 Ruolin Li
Ruolin Li Jiayi Yang
Jiayi Yang Jinfeng Ma
Jinfeng Ma Aimei Zhang
Aimei Zhang Hongfang Li
Hongfang Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 09 April 2024
Sec. Neurogenomics
Volume 15 - 2024 | https://doi.org/10.3389/fgene.2024.1373448
Introduction: Nuclear undecaprenyl pyrophosphate synthase 1 (NUS1) gene variants are associated with a range of phenotypes, including epilepsy, intellectual disability, cerebellar ataxia, Parkinson’s disease, dystonia, and congenital disorders of glycosylation. Additionally, cases describing genotypes and clinical features are rare.
Case Presentation: Herein, we report the case of a 23-year-old Chinese female patient who presented with tremors, intellectual disability, and epilepsy. A history of carbon monoxide exposure, brain trauma, or encephalitis was not present in this case. Trio whole-exome sequencing analysis revealed a de novo pathogenic variant of c.750del in exon 4, leading to p.Leu251* amino acid substitution. Genetic analysis failed to identify the identical mutations in the remaining family members who underwent screening. The patient was diagnosed with a rare congenital disease, “congenital glycosylation disorder, type 1aa, autosomal dominant, type 55, with seizures (MRD-55).”
Conclusion: We provide further evidence for the role of variants in NUS1 in the development of tremors, epilepsy, and intellectual disabilities. These findings expand our understanding of the clinical phenotypes of NUS1 variants.
Nuclear undecaprenyl pyrophosphate synthase 1 (NUS1) gene encodes the precursor of the N-terminal membrane of the Nogo-B receptor (NgBR) (Grabińska et al., 2017; Den et al., 2019; Riboldi et al., 2022), which acts as an essential subunit of the dehydrodolichyl diphosphate synthase (DHDDS) complex and plays a crucial role in maintaining glycosylation in mammals (Harrison et al., 2011; Park et al., 2014; Giladi et al., 2022).
The pathogenic variants of NUS1 were first identified in a family with a congenital disorder of glycosylation (CDG) (Park et al., 2014; Jaeken and Péanne, 2017). To date, 29 pathogenic or likely pathogenic mutations of NUS1 have been reported worldwide, 18 of which have traceable clinical phenotypes (according to the Human Mutation database) (Hu et al., 2023). The spectrum of phenotypes includes epilepsy, cerebellar ataxia, tremors, cortical myoclonus, intellectual disability, and psychomotor retardation (Ji et al., 2023). These variants are associated with dystonia, Parkinson’s disease, developmental and epileptic encephalopathy, and CDG (Monfrini et al., 2022). Therefore, different pathogenic variants may cause various diseases. Furthermore, cases describing genotypes and detailed clinical features are limited. Here, we report a case of a patient with a novel de novo pathogenic variant of the NUS1 gene (c.750del, p.Leu251*) who presented with tremors, epilepsy, and intellectual disability.
Our patient, a 23-year-old woman, was the second child born to nonconsanguineous healthy parents without any family history of intellectual disability or epilepsy. Her older sister is healthy. She was born spontaneously at full term without dystocia and had no history of carbon monoxide exposure, brain trauma, or encephalitis.
At the age of 6 years, it was noticed that she was intellectual disability, although her physical development (weight and height) was normal. She could walk and speak, and was not receive any medical intervention. At the age of 8 years, she presented with involuntary shaking and tremors of the head and neck, which spread to all her limbs, and were accompanied by an involuntary nod. The symptoms worsened during mood fluctuations. During this period, the patient was conscious with no binocular upward gaze, perioral bruising, or limb stiffness. She underwent brain magnetic resonance imaging (MRI) and electroencephalography (EEG). Brain MRI showed a left choroidal cyst and compression of the hippocampus.
On the basis of extensive slow waves of two–7 Hz, spike waves, sharp waves, and sharp-and-slow waves were observed in the bilateral temporal, parietal, and occipital lobes. Routine blood tests revealed no significant abnormalities. The possibility of epilepsy could not be excluded. The patient was treated with levetiracetam (500 mg, twice daily) and sodium valproate (500 mg, twice daily), which resulted in a slight improvement in the limb tremors. During this period, the patient experienced convulsions four times during quiet sleep; however, her family was unable to describe the symptoms in detail. Subsequently, it was decided to maintain the patient’s current treatment with only a dose adjustment.
