5. The details can be obtained in Supplementary Table S8.
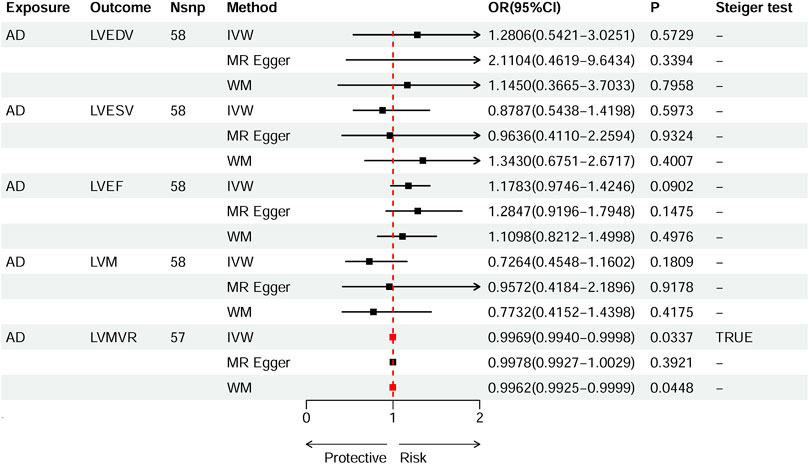
Figure 4. Causal effects of AD on left ventricular (LV) traits. Summary of the Mendelian randomization (MR) estimates derived from the inverse variance weighted (IVW), weighted median (WM), and MR-Egger methods. LVEDV, LV end-diastolic volume; LVESV, LV end-systolic volume; LVEF, LV ejection fraction; LVM, mass; LVMVR, left ventricular mass-to-end-diastolic volume ratio.
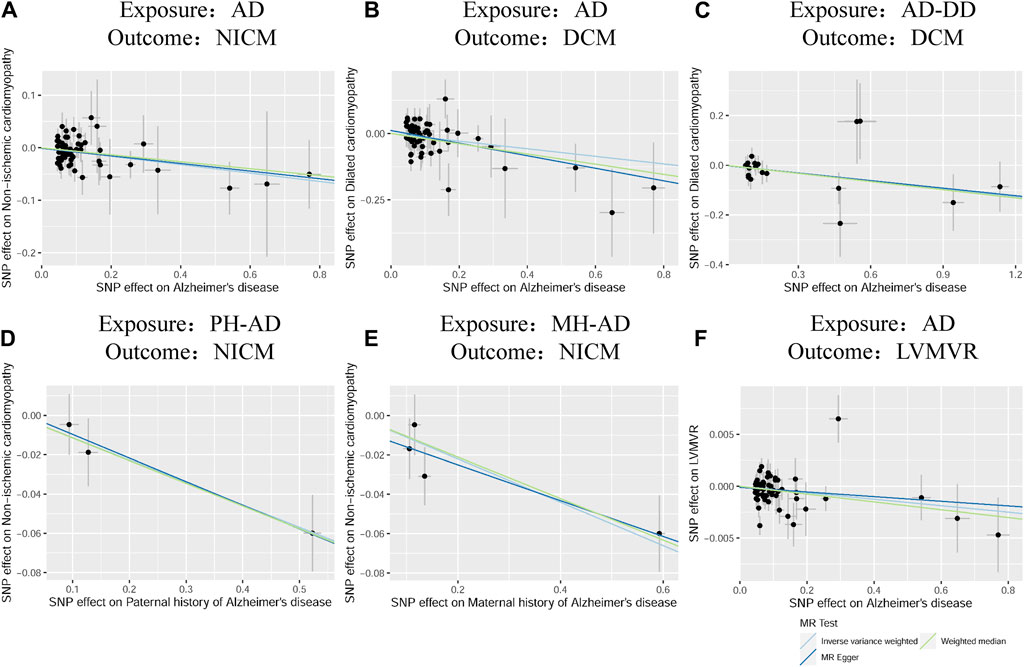
Figure 5. Scatter plots of the effect of AD on NICM, DCM, and left ventricular mass-to-end-diastolic volume ratio and scatter plots for the effect of MH-AD on NICM (A–F). AD, Alzheimer’s disease; AD-DD, different dataset for Alzheimer’s disease; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; LVMVR, left ventricular mass-to-end-diastolic volume ratio MH-AD, maternal history of Alzheimer’s disease; NICM, non-ischemic cardiomyopathy; PH-AD, paternal history of Alzheimer’s disease.
3.2.4 Sensitivity analyses
A series of sensitivity analyses were performed to confirm the robustness of the significant IVW estimates between exposures and outcomes, whether nominal or not (Table 2). The Cochran’s Q test and MR-Egger intercept showed no evidence of heterogeneity and unbalanced directional pleiotropy (all p-values >0.05). The MR-PRESSO global test did not detect any evidence of pleiotropic effects (p > 0.05) for most of the causal estimates. Due to the deficiency of SNPs (3) derived from a causal estimate of PH-AD on NICM, it was not available for the MR-PRESSO test. The funnel plots were symmetrical, and the leave-one-out analysis indicated that the causal estimates were not biased by any of the SNPs (Supplementary Figures S1–S12). The results of the Steiger test between exposures and outcomes were all TRUE, suggesting no inverse causal link (Figures 2–4).
