 Carmen Manso-Bazús1
Carmen Manso-Bazús1 Nino Spataro1
Nino Spataro1 Elisabeth Gabau2Viviana P. Beltrán-Salazar3Juan Pablo Trujillo-Quintero1Nuria Capdevila1
Elisabeth Gabau2Viviana P. Beltrán-Salazar3Juan Pablo Trujillo-Quintero1Nuria Capdevila1 Anna Brunet-Vega1
Anna Brunet-Vega1 Neus Baena1
Neus Baena1 A Arockia Jeyaprakash4,5Victor Martinez-Glez1
A Arockia Jeyaprakash4,5Victor Martinez-Glez1 Anna Ruiz1*
Anna Ruiz1*- 1Center for Genomic Medicine, Parc Taulí Hospital Universitari, Institut d’Investigació i Innovació Parc Taulí (I3PT-CERCA), Universitat Autònoma de Barcelona, Sabadell, Spain
- 2Paediatric Service, Parc Taulí Hospital Universitari, Institut d’Investigació i Innovació Parc Taulí (I3PT-CERCA), Universitat Autònoma de Barcelona, Sabadell, Spain
- 3Radiology Service, Parc Taulí Hospital Universitari, Institut d’investigación i innovació Parc Taulí (I3PT-CERCA), Universitat Autónoma de Barcelona, Sabadell, Spain
- 4Wellcome Centre for Cell Biology, University of Edinburgh, Edinburgh, United Kingdom
- 5The Gene Centre and Department of Biochemistry, Ludwig Maximilian Universität, München, Germany
Background: Moebius Syndrome (MBS) is a rare congenital neurological disorder characterized by paralysis of facial nerves, impairment of ocular abduction and other variable abnormalities. MBS has been attributed to both environmental and genetic factors as potential causes. Until now only two genes, PLXND1 and REV3L have been identified to cause MBS.
Results: We present a 9-year-old male clinically diagnosed with MBS, presenting facial palsy, altered ocular mobility, microglossia, dental anomalies and congenital torticollis. Radiologically, he lacks both abducens nerves and shows altered symmetry of both facial and vestibulocochlear nerves. Whole-exome sequence identified a de novo missense variant c.643G>A; p.Gly215Arg in CHN1, encoding the α2-chimaerin protein. The p.Gly215Arg variant is located in the C1 domain of CHN1 where other pathogenic gain of function variants have been reported. Bioinformatic analysis and molecular structural modelling predict a deleterious effect of the missense variant on the protein function.
Conclusion: Our findings support that pathogenic variants in the CHN1 gene may be responsible for different cranial congenital dysinnervation syndromes, including Moebius and Duane retraction syndromes. We propose to include CHN1 in the genetic diagnoses of MBS.
Introduction
Moebius syndrome (MBS) is a rare congenital neurological disease characterized by non-progressive facial palsy and impairment of ocular abduction, due to uni or bilateral paralysis or weakness of the facial (VII) and abducens (VI) cranial nerves. It can also be associated to paralysis of other cranial nerves (most notably cranial nerves V, IX, X, and XII). Other abnormalities include lingual hypoplasia, sensorineural hearing loss, craniofacial malformations (epicanthic folds, micrognathia), and abnormalities of the extremities (syndactyly, pes planus, valgus femur) (Terzis and Noah, 2002). Face and mouth functional anomalies implicate facial weakness, difficulties in speaking, eating, sucking and swallowing.
MBS is classified as a congenital cranial dysinnervation disorder (CCDD). These are disorders caused by developmental abnormalities of cranial nerves/nuclei resulting in primary or secondary dysinnervation. The CCDDs include Duane retraction syndrome (DRS), congenital fibrosis of the external ocular muscles (CFEOM), hereditary congenital facial palsy (HCFP), horizontal gaze palsy with progressive scoliosis (HGPPS) and MBS (Gutowski and Chilton, 2015; Oystreck, 2018; Jia et al., 2022).
The prevalence of MBS is estimated to be 1 in 10,000 to 1 in 2,500 live births with equal incidence in both sexes. Most patients present normal intelligence, while rare cases of autistic-like behaviours (0%–5%) and mild intellectual disability (9%–15%) have been reported (Picciolini et al., 2016).
