 Dorota M. Nowak-Malczewska
Dorota M. Nowak-Malczewska Joanna Swierkowska
Joanna Swierkowska Marzena Gajecka
Marzena Gajecka
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Genet. , 26 February 2024
Sec. Applied Genetic Epidemiology
Volume 15 - 2024 | https://doi.org/10.3389/fgene.2024.1301676
This article is part of the Research Topic Women in Applied Genetic Epidemiology View all 7 articles
Introduction: Keratoconus (KTCN) is a corneal ectasia, characterized by a progressive thinning and protrusion of the cornea, with a complex etiology involving genetic, behavioral, lifestyle, and environmental factors. Previous studies indicated that microRNAs (miRNAs) could be involved in KTCN pathogenesis. This in silico study aimed to identify precursor microRNAs (pre-miRNAs) differentially expressed in KTCN corneas and to characterize mature miRNAs and their target genes.
Materials and methods: Expression levels of pre-miRNAs were retrieved from our previously obtained RNA sequencing data of 25 KTCN and 25 non-KTCN human corneas (PMID:28145428, PMID:30994860). Differential expression with FDR ≤0.01 and ≥1.5-fold changes were considered significant. Lists of target genes (target score ≥90) of mature miRNAs were obtained from miRDB. Revealed up-/downregulated miRNAs and their target genes were assessed in databases and literature. Enrichment analyses were completed applying the DAVID database.
Results: From a total of 47 pre-miRNAs, six were remarkably upregulated (MIR184, MIR548I1, MIR200A, MIR6728, MIR429, MIR1299) and four downregulated (MIR6081, MIR27B, MIR23B, MIR23A) in KTCN corneas. Out of the 1,409 target genes, 220 genes with decreased and 57 genes with increased expression levels in KTCN samples vs non-KTCN samples were found. The extracellular matrix (ECM) organization, response to mechanical stimulus, regulation of cell shape, and signal transduction processes/pathways were identified as distinctive in enrichment analyses. Also, processes associated with the regulation of transcription and DNA binding were listed.
Conclusion: Indicated miRNAs and their target genes might be involved in KTCN pathogenesis via disruption of crucial molecular processes, including ECM organization and signal transduction.
Keratoconus (KTCN) is a corneal ectasia characterized by a progressive thinning and anterior protrusion of the cornea leading to reduced vision and changed refractive power (Reinstein et al., 2015). KTCN is typically bilateral but asymmetrical, with an onset usually occurring in late adolescence or early adulthood. KTCN is a multifactorial disease with a complex etiology involving multiple genes, as well as behavioral, lifestyle, and environmental factors (Karolak and Gajecka, 2017; Jaskiewicz et al., 2023c). Previously, we have performed RNA sequencing (RNA-seq) on corneas derived from 25 Polish patients with KTCN and 25 control individuals and revealed disruption of collagen synthesis and downregulation of core elements of the TGF-β, Hippo, and Wnt pathways in KTCN corneas (Kabza et al., 2017b). Then, we performed reanalysis of the RNA-seq dataset and have indicated 12 downregulated and 6 upregulated genes in KTCN that overlapped or were located in the near vicinity of the identified differentially methylated regions (Kabza et al., 2019). Moreover, analysis of the KTCN transcriptome allowed us to identify long non-coding RNAs that might be involved in KTCN etiology (Szczesniak et al., 2017).
Here, to complement both our in silico analysis of microRNAs (miRNAs) (Nowak and Gajecka, 2015) and our experimental transcriptomic study on KTCN and non-KTCN corneas (Kabza et al., 2017b; 2019), we retrieved differentially expressed precursor miRNAs (pre-miRNA) from our RNA-seq data. Mentioned miRNAs belong to a highly conserved class of small non-coding RNAs, about 22 nucleotides in length, which play substantial roles in regulating gene expression. The majority of miRNAs are transcribed from miRNA encoding genes by a RNA polymerase II to form primary miRNAs (pri-miRNAs). Then nuclear RNase III enzyme Drosha and its cofactor process pri-miRNAs into 60 nt length precursor miRNAs (pre-miRNAs) and further processing by RNase III enzyme Dicer leads to creation of mature miRNAs (Ghorai and Ghosh, 2014; Fuchs Wightman et al., 2018). Previously, miRNAs have been studied in KTCN mainly with the focus on miR-184 that was reported to be related to this disease (Hughes et al., 2011; Lechner et al., 2013; Bykhovskaya et al., 2015; Farzadfard et al., 2016; Stachon et al., 2022; Zhang et al., 2022).
The profile of miRNAs in KTCN has been already studied, however, there are substantial sequence variation and gene expression differences between populations and even families with KTCN (Karolak et al., 2011; 2016; Czugala et al., 2012), so the identified features characteristic for Polish patients, may, but do not have to, be a repetition of the already published results. As far as we are concerned, the miRNA profile in the corneas of KTCN patients from the Polish population has never been published. Thus, the purpose of our in silico study was to identify differentially expressed pre-miRNAs, and characterize mature miRNAs, their target genes and enriched molecular processes and signaling pathways to indicate those involved in KTCN pathogenesis.
Transcriptomic data from previously performed RNA-seq of 25 KTCN and 25 non-KTCN human corneas (Kabza et al., 2017b; Kabza et al., 2017a) was assessed. The 25 KTCN corneal tissues were derived from non-related Polish patients undergoing a keratoplasty procedure. The control 25 non-KTCN corneas were collected from patients, who were referred for corneal transplantation due to other reasons, such as bullous keratopathy, corneal scarring, perforations and ulcers. Medical examination of patients, clinical metadata, the handling of the corneas, and RNA extraction have been described elsewhere (Kabza et al., 2017b).
RNA-seq data was originally analyzed in 2017 (Kabza et al., 2017b) and then underwent reanalysis with the use of a modified pipeline in 2019 (Kabza et al., 2019). In the current study the data of the reanalysis (Kabza et al., 2019) was used.
Expression data for pre-miRNA was retrieved from the RNA-seq datasets that have been deposited in the Gene Expression Omnibus with the accession number GSE77938. An edgeR-limma workflow was employed to analyze RNA-seq data, utilizing gene-level counts as input (Robinson et al., 2010; Ritchie et al., 2015). The workflow encompasses pre-processing and exploratory data analysis, culminating in identifying differentially expressed genes and gene signatures. The analysis adhered to a previously established protocol (Law et al., 2016). Genes were deemed differentially expressed if they met the criteria of a false discovery rate (FDR) threshold value of 0.01 and a fold change of ≥1.5. The detailed bioinformatics workflow for this analysis was described in our previous publication (Kabza et al., 2019).