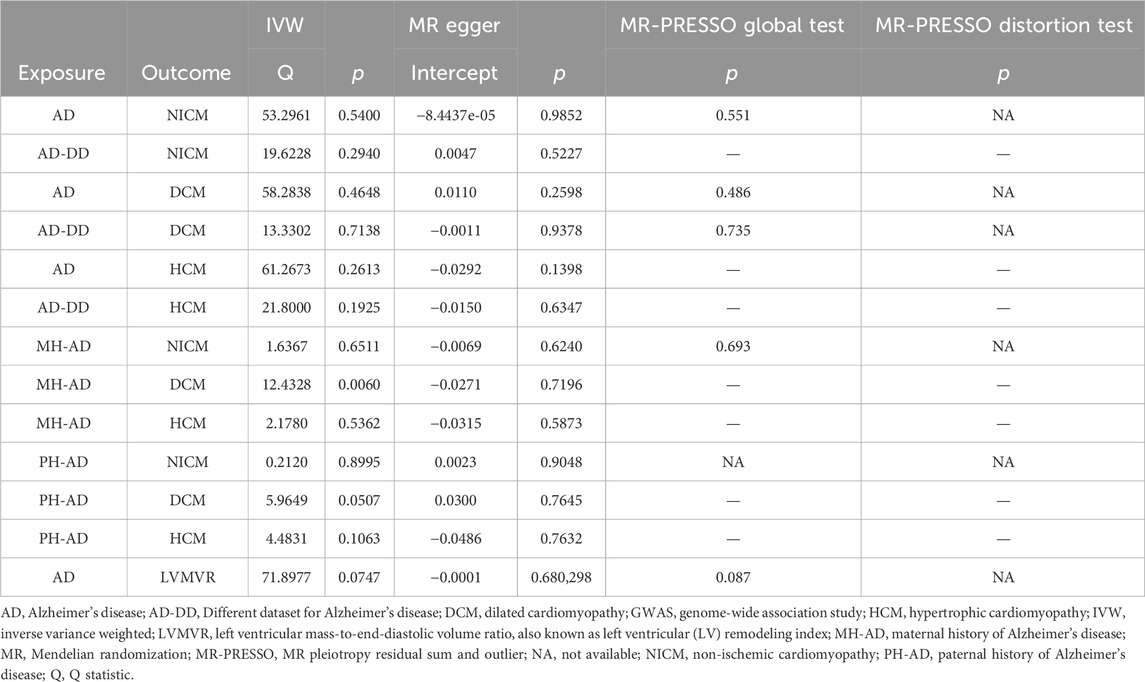
Table 2. Evaluation of heterogeneity and pleiotropy using different methods.
4 Discussion
This was the first two-sample MR analysis that systematically evaluated the potential causal effects of AD and FHAD on NICM and LV structure and function. We identified five main associations with evidence of causality, one of which showed a strong causal effect after multiple corrections. Before correction, AD was significantly associated with a lower risk of NICM and DCM, and PH-AD and MH-AD were significantly associated with a lower risk of NICM. Despite the small effect size (OR: 0.9969), AD was nominally associated with a decreased LVMVR. After multiple corrections, we found a strongly significant association between MH-AD and a lower risk of NICM, with a p-value smaller than 7.82 × 10−4. Sensitivity analyses, including tests of heterogeneity and pleiotropy, the leave-one-out test, and the Steiger test, confirmed the robustness of our findings.
Misfolded proteins play a crucial role in the pathogenesis of neurodegenerative diseases. Previous reviews have elucidated the link between AD and cardiac dysfunction from the perspective of proteotoxicity to cardiomyocytes (Willis and Patterson, 2013). Proteotoxicity refers to the process by which protein aggregates formed by misfolded proteins can lead to cell death. Misfolded soluble protein oligomers can disrupt cellular function through multiple pathways, and this process is not limited to neurodegenerative diseases (Sanbe et al., 2004). Cardiomyocytes are composed of highly specialized components, forming a syncytium with a unified contractile function. Their function depends on protein homeostasis, including protein synthesis, folding, and turnover. The ubiquitin-proteasome system and autophagy are responsible for the turnover of misfolded proteins (Willis and Patterson, 2013). Oligomeric protein deposits in cardiomyocytes are important factors contributing to pathologic cardiac hypertrophy and DCM. For example, soluble oligomers are detected in cardiomyocytes of patients with HCM and idiopathic DCM but not in healthy controls (Sanbe et al., 2004). Cytosolic aggregates immunoreactive for ubiquitin have also been found in cardiomyocytes from the hearts of patients with DCM (Kostin et al., 2003). On the other hand, similarities in preamyloid oligomers were found in αB-crystallin (CryAB)-mutation-associated cardiomyopathy and AD (Maloyan et al., 2007). CryAB is expressed in the heart and can function as part of larger molecular complexes containing proteins that need to go through the folding process (Fu and Liang, 2003; Kampinga et al., 2009). Moreover, voluntary exercise is beneficial for AD associated with misfolded proteins. Decreased deposition of cardiac amyloid oligomers was observed when CryAB transgenic mice exercised voluntarily (Maloyan et al., 2007). A small-sized case-control study including participants without cardiac or systemic diseases found that compared to the control group, patients with AD had greater interventricular septum, greater maximum wall thickness, and a 2-fold higher prevalence of diastolic dysfunction in echocardiography (Sanna et al., 2019). The proteotoxicity hypothesis extends the link between AD and NICM and provides a new genetic perspective on the pathogenesis of NICM. However, our findings suggest that AD may reduce the risk of NICM, which encourages us to investigate the possible mechanisms behind this phenomenon from another perspective.