Since the initial descriptions by von Graefe in 1880 (von Graefe, 1880) and by Moebius in 1888 (Möbius, 2008) it has been debated whether MBS has a genetic or a non-genetic aetiology. Most patients have a sporadic occurrence with a limited number of atypical familial cases (Tischfield et al., 2005; Schröder et al., 2013). Both intrauterine environmental factors and genetic causes have been proposed. Prenatal exposure to misoprostol or cocaine can lead to disruption of blood vessel migration during development and hindbrain hypoxia, resulting in cranial nerve dysfunction.
In 2015, de novo pathogenic variants affecting two genes, PLXND1 and REV3L were reported in six unrelated sporadic Moebius patients from a cohort of 110 patients. PLXND1 and REV3L are both involved in hindbrain development. Analysis of Plxnd1 and Rev3l mutant mice shows that disruption of these genes converge at the facial branchiomotor nucleus, affecting either motoneuron migration or proliferation (Tomas-Roca et al., 2015). The low frequency of pathogenic variants detected in these two genes as a cause of MBS suggests that de novo mutations in other genes may be responsible for MBS.
Here, we identified, using whole exome sequencing, a de novo novel heterozygous variant in the CHN1 gene in a patient diagnosed with MBS. Gain of function variants in the CHN1 gene have been previously shown to cause DRS, which is a congenital eye movement disorder characterised by variable horizontal duction deficits, with palpebral fissure narrowing and globe retraction on attempted adduction.
Material and methods
Patients and subjects
Patient genomic DNA was extracted from peripheral blood leukocytes from the patient and patient’s parents. Written informed consent was obtained from the patient’s parents.
Exome sequencing and data analysis
Exome capture was performed using the KAPA HyperExome probes (Roche) and sequenced on an Illumina Novaseq platform (Illumina, San Diego, CA, United States) at the National Centre of Genomic Analysis (CNAG-CRG, Barcelona, Spain), producing 2 × 150 nucleotides paired-end reads. Bioinformatics analysis was performed as described in Spataro et al., 2023. Sanger direct sequencing of candidate variants was done in the patient and parents to determine the inheritance pattern. Finally, variants were classified following the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) guidelines (Richards et al., 2015) and the recommendations provided by the Sequence Variant Interpretation working group at ClinGen (https://clinicalgenome.org/working-groups/sequence-variant-interpretation/).
Magnetic resonance imaging
Magnetic Resonance (MR) examination was performed using a 1.5 T Magnetic Resonance Imaging (MRI) scanner (Aera; Siemens Medical Solutions, Erlangen, Germany).
Structural modelling of the CHN1 p. Gly215Arg missense variant
The p. Gly215Arg variant was modelled using the published CHN1 crystal structure (3CXL) as a template using PyMOL, a molecular graphics program (The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC).
Results
Case clinical description
The proband is a 9-year-old male clinically diagnosed with MBS, son of healthy non-consanguineous parents.
He was born at term after uneventful pregnancy and delivery. After birth, he had significant difficulties in sucking, presenting microglossia, and remaining in hospital for a month. He was referred to a clinical genetics service at 2 months of age due to the microglossia (Figure 1A), limited tongue movements, left-winged scapula, and altered ocular mobility (Figure 1B). He presented bilateral convergent strabismus, bilateral abduction limitation and congenital torticollis (face to the right). He presented significant feeding and swallowing difficulties during his first 6 months, which improved later in life. Lack of facial expression was detected before 1 year of age.