Differentially expressed miRNA encoding genes were searched in miRBase1, Mouse Genome Informatics (MGI)2, and available literature data.
To assess sequence variation in miRNA encoding genes, variants derived from Ensembl exons were called by FreeBayes (Garrison and Marth, 2012). The obtained DNA sequence variants were filtered according to the Utah Genome Project Variant Calling Protocol (Garrison and Marth, 2012) and compared to a list of pre-miRNA coordinates by an in-home Python script.
An online database miRDB3 (Liu and Wang, 2019), containing predicted miRNAs’ targets in a human, was searched. Target genes were identified using the MirTarget2 algorithm, developed through the analysis of thousands of genes regulated by miRNAs using the support vector machines (Liu and Wang, 2019). A target score between 50 and 100 is assigned to each of the probable target genes, with higher scores indicating more reliable predictions of binding sites within the given genes for each miRNA (Liu and Wang, 2019). A score of ≥80, indicating the most likely real target genes, is recommended in the miRDB. Here, binding sites with a score value of 90 or greater were selected for further analysis to increase the prediction reliability.
The Kyoto Encyclopedia of Genes and Genomes (KEGG), REACTOME and Gene Ontology (GO) analyses were conducted applying the Database for Annotation, Visualization, and Integrated Discovery (DAVID) (Huang et al., 2009; Sherman et al., 2022). Genes with statistically significant increase and decrease in expression level were subjected to separate analyses in the aspect of serving as the targets for the analyzed miRNAs. This approach allowed for an exploration of the regulatory interactions between miRNAs and their corresponding target genes, shedding light on the mechanisms underlying gene expression in the cornea. The focus was given on biological process (BP), molecular function (MF), cellular component (CC) and KEGG/REACTOME pathways. p-value <0.05 and number of genes for each of identified GO terms/processes/pathways ≥3 were considered as the cutoff criteria in GO, REACTOME, and KEGG analyzes.
The obtained results were also compared to the results from our previously performed overrepresentation analysis of molecular pathways among genes upregulated and downregulated in KTCN (Kabza et al., 2017b).
A total of 47 pre-miRNAs in the studied corneal samples were identified (Supplementary Table S1). Six pre-miRNAs were found to be significantly upregulated, while four were significantly downregulated (Table 1), comparing KTCN vs non-KTCN samples. Those 10 pre-miRNAs are processed by Dicer into a total of 15 mature miRNAs, as five of these precursors form miRNAs from either the 3′or 5′end, as indicated in Table 1. Upregulation or downregulation of a pre-miRNA was indicated by the value of logFC. The pre-miRNA with the most altered expression was hsa-miR-184, which was found to be upregulated.
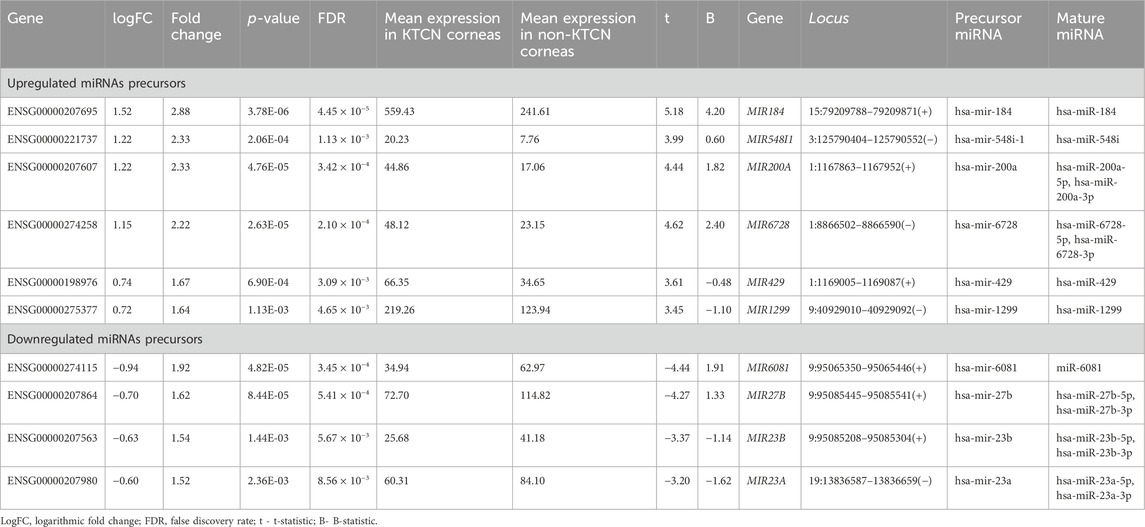
TABLE 1. Differentially expressed pre-miRNAs in KTCN corneas vs non-KTCN corneas. Upregulation and downregulation of miRNA is indicated by the value of logFC.
Of the analyzed pre-miRNAs, four are located intra-intronic. MIR6728 is located in the intron of the enolase 1 (ENO1) gene at chromosome 1. The mean expression of ENO1 was 2,649.65 transcripts per million (TPM) in KTCN corneas, and 1,251.51 TPM in non-KTCN corneas (2.06-fold change, FDR p-value = 2.47 × 10−7). MIR6081, MIR27B, and MIR23B genes (with levels of expression presented in Table 1) are located in introns of the aminopeptidase O gene (putative, AOPEP, expression level: 53.10 TPM in KTCN vs. 75.47 TPM in non-KTCN corneas) at chromosome 9. In addition, four genes MIR184, MIR548I1, MIR200A and MIR429 (with levels of expression shown in Table 1) are located within genes encoding lncRNAs, including ANKRD34C-AS1, LOC105374312, LOC124903818 and again in LOC124903818, respectively.
Searching the MGI database, we found that MIR23B, and MIR23A, were expressed in murine eyes at low and/or medium levels. MIR200A was found to be expressed at low level in RNA-seq, MIR184 and MIR27B were detected in RNA in situ/RT-PCR but not in RNA-seq, and MIR429 has not been expressed. All the mentioned MGI data is available at the following accession numbers: E-GEOD-33141, E-GEOD-63810, E-MTAB-6133, E-MTAB-9143; J:190177, J:124661, J:151925, J:124661, J:310450. There was no data available in the MGI for MIR548I1, MIR6728, MIR1299, and MIR6081 genes.