The brain-heart syndrome refers to the functional damage to the heart caused by brain disorders. Clinical and experimental studies have suggested that brain injuries, such as stroke (ischemic or hemorrhagic) and traumatic brain injury can lead to cardiac dysfunction and arrhythmias (Chen et al., 2017). Takotsubo cardiomyopathy, also known as stress cardiomyopathy, is characterized by transient left ventricular apical ballooning and is named after the Japanese octopus catcher pot (takotsubo) due to its similarity to the diseased heart (Tsuchihashi et al., 2001; Yoshimura et al., 2008). In patients with stroke, insular damage is a prominent predictor of takotsubo cardiomyopathy (Yoshimura et al., 2008). Decreased heart rate variability (HRV) has been observed in patients with an insular infarct, particularly among those with a right side infarct (Tokgözoglu et al., 1999). The autonomic nervous system consists of various interconnected areas in the telencephalon, diencephalon, and brainstem, including the insular cortex and hypothalamus (Wehrwein et al., 2016). The insular cortex appears to play a leading role in autonomic control of cardiac activity. The hypothalamus, locus coeruleus, cerebral neocortex, insular cortex, and brainstem related to the control of sympathetic and parasympathetic outflow are affected in AD. In addition, autonomic dysfunction in AD can be assessed by calculating HRV, which is closely related to cognitive dysfunction (Braak and Braak, 1995; Femminella et al., 2014; Giraldo et al., 2018). It can be inferred that central cholinergic malfunctioning may lead to autonomic dysfunction, establishing a link between higher cerebral and autonomic neural functions. Autonomic dysfunction of the heart was assessed by HRV in patients with AD, which was significantly correlated with the plasma activity of acetylcholinesterase (Giubilei et al., 1998). DCM is associated with decreased parasympathetic activity and enhanced sympathetic activity (Ajiki et al., 1993). Observational studies have also found a link between HRV and DCM (Hoffmann et al., 2000; Rashba et al., 2006; Grutter et al., 2012). Specifically, HRV values were significantly reduced in adult patients with DCM (Yi et al., 1997) and increased in male children with DCM who were less than 6 years of age (Grutter et al., 2012). In addition, adult patients with DCM and preserved HRV had a better prognosis (Yi et al., 1997; Rashba et al., 2006). However, HRV cannot help determine the type of autonomic dysfunction in patients with AD (inhibition or hyperfunction) (Mellingsæter et al., 2015; Belaidi and Bush, 2016; Santos et al., 2017). Therefore, we believe that HRV alone is insufficient to assess autonomic dysfunction in AD. Furthermore, we found that changes in HRV (reduced or increased) are opposite in patients with DCM at different ages (less than 6 years old or not) (Yi et al., 1997; Grutter et al., 2012), suggesting that different stages of AD (mild or severe) may have different effects on cardiac autonomic function.
Similarly, we considered stress cardiomyopathy as a result of neurological injury to understand the role of the sympathetic nervous system in the brain-heart interaction and attempted to link altered sympathetic function to AD. The hypothalamic paraventricular nucleus (HPN) controls sympathetic activity, while higher brain regions regulate sympathetic outflow to the heart through the HPA axis (Chen et al., 2017). Activation of the sympathetic nervous system can induce arrhythmias and myocardial necrosis when the hypothalamus is stimulated (Melville et al., 1963). In response to stress stimuli, the HPN secretes corticotropin-releasing factor that stimulates the pituitary gland to release adrenocorticotropic hormone, thereby increasing cortisol levels (Whitnall, 1993). Cortisol levels are related to stroke severity (Christensen et al., 2004). It has been found that during stressful conditions, structural and functional changes occur in brain regions associated with cognitive functions (Lupien et al., 2009). Additionally, higher cortisol levels in the cerebrospinal fluid (CSF) have been observed in patients with mild cognitive impairment compared to cognitively healthy populations (Ouanes and Popp, 2019). The catecholamine surge is widely believed to induce cardiac dysfunction after ischemic stroke. Increased sympathetic tone after brain injury manifests with a catecholamine surge. High levels of catecholamines led to cardiac hypertrophy or myocardial ischemia in animal experiments (van der Bilt et al., 2016; Chen et al., 2017). Elevated catecholamine levels have also been observed in patients with AD (Janssens et al., 2019). However, CSF or plasma levels of catecholamines dynamically change with AD progression (Janssens et al., 2019; Pillet et al., 2020; Henjum et al., 2022). Higher CSF levels of noradrenaline and adrenaline were found in patients in the early stages of AD (Henjum et al., 2022), but the CSF levels of norepinephrine decreased in more severe cases (Janssens et al., 2018). A similar trend was observed in the plasma concentrations of norepinephrine, which were significantly lower in control subjects and patients with advanced AD than in patients with early-stage AD (Pillet et al., 2020). Meanwhile, neuronal loss and shrinkage of the locus coeruleus (the primary site of noradrenaline synthesis) occur during AD progression (Takahashi et al., 2015; Kelly et al., 2017). We hypothesized that differences in the plasma levels of norepinephrine may be related to blood-brain barrier breakdown and locus coeruleus shrinkage, which weakens the central control of the sympathetic system. Autonomic failure is often characterized by reduced sympathetic outflow and is associated with neurodegenerative diseases. Compared to autonomic failure, sympathetic storms occur after catastrophic neurological injuries, and stress-induced cardiomyopathy arises after exposure to a catecholamine surge (Tahsili-Fahadan and Geocadin, 2017). As the disease progresses, autonomic failure may exceed sympathetic activation, leading to decreased norepinephrine levels in patients with advanced AD (Janssens et al., 2019). Based on these findings, we suggest that autonomic failure may explain how AD protects against NICM. Further studies are needed to confirm that autonomic failure characterized by decreased norepinephrine levels occurs in patients with advanced AD.