When the patient was 22 years old, she developed a generalized tremor due to fever, which could not be well controlled with the previous medications. She also walked unsteadily, sometimes stood unsteadily, and had intermittent blurred vision. Complete neurological examination showed generalized tremors, especially in the head and neck region, which were aggravated during activity but decreased at rest. Her limb muscle tone was increased, and ataxia was present. Intellectual disability was also observed. The brain MRI epilepsy sequence showed an abnormal signal near the temporal horn of the left lateral ventricle. A choroid plexus cyst and compression of the left hippocampus were considered (Figure 1). This was similar to the MRI findings obtained 8 years before. EEG revealed fast waves and low-to-moderate amplitude slow waves during wakefulness, poor regulation of the amplitude and wave rate, but with abnormal patterns during sleep. Blood and urine tests revealed no lactic acid, pyruvic acid, vitamin B12, thyroid function, ceruloplasmin, copper, or lipoprotein abnormalities. Trio whole-exome sequencing (WES) and mitochondrial gene sequencing were performed, and a novel NUS1 variant, c.750del (p.Leu251*), was identified. Clonazepam (0.25mg, twice daily) and levetiracetam (500 mg, twice daily) were remarkably effective in controlling the patient’s tremors and unsteady gait.
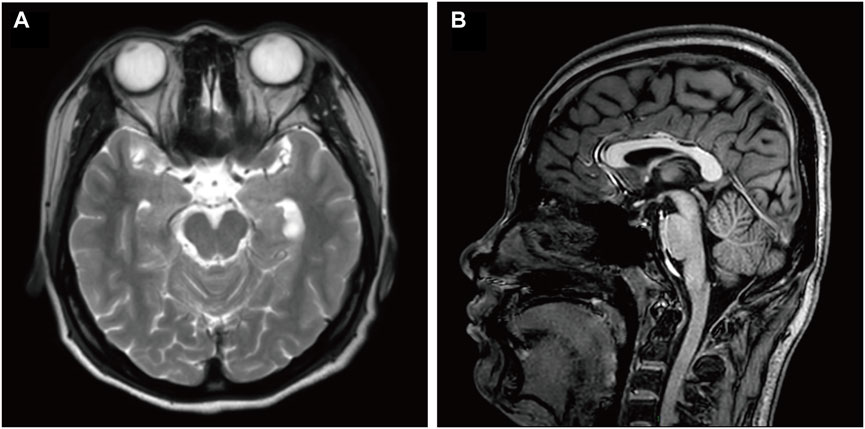
Figure 1. Brain MRI of the described case. (A) Axial T2 and (B) sagittal T1 brain MRI images of this patient, at an age of 22 years. Image A shows a compressed left hippocampus. Image B shows no cerebellar atrophy. MRI, magnetic resonance imaging; FLAIR, fluid-attenuated inversion recovery.
About 6 months later, the patient presented with whole-body tremors. She was unable to stand upright or walk. Before symptom development, she reported experiencing fatigue for several days. The physical examination results were comparable to those of the previous examinations. Haloperidol (1 mg, twice daily) and levodopa/benserazide (0.0625g, three times a day) were prescribed, which reduced the whole-body tremors. However, 5 months later, the body tremors recurred, sodium valproate and arotinolol hydrochloride were added to the regimen to control the tremors. Unfortunately, the patient continued to exhibit an unsteady gait, slow and slurred communication, and poor numeracy skills, along with deficiencies in logic and comprehension.