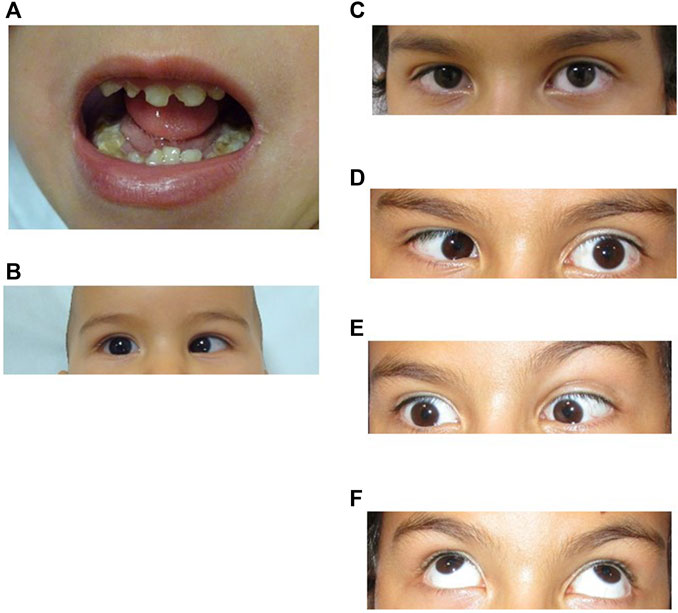
FIGURE 1. (A), Patient at 3 years 10 month old. Microglossia and dental anomalies. (B), Patient at 10 months of age showing limited outward gaze (abduction) of the right eye. (C–F). Patient at 9 years old. Ocular motility patterns. (C), Straight gaze showing mild esotropia of the right eye. (D), Horizontal left gaze showing limited abduction on the left eye. (E), Horizontal right gaze showing limited abduction on the right eye. (F), Full vertical eye movement. Exotropia in upper gaze.
At present, his cranial circumference, weight and height are within the normal range. He has been operated twice on both eyes to correct convergent strabismus (2.5 and 6 years) improving the torticollis. His left eye position has been centred but remains with limited abduction and his right eye remains with limited abduction and mild esotropia (Figures 1C–E). There is no limitation in his upper gaze (Figure 1F).
He presents hypomimia with reduced movements of the lower part of the forehead (Figure 2A). He also presents mild facial palsy and limited mouth movements. He cannot fully smile and when he smiles there is a mild asymmetry (Figure 2B) and he cannot blow (Figure 2C). According to family reports and medical health record, his facial expression has improved over the years. He still has a marked microglossia with altered movements, which limits language articulation (not swallowing). He presents malposition of the teeth (requiring orthodontics) and alterations of the dental enamel (presenting multiple cavities and needing corrective interventions since early childhood) (Figure 2D). Finally, his left winged scapula persists limiting abduction, with atrophy of the supraspinatus and dorsalis muscle on the same side.
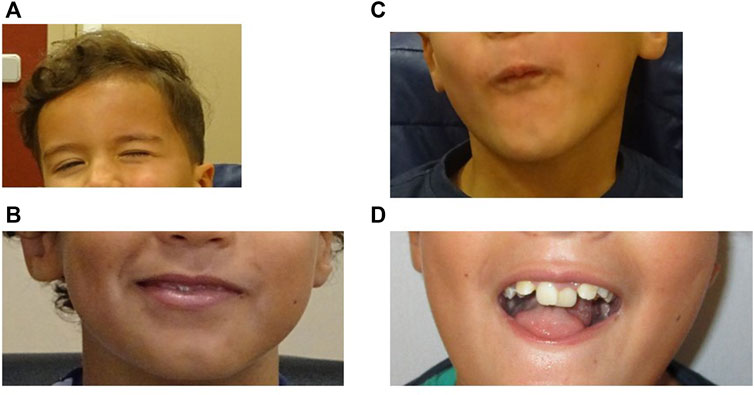
FIGURE 2. (A), Patient at 4 years 10 month old. Movements of the lower part of the forehead are reduced. (B,C), Patient at 9 years old showing mild facial palsy. (B), the patient cannot fully smile and when he smiles there is a mild asymmetry. (C), Limited mouth movements (he cannot blow). (D), Patient at 9 years old. Microglossia and dental anomalies.
Psychomotor development showed mild delay in the main items (crawling at around 14 months and autonomous walking at 20 months, due to hypotonia). Language development was slow because of difficulties in articulation, due to microglossia, which persist today. The patient showed no problems with receptive language. Social and learning development is within the normal range, with mild attentional symptoms that do not meet the diagnostic criteria for ADHD. Cognitive capacity is normal and there are no difficulties in acquiring basic and instrumental daily activities (such as dressing, eating, personal hygiene, transportation).