Assessing the DNA sequence variation, no variants were found within genes encoding the evaluated pre-miRNAs.
We further investigated the potential target genes (listed in Supplementary Table S2) for the selected 15 mature miRNAs (listed in Table 1, in the last column) using the miRDB database, and a total of 1,911 interactions were identified (Supplementary Table S2). Of these, 420 genes were regulated by more than one of the identified miRNAs, with five genes being targets of four of the miRNAs (TMEM135, DTNA, NDFIP2, NCOA2), 76 genes being targets of three miRNAs, and 339 genes being targets of two miRNAs. However, not all of these genes exhibited statistically significant altered expression levels. Among the genes regulated by these four miRNAs (miR-429, miR-27b-3p, miR-23b-3p, miR-23a-3p), only DTNA showed a statistically significant decrease in expression within the studied KTCN corneas. Specifically, the precursor coding miR-429 was detected with increased expression level, while the precursors for miR-27b-3p, miR-23b-3p, and miR-23a-3p displayed decreased expression levels in KTCN corneas.
Out of the 1,409 genes regulated by the analyzed miRNAs, 220 genes with significantly decreased expression and 57 with increased expression levels in KTCN samples were identified comparing KTCN to non-KTCN samples. Out of the 220 target genes displaying significantly decreased expression levels, 130 genes were found to interact with upregulated miRNAs, 75 genes with downregulated miRNAs, and 15 genes with both up- and downregulated miRNAs (Figure 1). Also, out of the 57 genes displaying increased expression, 18 genes were found to interact with upregulated miRNAs, 34 genes with downregulated miRNAs, and five genes with both up- and downregulated miRNAs (Figure 1).
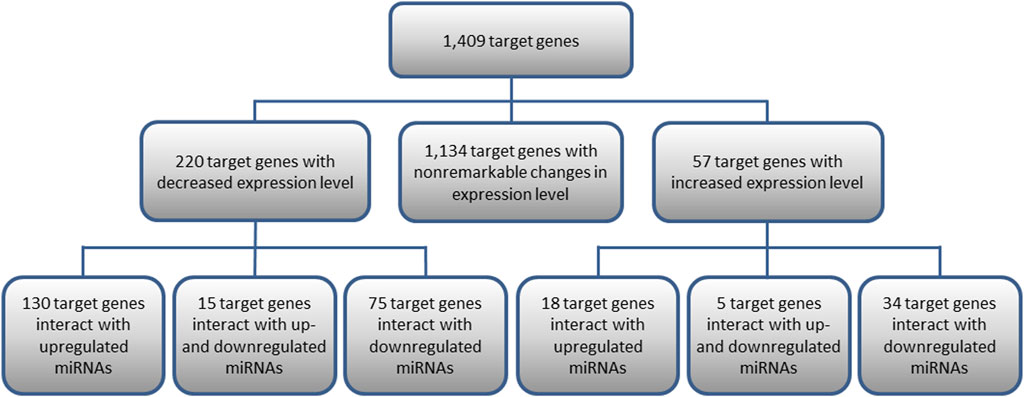
FIGURE 1. Target genes of 10 differentially expressed pre-miRNAs and their interactions with miRNAs. Presented are target genes with decreased or increased expression levels found to be statistically significant in KTCN corneas compared to non-KTCN corneas.
The enrichment analysis of miRNA’s target genes was conducted using the DAVID database and encompassed 220 genes with decreased and 57 with increased expression levels in KTCN corneas. Specifically, the downregulated genes were associated with 97 GO BP terms, 34 GO CC terms, and 30 MF terms. Furthermore, they were categorized into 24 processes/pathways within the KEGG database and 30 processes/pathways within the REACTOME database. Similarly, the upregulated target genes were linked to four GO BP terms, four GO CC terms, one KEGG pathway, and one REACTOME pathway (Supplementary Table S3).
Among the downregulated target genes, the three GO BP terms showing the lowest p-values were the ECM organization (13 genes, p-value = 5.95 × 10−7), response to mechanical stimulus (8 genes, p-value = 2.03 × 10−6), and regulation of cell shape (11 genes, p-value = 3.96 × 10−6). The GO MF indicated mainly molecular processes associated with the regulation of transcription and DNA binding (Figure 2). The REACTOME analysis pointed towards pathways associated with the ECM organization (15 genes, p-value = 3.38 × 10−5) and signal transduction (56 genes, p-value = 3.85 × 10−5). Compared to downregulated genes, fewer processes/pathways involving upregulated target genes were identified (Figure 2).
