 Fengji Cui
Fengji Cui Tuoya Wulan2
Tuoya Wulan2 Victor Wei Zhang
Victor Wei Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 06 March 2024
Sec. Neurogenomics
Volume 15 - 2024 | https://doi.org/10.3389/fgene.2024.1371282
Background: Developmental and epileptic encephalopathies (DEEs) are a group of heterogeneous neurodevelopmental diseases characterized mainly by developmental delay/intellectual disability and early-onset epilepsy. Researchers have identified variations in the KCNT2 gene (OMIM* 610044) as the cause of DEE type 57 (MIM# 617771).
Case presentation: We report in this study a 46-year-old woman who presented with early-onset epilepsy, intellectual disability, hypertrichosis, coarse facial features, and short stature. Besides, there were four other affected individuals in her family history, including two elder brothers, a younger brother, and their mother. We collected blood samples from the proband, her two affected brothers, and her clinically normal daughter for genetic analysis. Clinical exome sequencing revealed a novel heterozygous variant in the KCNT2 gene (NM_198503: c.188G>A, p.Arg63His) in the proband and her two affected brothers, while her daughter did not carry this variant. Furthermore, we reviewed all 25 patients identified in the literature with KCNT2 variants and compared their phenotypes.
Conclusion: Epilepsy and intellectual disability/developmental delay occur in almost all patients with KCNT2 variants. KCNT2-relevant DEEs partially overlap with the clinical phenotypes of KATP channel diseases, particularly in hypertrichosis and distinctive coarse facial features.
“Developmental and epileptic encephalopathy” (DEE), as defined by the International League Against Epilepsy (ILAE), refers to disorders influenced by developmental factors. These disorders frequently exhibit epileptic activity, significantly impacting brain development and functional capabilities (Scheffer et al., 2016; Scheffer et al., 2017). DEEs, characterized by their early onset and severity, often lead to long-term developmental and cognitive impairments. They are heterogeneous, with a high genetic etiology rate, and are among the most severe forms of epilepsy. DEEs typically appear in infancy or early childhood, with many cases presenting within the first year of life (Guerrini et al., 2023). Although each disease is relatively rare on an individual level, collectively, the overall incidence rate of DEE is about 1 in 340 children. Specifically, 1 in 590 children suffer from developmental and epileptic encephalopathy (DEE), and 1 in 800 children have both intellectual disability and epilepsy (ID + E) (Poke et al., 2023). DEEs severely affect the affected children’s quality of life and impose significant burdens on families and society (Palmer et al., 2021).
Numerous genes linked to epileptic encephalopathies are also involved in developmental impairments, indicating their dual role in these disorders. Thus, the underlying genetic cause may result in developmental delay (DD) and/or ID in its own right, with a superimposed epileptic encephalopathy further adversely affecting development and cognition. Recent genomic advances, especially in DNA sequencing, have identified an increasing number of variations in genes known to cause DEEs, particularly those encoding ion channels and neurotransmitter receptors (e.g., SCN2A, KCNA2, KCNB1, KCNQ2, KCNT1, KCNT2, and STXBP1) (Wild and Nelson, 2019; Kessi et al., 2020; Kim et al., 2020).
Potassium (K+) channels, the most diverse ion channel group, are vital for neuronal excitability and signaling. There are five types of K+ channels categorized based on the stimulus that activates them: voltage-gated (KV), calcium-activated (KCa), inwardly rectifying (Kir), ATP-sensitive (KATP), and sodium-activated (KNa) potassium channel sub-families (González et al., 2012; Li et al., 2018). In humans, two KNa channels have been described, Slack (also called Slo2.2) and Slick (also called Slo2.1), encoded by the KCNT1 gene (OMIM* 608167) and KCNT2 gene (OMIM* 610044), respectively. These channels are widely expressed in the central nervous system and are crucial in modulating membrane hyperpolarization resulting from repetitive firing and hetero-tetrameric channel formation in distinct brain regions (Chen et al., 2009; Rizzi et al., 2015).
The first reported heterozygous germline variant in the KCNT2 gene appeared in 2017 in a 4-year-old male with epileptic encephalopathy, characterized by neonatal hypotonia, intractable infantile-onset epilepsy, and profound DD (Gururaj et al., 2017). Recent studies have discovered pathogenic variants in the KCNT2 gene, leading to developmental and epileptic encephalopathy-57 (DEE57; MIM 617771) in 25 patients (Gururaj et al., 2017; Ambrosino et al., 2018; Alagoz et al., 2020; Inuzuka et al., 2020; Mao et al., 2020; Gong et al., 2021; Jackson et al., 2021; Cioclu et al., 2023). Advances in genomics, especially next-generation sequencing, have increasingly linked DEEs to genetic variations (Zhou et al., 2018). Treatment for DEEs focuses on symptomatic management through antiepileptic drugs and rehabilitation for DD. Recent studies show significant advancements in the treatment of DEEs. New therapeutic options, including precision therapies and repurposed drugs, are emerging, potentially improving seizure burden and neurological outcomes (Vasquez et al., 2022). Among them, these new medications include cannabidiol, everolimus, and repurposed drugs like fenfluramine, currently being used for the management of DEEs (Johannessen Landmark et al., 2021). Despite advances in treatment, the long-term prognosis of DEE is influenced by various factors, including the type of seizures, underlying causes, severity, and response to treatment, with many patients facing challenges, especially in their response to multiple Antiseizure Medications (ASMs) (Bravo et al., 2021; Samanta, 2021; Kienitz et al., 2022). Here, we reported a novel KCNT2 missense variant discovered in a family affected by developmental and epileptic encephalopathies.