Imaging analysis
Radiologically, MS is characterised by bilateral absent or hypoplasic cisternal and canalicular portion of the facial nerve, and cisternal portion of the abducens nerve (Razek et al., 2021). Patient’s MRI performed at 1 year of age did not identify the intracanicular and cisternal segments of the abducens nerves (VI) and showed an asymmetry in the normal position of the right VII and VIII nerves into the internal auditory channel (Figures 3A,B). However, the cisternal segment of bilateral facial nerve was present (Figure 3C). There was also microglossia (as seen clinically), but it was not possible to evaluate the lower cranial nerves. The trigeminal nerve was normal.
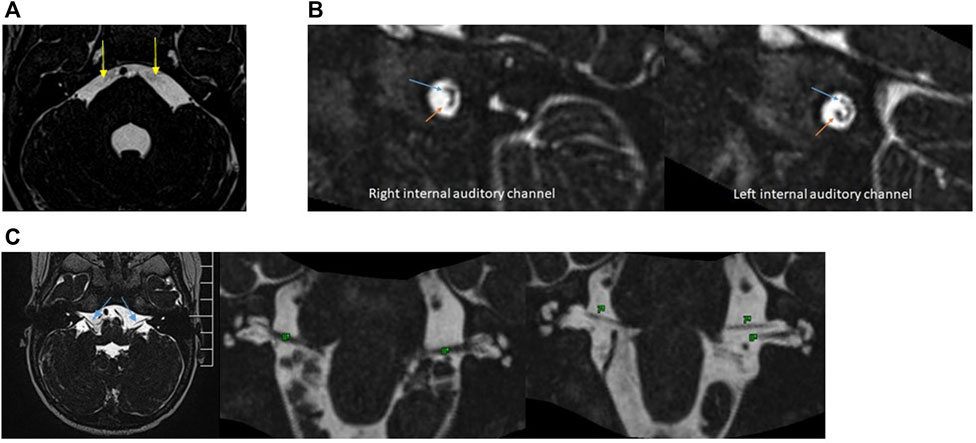
FIGURE 3. (A), 3D-constructive interference in steady State (CISS) sequences. Note the absence of bilateral VI nerves (yellow arrows). (B), 3D-CISS sequences with reconstructions perpendicular to the bilateral internal auditory channel show an asymmetry in the position of VII (blue arrows) and VIII nerves (orange arrows). The left side is normal. (C), 3D-CISS sequences in the axial plane with reconstructions in the coronal plane demonstrate the presence of a cisternal segment of bilateral facial (VII) (blue arrows) and vestibulocochlear nerve (VIII).
Genetic analysis
Using whole-exome sequencing (WES) a novel missense variant c.643G>A was detected in the CHN1 gene (NM_001822.7), which encodes the RacGAP signalling protein α2-chimaerin. The variant was confirmed by Sanger sequencing in the patient and was absent in both parents indicating that it has raised de novo (Figure 4A).
