 Silvia Riggio1
Silvia Riggio1 Marco Tolone2
Marco Tolone2 Gianluca Sottile3
Gianluca Sottile3 Serena Tumino4Baldassare Portolano1
Serena Tumino4Baldassare Portolano1 Anna Maria Sutera2
Anna Maria Sutera2 Maria Teresa Sardina1
Maria Teresa Sardina1 Alberto Cesarani5,6
Alberto Cesarani5,6 Salvatore Mastrangelo1*
Salvatore Mastrangelo1*- 1Dipartimento Scienze Agrarie, Alimentari e Forestali, University of Palermo, Palermo, Italy
- 2Dipartimento di Scienze Chimiche, Biologiche, Farmaceutiche, ed Ambientali, University of Messina, Messina, Italy
- 3Dipartimento Scienze Economiche, Aziendali e Statistiche, University of Palermo, Palermo, Italy
- 4Dipartimento di Agricoltura, Alimentazione e Ambiente, University of Catania, Catania, Italy
- 5Dipartimento di Agraria, University of Sassari, Sassari, Italy
- 6Department of Animal and Dairy Science, University of Georgia, Athens, GA, United States
Visna/Maedi virus (VMV) is lentiviral disease of sheep responsible for severe production losses. Multiple genomic regions associated with infection were reported indicating genetic complexity. In this study, a combined genome-wide approach using a high-density SNP array has been performed, comparing VMV-infected (n = 78) and non-infected (n = 66) individuals of the Valle del Belice breed. The serological tests showed a seroprevalence of 26%. The comparison among results from different approaches (GWAS, Fisher’s exact test and the FST analysis) revealed two association signals: on OAR03 close to the GRIN2B gene and on OAR05 close to the TMEM232 gene. To the best of our knowledge, there has been no previous association between these genes and lentiviral infection in any species. The GRIN2B gene plays a role in pain response, synaptic transmission, and receptor clustering, while TMEM232 is involved in the development of immune-related disorders. The results highlighted new aspects of the genetic complexity related to the resistance/susceptibility to VMV in sheep, confirming that studies on different breeds can lead to different results. The ideal approach for validation of the markers identified in our study is to use samples from a population independent from the discovery population with the same phenotype used in the discovery stage.
1 Introduction
Sheep and goats are infected by Visna/Maedi virus (VMV) and caprine arthritis encephalitis virus (CAEV) (respectively), which belong to the retroviridae family and are classified as small ruminant lentiviruses (SRLV). According to Minguijón et al. (2015), a wasting disease marked by persistent inflammation in carpal joints, udder, central nervous system, and lungs is caused by these viruses. The main productive and economic losses associated to the diseases are reduced milk production and ewe productivity, lower birth weights in offspring, early culling or death of the infected animals, cost of the control programs and export restrictions (Peterhans et al., 2004; Benavides et al., 2013; Gibson et al., 2018; Juste et al., 2020). The transmission of the disease can occur either through the ingestion of colostrum from infected ewes or through direct contact with the respiratory secretions of infected animals (Preziuso et al., 2004; Kalogianni et al., 2020). The VM shows a long incubation period, which is reported to be around three or 4 years, and a slow progressive and subclinical infection (Zink et al., 1987; Straub, 2004). Moreover, the slow seroconversion in the infected animals leads to an underdiagnosed and underreported disease rate and to high frequency of asymptomatic carriers (Illius et al., 2020).
Immune responses to infection result in antibodies that are typically detected throughout the entire life of the animals. Therefore, since there is no vaccination for SRLV, the control of this disease mainly relies on identifying seropositive animals (Kalogianni et al., 2020). Multiple diagnostic techniques can be used to detect SRLV infection and indirect methods have been proposed as the most appropriate to detect infected animals. In fact, the association between breeds and SRLV infection susceptibility patterns has been generally discovered using ELISA testing for the antibody’s detection (de Miguel et al., 2021).
In viral infections, without therapies or efficient vaccinations, genetic selection of resistant animals is of special interest (de Pablo-Maiso et al., 2018). The genome-wide approaches based on the Single Nucleotide Polymorphisms (SNPs) arrays created new opportunities to include disease control in the breeding programs. In particular, the genetic tools have been used to describe several candidate genes associated with SRLV seroreaction (Abendaño et al., 2021). The TMEM154 has been reported to be the major gene associated to VMV susceptibility by Heaton et al. (2012) and several subsequent independent studies (Molaee et al., 2018; Molaee et al., 2019; Yaman et al., 2019; Ramírez et al., 2021; Tumino et al., 2022; Riggio et al., 2023; Rodrigues et al., 2023). The polymorphism involved in resistance/susceptibility is located in the portion of the gene encoding the extracellular part of the protein and is characterised by the substitution, at position 35 of the protein, of glutamate (E) amino acid with lysine (K) one (Heaton et al., 2012). The resistance occurred only in homozygous KK animals, while heterozygous EK and homozygous EE were susceptible to disease. However, a genetic complexity of this disease can be supposed because of the several candidate genes associated with the resistance/susceptibility to VMV (Abendaño et al., 2021; Arcangeli et al., 2021), such as: CCR5 (White et al., 2009; Molaee et al., 2018), ovar-DRB1 (Larruskain et al., 2010), DPPA2/DPPA4 and SYTL3 (White et al., 2012), TLR9 and MYD88 (Sarafidou et al., 2013) and ZNF389 (White et al., 2014). Additionally, the disease can manifest differently depending on management systems, VMV strains, age, immune status, and sex of animals; furthermore, much evidence showed that susceptibility to SRLV infection exhibits significant variation across different breeds, contributing to the complexity of the trait (e.g., Keen et al., 1997; Molaee et al., 2019; Letko et al., 2021; Moretti et al., 2022).