The proband (Figure 1A), a 46-year-old woman of Han Chinese ethnicity, presented with early-onset epilepsy and ID, along with dysmorphic facial features (hypertrichosis and coarse facial features), and short stature (149.0 cm, below the third centile). A medical genetics outpatient clinic was consulted after her healthy 28-year-old daughter (Ⅲ-1) was advised to consider genetic testing before pregnancy due to the positive family history. The proband experienced initial epileptic seizures in infancy and occasional seizures during her childhood and adulthood, which were classified as focal to bilateral tonic-clonic seizures according to the 2017 ILAE seizure type classification. She had unpredictable seizure frequency, and the seizures usually resolved spontaneously within minutes. The latest seizure, triggered by emotional upset a year ago, involved limb stiffness and transient aphasia without loss of consciousness. The proband did not undergo brain MRI due to financial and geographical limitations. However, she had a normal head computed tomography (CT) scan, which excluded any major structural abnormalities. She had mild hypertrichosis and coarse facial features, such as thick hair and eyebrows, mildly downward-slanting eye corners, a broad nasal bridge, a relatively long philtrum, full and prominent lips, a wide mouth, and a slightly small lower jaw. Intellectually, she demonstrated lower cognitive function, with clear but slightly slow speech and a regular voice tone. She showed a limited capacity for deep thought and calculations. Nevertheless, she did not have any other comorbidities, such as cardiac, renal, or endocrine disorders. She also did not have any behavioral or psychiatric problems, such as autism spectrum disorder, attention deficit hyperactivity disorder and so on. Up to now, she has never received treatment with antiepileptic drugs.
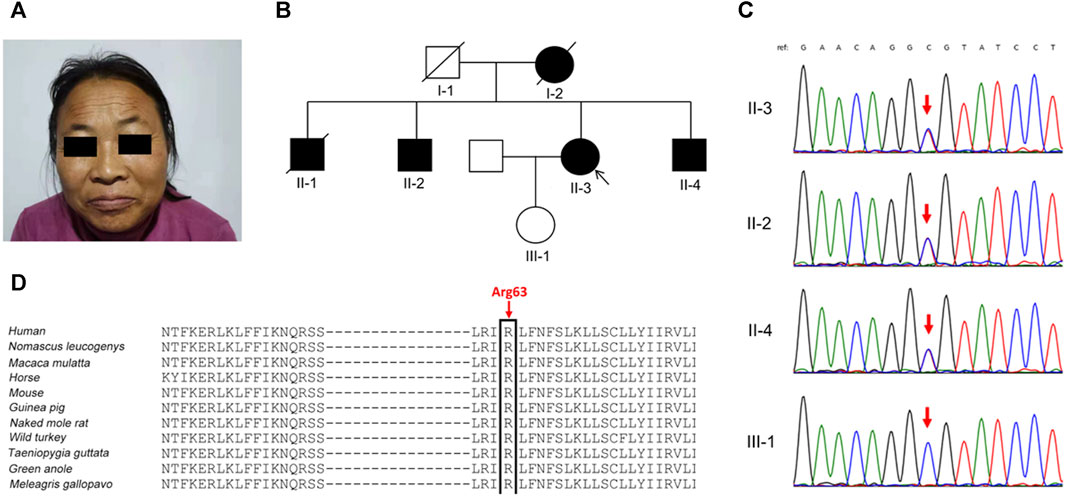
FIGURE 1. Clinical phenotypes and genetic analysis in the family. (A) The proband (Ⅱ-3) presented with prominent eyebrows, thick hair, and coarse facial features. (B) Pedigree of the family. (C) Sanger sequencing confirmed the KCNT2 variant c.188G>A (p.Arg63His) in the proband and her two brothers (Ⅱ-2 and Ⅱ-4), and not in her clinically normal daughter (red arrow). (D) The Arg63 residue is highly evolutionarily conserved among different species.
In the proband’s pedigree (Figure 1B), similar clinical phenotypes were observed in two elder brothers (Ⅱ-1 and Ⅱ-2), a younger brother (Ⅱ-4), and their mother (Ⅰ-2), who had passed away. The eldest brother (Ⅱ-1) had the most frequent epileptic seizures and died at 42 years old. The second eldest brother (Ⅱ-2) experienced fewer epileptic-like episodes, characterized by unclear consciousness, limb stiffness, and speech impairment, resembling a “shock” state during the most severe episodes. The youngest brother has no history of epilepsy but has an ID. Both the proband’s husband and daughter were clinically normal, and her daughter’s height was 165 cm.
Diagnostic clinical exome sequencing revealed a novel heterozygous missense variant in the KCNT2 gene (NM_198503: c.188G>A, p.Arg63His) in the proband and her two affected brothers, while her daughter did not carry this variant. Sanger sequencing confirmed the variant (Figure 1C). The variant p.Arg63His, found at a low frequency (2 out of 230,620 alleles) in the gnomAD database, is located at the junction of the N-terminal domain and S1 domain, showing high evolutionary conservation among different species (Figure 1D). The variant p.Arg63His was predicted to be damaging by various algorithms, including SIFT, Polyphen-2, MutationTaster, PROVEAN, and CADD. Based on these clinical and genetic characteristics, the affected individuals in this family were diagnosed with DEE caused by the KCNT2 variant.