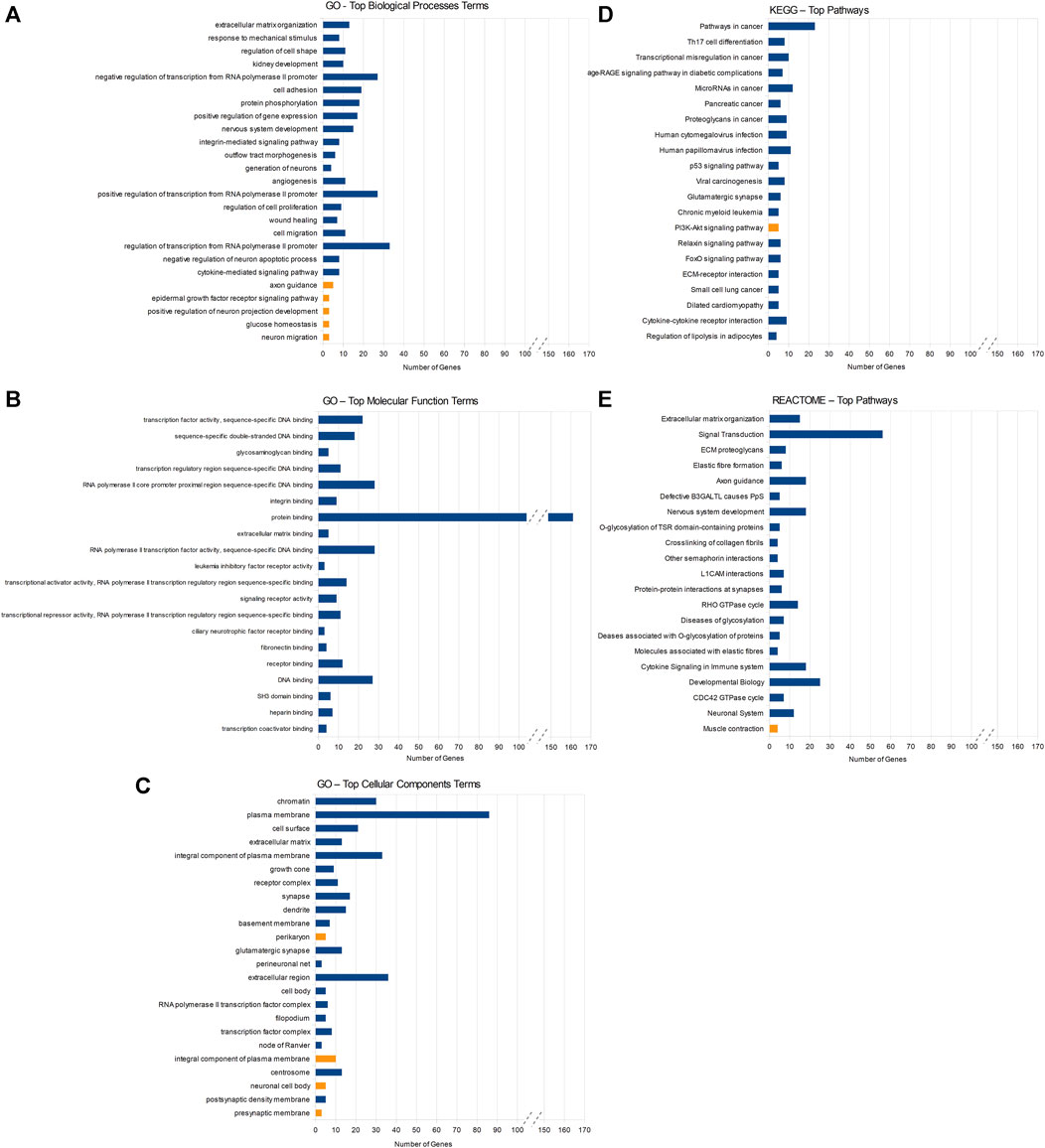
FIGURE 2. Results of DAVID enrichment analyses of miRNAs’ target genes in different databases: Gene Ontology (GO) for biological processes (A), molecular function (B), cellular component (C), the Kyoto Encyclopedia of Genes and Genomes (KEGG) (D), and REACTOME (E). Blue rectangles indicate pathways enriched with target genes of downregulated miRNAs and yellow rectangles stand for pathways enriched with target genes of upregulated miRNAs. Statistical significance decreases from top to bottom in all the figures.
We also compared the current data and results to the outcomes from our previous pathway overrepresentation analysis of the whole corneal transcriptome. In both, the analysis of the entire KTCN transcriptome and the analysis of the target genes for the miRNAs only, the majority of genes was assigned to the signal transduction pathway (Supplementary Table S4). Out of the genes found in the prominent pathways identified in the whole-transcriptome analysis, 34 genes exhibited regulation by the miRNAs assessed here. Among the significant elements in previously discussed pathways (TGF-β, Hippo, and Wnt pathways), two genes (TGFBR2 and FZD7) with decreased expression in KTCN corneas are controlled by miRNAs, whose precursors showed altered expression. TGFBR2 is a potential target gene of miR-23b-3p and miR-23a-3p, for which we observed decreased precursor expression in the analyzed samples. Whereas FZD7 is a target gene of miR-548i, which precursor exhibited significantly increased expression level.
We identified six significantly upregulated and four downregulated in KTCN pre-miRNAs in the RNA-seq data of the studied corneal samples derived from patients with KTCN and controls. The highest difference in expression between KTCN and non-KTCN corneas was reached by the upregulated miR-184, previously reported to be involved in KTCN and other corneal abnormalities (Hughes et al., 2011; Lechner et al., 2013; Bykhovskaya et al., 2015; Farzadfard et al., 2016). This miRNA has been found to be one of the most abundant miRNAs in the cornea and expressed in the corneal epithelium and stroma (Abu-Amero et al., 2015). The miR-184 regulates the expression of genes involved in cell proliferation, differentiation, and apoptosis, suggesting that it is a key regulator of corneal development and homeostasis (Abu-Amero et al., 2015). Previously, the c.57 C > T mutation in miR-184 has been identified to be responsible for inherited corneal and lens abnormalities in a five-generation family from Spain and in an autosomal-dominant form of severe anterior KTCN and early-onset anterior polar cataract in a Northern Irish family (Hughes et al., 2011; Bykhovskaya et al., 2015). On the contrary, in our miRDB search we identified only three miR-184 target genes with scores ≥90, namely, SETD9, NUS1, and CES2, and despite the considerable upregulation of pre-miR184, none of these genes showed altered expression in the analyzed samples. Therefore, upregulation of miR-184 might not have played a crucial role in the KTCN pathogenesis in our patients, especially that we found no sequence variants in the seed region of the MIR184 gene. Also no mutations were reported in the miR-184 seed region in Saudi Arabian patients with KTCN (Abu-Amero et al., 2015) and no association with sporadic KTCN in Greek patients has been revealed in genotyping of rs41280052 located in MIR184 (Moschos et al., 2016). It is worth emphasizing that although in our in silico analyses no target genes with altered expression for miR-184 were indicated, there are various genes that may be regulated by this miRNA. An example of such a gene is FZD7, which regulation by miRNA and, consequently, its impact on retinal neovascularization through the Wnt pathway, has been studied by Takahashi et al. (Takahashi et al., 2015). In our transcriptomic data, a significant decrease in FZD7 expression was observed in KTCN patients. It is possible that the increased level of mir-184 affects the Wnt pathway by regulating FZD7 expression. Therefore, it is important to consider the limitations of computer-based analyses, which may not always provide a comprehensive understanding of the investigated issue.