To date, few sheep GWAS studies aimed to identify additional potentially candidate genes involved in the resistance to VMV. On these premises, in this study, genome-wide approaches have been used to compare VMV-infected and non-infected genotyped individuals of the Valle del Belice sheep breed to identify candidate genomic regions potentially involved with the resistance to VMV infection.
2 Materials and methods
2.1 Animals and samples
Before collecting samples, a preliminary screening was carried out on 20 farms, to assess the prevalence status of Visna/Maedi in the Valle del Belìce breed within different herds. After this, the sampling was carried out in eight farms, in which the prevalence status of Visna/Maedi was at least 20%. Therefore, samples were collected from sheep flocks already known to be VM affected.
Following the recommendations of the European Union Directive 2010/63/EU, individual blood samples were collected from 300 ewes. The animals did not show clinical signs of the disease. From all involved ewes, an individual milk sample was also collected during the morning milking. All the farms raised only sheep and shared a similar management based on daytime grazing and night shelter. The epidemiological unrelatedness of farm was ensured by the lack of animal transfer among flocks. To allow for sufficient VMV exposure and subsequent seroconversion, age of sampled ewes was from 2 to 8 years (mean 4.42 ± 1.33).
2.2 Serological analyses by ELISA test
The milk samples were analyzed by the commercial ELISA assay ID Screen® MVV/CAEV Indirect Screening test (IDvet, Grabels, France), following the official instructions from the manufacturer. As the final phase of the process, ELISA plates were read at 450 nm wavelength using an ELISA plate reader. The cut-off value was defined based on the corrected optical density (OD) ratio between sample and positive control (S/P) at a wavelength of 450 nm. As suggested in the instructions provided by the manufacturer, samples were considered SRLVs negative (and defined control) or positive (and defined case) with S/p-value ≤ 50% or ≥60%, respectively.
To exclude false positives within samples, serological positive animals were tested twice using the ID Screen® MVV/CAEV. This retesting occurred at two distinct time points: first, during the initial screening to assess Visna/Maedi prevalence across all herds, and again 6 months later for the animals specifically chosen for the study.
2.3 DNA extraction, genotyping and quality control
Following the protocol of the PureLink Genomic DNA kit (Invitrogen), genomic DNA was extracted from 200 μL of blood. The DNA was quantified using the Nanodrop ND-1000 (Thermo Fisher Scientific).
All the individuals were genotyped at the TMEM154 gene by direct Sanger sequencing, to estimate the genotype frequencies of the E35K polymorphism (Riggio et al., 2023).
A subset of 144 animals including all seropositive (78 case group) and a reduced number of seronegative samples (66 control group), chosen from the same farm and sampled on the same day of cases, have been genotyped by Illumina Ovine SNP600K BeadChip array, which contains about 600,000 markers covering the whole ovine genome (Illumina, San Diego, California, United States). Markers positions and names were updated to the ARS-UI_Ramb_v2.0 assembly to retain those mapped on autosomal chromosomes only. The quality control was performed on both animals and variants using PLINK v1.9 (Chang et al., 2015) with the following criteria: missing genotypes rate lower than 2%, SNP call rate larger than 98%, minor allele frequency (MAF) > 0.05. Moreover, SNPs without Hardy-Weinberg equilibrium (p-value<0.001) were also discarded.
2.4 Genome-wide analyses
Pairwise identity-by-state distances among individuals were computed using PLINK v1.9 (Chang et al., 2015) and represented by multidimensional scaling analysis (MDS).
Univariate case/control GWAS was performed by the log-additive genetic model, implemented in the SNPassoc R package (González et al., 2007), considering the herd effect. The genome-wide significant threshold was fixed according to the Bonferroni correction method as 0.05/N (N is the number of tested SNPs). GenABEL (Aulchenko et al., 2007) was used to get the genomic inflation factor (λ) and quantile-quantile (Q-Q) plot to evaluate any potential systematic bias resulting from the population structure or analytical strategy. The p-value and the position of each SNP along the 26 Ovis aries chromosomes (OAR) were reported in the Manhattan plots.
PLINK v1.9 (Chang et al., 2015) was used to perform the genome-wide FST case-control analysis. SNPs were flagged as significant if their value was larger than the 0.9999 percentile (Mastrangelo et al., 2019). Finally, Fisher’s exact test was carried out through PLINK v1.9 (Chang et al., 2015). The potential bias from multiple testing was accounted by using the Bonferroni correction (p < 0.05, genome-wide).