In this study, we reported a novel KCNT2 gene variation (p.Arg63His) in a family and provided a comprehensive review of available literature on KCNT2 variations (Table 1) (Gururaj et al., 2017; Ambrosino et al., 2018; Alagoz et al., 2020; Inuzuka et al., 2020; Mao et al., 2020; Gong et al., 2021; Jackson et al., 2021; Cioclu et al., 2023). Among the 25 patients previously reported, 19 pathogenic KCNT2 variants were identified, including 16 missense variants, 1 in-frame deletion, 1 nonsense variant, and 1 frameshift variant. The only recurrent variants were observed at p190 position (R190H in patients #2, #3, #15, #16, #17; R190P in patients #18 and #19) (Ambrosino et al., 2018; Jackson et al., 2021; Álvarez-Mora et al., 2022). The location of each variant in the KCNT2 subunit is shown in Figure 2. According to the clinical phenotypes described in the literature, patients with KCNT2-relevant diseases usually present with early-onset epileptic seizures, intellectual impairment, infantile hypotonia, motor DD, dysmorphic features, and typical EEG. It is noteworthy that early onset epileptic seizures, ID/DD, infantile hypotonia, dysmorphic features, and typical EEG were reported in 14, 19, 13, 13, and 16 of the patients, respectively. Meanwhile, Ambrosino et al. (Ambrosino et al., 2018) reported missense KCNT2 variants (p.Arg190His and p.Arg190Pro) in two individuals, and they were both presented with epilepsy, intellectual disability, hypertrichosis, abnormal facial features, and short stature. However, Jackson et al., 2021 described two patients with the same variants, p.Arg190His and p.Arg190Pro, who had similar clinical phenotypes as described above but with no epilepsy. In this study, the proband and her four family members presented with similar clinical symptoms of dysmorphic features (hypertrichosis and coarse facial features), short stature, early-onset seizures, and intellectual disability.
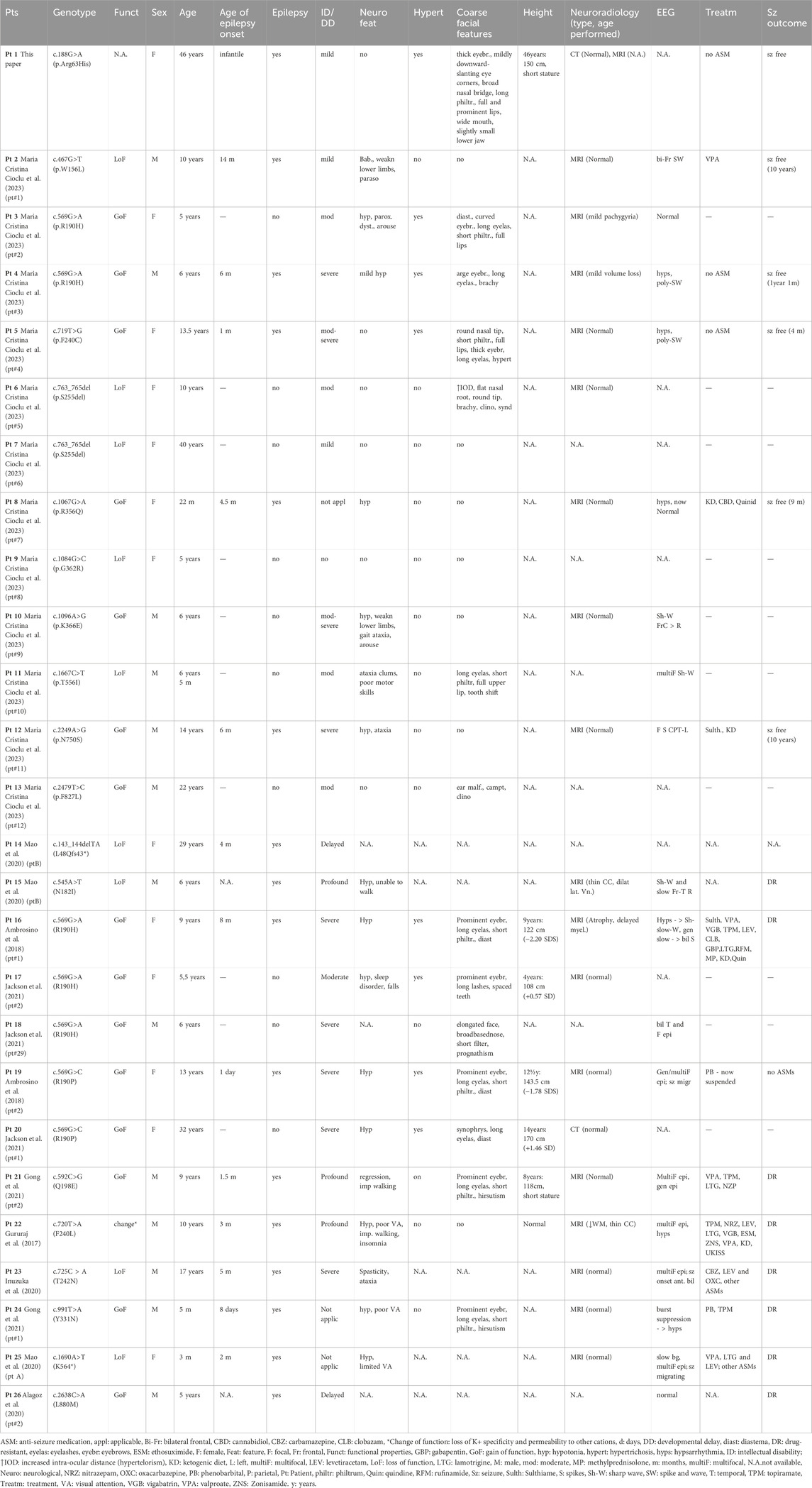
TABLE 1. Overview of the phenotypic and genetic findings of all identified patients with KCNT2-relevant diseases (n = 26).
