 Fatima Ouboukss1,2*
Fatima Ouboukss1,2* Zhour El Amrani1,2
Zhour El Amrani1,2 Hicham Bouchahta2
Hicham Bouchahta2 Ilham Ratbi1Aziza Sbiti2
Ilham Ratbi1Aziza Sbiti2 Thomas Liehr3Abdelaziz Sefiani1,2
Thomas Liehr3Abdelaziz Sefiani1,2 Abdelhafid Natiq1
Abdelhafid Natiq1- 1Faculty of Medicine and Pharmacy, Research Team in Genomics and Molecular Epidemiology of Genetic Diseases, Genomics Center of Human Pathologies, University Mohammed V in Rabat, Rabat, Morocco
- 2Department of Medical Genetics, National Institute of Health in Rabat, Rabat, Morocco
- 3Institute of Human Genetics, Jena University Hospital, Friedrich Schiller University, Jena, Germany
Introduction: The majority of small supernumerary marker chromosomes (sSMCs) are derived from one single chromosome. Complex sSMCs, on the other hand, consist of genetic material derived from more than one, normally two chromosomes. Complex sSMCs involving chromosomes 8 and 14 are rarely encountered.
Case presentation: We present here a 14-month-old boy born from an unrelated couple. At birth, the baby was hypotonic and had a cleft lip and palate, as well as ocular involvement. Throughout the course of development, the baby experienced feeding difficulties, stunted growth, and delayed psychomotor development. Banding together with molecular cytogenetics revealed a balanced maternal translocation t(8;14)(p22.3;q21)mat, leading due to meiotic 3:1 segregation to a partial trisomy of chromosomes 8 and 14 in the affected boy.
Discussion/Conclusion: This report highlights the importance of cytogenetics in diagnosis of rare genetic disorders, with impact on genetic counselling of patients and their families. There are three comparable cases in the literature involving both chromosomes 8 and 14, but with different breakpoints; the complex sSMC derived from chromosomes 8 and 14 in this case, characterized as der(14)t(8;14) (p22.3;q21)mat.
Introduction
Small supernumerary marker chromosomes (sSMCs) are structurally abnormal chromosomes that are difficult to identify or characterize unambiguously using conventional banding cytogenetics alone. These sSMCs are typically equal in size or smaller than chromosome 20 of the same metaphase spread (Liehr et al., 2004).
sSMCs have been found to be derived from any of the 24 human chromosomes, including both autosomes and gonosomes, with a predominant source being chromosome 15, followed by chromosome 22 (Liehr, 2023). sSMCs have been reported in various populations, with occurrences noted as follows: 0.043% in newborn infants, 0.077% in prenatal diagnosis cases, 0.433% in patients with intellectual disability, and 0.171% in individuals experiencing subfertility (Liehr and Weise, 2007). sSMCs may have different shapes and constitutions, like ring, centric minute and inverted duplication shape. Also, they may be continuous, discontinuous, single, multiple, neocentric, complex or form other rare subgroups as summarized in Liehr 2023. One of the smallest sSMC subgroups is constituted by the so-called complex sSMCs, they contain chromosomal material originating from more than one, normally two chromosomes (Trifonov et al., 2008; Liehr et al., 2013; Liehr, 2023).
The clinical presentation of sSMCs shows significant variability, and they are detected unexpectedly in routine karyotype analyses (Liehr et al., 2010). In our routine chromosome analysis, a 14-month-old boy was found to have an sSMC. R-banding technique showed 47,XY,+mar (ISCN, 2020). His father’s karyotype was 46,XY, while in his mother a balanced reciprocal translocation between chromosomes 8 and 14 was detected.
Case presentation
A 14-month-old infant, born to unrelated parents, the second child in a two-sibling family, with his older brother in good health is reported (Figure 1). The mother’s pregnancy was uneventful; she was 27 years old at the time of birth, while the father was 34. The patient’s birth weight was 3 kg, with a head circumference 32 cm (P50); he had an APGAR score of 8/10. At clinical evaluation at 14 months of age, the patient showed growth delay in terms of height and weight, delay in psychomotor development, and generalized hypotonia with feeding difficulties. The patient also exhibits dysmorphic features, including microcephaly, craniosynostosis, broad and prominent forehead, hypertelorism, microphthalmia, narrow palpebral fissures, arched eyebrows, flattened nose with wide nostrils, low-set ears, umbilical hernia and clinodactyly. Additionally, he had undergone surgery for a cleft lip and palate. His echocardiography, abdominal ultrasounds and brain MRI were normal.