Peripheral blood samples from the patient and her parents were collected for WES and mitochondrial genetic tests to identify the causal gene. WES results revealed a de novo variant of NUS1 (c.750del) (Figure 2) and a novel variant of UBTF (c.2026-3A>G) (Figure 3). Mitochondrial gene testing yielded negative results. The c.750del variant of NUS1 gene was an exon-deletion variant, and was not documented in the NHLBI Exome Sequencing Project, 1000G, and Exome Aggregation Consortium databases. Family-based WES confirmed that the c.750del variant was a de novo variant in this case. So according to the pathogenicity classification guidelines of the American College of Medical Genetics and Genomics (ACMG), the c.750del variant of NUS1 gene is classified as likely pathogenic (very strong evidence of pathogenicity [PVS1], strong evidence of pathogenicity [PS2] and moderate evidence of pathogenicity [PM2]). Moreover, a novel variant (c.2026-3A>G) of the UBTF gene on chromosome chr17:42284968 was also identified in our patient. Sequence reads were aligned against the HGMD Pro, PubMed, and ClinVar databases, and this variant had not yet been reported in the medical literature. The UBTF gene also revealed a heterozygous variant in the patient’s father. According to the pathogenicity classification guidelines of the ACMG, there is uncertain significance. Although the clinical features (epilepsy and intellectual disability) of our patient matched the phenotype of UBTF gene-related diseases, it is noteworthy that her father also carries the same variant without exhibiting any associated phenotype. This observation suggests that either the variant does not have significant clinical implications or that there may be a reduced penetrance of this variant in the family.
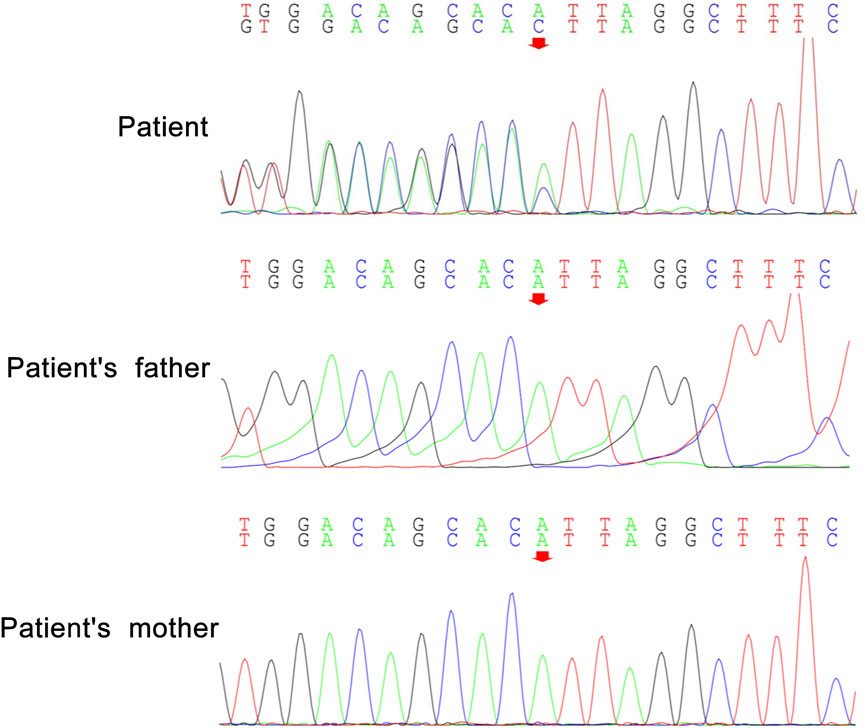
Figure 2. De novo heterozygous variants of NUS1 (c.750del) in our patient.
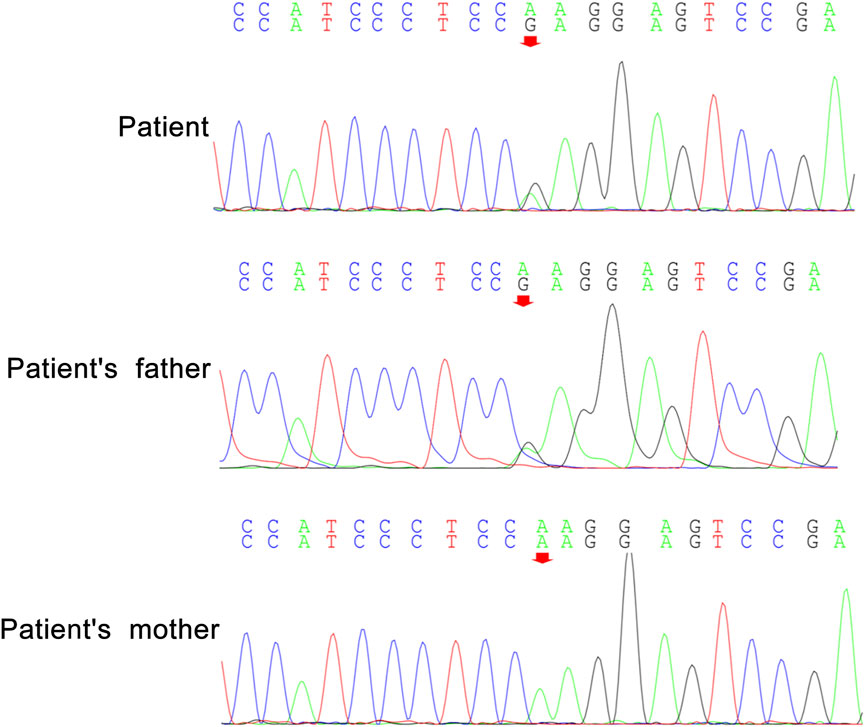
Figure 3. A novel variant c.2026-3A>G of UBTF in our patient.