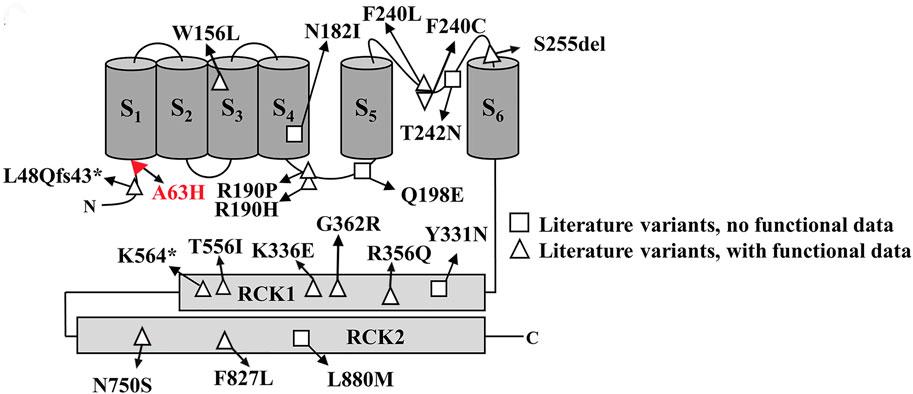
FIGURE 2. Schematic representative of the Slick channel with all published KCNT2 variations. The position of the variant Arg63His is located at the junction of the S1 domain and N-terminal domain, which is indicated in red.
Potassium channel dysfunction caused by genetic variations can lead to various neurological disorders. These disorders have clinical manifestations, including multiple forms of epilepsy, ID, and autism, among other conditions (Huang et al., 2018; Garrido, 2023). In the present study, five Chinese family members reported similar clinical symptoms of early-onset seizures, intellectual disability, hypertrichosis, coarse facial features, and short stature. Clinical exome sequencing revealed a novel heterozygous variant in the KCNT2 gene (c.188G>A, p.Arg63His) in the proband and her two affected brothers, while her healthy daughter did not carry this variant. We identified the p.Arg63His variant as the disease-causing variant in this family.
Numerous studies have shown that KNa channels contribute to slow afterhyperpolarization, the adaption of firing frequency in neurons, and the stabilization of resting membrane potential (Gao et al., 2008; Kaczmarek, 2013). Two known genes, KCNT1 and KCNT2, encode KNa channel subunits Slack and Slick. Slack and Slick’s channels contain six membrane-spanning domains (S1-S6), an intracellular N-terminus, and a long C-terminus (Kaczmarek, 2013). Slick’s amino acid sequence shows high homology with Slack’s, but the amino-terminal sequences display a notable difference. Previous functional studies of KCNT2 variants have shown that 10 variants (R190H, F240C, R356Q, K366E, N750S, F827L, R190P, Q198E, Y331N, L880M) exhibit gain-of-function (GoF) characteristics. In comparison, 8 variants (W156L, S255del, G362R, T556I, L48Qfs43*, N182I, T242N, K564*) show loss-of-function (LoF) phenotypic features (Gururaj et al., 2017; Ambrosino et al., 2018; Alagoz et al., 2020; Inuzuka et al., 2020; Mao et al., 2020; Gong et al., 2021; Jackson et al., 2021; Cioclu et al., 2023). Therefore, although the KCNT1 and KCNT2 channels are structurally and functionally similar, they seem to exhibit significant differences in the functional characteristics of pathogenic variants causing DEEs. In in vitro evaluations, KCNT1 variants almost always promote a GoF phenotype, whereas KCNT2 variants are almost evenly distributed between GoF and LoF (10 and 8, respectively). Moreover, the Slick channel uniquely regulates neuronal excitability, characterized by its rapid gating kinetics and sensitivity to intracellular ATP levels, in contrast to the Slack channel, which has been identified with potential sensitivity to intracellular chloride and sodium ions (Bhattacharjee et al., 2003; Xu et al., 2023). Research indicates that the mammalian central nervous system widely but heterogeneously distributes Slack and Slick’s channels, which are co-expressed by many types of central neurons (Rizzi et al., 2015; Rizzi et al., 2016). Within the dorsal root ganglia (DRGs), knockout of KCNT1 abolishes KNa current, and KCNT1 knockout mice exhibit enhanced itch and pain responses. However, the knockout of KCNT2 in the DRGs does not cancel KNa current (Martinez-Espinosa et al., 2015). A study from KCNT2 knockout mice revealed that Slick channels inhibit the excitability of calcitonin gene-related peptide (CGRP)-containing neurons, relieving pain after inflammation and injury (Tomasello et al., 2017). Until now, we still know very little about the clearly defined physiological function of the KCNT2 gene.