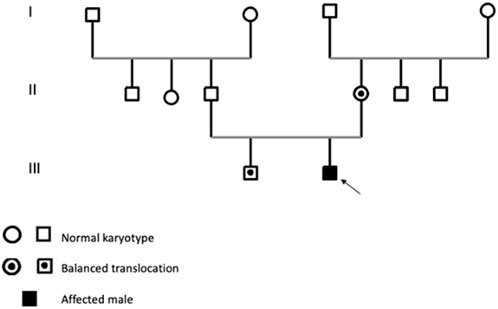
FIGURE 1. Pedigree balanced translocation in the mother’s family.
Materials and methods
Parents gave informed consent for the genetic analysis, which was performed in accordance with the Declaration of Helsinki protocols and approved by the local institutional review boards. Venous blood (3–5 ml) acquired in a heparinized tube was taken of the patient and his parents for RHG cytogenetic analysis (R-banding of human chromosomes by heat denaturation and Giemsa staining). About 0.4–0.8 ml of peripheral blood was incubated in complete lymphocyte culture medium for 72 h. Metaphases were harvested by adding KaryoMAX™ Colcemid™ solution for 50 min followed by hypotonic KCl (0.075M) treatment for 20 min and fixation using standard 3:1 methanol and acetic acid fixative (Dutrillaux and Couturier, 1981). Thereafter, fluorescence in situ hybridization (FISH) assay was performed to characterize the sSMC, it was performed on patient’s and his mother’s metaphases obtained from whole blood cultures. Multi-FISH applying three probes as whole chromosome painting (wcp) probes for chromosomes 8 and 14 (homemade, labelled in Cyanine 5 (yellow) and diethylaminocoumaine (DEAC) (as described in Liehr and Claussen, 2002), and the subtelomeric 8pter in Spectrum Green (D8S504, Abbott/Vysis, Germany) specific to the subtelomeric region 8p23.3. Chromosomes were counterstained by 4′,6-diamidino-2-phenylindole (DAPI). FISH technique was performed according to standard procedures (Liehr, 2002). Image acquisition was done using a Zeiss Axioplan microscope equipped with ISIS software (MetaSystems, Altlussheim, Germany) and 15 metaphases were analyzed.
Results
Karyotype analyses showed an sSMC suspected to be derived from chromosome 14 in the patient (Figure 2) and a balanced translocation involving the chromosomes 8 and 14 in the mother (Figure 3). FISH experiments did confirm this balanced translocation in the mother classified as 46,XX,t(8;14)(p22.3;q21).ish t(8;14)(wcp14+,wcp8+,D8S504-;wcp14+,wcp8+,D8S504+) and a complex sSMC in the son, (Figure 4). Thus, this patient had a partial trisomy of the 8p22.3-8pter regionand short-arm centromere region and proximal part of the long arm of chromosome 14 (Figure 5). The molecular cytogenetic result for the patient was as follows: 47,XY,+der(14)t(8;14)(p22.3;q21)mat.ish der(14)(wcp14+,wcp8+,D8S504+) (Figure 5). After parents’ request, cytogenetic investigation of the older brother, aged 5 years and unaffected, showed that he is a carrier of the balanced maternal (Figure 6).