A standard descriptive Linkage disequilibrium (LD) parameter, the squared correlation coefficient of allele frequencies at a pair of loci (r2), was obtained using PLINK v1.9 (Chang et al., 2015). The r2 value between significant SNPs was calculated. Finally, the genotypic frequencies were also estimated.
Candidate genes close to the significant markers were retrieved using the NCBI Genome Data Viewer (https://www.ncbi.nlm.nih.gov/genome/gdv/browser/genome/?id=GCF_016772045.1). The biological functions and phenotypes associated to the annotated genes were discovered through a comprehensive literature search.
3 Results
On a total of 300 sheep, the results of ELISA analysis identified 78 seropositive animals, which correspond to a seroprevalence of 26%. All these samples were genotyped for the amino acid substitution at position 35 of TMEM154. No homozygous animals for the resistant KK genotype were observed. The most frequent genotype was EE (75% of total animals), followed by EK (25% of total animals).
Subsequently, 78 seropositive animals (case group) and other 66 seronegative animals (control group) have been genotyped using the Ovine SNP600K array. Animals of the control group were chosen from the same farms as cases and were sampled on the same day. After quality control, the final number of markers used for the analyses was 353,100, whereas the total number of individuals was 142, since two animals (control group) were discarded because of poor quality genotyping.
To examine the relationships among individuals, MDS was performed (Supplementary Figure S1). The first two components, C1 and C2, accounted for 7.6% and 5.1% of the total variation, respectively. The MDS plot did not cluster the animals according to their different phenotypes (i.e., disease status). The absence of a well-defined population configuration was confirmed by the average inflation factor (λ) of 1.28, which indicated that the population structure did not influence the results of the GWAS.
In the latter, at the p < 0.05 Bonferroni corrected [−log10(P) = 6.85; Pnominal value = 1.42E−07], four associated markers were identified on three different chromosomes (Table 1). Figure 1 shows the Manhattan plot obtained in this GWAS.
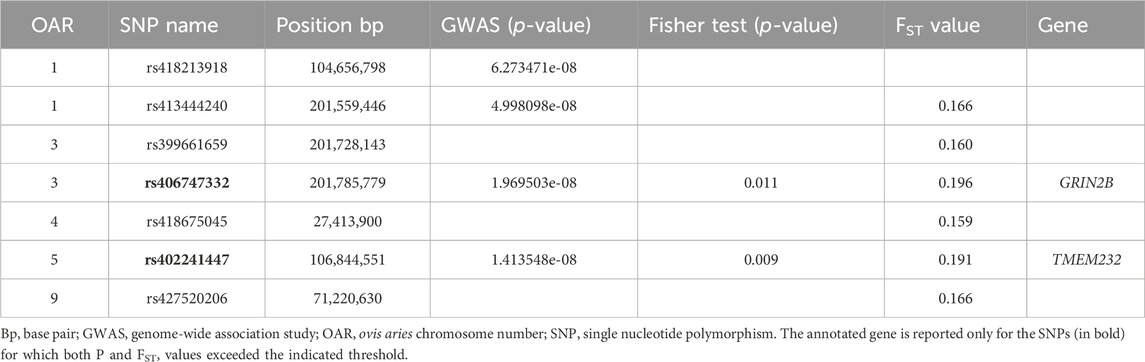
Table 1. Significant single nucleotide polymorphisms (SNPs) obtained using the genome-wide approaches (GWAS, Fisher’s exact test and the FST analysis) in the Valle del Belice sheep breed.
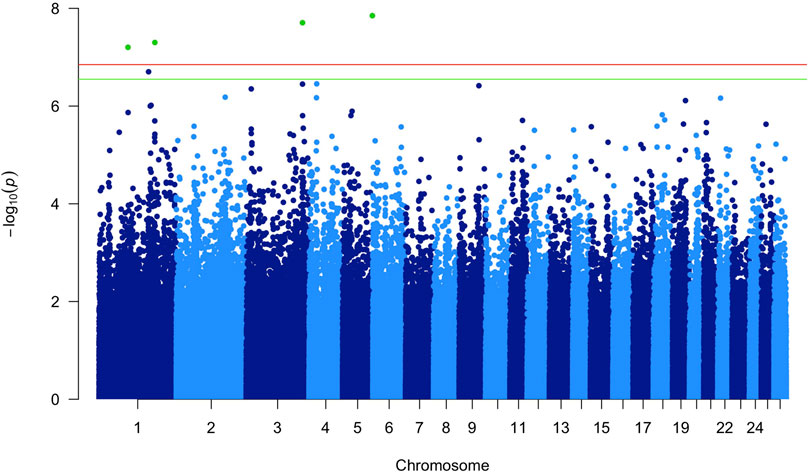
Figure 1. Manhattan plot of the p-values in the genome-wide association study (GWAS). The horizontal lines represent the genome-wide significance (red; p < 0.05) and suggestively significant (green; p < 0.10) single nucleotide polymorphisms (SNPs).
The Fisher’s exact test case-control association analysis identified only two of the four significant markers found with the GWAS: rs406747332 on OAR03 and rs402241447 on OAR05 (Table 1). To support these results, a genome-wide FST case-control analysis was also performed. The analysis showed a total of six SNPs above the selected threshold (FST = 0.160), located on five different chromosomes.