In 2017, Gururaj et al., 2017 first reported a heterozygous KCNT2 variation (Phe240Leu) in a patient with an early onset epileptic encephalopathy (EOEE), and relevant experiments confirmed a “change-of-function” effect of the variation by altering ion selectivity. The p.Phe190 residue of Slick is situated between helices S4 and S5, and variations in the KCNT2 gene affecting the Phe190 residue have been reported in four patients with DEE and dysmorphic features (Ambrosino et al., 2018; Jackson et al., 2021). Experimental results showed that Arg190His and Arg190Pro increase maximal K+ current densities and shift toward more negative membrane potentials, consistent with GoF effects (Ambrosino et al., 2018). Scientists have discovered two truncating alterations in KCNT2 (p.Leu48Glnfs43, p.Lys564). These alterations, predicted to be null variants, reduce the global current density of heteromeric channels, thereby impacting KNa function (Mao et al., 2020). Nonetheless, the harmful mechanism of truncating alterations is not due to haploinsufficiency, as the KCNT2 gene is likely to tolerate LoF alterations more (PLI = 0.04). Further research, including functional studies, is required to elucidate the pathogenic mechanism of the KCNT2 variant p.Arg63His in this study. Only a few studies have investigated patients with DEE due to KCNT2 variation, and the number of functional analyses performed is also limited. Thus, the establishment of genotype-phenotype correlations still needs to be completed.
Hypertrichosis and coarse facial features have been reported in several K+ channel opathies caused by variations in the KCNH1, KCNN3, KCNK4, and KCNJ8 genes (Cooper et al., 2014; Gripp et al., 2021; Apuril Velgara et al., 2022). Notably, KATP channels are uniquely evolved protein complexes that couple intracellular metabolism to the electrical activity by regulating plasma membrane K+ flux in response to changes in the intracellular concentrations of ATP and ADP, thus playing an essential role in the process physiological and pathophysiology (Driggers and Shyng, 2021). KATP channels are composed of an inwardly rectifying K+ channel subunit, either Kir6.1 (KCNJ8 gene) or Kir6.2 (KCNJ11 gene), plus a sulfonylurea receptor, either SUR1 (ABCC8 gene) or SUR2 (ABCC9 gene) that serve as the regulatory subunit. Research reports indicate that variations in KCNJ8 or ABCC9 cause Cantú syndrome. This syndrome features congenital hypertrichosis, distinctive facial characteristics such as a broad nasal bridge, long philtrum, a wide mouth with prominent lips, osteochondrodysplasia, and cardiovascular abnormalities. Studies demonstrate that pathogenic variants in ABCC9 or KCNJ8 increase the opening of the KATP channel resulting from decreased ATP-mediated inhibition, consistent with gain-of-function variations (Harakalova et al., 2012; McClenaghan et al., 2018). Minoxidil and diazoxide, which are KATP channel agonists, were initially used as antihypertensive drugs and commonly resulted in the side effects of hair overgrowth (Newfield, 2015; Suchonwanit et al., 2019).
Previous studies have shown that the Slick channel functions as a hybrid between two classes of K+ channels, named KNa channels and KATP channels (Bhattacharjee et al., 2003). The Slick channel can be activated via intracellular Na+ and Cl− and inhibited by intracellular ATP. Patients with KCNT1 variations have not reported hypertrichosis and coarse facial features. The clinical phenotypes of KCNT2 variations largely overlap with KATP channel diseases, which include epilepsy, ID/DD, hypertrichosis, and coarse facial features. Besides, patients carrying GoF KCNT2 variants often presented with more severe ID/DD, earlier epilepsy onset, and pronounced dysmorphisms, including hypertrichosis, compared to those with LoF variants (Cioclu et al., 2023). Hypertrichosis occurs in patients with K+ channelopathies that might be partial, local, or distributed over the whole body (Brownstein et al., 2013; Gripp et al., 2021). In this study, the proband presented with mild hypertrichosis characterized by thick scalp hair and prominent eyebrows, which improved gradually with age. This is consistent with the findings in the literature, where GoF variants were more frequently associated with severe ID/DD and earlier epilepsy onset (Cioclu et al., 2023). Short stature has been reported in three patients with KCNT2 variations (Ambrosino et al., 2018; Gong et al., 2021). In this study, all affected patients presented with short stature. However, the underlying mechanism of short stature by KCNT2 variations is unknown.
In our study, the proband is currently seizure-free and has no ASM treatment. While GoF KCNT2 variants are universally blocked by quinidine and fluoxetine, LoF variants like W156L or N182I exhibit a different pharmacological profile, being potentiated by loxapine or riluzole (Gururaj et al., 2017; Ambrosino et al., 2018; Alagoz et al., 2020; Inuzuka et al., 2020; Mao et al., 2020; Gong et al., 2021; Jackson et al., 2021; Cioclu et al., 2023). This implies that the same drug can have varying effects depending on the specific KCNT2 variant, necessitating more tailored pharmacological interventions based on the particular KCNT2 variant present in each patient.
In conclusion, our study, in light of recent findings, confirms the diverse clinical spectrum of KCNT2-related disorders and underscores the importance of comprehensive genetic and clinical evaluations for accurate diagnosis and management. However, our study faces limitations, including the inability to perform genetic testing on all key family members and the lack of comprehensive neuropsychiatric evaluations due to socioeconomic and geographical challenges. Moreover, establishing a clear genotype-phenotype correlation for KCNT2-related disorders requires more extensive studies and functional analyses of various KCNT2 variants.