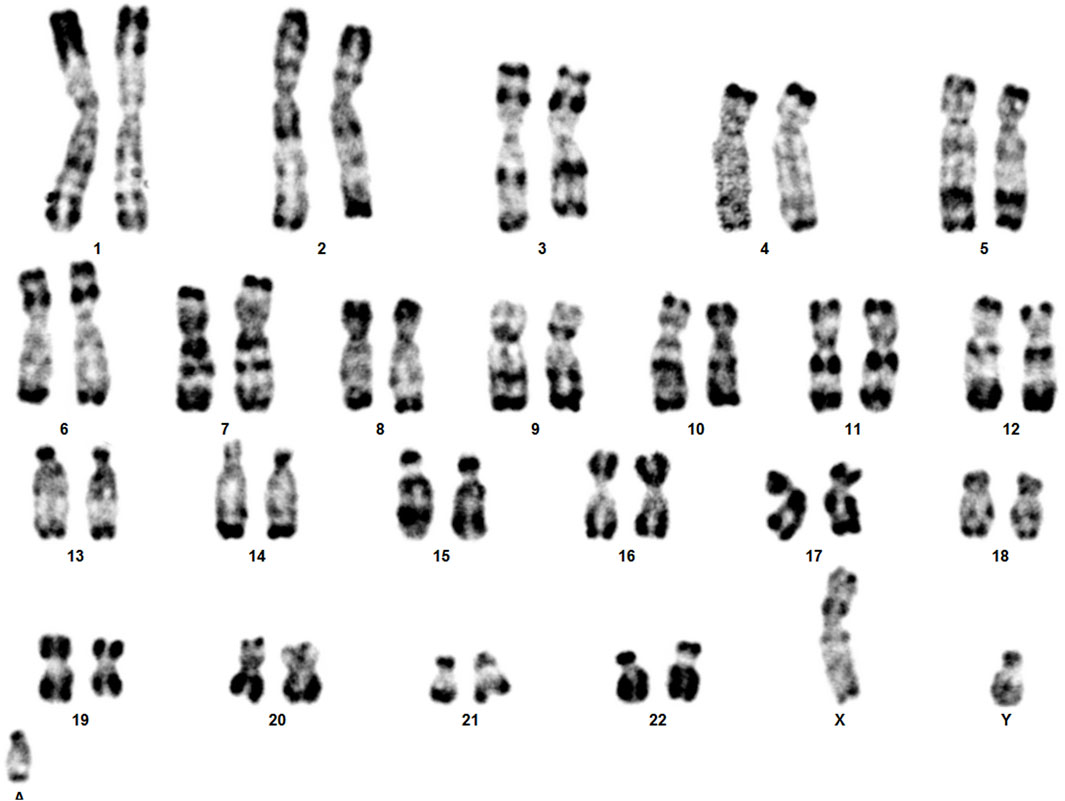
FIGURE 2. The karyotype of the patient showing sSMC.
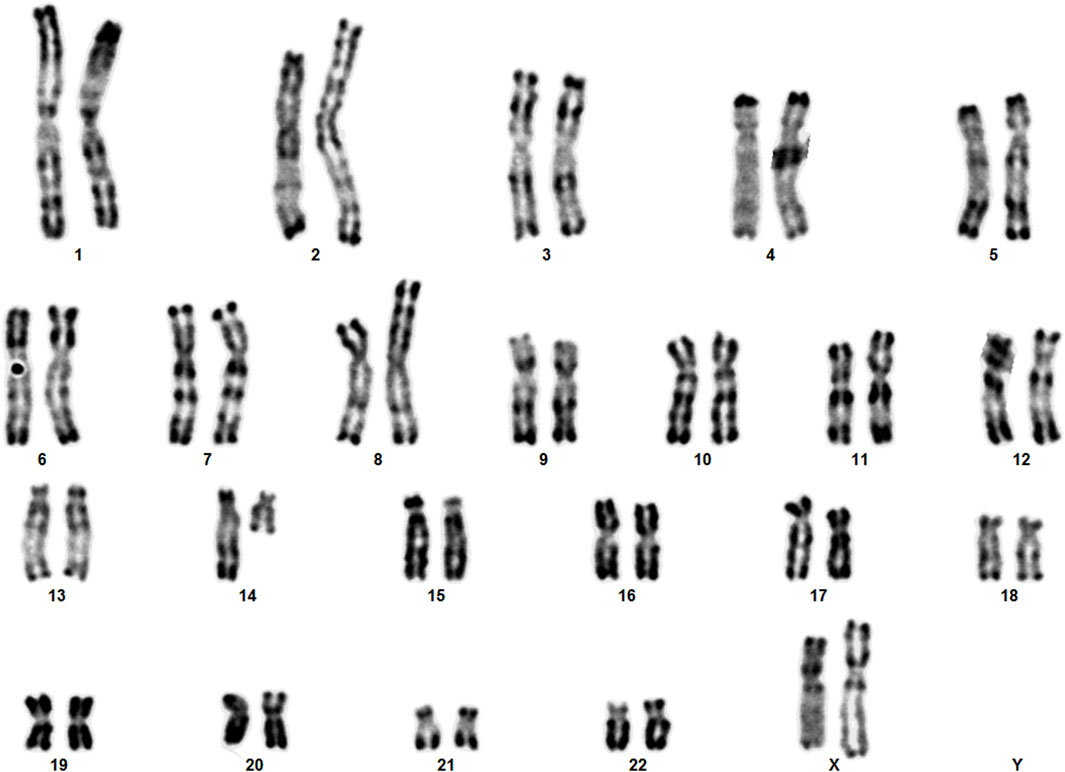
FIGURE 3. The karyotype of the mother showing the balanced translocation.