The identified MIR6728, MIR23B, MIR27B, and MIR6081 miRNA genes encode intronic miRNA that are transcribed together with ENO1 (MIR6728) and AOPEP (MIR23B, MIR27B, MIR6081) genes. MIR6728 is located in the locus 1p36.23–36.21 that has been identified in the linkage analysis in an Australian KTCN family (Burdon et al., 2008) and has been also discussed in our previous paper (Nowak and Gajecka, 2015). The ENO1 gene encodes enolase 1 (α-enolase) that functions as a glycolytic enzyme and also as a structural lens protein (tau-crystallin) in the monomeric form. In a previous study by Srivastava et al., relatively low or negligible levels of α-enolase and β-actin were detected in the epithelial cells of KTCN corneas compared to the normal levels observed in non-KTCN corneas (Srivastava et al., 2006). On the contrary, in our study the transcript of ENO1 reached a fold change of 2.06 with the higher expression level in KTCN corneas vs non-KTCN corneas. On the other hand, in the study by Burdon et al. no mutations were identified in ENO1 (Burdon et al., 2008). Statistically significant increased expression was also observed for the MIR6728 gene located within ENO1. Furthermore, we detected remarkable changes in the expression of target genes controlled by miR-6728-5p and/or miR-6728-3p, including the LOX gene, which is widely discussed in the context of KTCN. LOX encodes lysyl oxidase, an enzyme responsible for catalyzing the cross-linking of collagen and elastin fibers in the ECM. Previous studies have indicated that LOX expression is often decreased in the corneal tissue of individuals with KTCN (Dudakova et al., 2012; Shetty et al., 2015; Takaoka et al., 2016). Reduced LOX activity can lead to insufficient cross-linking of collagen and elastin fibers in the cornea, which in turn can result in corneal weakening and thinning (Dudakova et al., 2012). Our analyzes suggest that increased MIR6728 level, which encodes miR-6728-5p, might result in a substantial downregulation of LOX expression.
The second gene with an intronic miRNA encoding gene is AOPEP that encodes aminopeptidase O (putative) and is a member of the M1 zinc aminopeptidase family. MIR27B and MIR23B are in the miR-23b/27b/24–1 cluster. Two miR-23/27/24 clusters exist in the vertebrate genome: an intergenic miR-23a/27a/24–2 cluster and an intronic miR-23b/27b/24–1 cluster (Zhou et al., 2011). MiRNAs encoded by the miR-23/27/24 gene clusters are enriched in endothelial cells and highly vascularized tissues. Interestingly, one of our indicated miRNA encoding genes, MIR6081, is localized in a close proximity to this cluster. Inhibition of miR-23 and miR-27 represses angiogenesis in vitro, postnatal retinal vascular development and choroidal neovascularization in response to laser injury in a mouse model (Zhou et al., 2011). MiR-23 and miR-27 enhance angiogenesis by promoting angiogenic signaling through targeting Sprouty2 and Sema6A proteins (Zhou et al., 2011). Previously, expression levels of miR-23a-3p and miR-23b-3p have been found to be significantly upregulated in patients with senile cataract when compared with the control group (Kim et al., 2021). Overexpression of miR-23b-3p also promoted the proliferation, migration, and epithelial-mesenchymal transition of lens epithelial cells by targeting Sprouty2 (Liu et al., 2019). Furthermore, under oxidative stress, when the activation of apoptosis of lens epithelial cells may lead to the opacity of the lens and accelerate the progression of cataract, miR-23b-3p regulated apoptosis and autophagy via suppressing SIRT1 in these cells (Zhou et al., 2019). It has been indicated that miR-23a protects retinal pigment epithelium cells against oxidative damage through regulation of Fas cell surface death receptor or glutaminase-1 and glutamine uptake (Lin et al., 2011; Li et al., 2016; Zhou et al., 2020). High gene copy number of miR-23a was also associated with susceptibility to acute anterior uveitis (Yang et al., 2017). Then, miR-27b inhibited adipogenesis in orbital fibroblasts from patients with Graves’ orbitopathy (Jang et al., 2019). However, we did not observe changes in expression level of the mentioned SEMA6A, SPRY2, and SIRT1 genes in our data.
Considering other identified miRNAs, miR-429 plays an important role in hypoxia-induced retinopathy by inhibiting the HPSE-VEGF pathway and was downregulated in pterygium (Engelsvold et al., 2013; Xu H. et al., 2022). Also, members of the miR-200 family, including miR-200a, were downregulated and could be potential regulators of epithelial-mesenchymal transition in pterygium (Engelsvold et al., 2013). Also, miR-200a could play a protective role in the glaucoma-induced optical nerve injury by inhibiting the FGF7-mediated MAPK signaling pathway (Peng et al., 2019).
Performed pathway enrichment analyses highlighted the significance of our previously obtained results concerning the processes and signaling pathways involved in KTCN pathogenesis. Enrichment analyses of downregulated target genes pointed to several molecular processes and signaling pathways with the highest implication for ECM organization, response to mechanical stimulus, regulation of cell shape, and signal transduction. Also, GO terms enrichment analysis indicated mainly molecular processes associated with the regulation of transcription processes and DNA binding were revealed in the GO assessment. This is in line with our previous studies on KTCN corneas as we had identified differentially expressed genes that were involved in ECM organization, and signal transduction, including Wnt signaling pathways, disrupted in KTCN, assessing transcriptomic (Kabza et al., 2017b; Karolak et al., 2020b), genomic (Karolak et al., 2020a), and mitochondrial features (Nowak-Malczewska et al., 2021). Moreover, in our continuous study on additional corneal samples derived again from unrelated Polish patients with KTCN, we have identified both coding and non-coding sequence variation in genes that are involved, among others, in ECM and Wnt signaling (Jaskiewicz et al., 2023a). Cell-ECM interactions and ECM processing, being the critical elements of the wound healing process, were altered in corneal topographic regions of corneal epithelium from 23 patients with KTCN (Jaskiewicz et al., 2023b). Furthermore, we identified changes in DNA methylation level in WNT3 and WNT5A genes encoding Wnt ligands, which might be an explanation for the Wnt signaling pathway dysregulation in KTCN (Kabza et al., 2019). Several other studies support our results (Khaled et al., 2018; Xu L. et al., 2022; Dou et al., 2022; Akoto et al., 2023), including Bykhovskaya et al. claiming that the development of KTCN results in deregulation of gene expression of ECM and adhesion molecules (Bykhovskaya et al., 2016). The result highlighting the pathway of response to mechanical stimuli is consistent with the existing literature and our current studies on environmental factors and habits of patients with KTCN, which have pointed to repetitive mechanical corneal injuries caused by eye rubbing as a factor contributing to the development of KTCN, potentially through the alteration of gene expression in corneal epithelium (Najmi et al., 2019; Jaskiewicz et al., 2023c).
The limitation of our study is that the project was based on the RNA-seq data instead of the data of the small RNA sequencing. Thus, we analyzed expression levels of pre-miRNAs instead of the experimentally obtained mature miRNAs. Further experimental studies, including functional analyses were beyond the scope of this project. It remains possible that KTCN development might have led to changes in the expression level of these miRNAs and not the opposite. One possibility is the influence of the impaired wound healing process on the observed changes in pre-miRNA expression and, consequently, their influence on processes that are crucial in the development of KTCN.