In this study, we report a novel KCNT2 variant (p.Arg63His) in a family and comprehensively review available literature concerning KCNT2 variations. The KCNT2 variants can be classified into GoF or LoF, depending on their effects on channel current. Epilepsy or ID/DD occurs in almost all patients with KCNT2 variations, while hypertrichosis and distinctive coarse facial features are also commonly found. We observed that the GoF variants are associated with more severe epilepsy and DD, which may be due to an increase in channel activity and neuronal excitability, leading to hyperexcitability and seizures. Conversely, LoF variants are associated with milder epilepsy and variable developmental outcomes. The clinical phenotypes of KCNT2-relevant DEEs partially overlapped with KCNT1 variations and KATP channel diseases, mainly due to the hybrid function between KNa and KATP channels. We speculate that patients with GoF KCNT2 variations usually present with epilepsy, ID/DD, hypertrichosis, and coarse facial features; LoF KCNT2 variations are unlikely to lead to hypertrichosis and coarse facial features. However, further research and functional studies are necessary to make this conclusion. In conclusion, our study contributes to the evolving understanding of KCNT2-related disorders, highlighting the significance of GoF and LoF variants in determining the severity and range of clinical phenotypes, including ID/DD and hypertrichosis. This insight is crucial for tailoring appropriate therapeutic interventions and future research in this field.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
The studies involving humans were approved by the Chifeng Maternity Hospital ethics committee (ID: 2022-001-01). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
FC: Writing–original draft. TW: Data curation, Validation, Writing–review and editing. QZ: Formal Analysis, Methodology, Writing–review and editing. VZ: Project administration, Supervision, Writing–review and editing. YJ: Data curation, Validation, Writing–review and editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
We sincerely extend our appreciation to the proband and her families who participated in this study.
Author QZ and VZ were employed by company AmCare Genomics Lab.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alagoz, M., Kherad, N., Bozkurt, S., and Yuksel, A. (2020). New mutations in KCNT2 gene causing early infantile epileptic encephalopathy type 57: case study and literature review. Acta Biochim. Pol. 67 (3), 431–434. doi:10.18388/abp.2020_5364
Álvarez-Mora, M. I., Sánchez, A., Rodríguez-Revenga, L., Corominas, J., Rabionet, R., Puig, S., et al. (2022). Diagnostic yield of next-generation sequencing in 87 families with neurodevelopmental disorders. Orphanet J. Rare Dis. 17 (1), 60. doi:10.1186/s13023-022-02213-z
Ambrosino, P., Soldovieri, M. V., Bast, T., Turnpenny, P. D., Uhrig, S., Biskup, S., et al. (2018). De novo gain of function variants in KCNT2 as a novel cause of developmental and epileptic encephalopathy. Ann. neurology 83 (6), 1198–1204. doi:10.1002/ana.25248
Apuril Velgara, E. S., Mariani, M., Torella, A., Musacchia, F., consortium, T. U. D. P., Nigro, V., et al. (2022). Cantù syndrome: report of a patient with a novel variant in KCNJ8 and revision of literature. Am. J. Med. Genet. Part A 188 (6), 1661–1666. doi:10.1002/ajmg.a.62710
Bhattacharjee, A., Joiner, W. J., Wu, M., Yang, Y., Sigworth, F. J., and Kaczmarek, L. K. (2003). Slick (Slo2. 1), a rapidly-gating sodium-activated potassium channel inhibited by ATP. J. Neurosci. 23 (37), 11681–11691. doi:10.1523/JNEUROSCI.23-37-11681.2003
Bravo, P., Vaddiparti, A., and Hirsch, L. J. (2021). Pharmacotherapy for nonconvulsive seizures and nonconvulsive status epilepticus. Drugs 81 (7), 749–770. doi:10.1007/s40265-021-01502-4
Brownstein, C. A., Towne, M. C., Luquette, L. J., Harris, D. J., Marinakis, N. S., Meinecke, P., et al. (2013). Mutation of KCNJ8 in a patient with Cantu syndrome with unique vascular abnormalities–support for the role of K (ATP) channels in this condition. Eur. J. Med. Genet. 56 (12), 678–682. doi:10.1016/j.ejmg.2013.09.009
Chen, H., Kronengold, J., Yan, Y., Gazula, V.-R., Brown, M. R., Ma, L., et al. (2009). The N-terminal domain of Slack determines the formation and trafficking of Slick/Slack heteromeric sodium-activated potassium channels. J. Neurosci. 29 (17), 5654–5665. doi:10.1523/JNEUROSCI.5978-08.2009
Cioclu, M. C., Mosca, I., Ambrosino, P., Puzo, D., Bayat, A., Wortmann, S. B., et al. (2023). KCNT2-related disorders: phenotypes, functional and pharmacological properties. Ann. Neurology 94, 332–349. doi:10.1002/ana.26662
Cooper, P. E., Reutter, H., Woelfle, J., Engels, H., Grange, D. K., van Haaften, G., et al. (2014). Cantú syndrome resulting from activating mutation in the KCNJ 8 gene. Hum. Mutat. 35 (7), 809–813. doi:10.1002/humu.22555
Driggers, C. M., and Shyng, S.-L. (2021). “Production and purification of ATP-sensitive potassium channel particles for cryo-electron microscopy,” in Methods in enzymology (Amsterdam, Netherlands: Elsevier), 121–150.