Risk factors and protective factors associated with AD also affect NICM. Recently, Ewers et al. reported that soluble triggering receptor expressed on myeloid cell 2 (TREM2) is associated with milder cognitive decline in patients with AD (Ewers et al., 2019). The CSF concentrations of soluble TREM2 were found to increase with neurodegeneration and potentially reflect microglial activity. Microglia activated by the TREM2 signaling show greater chemotaxis and phagocytosis and may regulate the progression of amyloid plaques (Keren-Shaul et al., 2017; Krasemann et al., 2017). Using single-cell RNA sequencing and fate mapping, Fang et al. identified a unique subset of heart-resident macrophages (CD163+RETNLA+) in a mouse model of sepsis. This subset exhibited high expression of TREM2 and actively removed dysfunctional mitochondria excreted by cardiomyocytes. Conversely, macrophages lacking Trem2 were unable to self-renew, resulting in an excessive inflammatory response in cardiac tissue and more severe cardiac dysfunction (Zhang et al., 2023). ApoE was originally identified as a major ligand for low-density lipoprotein (LDL) and is involved in lipid metabolism. Increased levels of LDL, as a consequence of apoE2 and apoE4, increase the risk of heart disease. The fragments produced by structurally altered apoE4 undergoing neuron-specific proteolysis can lead to mitochondrial dysfunction and tau phosphorylation (Mahley, 2016). However, animal models of NICM have demonstrated that senile left ventricular hypertrophy and diastolic dysfunction develop in apoE-deficient mice in the absence of a high-fat diet, finally leading to heart failure (Liehn et al., 2021). Another cause of familial AD is the mutation of presenilin 1 (PSEN1) or presenilin 2 (PSEN2), which may be associated with DCM. Mutations of PSEN1 and PSEN2 have different effects on DCM. The former is associated with complete penetrance and advanced AD, whereas the latter exhibits partial penetrance and a better prognosis (Li et al., 2006).
LV dilation, systolic dysfunction, and decreased LVEF are the key features of DCM, making the assessment of LV size and LVEF crucial to the diagnosis, risk stratification, and treatment of DCM (Japp et al., 2016; Merlo et al., 2018). Ventricular remodeling is a major mechanism driving the progression of DCM to heart failure (Tobita et al., 2018). In this study, we investigated the causal effects of AD on five parameters representing the structure and function of the LV and found that AD was associated with decreased LVMVR (OR 0.9969, p-IVW = 0.0337). This finding is consistent with prior results regarding the protective effect of AD on NICM/DCM. LVMVR (also known as LV remodeling index) is a strong and independent predictor of diastolic function in patients with DCM (Buakhamsri et al., 2009; Xie et al., 2020). LVMVR has been shown to increase with age in both men and women (Peverill, 2021). A recent study demonstrated that higher LVMVR was significantly correlated with reduced LVEF in patients with type 2 diabetes (Xie et al., 2020). Although the effect size was small (OR: 0.9969), genetically predicted AD showed a negative effect on LVMVR, further supporting the reliability of the causal effect of AD on NICM/DCM.
Genetic factors play a significant role in both AD and DCM (Weintraub et al., 2017; Scheltens et al., 2021). In addition, we measured the causal effect of family history on NICM and found the substantial effect of genetic factors on the incidence of AD. Using MR analysis, we investigated the causal association between AD and NICM/DCM, providing a deeper insight into the involvement of genetic factors in the pathogenesis of NICM. Consistent with the results of MR, AD and DCM are genetically heterogeneous and cannot be solely attributed to individual DNA variants with large effects. Many variants with small effects on disease risk can result in noticeable phenotypes, and genetic or environmental modifications are increasingly important. Therefore, examination of more complex genetic factors can provide a deeper insight (Sierksma et al., 2020; Tayal et al., 2021).
We used GWAS summary statistics and MR analysis to investigate the causal effect of AD and FHAD on NICM/DCM and measure the causal effect of AD on LV traits. We addressed a novel issue that was not previously explored. Another advantage of this study was the use of MR analysis, which reduces potential biases caused by confounding factors and prevents reverse causation, commonly observed in traditional observational studies. Sensitivity analyses, including identification of heterogeneity and pleiotropy, leave-one-out analysis, and MR Steiger directionality test, were conducted to verify the robustness of our results. However, all participants were of European origin. Although it reduces the potential bias associated with demographics, it limits the generalizability of MR results to other populations. More importantly, although MR analysis suggested that AD may protect against NICM, further mechanistic and experimental studies are needed to confirm the genetic associations. The involvement of autonomic failure in the protective effect of AD on NICM is only a hypothesis based on the course of AD; thus, we need to treat this causality with caution.
5 Conclusion
In conclusion, our study uncovered the causal effects of AD on NICM/DCM and LV traits and revealed that AD and FHAD were associated with a decreased risk of NICM. Our findings implied that AD may be useful in etiological studies for NICM. Further studies are warranted to illuminate the underlying mechanisms linking AD to NICM.
Data availability statement
The data presented in the study are deposited in https://www.ebi.ac.uk/gwas/, accession number (PMID): 35379992/30820047/29777097/34594039/33495596; and https://gwas.mrcieu.ac.uk/, accession number (GWAS ID): finn-b-I9_NONISCHCARDMYOP/finn-b-I9_HYPERTROCARDMYOP.
Ethics statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author contributions
ZL: Supervision, Validation, Writing–original draft, Writing–review and editing. XC: Conceptualization, Formal Analysis, Validation, Writing–review and editing. WH: Conceptualization, Software, Validation, Writing–review and editing. HC: Methodology, Validation, Writing–review and editing. DC: Supervision, Validation, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We would like to thank all of the participants and investigators of the studies that were used throughout this MR research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1379865/full#supplementary-material
Abbreviations
AD, Alzheimer’s disease; AD-DD, Different dataset for Alzheimer’s disease; apoE, apolipoprotein E; DCM, dilated cardiomyopathy; FHAD, family history of Alzheimer’s disease; GWAS, genome-wide association study; HCM, hypertrophic cardiomyopathy; LV, left ventricular; LVEDV, LV end-diastolic volume; LVESV, LV end-systolic volume; LVEF, LV ejection fraction; LVM, mass; LVMVR, left ventricular mass-to-end-diastolic volume ratio; MH-AD, maternal history of Alzheimer’s disease; MR, Mendelian randomization; MR-PRESSO, MR pleiotropy residual sum and outlier; NA, Not Applicable; NICM, non-ischemic cardiomyopathy; PH-AD, Paternal history of Alzheimer’s disease; SNPs, single nucleotide polymorphisms.