The NUS1 gene is located on chromosome 6q22.1 (NM_138,459.4 and NP_612468.1) and encodes NgBR, with a length of 293 amino acids (Harrison et al., 2011; Szafranski et al., 2015). As a subunit of cis-prenyltransferase, NgBR interacts with DHDDS to promote isoprenyltransferase activity (Harrison et al., 2011; Park et al., 2014; Giladi et al., 2022). The absence of epilepsy and ataxia are more common in the 6q22 mutation than in the 6q16 and 6q21 mutations (Araki et al., 2020). The NUS1 mutation in 6q22, as a non-ion channel gene, may contribute to the pathogenesis of epilepsy and present as a syndrome characterized by cerebellar ataxia and tremors. Herein, we report a patient presented with tremors, epilepsy, and intellectual disability, attributed to NUS1 variant.
A novel variant c.750del (p.Leu251*) of NUS1 (NM_138,459) on chromosome chr6:118024826 was identified in our patient via WES analysis, and it has not been reported in previous cases with heterozygous NUS1 gene mutations. A novel heterozygous deletion in exon four leads to premature protein termination. The genetic variation site located in the C-terminal structural domain of NgBR is essential for its interaction with the DHDSS. Due to the C-terminal transformation, the NgBR is unable to interact with DHDDS or the substrates, limiting the enzyme activity of the cis-prenyltransferase and negatively affecting its functionality, ultimately leads to tremors, epilepsy, intellectual disability, and ataxia (Harrison et al., 2011). The majority of the pathogenic variants of NUS1 are severe deleterious mutations (frameshift, stop, splice-disruptive, exon deletions, and chromosomal deletions), resulting in the loss of complete proteins (Araki et al., 2020). The pathogenic features of the identified nonsense mutation consistent with the previously documented haploinsufficiency mechanism associated with this gene (Araki et al., 2020). Further experiments are needed to confirm this observation, and the potential correlation between the genotypic phenotype and the clinical phenotype of this novel variant necessitates further investigation.
Since the first identification of a NUS1 mutation in 2014, a total of 29 cases with confirmed causative mutations in NUS1 have been reported, eighteen of which have detailed clinical phenotypes (Park et al., 2014; Riboldi et al., 2022; Hu et al., 2023). NUS1 variants are rare, and result in various clinical phenotypes, include CDG, MRD55, Parkinson’s disease, dystonia and others (Ji et al., 2023). A comparative analysis was conducted on the symptoms exhibited in 18 previously reported cases of NUS1 mutations and those observed in our case study, and found varying degrees of overlap in phenotypes. Over half of the cases demonstrated symptoms consistent with those described in our case, including intellectual disability, cerebellar ataxia, tremors, and epileptic seizures. Fever-induced seizures and other symptoms were less common, affecting only two individuals (Hu et al., 2023). The majority of the cases exhibited normal muscular tone, only two patients showed hypotonia. Of note, 84.2% of cases were diagnosed with epilepsy-related disorders, indicating a significant association between NUS1 variants and epilepsy. In the majority of patients with tremors accompanied by epilepsy and cerebellar ataxia syndrome, MRI has revealed varying degrees of cerebellar atrophy (Den et al., 2019). The brain MRI epilepsy sequences of our patient revealed compression of the left hippocampus. The hippocampus is primarily responsible for short-term memory storage, conversion, and orientation. Indeed, our patient also presented with learning and memory impairment. Although our patient did not show significant cerebellar atrophy on MRI, she did show the clinical phenotype of cerebellar ataxia. This difference may be associated with genetic variations at different gene sites.