The two markers with the highest FST values (>0.190) between groups overlapped with the most significant SNPS identified in the GWAS [rs406747332 (−log10(P) = 7.71] and rs402241447 [−log10(P) = 7.85] (Table 1) and with the Fisher’s exact test (Table 1).
According to the LD analysis results, these two significant SNPs showed low r2 value (0.117). The genotypic frequencies of these two SNPs exhibited variations between the two groups (Supplementary Table S1). For example, for rs406747332, the seropositive animals had a higher frequency of CC homozygous genotype (49%) than seronegative individuals, mostly heterozygous (55%). A total of two candidate genes (Table 1) were found in the surroundings (200 kb upstream and 200 kb downstream) of the markers identified with the three approaches.
4 Discussion
In this study, genome-wide approaches have been used to identify candidate genomic regions potentially involved with the resistance to VMV infection in the Valle del Belice sheep breed.
The serological tests showed an average seroprevalence of 26% in the Valle del Belice sheep breed. The seroprevalence presence is an important aspect, since that in herds with very low prevalence values, negative animals could be false negative due to their lower probability of infection. Consistent with our results, previous studies reported similar seroprevalence in other Italian sheep breeds (Moretti et al., 2022; Tumino et al., 2022).
The genetic involvement in susceptibility to VMV has been suggested by the breed differences reported in the literature. However, mutations in the gene TMEM154 have been consistently associated with VMV susceptibility (Heaton et al., 2012); unfortunately, those mutations are not associated with complete resistance. White et al. (2012) conducted a GWAS on various sheep breeds, validating the findings concerning the TMEM154 gene. Additionally, the authors identified twelve other genomic regions that could potentially be linked to the infection. Recently, Letko et al. (2021) reported no association between genotypes at the TMEM154 gene and VMV susceptibility in the Valais Blacknose sheep, showing that this gene does not affect individual susceptibility to infection in this breed. Also, Ramírez et al. (2021) suggested that the relationship between TMEM154 E35K genotyping and SRLV susceptibility patterns could be breed-specific and reported a positive association only in one of the four investigated sheep breeds. In addition, the high frequency of serologically positive animals (55%) carrying the less susceptible genotype KK in the Merinoland breed reported by Molaee et al. (2019) implies that the correlation between the TMEM154 E35K and disease susceptibility in sheep needs to be approached with caution. This suggests the need for further studies to identify other genetic factors involved in developing VMV infection. Indeed, this is the first genome-wide study using a high-density SNP array aimed to identify additional candidate genomic regions potentially involved with susceptibility to VMV infection in an Italian sheep breed.
Unravelling the genetic basis of a complex trait, such as the resistance to diseases, requires careful and rigorous analysis. Based on the increased analytical power in reducing the number of false positive signals when multiple methodologies are adopted in parallel (Mastrangelo et al., 2018), this study used three distinct approaches in conjunction with the use of high-density SNP data. Despite the moderate sample size of our study, like previous GWASs in small ruminants (Mastrangelo et al., 2018; Cecchi et al., 2019; Lei et al., 2020; Letko et al., 2021; Minozzi et al., 2023), this combined approach can boost the detection accuracy and reduce unknown biases (Schiavo et al., 2018). The identification of candidate genomic regions by more than one methodology may be seen as strong evidence of the activity on a particular phenotype. This also allowed to exclude other regions that reached or were close to the defined thresholds derived by several factors that could not be better managed (i.e., genetic drift, population structure, ascertain bias of the SNP chip tool) (Schiavo et al., 2018). Therefore, the present study is sufficiently powered to find genome-wide significant SNPs related with VMV susceptibility under the hypothesis that we had ewes sampled from the same breed and there was no phenotype misclassification. Among positive and negative serological individuals, no homozygous resistant KK genotypes at the TMEM154 gene were identified in the breed. Therefore, on the analyzed sample, this gene would not seem to affect the individual susceptibility to infection.
The results from the three different approaches highlighted the presence of shared genomic regions on OAR03 and OAR05 that are potentially associated with the observed phenotypic differences (Table 1). In fact, only two SNPs were identified associated with the analyzed trait and genome-wide approaches with few outlier markers, and in the absence of other signals, can be considered as biologically relevant (Kijas et al., 2013; Mastrangelo et al., 2019). Linkage disequilibrium between the two SNPs showed a moderate value (∼0.1). This agreed with previous studies that reported from moderate to small r2 value for non-syntenic pairs. For example, in cattle, several authors noted that all r2 values for non-syntenic pairs were very small (generally a low percentage has a r2 value of more than 0.1) (Khatkar et al., 2008; Beghain et al., 2013).