Summarizing, in this study on miRNAs in corneas of KTCN patients from the Polish population we identified pre-miRNAs expressed in KTCN corneas and characterized in silico the mature miRNAs, their target genes and molecular processes/signaling pathways that could be involved in KTCN pathogenesis. The identified differentially expressed miRNAs might contribute to KTCN pathogenesis via disruption of ECM organization and signal transduction pathways that we have already indicated as causative in our previous KTCN research.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The studies involving humans were approved by the Institutional Review Boards at Poznan University of Medical Sciences in Poland. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
DN-M: Investigation, Methodology, Writing–original draft. JS: Investigation, Methodology, Writing–original draft. MG: Funding acquisition, Supervision, Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Supported by the National Science Centre in Poland (2018/31/B/NZ5/03280 to MG).
We are grateful to our team members, Drs. Michal Kabza, Justyna A. Karolak, Malgorzata Rydzanicz, and Rafal Ploski for processing experimental samples and raw data in our previous projects that the current work is based on.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1301676/full#supplementary-material
BP, biological process; CC, cellular component; ECM, extracellular matrix; FDR, false discovery rate; KTCN, keratoconus; MF, molecular function; miRNAs, microRNAs; pre-miRNAs, precursor miRNAs; pri-miRNAs, primary miRNAs; TPM, transcripts per million.
2http://www.informatics.jax.org/
Abu-Amero, K. K., Helwa, I., Al-Muammar, A., Strickland, S., Hauser, M. A., Allingham, R. R., et al. (2015). Screening of the seed region of MIR184 in keratoconus patients from Saudi arabia. Biomed. Res. Int. 2015, 604508. doi:10.1155/2015/604508
Akoto, T., Cai, J., Nicholas, S., McCord, H., Estes, A. J., Xu, H., et al. (2023). Unravelling the impact of cyclic mechanical stretch in keratoconus-A transcriptomic profiling study. Int. J. Mol. Sci. 24, 7437. doi:10.3390/ijms24087437
Burdon, K. P., Coster, D. J., Charlesworth, J. C., Mills, R. A., Laurie, K. J., Giunta, C., et al. (2008). Apparent autosomal dominant keratoconus in a large Australian pedigree accounted for by digenic inheritance of two novel loci. Hum. Genet. 124, 379–386. doi:10.1007/s00439-008-0555-z
Bykhovskaya, Y., Caiado Canedo, A. L., Wright, K. W., and Rabinowitz, Y. S. (2015). C.57 C > T mutation in MIR 184 is responsible for congenital cataracts and corneal abnormalities in a five-generation family from galicia, Spain. Ophthalmic Genet. 36, 244–247. doi:10.3109/13816810.2013.848908
Bykhovskaya, Y., Gromova, A., Makarenkova, H. P., and Rabinowitz, Y. S. (2016). Abnormal regulation of extracellular matrix and adhesion molecules in corneas of patients with keratoconus. Int. J. Keratoconus Ectatic Corneal Dis. 5, 63–70. doi:10.5005/jp-journals-10025-1123
Czugala, M., Karolak, J. A., Nowak, D. M., Polakowski, P., Pitarque, J., Molinari, A., et al. (2012). Novel mutation and three other sequence variants segregating with phenotype at keratoconus 13q32 susceptibility locus. Eur. J. Hum. Genet. 20, 389–397. doi:10.1038/ejhg.2011.203
Dou, S., Wang, Q., Zhang, B., Wei, C., Wang, H., Liu, T., et al. (2022). Single-cell atlas of keratoconus corneas revealed aberrant transcriptional signatures and implicated mechanical stretch as a trigger for keratoconus pathogenesis. Cell Discov. 8, 66. doi:10.1038/s41421-022-00397-z
Dudakova, L., Liskova, P., Trojek, T., Palos, M., Kalasova, S., and Jirsova, K. (2012). Changes in lysyl oxidase (LOX) distribution and its decreased activity in keratoconus corneas. Exp. Eye Res. 104, 74–81. doi:10.1016/j.exer.2012.09.005
Engelsvold, D. H., Utheim, T. P., Olstad, O. K., Gonzalez, P., Eidet, J. R., Lyberg, T., et al. (2013). miRNA and mRNA expression profiling identifies members of the miR-200 family as potential regulators of epithelial-mesenchymal transition in pterygium. Exp. Eye Res. 115, 189–198. doi:10.1016/j.exer.2013.07.003
Farzadfard, A., Nassiri, N., Moghadam, T. N., Paylakhi, S. H., and Elahi, E. (2016). Screening for MIR184 mutations in Iranian patients with keratoconus. J. Ophthalmic Vis. Res. 11, 3–7. doi:10.4103/2008-322X.180715
Fuchs Wightman, F., Giono, L. E., Fededa, J. P., and de la Mata, M. (2018). Target RNAs strike back on MicroRNAs. Front. Genet. 9, 435. doi:10.3389/fgene.2018.00435
Garrison, E., and Marth, G. (2012). Haplotype-based variant detection from short-read sequencing. arXiv:1207.3907 [q-bio]. Available at: http://arxiv.org/abs/1207.3907 (Accessed May 17, 2021).