FIGURE 4. Result of molecular cytogenetics for the patient and his mother. For each (derivative) chromosome left inverted DAPI-banding, and right the FISH-results are shown.
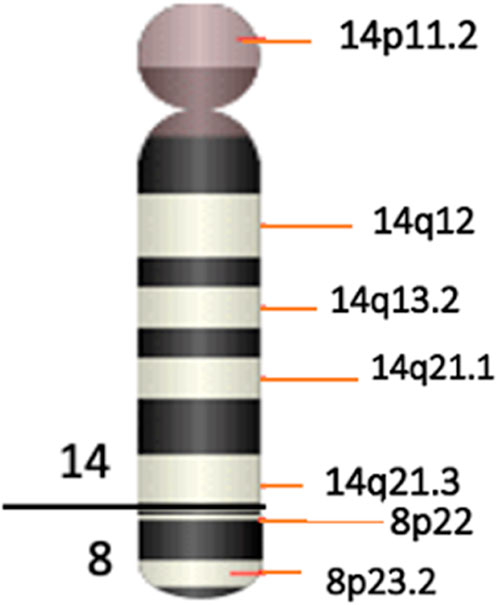
FIGURE 5. Ideogram of the sSMC(14) as a der(14)t(8;14)(p22.3;q21).
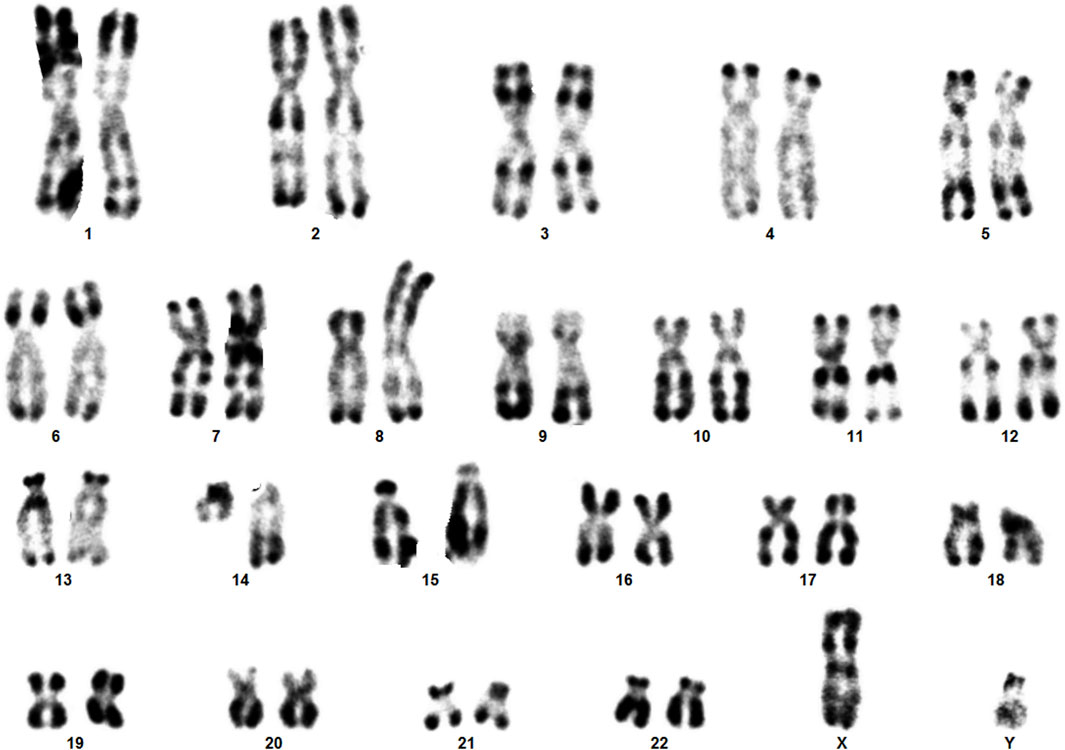
FIGURE 6. The karyotype of brother showing the balanced translocation.