One of the two significant markers (rs402241447) was located near the GRIN2B gene, whereas the other SNP located on OAR05 (rs402241447) was close to the TMEM232 gene. These markers were not identified in previous studies as associated with the investigated trait. However, there is some correspondence between the results of this study and previous GWAS in sheep, as the best-associated SNP on chromosome nine at 69843937 bp, identified by Letko et al. (2021) was near to one of the six markers identified in this study using FST analysis (Table 1). The GRIN2B gene plays a role in receptor clustering, synaptic transmission, and pain sensitivity. The protein encoded by this gene is a NMDA receptor channel subunit for neuronal communication (Paoletti et al., 2013). Hu et al. (2016) identified the GRIN2B gene as potentially involved in the neurodevelopmental disorders in humans. In livestock, this gene is associated with regulating calcium channels, which are essential for several biological functions, including vasoconstriction (Witt and Huerta-Sánchez, 2019). An epigenome-wide association research found that the differentially methylated areas of the TMEM232 gene are correlated with multiple sclerosis in humans (Souren et al., 2019). Changes in the same gene have also been associated with allergic rhinitis (Ramasamy et al., 2011). A possible involvement of the TMEM232 gene in the development of immune diseases has also been suggested (Han et al., 2023). In cattle, it was reported as a differentially expressed gene between lactation stages (Bhat et al., 2019), and involved in behavior traits (Friedrich et al., 2016). To our knowledge, these genes have not previously been associated with lentiviral infection in any species. In a previous GWAS, White et al. (2012) reported different candidate genes, but as in our study, those genes were not previously identified. After all, before the study of Heaton et al. (2012), even the function of the major gene TMEM154 had never been reported for any species. In fact, a previous study identified TMEM154 in a human GWAS for asthma severity (Slager et al., 2011), suggesting that this gene may play a role in immune responses, with a function similar to the TMEM232 here identified as a new potential candidate gene. Moreover, it is noteworthy that both genes belonging to the TMEM family, code for proteins that are predicted to be components of diverse cell membranes, such as mitochondrial, endoplasmic reticulum, lysosome, and Golgi membranes. TMEMs are widely present across different cell types and play key roles in essential functions such as epidermal keratinization, autophagy, and smooth muscle contraction. Additionally, certain TMEM proteins are critical in immune response, and they are key components of inflammatory signaling pathways, facilitating the production of pro-inflammatory cytokines (Schmit and Michiels, 2018).
5 Conclusion
Genetic selection of resistant animals is of particular interest in viral diseases lacking effective treatments or vaccines. Our study suggests new aspects of the genetic complexity related to the resistance/susceptibility to SRLV in sheep species. Moreover, it describes for the first time the involvement in susceptibility/resistance to VMV of some genes never identified before, confirming that studies on different breeds can lead to different results. The ideal approach for validation of the markers identified in our study is to use samples from a population independent from the discovery population with the same phenotype used in the discovery stage. Additional analysis based on sequencing and increased number of genotyped sheep would help confirm our findings, refine these results, and better understand the genetic basis of VMV.
Data availability statement
The data presented in the study are deposited in the figshare repository, accession number 10.6084/m9.figshare.25858267. This data can be found here: https://doi.org/10.6084/m9.figshare.25858267.v1
Ethics statement
The animal study was approved by The animal study protocol was approved by the Bioethics Committee of the University of Palermo: protocol code UNPA-CLE–98597, in agreement with the recommendations of the European Union (EU) Directive 2010/63/EU to ensure appropriate animal care. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
SR: Formal Analysis, Investigation, Writing–review and editing. MT: Data curation, Formal Analysis, Investigation, Writing–review and editing. GS: Formal Analysis, Investigation, Visualization, Writing–review and editing. ST: Data curation, Investigation, Visualization, Writing–review and editing. BP: Funding acquisition, Supervision, Visualization, Writing–review and editing. AS: Investigation, Methodology, Visualization, Writing–review and editing. MS: Data curation, Funding acquisition, Investigation, Resources, Visualization, Writing–review and editing. AC: Data curation, Formal Analysis, Investigation, Methodology, Visualization, Writing–review and editing. SM: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Resources, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was financed by LEO: Livestock Environment Opendata, PSRN 2014-2020 -Sottomisura: 16.2, Project number PRJ-0185, CUP: J84I18000090007.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1376883/full#supplementary-material
Supplementary Table S1 | Genotypic frequencies of the best associated markers for case/control samples.
Supplementary Figure S2 | Multidimensional scaling (MDS) plot of seropositive (red) and seronegative (blue) individuals.