References
Ajiki, K., Murakawa, Y., Yanagisawa-Miwa, A., Usui, M., Yamashita, T., Oikawa, N., et al. (1993). Autonomic nervous system activity in idiopathic dilated cardiomyopathy and in hypertrophic cardiomyopathy. Am. J. Cardiol. 71(15), 1316–1320. doi:10.1016/0002-9149(93)90547-p
Armstrong, R. A. (2014). When to use the Bonferroni correction. Ophthalmic Physiol. Opt. 34 (5), 502–508. doi:10.1111/opo.12131
Belaidi, A. A., and Bush, A. I. (2016). Iron neurochemistry in Alzheimer's disease and Parkinson's disease: targets for therapeutics. J. Neurochem. 139 (Suppl. 1), 179–197. doi:10.1111/jnc.13425
Bellenguez, C., Küçükali, F., Jansen, I. E., Kleineidam, L., Moreno-Grau, S., Amin, N., et al. (2022). New insights into the genetic etiology of Alzheimer's disease and related dementias. Nat. Genet. 54 (4), 412–436. doi:10.1038/s41588-022-01024-z
Bowden, J., Davey Smith, G., and Burgess, S. (2015). Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44 (2), 512–525. doi:10.1093/ije/dyv080
Bowden, J., Davey Smith, G., Haycock, P. C., and Burgess, S. (2016). Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40 (4), 304–314. doi:10.1002/gepi.21965
Bowden, J., Del Greco, M. F., Minelli, C., Zhao, Q., Lawlor, D. A., Sheehan, N. A., et al. (2019). Improving the accuracy of two-sample summary-data Mendelian randomization: moving beyond the NOME assumption. Int. J. Epidemiol. 48 (3), 728–742. doi:10.1093/ije/dyy258
Braak, H., and Braak, E. (1995). Staging of Alzheimer's disease-related neurofibrillary changes. Neurobiol. Aging 16 (3), 271–278. doi:10.1016/0197-4580(95)00021-6
Buakhamsri, A., Popovic, Z. B., Lin, J., Lim, P., Greenberg, N. L., Borowski, A. G., et al. (2009). Impact of left ventricular volume/mass ratio on diastolic function. Eur. Heart J. 30 (10), 1213–1221. doi:10.1093/eurheartj/ehp084
Burgess, S., Dudbridge, F., and Thompson, S. G. (2016). Combining information on multiple instrumental variables in Mendelian randomization: comparison of allele score and summarized data methods. Stat. Med. 35 (11), 1880–1906. doi:10.1002/sim.6835
Burgess, S., and Thompson, S. G.CRP CHD Genetics Collaboration (2011). Avoiding bias from weak instruments in Mendelian randomization studies. Int. J. Epidemiol. 40 (3), 755–764. doi:10.1093/ije/dyr036
Chen, Z., Venkat, P., Seyfried, D., Chopp, M., Yan, T., and Chen, J. (2017). Brain-heart interaction: cardiac complications after stroke. Circ. Res. 121 (4), 451–468. doi:10.1161/circresaha.117.311170
Christensen, H., Boysen, G., and Johannesen, H. H. (2004). Serum-cortisol reflects severity and mortality in acute stroke. J. Neurol. Sci. 217 (2), 175–180. doi:10.1016/j.jns.2003.09.013
Clarke, L., Zheng-Bradley, X., Smith, R., Kulesha, E., Xiao, C., Toneva, I., et al. (2012). The 1000 Genomes Project: data management and community access. Nat. Methods 9 (5), 459–462. doi:10.1038/nmeth.1974
Davies, N. M., Holmes, M. V., and Davey Smith, G. (2018). Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. Bmj 362, k601. doi:10.1136/bmj.k601
Elliott, P., Andersson, B., Arbustini, E., Bilinska, Z., Cecchi, F., Charron, P., et al. (2008). Classification of the cardiomyopathies: a position statement from the European society of cardiology working group on myocardial and pericardial diseases. Eur. Heart J. 29 (2), 270–276. doi:10.1093/eurheartj/ehm342
Ewers, M., Franzmeier, N., Suárez-Calvet, M., Morenas-Rodriguez, E., Caballero, M. A. A., Kleinberger, G., et al. (2019). Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer's disease. Sci. Transl. Med. 11 (507), eaav6221. doi:10.1126/scitranslmed.aav6221
Femminella, G. D., Rengo, G., Komici, K., Iacotucci, P., Petraglia, L., Pagano, G., et al. (2014). Autonomic dysfunction in Alzheimer's disease: tools for assessment and review of the literature. J. Alzheimers Dis. 42 (2), 369–377. doi:10.3233/jad-140513
Fu, L., and Liang, J. J. (2003). Alteration of protein-protein interactions of congenital cataract crystallin mutants. Invest. Ophthalmol. Vis. Sci. 44 (3), 1155–1159. doi:10.1167/iovs.02-0950
Giraldo, D. L., García-Arteaga, J. D., Cárdenas-Robledo, S., and Romero, E. (2018). Characterization of brain anatomical patterns by comparing region intensity distributions: applications to the description of Alzheimer's disease. Brain Behav. 8 (4), e00942. doi:10.1002/brb3.942
Giubilei, F., Strano, S., Imbimbo, B. P., Tisei, P., Calcagnini, G., Lino, S., et al. (1998). Cardiac autonomic dysfunction in patients with Alzheimer disease: possible pathogenetic mechanisms. Alzheimer Dis. Assoc. Disord. 12 (4), 356–361. doi:10.1097/00002093-199812000-00017