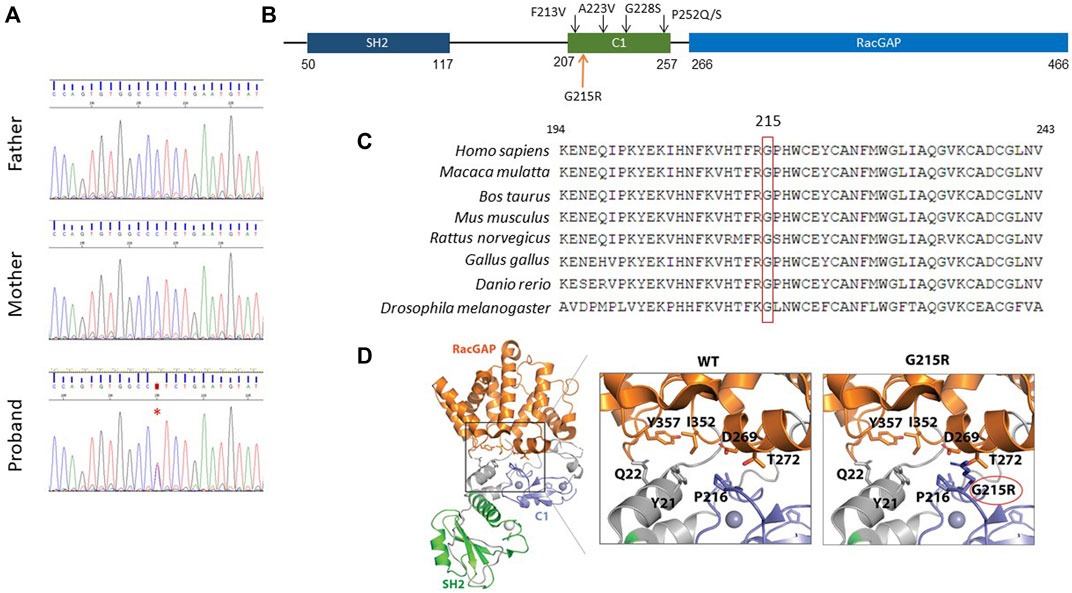
FIGURE 4. Molecular characterisation of the c.643G>A; p. Gly215Arg variant. (A), De novo inheritance pattern of CHN1 c.643G>A variant. A red asterisk indicates the variant. (B), Schematic structure of the human CHN1 protein. The upper black arrows indicate previously reported pathogenic variants in the C1 domain. (C), Multiple-sequence alignment showing the conservation of Gly215 residue in CHN1 across evolution. (D), Cartoon representation of the protein structure CHN1. Insets show close-up views of (left) the interaction mediated by CHN1 Gly215 with nearby amino acid residues in stick representation.
This c.643G>A variant replaces a glycine residue for an arginine in the C1 domain of the protein (p.Gly215Arg) (Figure 4B). The amino acid sequence alignment analysis indicated that Gly215 in the CHN1 protein is highly conserved throughout evolution (Figure 4C). The p.Gly215Arg variant is considered damaging or potentially disease-causing by most in silico predictors used (Supplementary Table S1) and is not present in the population databases gnomAD (Karczewski et al., 2020). In addition, prediction of protein stability determined by different software showed protein destabilisation indicated by a decrease in Gibbs free energy (Supplementary Table S2).
The α2-chimaerin longest isoform has an N-terminal SH2 domain (50-117aa), a C1 domain (207-255aa) that binds to diacylglycerol and a RacGAP domain (266-456aa) that interacts and downregulates Rac activity. α2-chimaerin is located in the cytoplasm in an inactive closed conformation. It unfolds and translocates to the membrane in response to DAG signalling, exposing the RacGAP domain and inactivating Rac. Previously identified variants have been shown to hyper-activate α2-chimaerin RacGAP activity either by destabilizing its closed conformation or by directly altering DAG or Rac binding (Miyake et al., 2008).
Gly 215 is located within the C1 domain in a loop that forms the interaction interface with the Rac GAP domain. Structural modelling of the p.Gly215Arg substitution showed steric clashes between amino acid Arg215 and RacGAP domain residues Thr272 and Asp269. This suggests that the p.Gly215Arg might alter the interface between the C1 and the RacGAP binding domain, changing their relative orientation and leading to an ‘open’ conformation, facilitating its translocation to the membrane (Figure 4D).
The c.643G>A (p. Gly215Arg) in the CHN1 gene was classified as likely pathogenic according to the American College of Medical Genetics (ACMG) guidelines, meeting the following criteria: 1) PM1, located in a functional domain of the protein; 2) PM6, de novo; 3) PM2_supporting, absent in population databases; 4) PP2; Missense variant in a gene with low rate of benign missense mutations and for which missense mutation is a common mechanism of a disease and 5) PP3, computational evidence shows a deleterious effect. The variant has been submitted to the ClinVar (Landrum et al., 2018) database (ref. VCV001708248.1).
Discussion
Pathogenic variants in the CHN1 gene have been mostly associated with Duane retraction syndrome (Miyake et al., 2008). Although Duane syndrome is commonly a sporadic disorder, 10% of cases are familial inherited cases. At least two other genes have been associated to Duane syndrome: MAFB (Park et al., 2016) and SALL4 (Al-Baradie et al., 2002).