References
Abendaño, N., Esparza-Baquer, A., Bernales, I., Reina, R., de Andrés, D., and Jugo, B. M. (2021). Gene expression profiling reveals new pathways and genes associated with Visna/Maedi viral disease. Animals 11 (6), 1785. doi:10.3390/ani11061785
Arcangeli, C., Lucarelli, D., Torricelli, M., Sebastiani, C., Ciullo, M., Pellegrini, C., et al. (2021). First survey of SNPs in TMEM154, TLR9, MYD88 and CCR5 genes in sheep reared in Italy and their association with resistance to SRLVs infection. Viruses 13 (7), 1290. doi:10.3390/v13071290
Aulchenko, Y. S., Ripke, S., Isaacs, A., and Van Duijn, C. M. (2007). GenABEL: an R library for genome-wide association analysis. Bioinformatics 23 (10), 1294–1296. doi:10.1093/bioinformatics/btm108
Beghain, J., Boitard, S., Weiss, B., Boussaha, M., Gut, I., and Rocha, D. (2013). Genome-wide linkage disequilibrium in the Blonde d'Aquitaine cattle breed. J. Anim. Breed. Genet. 130 (4), 294–302. doi:10.1111/j.1439-0388.2012.01020.x
Benavides, J., Fuertes, M., García-Pariente, C., Otaola, J., Delgado, L., Pérez, V., et al. (2013). Impact of maedi-visna in intensively managed dairy sheep. Veterinary J. 197, 607–612. doi:10.1016/j.tvjl.2013.03.031
Bhat, S. A., Ahmad, S. M., Ibeagha-Awemu, E. M., Bhat, B. A., Dar, M. A., Mumtaz, P. T., et al. (2019). Comparative transcriptome analysis of mammary epithelial cells at different stages of lactation reveals wide differences in gene expression and pathways regulating milk synthesis between Jersey and Kashmiri cattle. PLoS One 14, e0211773. doi:10.1371/journal.pone.0211773
Cecchi, F., Dadousis, C., Bozzi, R., Fratini, F., Russo, C., Bandecchi, P., et al. (2019). Genome scan for the possibility of identifying candidate resistance genes for goat lentiviral infections in the Italian Garfagnina goat breed. Trop. Anim. Health Prod. 51, 729–733. doi:10.1007/s11250-018-1728-y
Chang, C. C., Chow, C. C., Tellier, L. C., Vattikuti, S., Purcell, S. M., and Lee, J. J. (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4 (1), 7–015. doi:10.1186/s13742-015-0047-8
de Miguel, R., Arrieta, M., Rodríguez-Largo, A., Echeverría, I., Resendiz, R., Pérez, E., et al. (2021). Worldwide prevalence of small ruminant lentiviruses in sheep: a systematic review and meta-analysis. Animals 11 (3), 784. doi:10.3390/ani11030784
de Pablo-Maiso, L., Doménech, A., Echeverría, I., Gómez-Arrebola, C., De Andrés, D., Rosati, S., et al. (2018). Prospects in innate immune responses as potential control strategies against non-primate lentiviruses. Viruses 10, 435. doi:10.3390/v10080435
Friedrich, J., Brand, B., Ponsuksili, S., Graunke, K. L., Langbein, J., Knaust, J., et al. (2016). Detection of genetic variants affecting cattle behaviour and their impact on milk production: a genome-wide association study. Anim. Genet. 47, 12–18. doi:10.1111/age.12371
Gibson, L., Dun, K., and Baird, A. (2018). The true cost of maedi visna virus (MVV) infection in a commercial flock. Livestock 23, 238–243. doi:10.12968/live.2018.23.5.238
González, J. R., Armengol, L., Solé, X., Guinó, E., Mercader, J. M., Estivill, X., et al. (2007). SNPassoc: an R package to perform whole genome association studies. Bioinformatics 23 (5), 644–645. doi:10.1093/bioinformatics/btm025
Han, J., Cai, X., Qin, S., Zhang, Z., Wu, Y., Shi, Y., et al. (2023). TMEM232 promotes the inflammatory response in atopic dermatitis via NF-κB and STAT3 signaling pathways. Br. J. Dermatol 189, 195–209. doi:10.1093/bjd/ljad078
Heaton, M. P., Clawson, M. L., Chitko-Mckown, C. G., Leymaster, K. A., Smith, T. P., Harhay, G. P., et al. (2012). Reduced lentivirus susceptibility in sheep with TMEM154 mutations. PLoS Genet. 8, e1002467. doi:10.1371/journal.pgen.1002467
Hu, C., Chen, W., Myers, S. J., Yuan, H., and Traynelis, S. F. (2016). Human GRIN2B variants in neurodevelopmental disorders. J. Pharmacolol. Sci. 132, 115–121. doi:10.1016/j.jphs.2016.10.002
Illius, A. W., Lievaart-Peterson, K., McNeilly, T. N., and Savill, N. J. (2020). Epidemiology and control of maedi-visna virus: curing the flock. PLoS One 15 (9), e0238781. doi:10.1371/journal.pone.0238781
Juste, R. A., Villoria, M., Leginagoikoa, I., Ugarte, E., and Minguijon, E. (2020). Milk production losses in Latxa dairy sheep associated with small ruminant lentivirus infection. Prev. Vet. Med. 176, 104886. doi:10.1016/j.prevetmed.2020.104886
Kalogianni, A. I., Bossis, I., Ekateriniadou, L. V., and Gelasakis, A. I. (2020). Etiology, epizootiology and control of maedi-visna in dairy sheep: a review. Animals 10, 616. doi:10.3390/ani10040616