Grutter, G., Giordano, U., Alfieri, S., Iodice, F., Drago, F., Ravà, L., et al. (2012). Heart rate variability abnormalities in young patients with dilated cardiomyopathy. Pediatr. Cardiol. 33 (7), 1171–1174. doi:10.1007/s00246-012-0277-9
Hemani, G., Tilling, K., and Davey Smith, G. (2017). Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 13 (11), e1007081. doi:10.1371/journal.pgen.1007081
Henjum, K., Watne, L. O., Godang, K., Halaas, N. B., Eldholm, R. S., Blennow, K., et al. (2022). Cerebrospinal fluid catecholamines in Alzheimer's disease patients with and without biological disease. Transl. Psychiatry 12 (1), 151. doi:10.1038/s41398-022-01901-5
Hoffmann, J., Grimm, W., Menz, V., Müller, H. H., and Maisch, B. (2000). Heart rate variability and baroreflex sensitivity in idiopathic dilated cardiomyopathy. Heart 83 (5), 531–538. doi:10.1136/heart.83.5.531
Janssens, J., Atmosoerodjo, S. D., Vermeiren, Y., Absalom, A. R., den Daas, I., and De Deyn, P. P. (2019). Sampling issues of cerebrospinal fluid and plasma monoamines: investigation of the circadian rhythm and rostrocaudal concentration gradient. Neurochem. Int. 128, 154–162. doi:10.1016/j.neuint.2019.04.015
Janssens, J., Vermeiren, Y., Fransen, E., Aerts, T., Van Dam, D., Engelborghs, S., et al. (2018). Cerebrospinal fluid and serum MHPG improve Alzheimer's disease versus dementia with Lewy bodies differential diagnosis. Alzheimers Dement. (Amst) 10, 172–181. doi:10.1016/j.dadm.2018.01.002
Japp, A. G., Gulati, A., Cook, S. A., Cowie, M. R., and Prasad, S. K. (2016). The diagnosis and evaluation of dilated cardiomyopathy. J. Am. Coll. Cardiol. 67 (25), 2996–3010. doi:10.1016/j.jacc.2016.03.590
Kamat, M. A., Blackshaw, J. A., Young, R., Surendran, P., Burgess, S., Danesh, J., et al. (2019). PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinformatics 35 (22), 4851–4853. doi:10.1093/bioinformatics/btz469
Kampinga, H. H., Hageman, J., Vos, M. J., Kubota, H., Tanguay, R. M., Bruford, E. A., et al. (2009). Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 14 (1), 105–111. doi:10.1007/s12192-008-0068-7
Kelly, S. C., He, B., Perez, S. E., Ginsberg, S. D., Mufson, E. J., and Counts, S. E. (2017). Locus coeruleus cellular and molecular pathology during the progression of Alzheimer's disease. Acta Neuropathol. Commun. 5 (1), 8. doi:10.1186/s40478-017-0411-2
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., et al. (2017). A unique microglia type associated with restricting development of Alzheimer's disease. Cell 169 (7), 1276–1290. doi:10.1016/j.cell.2017.05.018
Kostin, S., Pool, L., Elsässer, A., Hein, S., Drexler, H. C., Arnon, E., et al. (2003). Myocytes die by multiple mechanisms in failing human hearts. Circ. Res. 92 (7), 715–724. doi:10.1161/01.Res.0000067471.95890.5c
Krasemann, S., Madore, C., Cialic, R., Baufeld, C., Calcagno, N., El Fatimy, R., et al. (2017). The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47 (3), 566–581. doi:10.1016/j.immuni.2017.08.008
Kunkle, B. W., Grenier-Boley, B., Sims, R., Bis, J. C., Damotte, V., Naj, A. C., et al. (2019). Genetic meta-analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51 (3), 414–430. doi:10.1038/s41588-019-0358-2
Kurki, M. I., Karjalainen, J., Palta, P., Sipilä, T. P., Kristiansson, K., Donner, K. M., et al. (2023). FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 613 (7944), 508–518. doi:10.1038/s41586-022-05473-8
Kusumoto, F. M., Calkins, H., Boehmer, J., Buxton, A. E., Chung, M. K., Gold, M. R., et al. (2014). HRS/ACC/AHA expert consensus statement on the use of implantable cardioverter-defibrillator therapy in patients who are not included or not well represented in clinical trials. J. Am. Coll. Cardiol. 64 (11), 1143–1177. doi:10.1016/j.jacc.2014.04.008
Lawlor, D. A. (2016). Commentary: two-sample Mendelian randomization: opportunities and challenges. Int. J. Epidemiol. 45 (3), 908–915. doi:10.1093/ije/dyw127
Lawlor, D. A., Harbord, R. M., Sterne, J. A., Timpson, N., and Davey Smith, G. (2008). Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat. Med. 27 (8), 1133–1163. doi:10.1002/sim.3034
Li, D., Parks, S. B., Kushner, J. D., Nauman, D., Burgess, D., Ludwigsen, S., et al. (2006). Mutations of presenilin genes in dilated cardiomyopathy and heart failure. Am. J. Hum. Genet. 79 (6), 1030–1039. doi:10.1086/509900
Liehn, E. A., Lupan, A. M., Diaconu, R., Ioana, M., Streata, I., Manole, C., et al. (2021). Heart function assessment during aging in apolipoprotein E knock-out mice. Discov. (Craiova) 9 (3), e136. doi:10.15190/d.2021.15