Considering the epileptic seizures, intellectual disability, tremors, and identified pathogenic NUS1 variant, the patient was diagnosed with “congenital glycosylation disorder, type 1aa, autosomal dominant, type 55, with seizures (MRD-55)”, which is a rare congenital disease. It was first reported in three unrelated patients who presented with myoclonic seizures and harbored heterozygous de novo pathogenic variants of the NUS1 gene (Zhang et al., 2021). A study of five familial patients with epilepsy, cerebellar ataxia, and tremors showed a novel heterozygous NUS1 variant (Hamdan et al., 2017; Zhang et al., 2021). All individuals exhibited cerebellar ataxia and tremors, with three presented with intellectual disabilities but no seizures, one showed generalized epilepsy, and one presented parkinsonism without epilepsy.
Based on previous reports on the treatment of patients with NUS1 gene mutations, oxcarbazepine has been shown to improve speech and motor functions. Furthermore, baclofen demonstrated a notable reduction in myoclonus and a modest improvement in gait disturbances in the patient (Den et al., 2019; Zhang et al., 2021). Valproate has demonstrated efficacy in managing epilepsy and tremors in patients with NUS1 mutations in comparison with a control (Haginoya et al., 2021). In a patient who presented with bradykinesia and resting tremors, zonisamide treatment effectively controlled the symptoms; nevertheless, the symptoms were levodopa-resistant (Araki et al., 2020). Our patient was administered with levetiracetam, clonazepam, levodopa, and benserazide hydrochloride. Her symptoms were alleviated and effectively controlled throughout the follow-up phase.
At present, the prevalence of cases exhibiting NUS1 pathogenic variants remains limited, and the absence of thorough documentation and characterization for a substantial proportion of these cases, hinders the delineation of a definitive genotype-phenotype correlation, especially given the wide spectrum of diseases associated with NUS1 pathogenic variants. Otherwise, a clear pattern of drug efficacy is not apparent in the cases that underwent drug treatment. The precise molecular mechanisms by which NUS1 pathogenic variants contribute to the manifestation of various diseases in affected patients remain unclear. Additional in-depth case studies are necessary to clarify the potential correlation between NUS1 variants and disease progression. Further investigation and functional analyses are required to improve understanding of the NUS1 gene and its related disorders.
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by The Ethics Committee of Affiliated Hospital of Jining Medical University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
RL: Investigation, Writing–original draft, Writing–review and editing. JY: Writing–review and editing, Data curation. JM: Data curation, Writing–review and editing. AZ: Data curation, Writing–review and editing. HL: Funding acquisition, Project administration, Supervision, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (grant number: 82301429), Key Research and Development Plan of Jining City (grant number: 2022YXNS051), PhD Research Foundation of Affiliated Hospital of Jining Medical University (2021-BS-004) and Affiliated Hospital of Jining Medical University Special Clinical Research Plan for the Attending Physician Team (grant number: ZZTD-MS-2023-03).
The authors thank the patient and her family for granting permission to publish this information.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Araki, K., Nakamura, R., Ito, D., Kato, K., Iguchi, Y., Sahashi, K., et al. (2020). NUS1 mutation in a family with epilepsy, cerebellar ataxia, and tremor. Epilepsy Res. 164, 106371. doi:10.1016/j.eplepsyres.2020.106371
Den, K., Kudo, Y., Kato, M., Watanabe, K., Doi, H., Tanaka, F., et al. (2019). Recurrent NUS1 canonical splice donor site mutation in two unrelated individuals with epilepsy, myoclonus, ataxia and scoliosis - a case report. BMC Neurol. 19 (1), 253. doi:10.1186/s12883-019-1489-x