Keen, I. E., Hungerford, L. L., Wittum, T. E., Kwang, J., and Littledike, E. T. (1997). Risk factors for seroprevalence of ovine lentivirus in breeding Ewe flocks in Nebraska, USA. Prev. Vet. Med. 30, 81–94. doi:10.1016/s0167-5877(96)01121-x
Khatkar, M. S., Nicholas, F. W., Collins, A. R., Zenger, K. R., Cavanagh, J. A., Barris, W., et al. (2008). Extent of genome-wide linkage disequilibrium in Australian Holstein-Friesian cattle based on a high-density SNP panel. BMC Genomics 9 (1), 187–218. doi:10.1186/1471-2164-9-187
Kijas, J. W., Serrano, M., McCulloch, R., Li, Y., Salces Ortiz, J., Calvo, J. H., et al. (2013). Genome wide association for a dominant pigmentation gene in sheep. J. Anim. Breed. Genet. 130, 468–475. doi:10.1111/jbg.12048
Larruskain, A., Minguijón, E., García-Etxebarria, K., Moreno, B., Arostegui, I., Juste, R. A., et al. (2010). MHC class II DRB1 gene polymorphism in the pathogenesis of Maedi–Visna and pulmonary adenocarcinoma viral diseases in sheep. Immunogenetics 62, 75–83. doi:10.1007/s00251-009-0419-2
Lei, H., Yang, T., Mahmood, S., Abo-Ismail, M., Roy, B. C., Li, C., et al. (2020). A genome-wide case-control association study of dark cutting in beef cattle. Can. J. Animal Sci. 101 (1), 158–167. doi:10.1139/cjas-2019-0039
Letko, A., Bützberger, C., Hirter, N., Paris, J. M., Abril, C., and Drögemüller, C. (2021). Genetic evaluation of small ruminant lentivirus susceptibility in Valais blacknose sheep. Anim. Genet. 52, 781–782. doi:10.1111/age.13108
Mastrangelo, S., Sottile, G., Sardina, M. T., Sutera, A. M., Tolone, M., Di Gerlando, R., et al. (2019). A combined genome-wide approach identifies a new potential candidate marker associated with the coat color sidedness in cattle. Livest. Sci. 225, 91–95. doi:10.1016/j.livsci.2019.05.009
Mastrangelo, S., Sottile, G., Sutera, A. M., Di Gerlando, R., Tolone, M., Moscarelli, A., et al. (2018). Genome-wide association study reveals the locus responsible for microtia in Valle del Belice sheep breed. Anim. Genet. 49, 636–640. doi:10.1111/age.12719
Minguijón, E., Reina, R., Pérez, M., Polledo, L., Villoria, M., Ramírez, H., et al. (2015). Small ruminant lentivirus infections and diseases. Vet. Microbiol. 181, 75–89. doi:10.1016/j.vetmic.2015.08.007
Minozzi, G., De Iorio, M. G., Palazzo, F., Gandini, G., Biffani, S., Paolillo, G., et al. (2023). Genome-wide association study for antibody response to Mycobacterium avium ssp. paratubeculosis in goats. Anim. Genet. 54 (1), 78–81. doi:10.1111/age.13271
Molaee, V., Eltanany, M., and Lühken, G. (2018). First survey on association of TMEM154 and CCR5 variants with serological maedi-visna status of sheep in German flocks. Vet. Res. 49, 36–10. doi:10.1186/s13567-018-0533-y
Molaee, V., Otarod, V., Abdollahi, D., and Lühken, G. (2019). Lentivirus susceptibility in Iranian and German sheep assessed by determination of TMEM154 E35K. Animals 9, 685. doi:10.3390/ani9090685
Moretti, R., Sartore, S., Colitti, B., Profiti, M., Chessa, S., Rosati, S., et al. (2022). Susceptibility of different TMEM154 genotypes in three Italian sheep breeds infected by different SRLV genotypes. Vet. Res. 53, 60–67. doi:10.1186/s13567-022-01079-0
Paoletti, P., Bellone, C., and Zhou, Q. (2013). NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 14, 383–400. doi:10.1038/nrn3504
Peterhans, E., Greenland, T., Badiola, J., Harkiss, G., Bertoni, G., Amorena, B., et al. (2004). Routes of transmission and consequences of small ruminant lentiviruses (SRLVs) infection and eradication schemes. Vet. Res. 35, 257–274. doi:10.1051/vetres:2004014
Preziuso, S., Renzoni, G., Allen, T. E., Taccini, E., Rossi, G., Demartini, J. C., et al. (2004). Colostral transmission of maedi visna virus: sites of viral entry in lambs born from experimentally infected ewes. Vet. Microbiol. 104, 157–164. doi:10.1016/j.vetmic.2004.09.010
Ramasamy, A., Curjuric, I., Coin, L. J., Kumar, A., McArdle, W. L., Imboden, M., et al. (2011). A genome-wide meta-analysis of genetic variants associated with allergic rhinitis and grass sensitization and their interaction with birth order. J. Allergy Clin. Immunol. 128, 996–1005. doi:10.1016/j.jaci.2011.08.030
Ramírez, H., Echeverría, I., Benito, A. A., Glaria, I., Benavides, J., Pérez, V., et al. (2021). Accurate diagnosis of small ruminant lentivirus infection is needed for selection of resistant sheep through TMEM154 E35K genotyping. Pathogens 10, 83. doi:10.3390/pathogens10010083