Here, we describe a patient diagnosed with Moebius syndrome, carrier of a novel de novo missense variant in the CHN1 gene. CHN1 encodes the signalling protein alpha2-chimaerin, a crucial regulator of axon guidance in the ocular motor system (Miyake et al., 2008; Chilton and Guthrie, 2017). Pathogenic variants in the CHN1 gene have been shown to hyperactivate a2-chimerin RacGAP activity by either destabilizing the protein inactive closed conformation or by directly altering DAG or Rac binding resulting in aberrant cranial motor neuron development (Miyake et al., 2008). Moreover, both heterozygous and homozygous knockin mice harbouring a Duane syndrome gain-of-function missense mutation show eye movement abnormalities and unilateral or bilateral globe retraction. α2-chimaerin mutation alter the primary development of the abducens, trochlear, and C1 nerves. Mechanistically, these three different motor neuron populations use ephrin/EphA4-mediated signaling pathways upstream of mutant α2-chimaerin in distinct manners to guide developing axons (Nugent et al., 2017).
Bioinformatic analysis and structural modelling of the p.Gly215Arg variant supports a putative gain of function effect, where the p.Gly215Arg substitution will result in the destabilization of the closed conformation of the CHN1 protein, increasing its RacGAP activity as has been observed for other variants in the C1 domain of the CHN1 gene. Variants located in the C1 domain such as Phe213Val, Ala223Val, and Pro252/Gln/Ser have been shown to enhance Rac-GAP activity by destabilizing the closed conformation of α2-chimaerin and enhancing membrane translocation (Miyake et al., 2008).
Here we show that the novel p.Gly215Arg variant in the CHN1 gene is associated with MBS. The patient presents facial palsy, altered ocular mobility, microglossia and congenital torticollis. Radiologically, he lacks both abducens nerves and shows altered symmetry of both facial and vestibulocochlear nerves. Variants in CHN1 have been postulated to be mainly associated with bilateral DRS, often with some abnormalities in vertical gaze and other rare somatic disturbances (Miyake et al., 2011). However, a recent report describes a missense variant in the CHN1 gene (p.Tyr221His) in a child and his father with DRS, associated with swallowing difficulties and unilateral trapeze aplasia (Angelini et al., 2021). All symptoms are related to anomalies in different cranial nerves: VI, IX, X, XI, and XII suggesting that variants in the CHN1 gene affect the development of other cranial nerves in addition to the oculomotor system (Angelini et al., 2021). It has been shown that α2-chimaerin is expressed in all developing cranial motor neurons, including the abducens, trochlear and oculomotor, as well as in most developing neurons throughout the central and peripheral nervous system. Our data support that the gain of function variants in the CHN1 gene may be responsible for a spectrum of phenotypes, from DRS to MBS, affecting the normal development of cranial motor neurons. Further analyses are needed to show whether variants in CHN1 may cause other congenital cranial dysinnervation syndromes and to characterize their degree of variability in clinical expressivity.
To date, only two genes have been proposed to cause MBS (PLXND1 and REV3L). Animal models indicate that PLXND1 and REV3L cause hypoplasia of the facial motor nucleus while CHN1 knock-in mice present alteration in the primary development of the abducens, trochlear, and C1 nerves (Tomas-Roca et al., 2015; Nugent et al., 2017). Apparently, the three genes act by different molecular pathways converging in defects of motoneuron migration, proliferation and axon guidance (Tomas-Roca et al., 2015; Nugent et al., 2017).