Giladi, M., Lisnyansky Bar-El, M., Vaňková, P., Ferofontov, A., Melvin, E., Alkaderi, S., et al. (2022). Structural basis for long-chain isoprenoid synthesis by cis-prenyltransferases. Sci. Adv. 8 (20), eabn1171. doi:10.1126/sciadv.abn1171
Grabińska, K. A., Edani, B. H., Park, E. J., Kraehling, J. R., and Sessa, W. C. (2017). A conserved C-terminal RXG motif in the NgBR subunit of cis-prenyltransferase is critical for prenyltransferase activity. J. Biol. Chem. 292 (42), 17351–17361. doi:10.1074/jbc.M117.806034
Haginoya, K., Sekiguchi, F., Munakata, M., Yokoyama, H., Hino-Fukuyo, N., Uematsu, M., et al. (2021). A patient with a 6q22.1 deletion and a phenotype of non-progressive early-onset generalized epilepsy with tremor. Epilepsy Behav. Rep. 15, 100405. doi:10.1016/j.ebr.2020.100405
Hamdan, F. F., Myers, C. T., Cossette, P., Lemay, P., Spiegelman, D., Laporte, A. D., et al. (2017). High rate of recurrent de novo mutations in developmental and epileptic encephalopathies. Am. J. Hum. Genet. 101 (5), 664–685. doi:10.1016/j.ajhg.2017.09.008
Harrison, K. D., Park, E. J., Gao, N., Kuo, A., Rush, J. S., Waechter, C. J., et al. (2011). Nogo-B receptor is necessary for cellular dolichol biosynthesis and protein N-glycosylation. Embo J. 30 (12), 2490–2500. doi:10.1038/emboj.2011.147
Hu, Y., Huang, M., Wen, J., Gao, J., Long, W., Shen, Y., et al. (2023). Case report: splicing effect of a novel heterozygous variant of the NUS1 gene in a child with epilepsy. Front. Genet. 14, 1224949. doi:10.3389/fgene.2023.1224949
Jaeken, J., and Péanne, R. (2017). What is new in CDG? J. Inherit. Metab. Dis. 40 (4), 569–586. doi:10.1007/s10545-017-0050-6
Ji, C., Zhao, J., Zhang, J., and Wang, K. (2023). Novel NUS1 variant in a Chinese patient with progressive myoclonus epilepsy: a case report and systematic review. Neurol. Sci. 44 (10), 3495–3498. doi:10.1007/s10072-023-06851-4
Miller, D. T., Lee, K., Abul-Husn, N. S., Amendola, L. M., Brothers, K., Chung, W. K., et al. (2023). ACMG SF v3.2 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 25 (8), 100866. doi:10.1016/j.gim.2023.100866
Monfrini, E., Miller, C., Frucht, S. J., Di Fonzo, A., and Riboldi, G. M. (2022). Progressive myoclonus without epilepsy due to a NUS1 frameshift insertion: dyssynergia cerebellaris myoclonica revisited. Park. Relat. Disord. 98, 53–55. doi:10.1016/j.parkreldis.2022.03.016
Park, E. J., Grabińska, K. A., Guan, Z., Stránecký, V., Hartmannová, H., Hodaňová, K., et al. (2014). Mutation of Nogo-B receptor, a subunit of cis-prenyltransferase, causes a congenital disorder of glycosylation. Cell Metab. 20 (3), 448–457. doi:10.1016/j.cmet.2014.06.016
Riboldi, G. M., Monfrini, E., Stahl, C., and Frucht, S. J. (2022). NUS1 and epilepsy-myoclonus-ataxia syndrome: an under-recognized entity? Tremor Other Hyperkinet Mov. (N Y) 12, 21. doi:10.5334/tohm.696
Szafranski, P., Von Allmen, G. K., Graham, B. H., Wilfong, A. A., Kang, S. H. L., Ferreira, J. A., et al. (2015). 6q22.1 microdeletion and susceptibility to pediatric epilepsy. Eur. J. Hum. Genet. 23 (2), 173–179. doi:10.1038/ejhg.2014.75
Keywords: amino acid substitution, case report, exome sequencing, intellectual disability, epilepsy, tremor, undecaprenyl pyrophosphate synthetase, young adult
Citation: Li R, Yang J, Ma J, Zhang A and Li H (2024) Case report: Novel NUS1 variant in a Chinese patient with tremors and intellectual disability. Front. Genet. 15:1373448. doi: 10.3389/fgene.2024.1373448
Received: 19 January 2024; Accepted: 22 March 2024;
Published: 09 April 2024.
Edited by:
Ivano Di Meo, IRCCS Carlo Besta Neurological Institute Foundation, ItalyReviewed by:
Giulietta Maria Riboldi, New York University, United StatesCopyright © 2024 Li, Yang, Ma, Zhang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongfang Li, bWFsaW4tbGhmQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.