Discussion
Genomic imbalances, encompassing deletions, duplications, triplications, or amplifications, have the potential to cause a spectrum of conditions including mental retardation and a range of congenital anomalies, with the specific outcomes’ contingent upon the source and type of genetic material involved. Such imbalances can also go undetected in case of being too small, and beyond the detection capability of banding cytogenetics (Liehr, 2009).
Here we have identified a rare complex sSMC originating from chromosomes 8 and 14. This case shows parallels with the first reported complex sSMC(14) described by (Guilherme et al., 2013) in 2013, which also resulted from a translocation between chromosomes 8 and 14 (Table 1). Moreover, two additional cases of complex sSMCs from translocations involving chromosomes 8 and 14 have been documented (Soudek et al., 1978; Moore et al., 1992), but the breakpoint locations differ from those in our patient, specifically at 8p and 14q [see also cases 14-Uc-10, 14-Uc-11, 14-Uc-26 (Liehr, 2023)]. There are overall 27 complex sSMCs derived from chromosome 14 (Liehr, 2023).
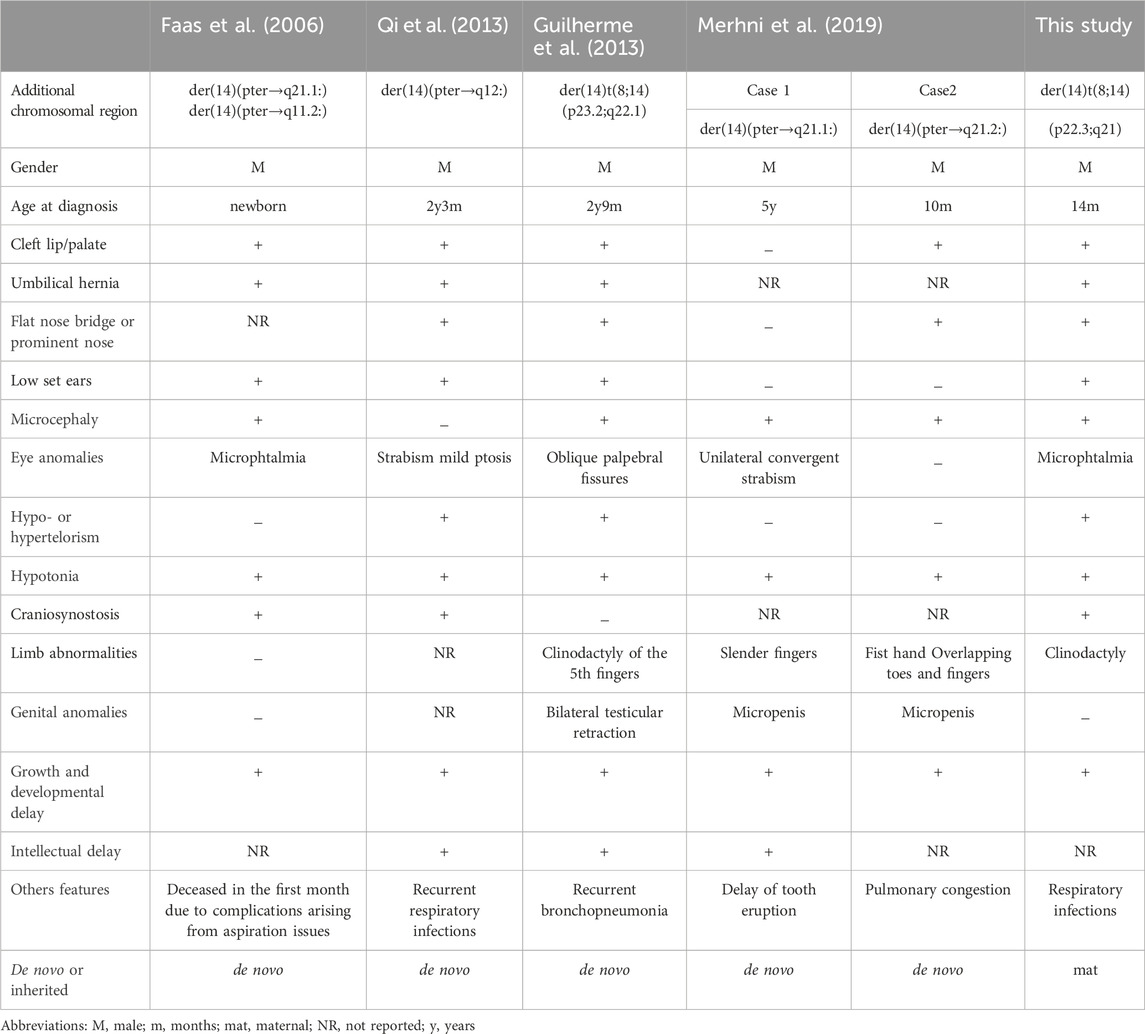
TABLE 1. Features of five patients with partial 14 trisomy.