We propose that CHN1 should be included in the genetic diagnoses of MBS as well as other CCDDs. CCDDs represent a group of developmental disorders that commonly involve disturbances to ocular motility. There is phenotypic variability between CCDDs subtypes with overlap between entities. Some conditions can be distinguished by the presence of other associated features. For instance, HGPPS individuals are expected to have scoliosis while patients with MBS have facial weakness. While it can be difficult to a clinician to distinguish the type and degree of the ocular phenotype, genetic diagnosis may contribute to diagnosis confirmation.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by the Institutional Ethics Committee of Institut d’Investigació i Innovació Parc Taulí I3PT. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
CM-B: Writing–original draft, Writing–review and editing, Conceptualization, Data curation, Methodology. NS: Writing–review and editing, Data curation, Methodology, Software. EG: Writing–review and editing, Conceptualization, Data curation. VB-S: Writing–review and editing, Data curation, Methodology. JT-Q: Writing–review and editing. NC: Writing–review and editing. AB-V: Writing–review and editing. NB: Writing–review and editing. AJ: Investigation, Writing–review and editing, Data curation, Formal Analysis. VM-G: Writing–review and editing. AR: Project administration, Writing–original draft, Conceptualization, Data curation, Methodology, Supervision, Validation, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was partially founded by Institut d’Investigació i innovació Parc Taulí I3PT (AR, CIR2020/021), and CERCA Programme/Generalitat de Catalunya. This work was supported by grants from Instituto de Salud Carlos III-FEDER, (PI19/01902).
Acknowledgments
The authors are grateful to the patient and his parents for their collaboration.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1291063/full#supplementary-material
References
Al-Baradie, R., Yamada, K., St Hilaire, C., Chan, W.-M., Andrews, C., McIntosh, N., et al. (2002). Duane radial ray syndrome (Okihiro syndrome) maps to 20q13 and results from mutations in SALL4, a new member of the SAL family. Am. J. Hum. Genet. 71 (5), 1195–1199. doi:10.1086/343821
Angelini, C., Trimouille, A., Arveiler, B., Espil-Taris, C., Ichinose, N., Lasseaux, E., et al. (2021). CHN1 and duane retraction syndrome: expanding the phenotype to cranial nerves development disease. Eur. J. Med. Genet. 64 (4), 104188. doi:10.1016/j.ejmg.2021.104188
Chilton, J. K., and Guthrie, S. (2017). Axons get ahead: insights into axon guidance and congenital cranial dysinnervation disorders. Dev. Neurobiol. 77 (7), 861–875. doi:10.1002/dneu.22477
Gutowski, N. J., and Chilton, J. K. (2015). The congenital cranial dysinnervation disorders. Archives Dis. Child. 100 (7), 678–681. doi:10.1136/archdischild-2014-307035
Jia, H., Ma, Q., Liang, Y., Wang, D., Chang, Q., Zhao, B., et al. (2022). Clinical and genetic characteristics of Chinese patients with congenital cranial dysinnervation disorders. Orphanet J. Rare Dis. 17 (1), 431. doi:10.1186/s13023-022-02582-5
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581 (7809), 434–443. doi:10.1038/s41586-020-2308-7
Landrum, M. J., Lee, J. M., Benson, M., Brown, G. R., Chao, C., Chitipiralla, S., et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46 (D1), D1062–D1067. doi:10.1093/nar/gkx1153
Miyake, N., Chilton, J., Psatha, M., Cheng, L., Andrews, C., Chan, W.-M., et al. (2008). Human CHN1 mutations hyperactivate alpha2-chimaerin and cause Duane’s retraction syndrome. Sci. (New York, N.Y.) 321 (5890), 839–843. doi:10.1126/science.1156121
Miyake, N., Demer, J. L., Shaaban, S., Andrews, C., Chan, W.-M., Christiansen, S. P., et al. (2011). Expansion of the CHN1 strabismus phenotype. Investigative Ophthalmol. Vis. Sci. 52 (9), 6321–6328. doi:10.1167/iovs.11-7950