Ghorai, A., and Ghosh, U. (2014). miRNA gene counts in chromosomes vary widely in a species and biogenesis of miRNA largely depends on transcription or post-transcriptional processing of coding genes. Front. Genet. 5, 100. doi:10.3389/fgene.2014.00100
Huang, D. W., Sherman, B. T., and Lempicki, R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. doi:10.1038/nprot.2008.211
Hughes, A. E., Bradley, D. T., Campbell, M., Lechner, J., Dash, D. P., Simpson, D. A., et al. (2011). Mutation altering the miR-184 seed region causes familial keratoconus with cataract. Am. J. Hum. Genet. 89, 628–633. doi:10.1016/j.ajhg.2011.09.014
Jang, S. Y., Chae, M. K., Lee, J. H., Lee, E. J., and Yoon, J. S. (2019). MicroRNA-27 inhibits adipogenic differentiation in orbital fibroblasts from patients with Graves’ orbitopathy. PLoS One 14, e0221077. doi:10.1371/journal.pone.0221077
Jaskiewicz, K., Maleszka-Kurpiel, M., Kabza, M., Karolak, J. A., and Gajecka, M. (2023a). Sequence variants contributing to dysregulated inflammatory responses across keratoconic cone surface in adolescent patients with keratoconus. Front. Immunol. 14, 1197054. doi:10.3389/fimmu.2023.1197054
Jaskiewicz, K., Maleszka-Kurpiel, M., Matuszewska, E., Kabza, M., Rydzanicz, M., Malinowski, R., et al. (2023b). The impaired wound healing process is a major factor in remodeling of the corneal epithelium in adult and adolescent patients with keratoconus. Invest. Ophthalmol. Vis. Sci. 64, 22. doi:10.1167/iovs.64.2.22
Jaskiewicz, K., Maleszka-Kurpiel, M., Michalski, A., Ploski, R., Rydzanicz, M., and Gajecka, M. (2023c). Non-allergic eye rubbing is a major behavioral risk factor for keratoconus. PLoS One 18, e0284454. doi:10.1371/journal.pone.0284454
Kabza, M., Karolak, J., Rydzanicz, M., Szcześniak, M., Nowak, D., Ginter-Matuszewska, B., et al. (2017a). Transcriptome profiling of human keratoconus corneas through RNA sequencing identifies collagen synthesis disruption and downregulation of core elements of TGF-β, Hippo, and Wnt pathways.
Kabza, M., Karolak, J. A., Rydzanicz, M., Szcześniak, M. W., Nowak, D. M., Ginter-Matuszewska, B., et al. (2017b). Collagen synthesis disruption and downregulation of core elements of TGF-β, Hippo, and Wnt pathways in keratoconus corneas. Eur. J. Hum. Genet. 25, 582–590. doi:10.1038/ejhg.2017.4
Kabza, M., Karolak, J. A., Rydzanicz, M., Udziela, M., Gasperowicz, P., Ploski, R., et al. (2019). Multiple differentially methylated regions specific to keratoconus explain known keratoconus linkage loci. Invest. Ophthalmol. Vis. Sci. 60, 1501–1509. doi:10.1167/iovs.18-25916
Karolak, J. A., and Gajecka, M. (2017). Genomic strategies to understand causes of keratoconus. Mol. Genet. Genomics 292, 251–269. doi:10.1007/s00438-016-1283-z
Karolak, J. A., Gambin, T., Pitarque, J. A., Molinari, A., Jhangiani, S., Stankiewicz, P., et al. (2016). Variants in SKP1, PROB1, and IL17B genes at keratoconus 5q31.1-q35.3 susceptibility locus identified by whole-exome sequencing. Eur. J. Hum. Genet. 25, 73–78. doi:10.1038/ejhg.2016.130
Karolak, J. A., Gambin, T., Rydzanicz, M., Polakowski, P., Ploski, R., Szaflik, J. P., et al. (2020a). Accumulation of sequence variants in genes of Wnt signaling and focal adhesion pathways in human corneas further explains their involvement in keratoconus. PeerJ 8, e8982. doi:10.7717/peerj.8982
Karolak, J. A., Ginter-Matuszewska, B., Tomela, K., Kabza, M., Nowak-Malczewska, D. M., Rydzanicz, M., et al. (2020b). Further evaluation of differential expression of keratoconus candidate genes in human corneas. PeerJ 8, e9793. doi:10.7717/peerj.9793
Karolak, J. A., Kulinska, K., Nowak, D. M., Pitarque, J. A., Molinari, A., Rydzanicz, M., et al. (2011). Sequence variants in COL4A1 and COL4A2 genes in Ecuadorian families with keratoconus. Mol. Vis. 17, 827–843.
Khaled, M. L., Bykhovskaya, Y., Yablonski, S. E. R., Li, H., Drewry, M. D., Aboobakar, I. F., et al. (2018). Differential expression of coding and long noncoding RNAs in keratoconus-affected corneas. Invest. Ophthalmol. Vis. Sci. 59, 2717–2728. doi:10.1167/iovs.18-24267
Kim, Y. J., Lee, W. J., Ko, B.-W., Lim, H. W., Yeon, Y., Ahn, S. J., et al. (2021). Investigation of MicroRNA expression in anterior lens capsules of senile cataract patients and MicroRNA differences according to the cataract type. Transl. Vis. Sci. Technol. 10, 14. doi:10.1167/tvst.10.2.14
Law, C. W., Alhamdoosh, M., Su, S., Dong, X., Tian, L., Smyth, G. K., et al. (2016). RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Res 5, 1408. ISCB Comm J-1408. doi:10.12688/f1000research.9005.1
Lechner, J., Bae, H. A., Guduric-Fuchs, J., Rice, A., Govindarajan, G., Siddiqui, S., et al. (2013). Mutational analysis of MIR184 in sporadic keratoconus and myopia. Invest. Ophthalmol. Vis. Sci. 54, 5266–5272. doi:10.1167/iovs.13-12035
Li, D.-D., Zhong, B.-W., Zhang, H.-X., Zhou, H.-Y., Luo, J., Liu, Y., et al. (2016). Inhibition of the oxidative stress-induced miR-23a protects the human retinal pigment epithelium (RPE) cells from apoptosis through the upregulation of glutaminase and glutamine uptake. Mol. Biol. Rep. 43, 1079–1087. doi:10.1007/s11033-016-4041-8
Lin, H., Qian, J., Castillo, A. C., Long, B., Keyes, K. T., Chen, G., et al. (2011). Effect of miR-23 on oxidant-induced injury in human retinal pigment epithelial cells. Invest. Ophthalmol. Vis. Sci. 52, 6308–6314. doi:10.1167/iovs.10-6632
Liu, W., and Wang, X. (2019). Prediction of functional microRNA targets by integrative modeling of microRNA binding and target expression data. Genome Biol. 20, 18. doi:10.1186/s13059-019-1629-z
Liu, W., Yang, Y., Yan, J., and Wang, L. (2019). MicroRNA-23b-3p promotes the proliferation, migration, and epithelial-mesenchymal transition of lens epithelial cells by targeting Sprouty2. Acta histochem. 121, 704–711. doi:10.1016/j.acthis.2019.05.007