As shown in Table 1 several clinical features such as growth delay, psychomotor development delay, hypotonia, facial dysmorphia, microcephaly, umbilical hernia were found in our proband and in patients with partial trisomy 8 and 14. We suggest that the predominant clinical features observed in our patient result mainly from trisomy 14q, as they are similar to pure proximal partial trisomy 14q cases (Merhni et al., 2019) also included in Table 1. Parts of clinical variability noted between the five patients could be due to the additional abnormalities observed, like trisomy 8p. Also, in case reported by (Faas et al., 2006) in 2006 there was an additional derivative chromosome 14 in a mosaic state.
The duplication of the genomic region 8p22p23.3 has been linked to instances where individuals are clinically normal or have a mild intellectual disability without notable dysmorphic features. This suggests that the genes in this region may have a limited influence on neurobehavioral and physical development (Brooks et al., 1998).
Thereby, we assume that the predominant clinical features observed in our patient result mainly from trisomy 14q. According to Mapview (https://www.ncbi.nlm.nih.gov/mapview), the duplicated region 14q11-q21 contains several genes, including 137 identified in the OMIM database. Among these, the genes FOXG1 (forkhead box G1), that plays a critical role in brain development. Studies have shown it to be dose-sensitive, meaning that variations in its expression levels can have significant impacts. FOXG1 is important both in the development of the brain during prenatal stages and in postnatal neurogenesis (Hettige and Ernst, 2019). The duplication of the FOXG1 gene has been linked to severe developmental retardation, as evidenced in various studies (Brunetti-Pierri et al., 2011). Meanwhile, the role of additional genes in the large sized involved regions cannot be excluded. Table 2 presents an overview of certain genes implicated in the symptoms exhibited by our patient.

TABLE 2. Candidate genes possibly being responsible for parts of phenotype from 14q11.2 to 14q21 region.
Conclusion
As a patient with sSMC(14) was indicative to identify a maternal balanced translocation, this case is a good example for high risk of recurrence in such familial cases. Thus, chromosomal analysis of patients and their parents and other relatives (like here the brother) are essential for correct genetic counseling.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by the comité d’éthique: Institutional Review Board Affiliation: National Institute of Health, Rabat, Morocco. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
FO: Investigation, Writing–original draft, Methodology. ZE: Writing–original draft. HB: Writing–original draft. IR: Writing–review and editing, Validation. ASb: Writing–review and editing. TL: Formal Analysis, Writing–review and editing. ASe: Writing–review and editing. AN: Validation, Writing–review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We thank the patient and his family.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Brooks, S. S., Genovese, M., Gu, H., Duncan, C. J., Shanske, A., and Jenkins, E. C. (1998). Normal adaptive function with learning disability in duplication 8p including band p22. Am. J. Med. Genet. 78 (2), 114–117. doi:10.1002/(sici)1096-8628(19980630)78:2<114::aid-ajmg3>3.3.co;2-6
Brunetti-Pierri, N., Paciorkowski, A. R., Ciccone, R., Mina, E. D., Bonaglia, M. C., Borgatti, R., et al. (2011). Duplications of FOXG1 in 14q12 are associated with developmental epilepsy, mental retardation, and severe speech impairment. Eur. J. Hum. Genet. 19 (1), 102–107. doi:10.1038/ejhg.2010.142
Faas, B. H., Van, D. D. J., Wunderink, M. I., Merkx, G., and Brunner, H. G. (2006). Multiple congenital abnormalities in a newborn with two supernumerary marker chromosomes derived from chromosome 14. Genet. Couns. 17, 349–357.