Gao, S.-b., Wu, Y., Lü, C.-x., Guo, Z.-h., Li, C.-h., and Ding, J.-p. (2008). Slack and Slick KNa channels are required for the depolarizing afterpotential of acutely isolated, medium diameter rat dorsal root ganglion neurons. Acta Pharmacol. Sin. 29 (8), 899–905. doi:10.1111/j.1745-7254.2008.00842.x
Garrido, J. J. (2023). Contribution of axon initial segment structure and channels to brain pathology. Cells 12 (8), 1210. doi:10.3390/cells12081210
Gong, P., Jiao, X., Yu, D., and Yang, Z. (2021). Case report: causative de novo variants of KCNT2 for developmental and epileptic encephalopathy. Front. Genet. 12, 649556. doi:10.3389/fgene.2021.649556
González, C., Baez-Nieto, D., Valencia, I., Oyarzún, I., Rojas, P., Naranjo, D., et al. (2012). K+ channels: function-structural overview. Compr. Physiol. 2 (3), 2087–2149. doi:10.1002/cphy.c110047
Gripp, K. W., Smithson, S. F., Scurr, I. J., Baptista, J., Majumdar, A., Pierre, G., et al. (2021). Syndromic disorders caused by gain-of-function variants in KCNH1, KCNK4, and KCNN3—a subgroup of K+ channelopathies. Eur. J. Hum. Genet. 29 (9), 1384–1395. doi:10.1038/s41431-021-00818-9
Guerrini, R., Conti, V., Mantegazza, M., Balestrini, S., Galanopoulou, A. S., and Benfenati, F. (2023). Developmental and epileptic encephalopathies: from genetic heterogeneity to phenotypic continuum. Physiol. Rev. 103 (1), 433–513. doi:10.1152/physrev.00063.2021
Gururaj, S., Palmer, E. E., Sheehan, G. D., Kandula, T., Macintosh, R., Ying, K., et al. (2017). A de novo mutation in the sodium-activated potassium channel KCNT2 alters ion selectivity and causes epileptic encephalopathy. Cell Rep. 21 (4), 926–933. doi:10.1016/j.celrep.2017.09.088
Harakalova, M., Van Harssel, J. J., Terhal, P. A., Van Lieshout, S., Duran, K., Renkens, I., et al. (2012). Dominant missense mutations in ABCC9 cause Cantu syndrome. Nat. Genet. 44 (7), 793–796. doi:10.1038/ng.2324
Huang, Y., Tao, J., and Zhao, R. (2018). Potassium channels and CNS diseases. CNS Neurological Disorders-Drug Targets 17 (4), 245–247. doi:10.2174/187152731704180706105155
Inuzuka, L. M., Macedo-Souza, L. I., Della-Ripa, B., Monteiro, F. P., Ramos, L., Kitajima, J. P., et al. (2020). Additional observation of a de novo pathogenic variant in KCNT2 leading to epileptic encephalopathy with clinical features of frontal lobe epilepsy. Brain Dev. 42 (9), 691–695. doi:10.1016/j.braindev.2020.05.003
Jackson, A., Banka, S., Stewart, H., Consortium, G. E. R., Robinson, H., Lovell, S., et al. (2021). Recurrent KCNT2 missense variants affecting p. Arg190 result in a recognizable phenotype. Am. J. Med. Genet. Part A 185 (10), 3083–3091. doi:10.1002/ajmg.a.62370
Johannessen Landmark, C., Potschka, H., Auvin, S., Wilmshurst, J. M., Johannessen, S. I., Kasteleijn-Nolst Trenité, D., et al. (2021). The role of new medical treatments for the management of developmental and epileptic encephalopathies: novel concepts and results. Epilepsia 62 (4), 857–873. doi:10.1111/epi.16849
Kaczmarek, L. K. (2013). Slack, slick, and sodium-activated potassium channels. Int. Sch. Res. Notices 2013, 354262. doi:10.1155/2013/354262
Kessi, M., Chen, B., Peng, J., Tang, Y., Olatoutou, E., He, F., et al. (2020). Intellectual disability and potassium channelopathies: a systematic review. Front. Genet. 11, 614. doi:10.3389/fgene.2020.00614
Kienitz, R., Kay, L., Beuchat, I., Gelhard, S., von Brauchitsch, S., Mann, C., et al. (2022). Benzodiazepines in the management of seizures and status epilepticus: a review of routes of delivery, pharmacokinetics, efficacy, and tolerability. CNS drugs 36 (9), 951–975. doi:10.1007/s40263-022-00940-2
Kim, H. J., Yang, D., Kim, S. H., Kim, B., Kim, H. D., Lee, J. S., et al. (2020). The phenotype and treatment of SCN2A-related developmental and epileptic encephalopathy. Epileptic Disord. 22 (5), 563–570. doi:10.1684/epd.2020.1199
Li, Y., Xu, J., Xu, Y., Zhao, X.-Y., Liu, Y., Wang, J., et al. (2018). Regulatory effect of general anesthetics on activity of potassium channels. Neurosci. Bull. 34, 887–900. doi:10.1007/s12264-018-0239-1
Mao, X., Bruneau, N., Gao, Q., Becq, H., Jia, Z., Xi, H., et al. (2020). The epilepsy of infancy with migrating focal seizures: identification of de novo mutations of the KCNT2 gene that exert inhibitory effects on the corresponding heteromeric KNa1. 1/KNa1. 2 potassium channel. Front. Cell. Neurosci. 14, 1. doi:10.3389/fncel.2020.00001