Moschos, M. M., Droutsas, K., Sioziou, A., Dettoraki, M., and Gazouli, M. (2016). Mutational analysis of pre-miR-184 and hsa-mir-568 in Greek patients with sporadic keratoconus. Cornea 35, 631–633. doi:10.1097/ICO.0000000000000769
Najmi, H., Mobarki, Y., Mania, K., Altowairqi, B., Basehi, M., Mahfouz, M. S., et al. (2019). The correlation between keratoconus and eye rubbing: a review. Int. J. Ophthalmol. 12, 1775–1781. doi:10.18240/ijo.2019.11.17
Nowak, D. M., and Gajecka, M. (2015). Nonrandom distribution of miRNAs genes and single nucleotide variants in keratoconus loci. PLoS One 10, e0132143. doi:10.1371/journal.pone.0132143
Nowak-Malczewska, D. M., Karolak, J. A., Swierkowska, J., Jaworska, M. M., Kulinska, K. I., Polakowski, P., et al. (2021). Changes in nuclear gene expression related to mitochondrial function affect extracellular matrix, collagens, and focal adhesion in keratoconus. Transl. Vis. Sci. Technol. 10, 6. doi:10.1167/tvst.10.11.6
Peng, H., Sun, Y.-B., Hao, J.-L., Lu, C.-W., Bi, M.-C., and Song, E. (2019). Neuroprotective effects of overexpressed microRNA-200a on activation of glaucoma-related retinal glial cells and apoptosis of ganglion cells via downregulating FGF7-mediated MAPK signaling pathway. Cell Signal 54, 179–190. doi:10.1016/j.cellsig.2018.11.006
Reinstein, D. Z., Archer, T. J., Urs, R., Gobbe, M., RoyChoudhury, A., and Silverman, R. H. (2015). Detection of keratoconus in clinically and algorithmically topographically normal fellow eyes using epithelial thickness analysis. J. Refract Surg. 31, 736–744. doi:10.3928/1081597X-20151021-02
Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W., et al. (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. doi:10.1093/nar/gkv007
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi:10.1093/bioinformatics/btp616
Sherman, B. T., Hao, M., Qiu, J., Jiao, X., Baseler, M. W., Lane, H. C., et al. (2022). DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221. doi:10.1093/nar/gkac194
Shetty, R., Sathyanarayanamoorthy, A., Ramachandra, R. A., Arora, V., Ghosh, A., Srivatsa, P. R., et al. (2015). Attenuation of lysyl oxidase and collagen gene expression in keratoconus patient corneal epithelium corresponds to disease severity. Mol. Vis. 21, 12–25.
Srivastava, O. P., Chandrasekaran, D., and Pfister, R. R. (2006). Molecular changes in selected epithelial proteins in human keratoconus corneas compared to normal corneas. Mol. Vis. 12, 1615–1625.
Stachon, T., Nastaranpour, M., Seitz, B., Meese, E., Latta, L., Taneri, S., et al. (2022). Altered regulation of mRNA and miRNA expression in epithelial and stromal tissue of keratoconus corneas. Invest. Ophthalmol. Vis. Sci. 63, 7. doi:10.1167/iovs.63.8.7
Szczesniak, M. W., Kabza, M., Karolak, J. A., Rydzanicz, M., Nowak, D. M., Ginter-Matuszewska, B., et al. (2017). KTCNlncDB-a first platform to investigate lncRNAs expressed in human keratoconus and non-keratoconus corneas. Database (Oxford) 2017, baw168. doi:10.1093/database/baw168
Takahashi, Y., Chen, Q., Rajala, R. V. S., and Ma, J.-X. (2015). MicroRNA-184 modulates canonical Wnt signaling through the regulation of frizzled-7 expression in the retina with ischemia-induced neovascularization. FEBS Lett. 589, 1143–1149. doi:10.1016/j.febslet.2015.03.010
Takaoka, A., Babar, N., Hogan, J., Kim, M., Price, M. O., Price, F. W., et al. (2016). An evaluation of lysyl oxidase-derived cross-linking in keratoconus by liquid chromatography/mass spectrometry. Invest. Ophthalmol. Vis. Sci. 57, 126–136. doi:10.1167/iovs.15-18105
Xu, H., Yang, B., Ren, Z., Wu, D., Hu, A., and Hu, J. (2022a). miR-429 negatively regulates the progression of hypoxia-induced retinal neovascularization by the HPSE-VEGF pathway. Exp. Eye Res. 223, 109196. doi:10.1016/j.exer.2022.109196
Xu, L., Yang, K., Yin, S., Gu, Y., Fan, Q., Wang, Y., et al. (2022b). Family-based exome sequencing identifies candidate genes related to keratoconus in Chinese families. Front. Genet. 13, 988620. doi:10.3389/fgene.2022.988620
Yang, L., Du, L., Yue, Y., Huang, Y., Zhou, Q., Cao, S., et al. (2017). miRNA copy number variants confer susceptibility to acute anterior uveitis with or without ankylosing spondylitis. Invest. Ophthalmol. Vis. Sci. 58, 1991–2001. doi:10.1167/iovs.16-21047
Zhang, Y., Che, D., Cao, Y., Yue, Y., He, T., Zhu, Y., et al. (2022). MicroRNA profiling in the aqueous humor of keratoconus eyes. Transl. Vis. Sci. Technol. 11, 5. doi:10.1167/tvst.11.12.5
Zhou, Q., Gallagher, R., Ufret-Vincenty, R., Li, X., Olson, E. N., and Wang, S. (2011). Regulation of angiogenesis and choroidal neovascularization by members of microRNA-23∼27∼24 clusters. Proc. Natl. Acad. Sci. U. S. A. 108, 8287–8292. doi:10.1073/pnas.1105254108
Zhou, W., Xu, J., Wang, C., Shi, D., and Yan, Q. (2019). miR-23b-3p regulates apoptosis and autophagy via suppressing SIRT1 in lens epithelial cells. J. Cell Biochem. 120, 19635–19646. doi:10.1002/jcb.29270
Keywords: pre-miRNAs, microRNAs’ target genes, corneal samples, RNA-seq, extracellular matrix organization, signal transduction, miR-184
Citation: Nowak-Malczewska DM, Swierkowska J and Gajecka M (2024) Differentially expressed microRNAs targeting genes in key pathways in keratoconus. Front. Genet. 15:1301676. doi: 10.3389/fgene.2024.1301676
Received: 25 September 2023; Accepted: 12 February 2024;
Published: 26 February 2024.
Edited by:
Kristina Allen-Brady, The University of Utah, United StatesCopyright © 2024 Nowak-Malczewska, Swierkowska and Gajecka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marzena Gajecka, Z2FtYXJAbWFuLnBvem5hbi5wbA==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.