Lupien, S. J., McEwen, B. S., Gunnar, M. R., and Heim, C. (2009). Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat. Rev. Neurosci. 10 (6), 434–445. doi:10.1038/nrn2639
Mahley, R. W. (2016). Apolipoprotein E: from cardiovascular disease to neurodegenerative disorders. J. Mol. Med. Berl. 94 (7), 739–746. doi:10.1007/s00109-016-1427-y
Maloyan, A., Gulick, J., Glabe, C. G., Kayed, R., and Robbins, J. (2007). Exercise reverses preamyloid oligomer and prolongs survival in alphaB-crystallin-based desmin-related cardiomyopathy. Proc. Natl. Acad. Sci. U. S. A. 104 (14), 5995–6000. doi:10.1073/pnas.0609202104
Marioni, R. E., Harris, S. E., Zhang, Q., McRae, A. F., Hagenaars, S. P., Hill, W. D., et al. (2018). GWAS on family history of Alzheimer's disease. Transl. Psychiatry 8 (1), 99. doi:10.1038/s41398-018-0150-6
Mellingsæter, M. R., Wyller, T. B., Ranhoff, A. H., Bogdanovic, N., and Wyller, V. B. (2015). Reduced sympathetic response to head-up tilt in subjects with mild cognitive impairment or mild Alzheimer's dementia. Dement. Geriatr. Cogn. Dis. Extra 5 (1), 107–115. doi:10.1159/000375297
Melville, K. I., Blum, B., Shister, H. E., and Silver, M. D. (1963). Cardiac ischemic changes and arrhythmias induced by hypothalamic stimulation. Am. J. Cardiol. 12, 781–791. doi:10.1016/0002-9149(63)90281-9
Merlo, M., Cannatà, A., Gobbo, M., Stolfo, D., Elliott, P. M., and Sinagra, G. (2018). Evolving concepts in dilated cardiomyopathy. Eur. J. Heart Fail 20 (2), 228–239. doi:10.1002/ejhf.1103
Ouanes, S., and Popp, J. (2019). High cortisol and the risk of dementia and Alzheimer's disease: a review of the literature. Front. Aging Neurosci. 11, 43. doi:10.3389/fnagi.2019.00043
Peverill, R. E. (2021). Changes in left ventricular size, geometry, pump function and left heart pressures during healthy aging. Rev. Cardiovasc Med. 22 (3), 717–729. doi:10.31083/j.rcm2203079
Pillet, L. E., Taccola, C., Cotoni, J., Thiriez, H., André, K., and Verpillot, R. (2020). Correlation between cognition and plasma noradrenaline level in Alzheimer's disease: a potential new blood marker of disease evolution. Transl. Psychiatry 10 (1), 213. doi:10.1038/s41398-020-0841-7
Pimentel, M., Rohde, L. E., Zimerman, A., and Zimerman, L. I. (2016). Sudden cardiac death markers in non-ischemic cardiomyopathy. J. Electrocardiol. 49 (3), 446–451. doi:10.1016/j.jelectrocard.2016.03.012
Rashba, E. J., Estes, N. A., Wang, P., Schaechter, A., Howard, A., Zareba, W., et al. (2006). Preserved heart rate variability identifies low-risk patients with nonischemic dilated cardiomyopathy: results from the DEFINITE trial. Heart rhythm. 3 (3), 281–286. doi:10.1016/j.hrthm.2005.11.028
Sakaue, S., Kanai, M., Tanigawa, Y., Karjalainen, J., Kurki, M., Koshiba, S., et al. (2021). A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 53 (10), 1415–1424. doi:10.1038/s41588-021-00931-x
Sanbe, A., Osinska, H., Saffitz, J. E., Glabe, C. G., Kayed, R., Maloyan, A., et al. (2004). Desmin-related cardiomyopathy in transgenic mice: a cardiac amyloidosis. Proc. Natl. Acad. Sci. U. S. A. 101 (27), 10132–10136. doi:10.1073/pnas.0401900101
Sanna, G. D., Nusdeo, G., Piras, M. R., Forteleoni, A., Murru, M. R., Saba, P. S., et al. (2019). Cardiac abnormalities in alzheimer disease: clinical relevance beyond pathophysiological rationale and instrumental findings? JACC Heart Fail 7 (2), 121–128. doi:10.1016/j.jchf.2018.10.022
Santos, C. Y., Machan, J. T., Wu, W. C., and Snyder, P. J. (2017). Autonomic cardiac function in preclinical Alzheimer's disease. J. Alzheimers Dis. 59 (3), 1057–1065. doi:10.3233/jad-170217
Scheltens, P., De Strooper, B., Kivipelto, M., Holstege, H., Chételat, G., Teunissen, C. E., et al. (2021). Alzheimer's disease. Lancet 397 (10284), 1577–1590. doi:10.1016/s0140-6736(20)32205-4
Sierksma, A., Escott-Price, V., and De Strooper, B. (2020). Translating genetic risk of Alzheimer's disease into mechanistic insight and drug targets. Science 370 (6512), 61–66. doi:10.1126/science.abb8575
Stamatelopoulos, K., Pol, C. J., Ayers, C., Georgiopoulos, G., Gatsiou, A., Brilakis, E. S., et al. (2018). Amyloid-beta (1-40) peptide and subclinical cardiovascular disease. J. Am. Coll. Cardiol. 72 (9), 1060–1061. doi:10.1016/j.jacc.2018.06.027
Tadros, R., Francis, C., Xu, X., Vermeer, A. M. C., Harper, A. R., Huurman, R., et al. (2021). Shared genetic pathways contribute to risk of hypertrophic and dilated cardiomyopathies with opposite directions of effect. Nat. Genet. 53 (2), 128–134. doi:10.1038/s41588-020-00762-2