Möbius, P. J. (2008). About congenital bilateral abducens and facialis palsy (1888). Strabismus 16 (1), 39–44. doi:10.1080/09273970801946737
Nugent, A. A., Park, J. G., Wei, Y., Tenney, A. P., Gilette, N. M., DeLisle, M. M., et al. (2017). Mutant α2-chimaerin signals via bidirectional ephrin pathways in Duane retraction syndrome. J. Clin. Investigation 127 (5), 1664–1682. doi:10.1172/JCI88502
Oystreck, D. T. (2018). Ophthalmoplegia and congenital cranial dysinnervation disorders. J. Binocul. Vis. Ocular Motil. 68 (1), 31–33. doi:10.1080/2576117X.2017.1416242
Park, J. G., Tischfield, M. A., Nugent, A. A., Cheng, L., Di Gioia, S. A., Chan, W.-M., et al. (2016). Loss of MAFB function in humans and mice causes duane syndrome, aberrant extraocular muscle innervation, and inner-ear defects. Am. J. Hum. Genet. 98 (6), 1220–1227. doi:10.1016/j.ajhg.2016.03.023
Picciolini, O., Porro, M., Cattaneo, E., Castelletti, S., Masera, G., Mosca, F., et al. (2016). Moebius syndrome: clinical features, diagnosis, management and early intervention. Italian J. Pediatr. 42, 56. doi:10.1186/s13052-016-0256-5
Razek, A. A. K. A., Maher, H., Kasem, M. A., and Helmy, E. (2021). Imaging of congenital cranial dysinnervation disorders: what radiologist wants to know? Clin. Imaging 71, 106–116. doi:10.1016/j.clinimag.2020.10.055
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., and Gastier-Foster, J.ACMG Laboratory Quality Assurance Committee (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular Pathology. Genet. Med. Official J. Am. Coll. Med. Genet. 17 (5), 405–424. doi:10.1038/gim.2015.30
Schröder, J. C., Läßig, A. K., Galetzka, D., Peters, A., Castle, J. C., Diederich, S., et al. (2013). A boy with homozygous microdeletion of NEUROG1 presents with a congenital cranial dysinnervation disorder [Moebius syndrome variant]. Behav. Brain Funct. BBF 9, 7. doi:10.1186/1744-9081-9-7
Spataro, N., Trujillo-Quintero, J. P., Manso, C., Gabau, E., Capdevila, N., Martinez-Glez, V., et al. (2023). High performance of a dominant/X-linked gene panel in patients with neurodevelopmental disorders. Genes 14 (3), 708. doi:10.3390/genes14030708
Terzis, J. K., and Noah, E. M. (2002). Möbius and Möbius-like patients: etiology, diagnosis, and treatment options. Clin. Plastic Surg. 29 (4), 497–514. doi:10.1016/s0094-1298(02)00019-6
Tischfield, M. A., Bosley, T. M., Salih, M. A. M., Alorainy, I. A., Sener, E. C., Nester, M. J., et al. (2005). Homozygous HOXA1 mutations disrupt human brainstem, inner ear, cardiovascular and cognitive development. Nat. Genet. 37 (10), 1035–1037. doi:10.1038/ng1636
Tomas-Roca, L., Tsaalbi-Shtylik, A., Jansen, J. G., Singh, M. K., Epstein, J. A., Altunoglu, U., et al. (2015). De novo mutations in PLXND1 and REV3L cause Möbius syndrome. Nat. Commun. 6, 7199. doi:10.1038/ncomms8199
Keywords: moebius syndrome, genetic diagnosis, CHN1, novel variant, congenital dysinnervation syndromes
Citation: Manso-Bazús C, Spataro N, Gabau E, Beltrán-Salazar VP, Trujillo-Quintero JP, Capdevila N, Brunet-Vega A, Baena N, Jeyaprakash AA, Martinez-Glez V and Ruiz A (2024) Case report: Identification of a novel variant p.Gly215Arg in the CHN1 gene causing Moebius syndrome. Front. Genet. 15:1291063. doi: 10.3389/fgene.2024.1291063
Received: 08 September 2023; Accepted: 15 January 2024;
Published: 31 January 2024.
Edited by:
Antonio Pizzuti, Sapienza University of Rome, ItalyReviewed by:
Ariadna Gonzalez-Del Angel, National Institute of Pediatrics, MexicoMary Whitman, Harvard Medical School, United States
Copyright © 2024 Manso-Bazús, Spataro, Gabau, Beltrán-Salazar, Trujillo-Quintero, Capdevila, Brunet-Vega, Baena, Jeyaprakash, Martinez-Glez and Ruiz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Ruiz, YXJ1aXpuQHRhdWxpLmNhdA==