Martinez-Espinosa, P. L., Wu, J., Yang, C., Gonzalez-Perez, V., Zhou, H., Liang, H., et al. (2015). Knockout of Slo2. 2 enhances itch, abolishes KNa current, and increases action potential firing frequency in DRG neurons. Elife 4, e10013. doi:10.7554/eLife.10013
McClenaghan, C., Hanson, A., Sala-Rabanal, M., Roessler, H. I., Josifova, D., Grange, D. K., et al. (2018). Cantu syndrome–associated SUR2 (ABCC9) mutations in distinct structural domains result in KATP channel gain-of-function by differential mechanisms. J. Biol. Chem. 293 (6), 2041–2052. doi:10.1074/jbc.RA117.000351
Newfield, R. S. (2015). Topical sulfonylurea as a novel therapy for hypertrichosis secondary to diazoxide, and potentially for other conditions with excess hair growth. Med. hypotheses 85 (6), 969–971. doi:10.1016/j.mehy.2015.08.025
Palmer, E. E., Howell, K., and Scheffer, I. E. (2021). Natural history studies and clinical trial readiness for genetic developmental and epileptic encephalopathies. Neurotherapeutics 18, 1432–1444. doi:10.1007/s13311-021-01133-3
Poke, G., Stanley, J., Scheffer, I. E., and Sadleir, L. G. (2023). Epidemiology of developmental and epileptic encephalopathy and of intellectual disability and epilepsy in children. Neurology 100 (13), e1363–e1375. doi:10.1212/WNL.0000000000206758
Rizzi, S., Knaus, H. G., and Schwarzer, C. (2016). Differential distribution of the sodium-activated potassium channels slick and slack in mouse brain. J. Comp. Neurology 524 (10), 2093–2116. doi:10.1002/cne.23934
Rizzi, S., Schwarzer, C., Kremser, L., Lindner, H. H., and Knaus, H.-G. (2015). Identification of potential novel interaction partners of the sodium-activated potassium channels Slick and Slack in mouse brain. Biochem. biophysics Rep. 4, 291–298. doi:10.1016/j.bbrep.2015.09.024
Samanta, D. (2021). Rescue therapies for seizure emergencies: current and future landscape. Neurol. Sci. 42, 4017–4027. doi:10.1007/s10072-021-05468-9
Scheffer, I. E., Berkovic, S., Capovilla, G., Connolly, M. B., French, J., Guilhoto, L., et al. (2017). ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia 58 (4), 512–521. doi:10.1111/epi.13709
Scheffer, I. E., French, J., Hirsch, E., Jain, S., Mathern, G. W., Moshé, S. L., et al. (2016). Classification of the epilepsies: new concepts for discussion and debate—special report of the ILAE Classification Task Force of the Commission for Classification and Terminology. Epilepsia open 1 (1-2), 37–44. doi:10.1002/epi4.5
Suchonwanit, P., Thammarucha, S., and Leerunyakul, K. (2019). Minoxidil and its use in hair disorders: a review. Drug Des. Dev. Ther. 13, 2777–2786. doi:10.2147/DDDT.S214907
Tomasello, D. L., Hurley, E., Wrabetz, L., and Bhattacharjee, A. (2017). Slick (Kcnt2) sodium-activated potassium channels limit peptidergic nociceptor excitability and hyperalgesia. J. Exp. Neurosci. 11, 1179069517726996. doi:10.1177/1179069517726996
Vasquez, A., Buraniqi, E., and Wirrell, E. C. (2022). New and emerging pharmacologic treatments for developmental and epileptic encephalopathies. Curr. Opin. Neurology 35 (2), 145–154. doi:10.1097/wco.0000000000001029
Wild, B., and Nelson, S. L. (2019). STXBP1-Related developmental and epileptic encephalopathy. Pediatr. Neurol. Briefs 33, 6. doi:10.15844/pedneurbriefs-33-6
Xu, J., Lv, Y.-T., Zhao, X.-Y., Wang, J.-J., Shen, Z.-S., Li, J., et al. (2023). Identification of sodium-and chloride-sensitive sites in the Slack channel. J. Neurosci. 43 (15), 2665–2681. doi:10.1523/JNEUROSCI.1365-22.2023
Keywords: DEE, epilepsy, intellectual disability, KCNT2, KATP channel
Citation: Cui F, Wulan T, Zhang Q, Zhang VW and Jiang Y (2024) Identification of a novel KCNT2 variant in a family with developmental and epileptic encephalopathies: a case report and literature review. Front. Genet. 15:1371282. doi: 10.3389/fgene.2024.1371282
Received: 16 January 2024; Accepted: 20 February 2024;
Published: 06 March 2024.
Edited by:
Salvatore Gallone, University of Turin, ItalyReviewed by:
Jianxiang Liao, Shenzhen Children’s Hospital, ChinaCopyright © 2024 Cui, Wulan, Zhang, Zhang and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuhua Jiang, anloMjY3NDE2MDk3NEAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.