Guilherme, R. S., Dutra, A. R. N., Perez, A. B. A., Takeno, S. S., Oliveira, M. M., Kulikowski, L. D., et al. (2013). First Report of a Small Supernumerary der(8;14) Marker Chromosome. Cytogenet Genome Res. 139 (4), 284–288. doi:10.1159/000348743
Hettige, N. C., and Ernst, C. (2019). FOXG1 dose in brain development. Front. Pediatr. 7, 482. doi:10.3389/fped.2019.00482
Liehr, T. (2009). Small supernumerary marker chromosomes (sSMCs): a spotlight on some nomenclature problems. J. Histochem Cytochem 57, 991–993. doi:10.1369/jhc.2009.954370
Liehr, T. (2023). Small supernumerary marker chromosomes. Avaliable at: https://cs-tl.de/DB/CA/sSMC/0-Start.html (Accessed 12 December, 2023).
Liehr, T., Cirkovic, S., Lalic, T., Guc-Scekic, M., de Almeida, C., Weimer, J., et al. (2013). Complex small supernumerary marker chromosomes–an update. Mol. Cytogenet. 6 (1), 46–6. doi:10.1186/1755-8166-6-46
Liehr, T., and Claussen, U. (2002). Current developments in human molecular cytogenetic techniques. Curr. Mol. Med. 2 (3), 283–297. doi:10.2174/1566524024605725
Liehr, T., Claussen, U., and Starke, H. (2004). Small supernumerary marker chromosomes (sSMC) in humans. Cytogenet Genome Res. 107 (1–2), 55–67. doi:10.1159/000079572
Liehr, T., Karamysheva, T., Merkas, M., Brecevic, L., Hamid, A. B., Ewers, E., et al. (2010). Somatic mosaicism in cases with small supernumerary marker chromosomes. Curr. Genomics. Sep. 11 (6), 432–439. doi:10.2174/138920210793176029
Liehr, T., and Weise, A. (2007). Frequency of small supernumerary marker chromosomes in prenatal, newborn, developmentally retarded and infertility diagnostics. Int. J. Mol. Med. 19 (5), 719–731. doi:10.3892/ijmm.19.5.719
Merhni, H., Zerkaoui, M., Natiq, A., Sbiti, A., Liehr, T., and Sefiani, A. (2019). Constitutional partial proximal trisomy 14q11. 2 to 14q21: two new Moroccan cases and review of the literature. OBM Genet. 3 (3), 1–17. doi:10.21926/obm.genet.1903085
Moore, C. M., Barnum, K., Kaye, C. I., Kagan-Hallett, K. S., and Liang, J. C. (1992). Trisomy 8p: unusual origin detected by fluorescence in situ hybridization. Hum. Genet. 89, 307–310. doi:10.1007/BF00220547
Qi, M., Zhao, Y., and Wang, Y. (2013). A new small supernumerary marker chromosome involving 14pter→ q12 in a child with severe neurodevelopmental retardation: case report and literature review. Gene 531 (2), 457–461. doi:10.1016/j.gene.2013.08.084
Soudek, D., Hunter, P., O'shaughnessy, S., Simpson, N. E., and Soudek, V. (1978). Familial translocation t (8; 14) with a case of tertiary trisomy,+ 14q. Birth Defects Orig. Artic. Ser. 14 (6C), 309–315.
Keywords: complex small supernumerary marker chromosome (sSMC), partial trisomy, molecular cytogenetics, clinical features, chromosome 8 and 14
Citation: Ouboukss F, El Amrani Z, Bouchahta H, Ratbi I, Sbiti A, Liehr T, Sefiani A and Natiq A (2024) A maternally derived complex small supernumerary marker chromosome involving chromosomes 8 and 14: case report and review of the literature. Front. Genet. 15:1331676. doi: 10.3389/fgene.2024.1331676
Received: 01 November 2023; Accepted: 23 January 2024;
Published: 23 February 2024.
Edited by:
Paulo Ricardo Gazzola Zen, Federal University of Health Sciences of Porto Alegre, BrazilReviewed by:
Patricia Trevisan, University of Colorado, United StatesRafaella Mergener, Federal University of Health Sciences of Porto Alegre, Brazil
Copyright © 2024 Ouboukss, El Amrani, Bouchahta, Ratbi, Sbiti, Liehr, Sefiani and Natiq. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fatima Ouboukss, ZmF0aW1hb3Vib3Vrc3NAZ21haWwuY29t