Tahsili-Fahadan, P., and Geocadin, R. G. (2017). Heart-brain Axis: effects of neurologic injury on cardiovascular function. Circ. Res. 120 (3), 559–572. doi:10.1161/circresaha.116.308446
Takahashi, J., Shibata, T., Sasaki, M., Kudo, M., Yanezawa, H., Obara, S., et al. (2015). Detection of changes in the locus coeruleus in patients with mild cognitive impairment and Alzheimer's disease: high-resolution fast spin-echo T1-weighted imaging. Geriatr. Gerontol. Int. 15 (3), 334–340. doi:10.1111/ggi.12280
Tayal, U., Ware, J. S., Lakdawala, N. K., Heymans, S., and Prasad, S. K. (2021). Understanding the genetics of adult-onset dilated cardiomyopathy: what a clinician needs to know. Eur. Heart J. 42 (24), 2384–2396. doi:10.1093/eurheartj/ehab286
Tobita, T., Nomura, S., Fujita, T., Morita, H., Asano, Y., Onoue, K., et al. (2018). Genetic basis of cardiomyopathy and the genotypes involved in prognosis and left ventricular reverse remodeling. Sci. Rep. 8 (1), 1998. doi:10.1038/s41598-018-20114-9
Tokgözoglu, S. L., Batur, M. K., Topçuoglu, M. A., Saribas, O., Kes, S., and Oto, A. (1999). Effects of stroke localization on cardiac autonomic balance and sudden death. Stroke 30 (7), 1307–1311. doi:10.1161/01.str.30.7.1307
Tsuchihashi, K., Ueshima, K., Uchida, T., Oh-mura, N., Kimura, K., Owa, M., et al. (2001). Transient left ventricular apical ballooning without coronary artery stenosis: a novel heart syndrome mimicking acute myocardial infarction. Angina Pectoris-Myocardial Infarction Investigations in Japan. J. Am. Coll. Cardiol. 38 (1), 11–18. doi:10.1016/s0735-1097(01)01316-x
Tublin, J. M., Adelstein, J. M., Del Monte, F., Combs, C. K., and Wold, L. E. (2019). Getting to the heart of alzheimer disease. Circ. Res. 124 (1), 142–149. doi:10.1161/circresaha.118.313563
van der Bilt, I. A., Vendeville, J. P., van de Hoef, T. P., Begieneman, M. P., Lagrand, W. K., Kros, J. M., et al. (2016). Myocarditis in patients with subarachnoid hemorrhage: a histopathologic study. J. Crit. Care 32, 196–200. doi:10.1016/j.jcrc.2015.12.005
Verbanck, M., Chen, C. Y., Neale, B., and Do, R. (2018). Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 50 (5), 693–698. doi:10.1038/s41588-018-0099-7
Wang, Y., Jia, H., and Song, J. (2023). Accurate classification of non-ischemic cardiomyopathy. Curr. Cardiol. Rep. 25 (10), 1299–1317. doi:10.1007/s11886-023-01944-0
Wehrwein, E. A., Orer, H. S., and Barman, S. M. (2016). Overview of the anatomy, physiology, and pharmacology of the autonomic nervous system. Compr. Physiol. 6 (3), 1239–1278. doi:10.1002/cphy.c150037
Weintraub, R. G., Semsarian, C., and Macdonald, P. (2017). Dilated cardiomyopathy. Lancet 390 (10092), 400–414. doi:10.1016/s0140-6736(16)31713-5
Whitnall, M. H. (1993). Regulation of the hypothalamic corticotropin-releasing hormone neurosecretory system. Prog. Neurobiol. 40 (5), 573–629. doi:10.1016/0301-0082(93)90035-q
Willis, M. S., and Patterson, C. (2013). Proteotoxicity and cardiac dysfunction--Alzheimer's disease of the heart? N. Engl. J. Med. 368 (5), 455–464. doi:10.1056/NEJMra1106180
Xie, L. J., Dong, Z. H., Yang, Z. G., Deng, M. Y., Gao, Y., Jiang, L., et al. (2020). Assessment of left ventricular deformation in patients with type 2 diabetes mellitus by cardiac magnetic resonance tissue tracking. Sci. Rep. 10 (1), 13126. doi:10.1038/s41598-020-69977-x
Yi, G., Goldman, J. H., Keeling, P. J., Reardon, M., McKenna, W. J., and Malik, M. (1997). Heart rate variability in idiopathic dilated cardiomyopathy: relation to disease severity and prognosis. Heart 77 (2), 108–114. doi:10.1136/hrt.77.2.108
Yoshimura, S., Toyoda, K., Ohara, T., Nagasawa, H., Ohtani, N., Kuwashiro, T., et al. (2008). Takotsubo cardiomyopathy in acute ischemic stroke. Ann. Neurol. 64 (5), 547–554. doi:10.1002/ana.21459
Keywords: Alzheimer’s disease, non-ischemic cardiomyopathy, dilated cardiomyopathy, left ventricular structure and function, Mendelian randomization
Citation: Li Z, Chen X, He W, Chen H and Chen D (2024) The causal effect of Alzheimer’s disease and family history of Alzheimer’s disease on non-ischemic cardiomyopathy and left ventricular structure and function: a Mendelian randomization study. Front. Genet. 15:1379865. doi: 10.3389/fgene.2024.1379865
Received: 31 January 2024; Accepted: 15 May 2024;
Published: 05 June 2024.
Edited by:
Javed Iqbal, University of Sargodha, PakistanReviewed by:
Fuwen Yao, Stanford University, United StatesXiao Han, Howard Hughes Medical Institute (HHMI), United States
Copyright © 2024 Li, Chen, He, Chen and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dehai Chen, ZGVoYWljaGVuMjAyM0AxMjYuY29t