Riggio, S., Di Gerlando, R., Mastrangelo, S., Rizzuto, I., Tolone, M., and Sardina, M. T. (2023). Maedi Visna virus infection and TMEM154 genotypes in Valle del Belìce sheep breed. Ital. J. Anim. Sci. 22 (1), 754–759. doi:10.1080/1828051x.2023.2243095
Rodrigues, C. S., de Faria, D. A., Lacerda, T. S. A., Paiva, S. R., Caetano, A. R., Blackburn, H., et al. (2023). Lentivirus susceptibility in Brazilian and US sheep with TMEM154 mutations. Genes 14, 70. doi:10.3390/genes14010070
Sarafidou, T., Stamatis, C., Kalozoumi, G., Spyrou, V., Fthenakis, G. C., Billinis, C., et al. (2013). Toll like Receptor 9 (TLR9) polymorphism G520R in sheep is associated with seropositivity for small ruminant lentivirus. PLoS ONE 8, e63901. doi:10.1371/journal.pone.0063901
Schiavo, G., Bertolini, F., Utzeri, V. J., Ribani, A., Geraci, C., Santoro, L., et al. (2018). Taking advantage from phenotype variability in a local animal genetic resource: identification of genomic regions associated with the hairless phenotype in Casertana pigs. Anim. Genet. 49, 321–325. doi:10.1111/age.12665
Schmit, K., and Michiels, C. (2018). TMEM proteins in cancer: a review. Front. Pharmacol. 9, 1345. doi:10.3389/fphar.2018.01345
Slager, R. E., Li, X., Meyers, D. A., and Bleecker, E. R. (2011). “Recent developments in the genetics of asthma susceptibility and severity.” in Difficult-to-Treat Severe Asthma, Editors Chung K.F., Bel E.H., and Wenzel S.E. (Plymouth, UK: European Respiratory Society), 82–96.
Souren, N. Y., Gerdes, L. A., Lutsik, P., Gasparoni, G., Beltrán, E., Salhab, A., et al. (2019). DNA methylation signatures of monozygotic twins clinically discordant for multiple sclerosis. Nat. Commun. 10, 2094. doi:10.1038/s41467-019-09984-3
Straub, O. C. (2004). Maedi–Visna virus infection in sheep. History and present knowledge. Comp. Immunol. Microbiol. Infect. Dis. 27 (1), 1–5. doi:10.1016/S0147-9571(02)00078-4
Tumino, S., Tolone, M., Galluzzo, P., Migliore, S., Sechi, T., Bordonaro, S., et al. (2022). Alternative molecular tools for the fight against infectious diseases of small ruminants: native Sicilian sheep breeds and maedi-visna genetic susceptibility. Animals 12, 1630. doi:10.3390/ani12131630
White, S. N., Mousel, M. R., Herrmann-Hoesing, L. M., Reynolds, J. O., Leymaster, K. A., Neibergs, H. L., et al. (2012). Genome-wide association identifies multiple genomic regions associated with susceptibility to and control of ovine lentivirus. PLoS ONE 7, e47829. doi:10.1371/journal.pone.0047829
White, S. N., Mousel, M. R., Reynolds, J. O., Herrmann-Hoesing, L. M., and Knowles, D. P. (2014). Deletion variant near ZNF389 is associated with control of ovine lentivirus in multiple sheep flocks. Anim. Genet. 45, 297–300. doi:10.1111/age.12107
White, S. N., Mousel, M. R., Reynolds, J. O., Lewis, G. S., and Herrmann-Hoesing, L. M. (2009). Common promoter deletion is associated with 3.9-fold differential transcription of ovine CCR5 and reduced proviral level of ovine progressive pneumonia virus. Anim. Genet. 40, 583–589. doi:10.1111/j.1365-2052.2009.01882.x
Witt, K. E., and Huerta-Sánchez, E. (2019). Convergent evolution in human and domesticate adaptation to high-altitude environments. Philos. Trans. R. Soc. B 374, 20180235. doi:10.1098/rstb.2018.0235
Yaman, Y., Keles, M., Aymaz, R., Sevim, S., Sezenler, T., Onaldi, T., et al. (2019). Association of TMEM154 variants with visna/maedi virus infection in Turkish sheep. Small Rumin. Res. 177, 61–67. doi:10.1016/j.smallrumres.2019.06.006
Keywords: small ruminant lentiviruses, local sheep, genome-wide approaches, high density array, candidate genes
Citation: Riggio S, Tolone M, Sottile G, Tumino S, Portolano B, Sutera AM, Sardina MT, Cesarani A and Mastrangelo S (2024) A high-density genome-wide approach reveals novel genetic markers linked to small ruminant lentivirus susceptibility in sheep. Front. Genet. 15:1376883. doi: 10.3389/fgene.2024.1376883
Received: 26 January 2024; Accepted: 10 May 2024;
Published: 06 June 2024.
Edited by:
Kefei Chen, Curtin University, AustraliaReviewed by:
Stefano Biffani, National Research Council (CNR), ItalyAlfredo Pauciullo, University of Turin, Italy
Copyright © 2024 Riggio, Tolone, Sottile, Tumino, Portolano, Sutera, Sardina, Cesarani and Mastrangelo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Salvatore Mastrangelo, c2FsdmF0b3JlLm1hc3RyYW5nZWxvQHVuaXBhLml0