 Sk Imran Ali
Sk Imran Ali Mohammad J. Najaf-Panah1
Mohammad J. Najaf-Panah1 F. Ester Lujan
F. Ester Lujan Amanda K. Ashley
Amanda K. Ashley- 1Department of Chemistry and Biochemistry, New Mexico State University, Las Cruces, NM, United States
- 2National Center for Genome Resources, Santa Fe, NM, United States
Background: Maintenance of the genome is essential for cell survival, and impairment of the DNA damage response is associated with multiple pathologies including cancer and neurological abnormalities. DNA-PKcs is a DNA repair protein and a core component of the classical nonhomologous end-joining pathway, but it also has roles in modulating gene expression and thus, the overall cellular response to DNA damage.
Methods: Using cells producing either wild-type (WT) or kinase-inactive (KR) DNA-PKcs, we assessed global alterations in gene expression in the absence or presence of DNA damage. We evaluated differential gene expression in untreated cells and observed differences in genes associated with cellular adhesion, cell cycle regulation, and inflammation-related pathways. Following exposure to etoposide, we compared how KR versus WT cells responded transcriptionally to DNA damage.
Results: Downregulated genes were mostly involved in protein, sugar, and nucleic acid biosynthesis pathways in both genotypes, but enriched biological pathways were divergent, again with KR cells manifesting a more robust inflammatory response compared to WT cells. To determine what major transcriptional regulators are controlling the differences in gene expression noted, we used pathway analysis and found that many master regulators of histone modifications, proinflammatory pathways, cell cycle regulation, Wnt/β-catenin signaling, and cellular development and differentiation were impacted by DNA-PKcs status. Finally, we have used qPCR to validate selected genes among the differentially regulated pathways to validate RNA sequence data.
Conclusion: Overall, our results indicate that DNA-PKcs, in a kinase-dependent fashion, decreases proinflammatory signaling following genotoxic insult. As multiple DNA-PK kinase inhibitors are in clinical trials as cancer therapeutics utilized in combination with DNA damaging agents, understanding the transcriptional response when DNA-PKcs cannot phosphorylate downstream targets will inform the overall patient response to combined treatment.
Introduction
Eukaryotic cells frequently experience genotoxic stress from various exogenous and endogenous sources including exposure to UV radiation, reactive oxygen/nitrogen species formed during metabolism, and genotoxic chemicals, inducing myriad types of DNA damage (Lindahl and Barnes, 2000). DNA double-strand breaks (DSB) are deleterious and if left unrepaired or misrepaired can cause genomic instability associated with multiple diseases, including cancer. In response to DSB, cells initiate a complex, coordinated set of pathways, collectively known as DNA damage response (DDR), which involves damage sensing, transferring signals via signal transducers, and the response of many effector proteins to damage. Ideally, the DDR serves to repair DNA damage, when possible, through activation of repair proteins and upregulating transcription of additional genes needed for a full response. However, if repair is not feasible, the DDR initiates apoptotic death. In eukaryotes, the majority of DSB are repaired by the non-homologous end-joining (NHEJ) and homologous recombination (HR) pathways. The DNA-dependent protein kinase catalytic subunit (DNA-PKcs), a member of the phosphatidyl inositol 3-kinase-related kinase (PIKK) family of proteins, including ATM and ATR, is directly involved in NHEJ-mediated repair (Blackford and Jackson, 2017). DNA-PKcs is recruited to DSB by the Ku 70/80 heterodimer, forming the catalytically active DNA-PK holoenzyme, which in turn results in DNA-PKcs autophosphorylation and phosphorylation of other proteins, including Ku, H2AX, and RPA32. Additionally, DNA-PKcs serves to tether the two ends of the broken DNA (Yoo and Dynan, 1999) and activates other NHEJ proteins, XRCC4, XLF, Lig4 and PAXX, to facilitate DNA end-processing and ultimately ligation (Neal and Meek, 2011; Xing and Oksenych, 2019). DNA-PKcs regulates proteins involved in HR, including ATM, WRN, RPA32, cAbl and SMC1, facilitating crosstalk between the DDR proteins and two dominant DSB repair pathways (Karmakar et al., 2002; Collis et al., 2005; Shrivastav et al., 2008; Liu et al., 2012; Ashley et al., 2014; Blackford and Jackson, 2017). DNA-PKcs phosphorylates KAP-1 immediately after DSB and promotes chromatin decondensation to facilitate the recruitment of DDR proteins (Lu et al., 2019). Clearly, DNA-PKcs has multifaceted roles in regulating the cellular response to DSB.
DNA-PKcs also partakes in regulating proteins involved in cellular transcription. DNA-PKcs phosphorylates Snail1, a zinc-finger transcription factor involved in the epithelial-to-mesenchymal transition, which promotes its stability and function potentiation (Goodwin and Knudsen, 2014). DNA-PKcs is also involved in the activation of several metabolic genes. DNA-PKcs activation in the hypoxic condition protects HIF1α from degradation, which promotes the expression of GluT1, one of its target genes (Bouquet et al., 2011). DNA-PKcs promotes fatty acid biosynthesis in an insulin-dependent manner by activating the transcription factor USF-1 (Wong et al., 2009). During aging, DNA-PKcs phosphorylation of the heat-shock protein HSP90α decreases its mitochondrial function in skeletal muscles, thus decreasing overall metabolism and fitness (Park et al., 2017). DNA-PKcs was originally isolated as a component of the SP1 transcriptional complex as well as co-eluted with the largest subunit of RNA polymerase II (RNAPII) (Jackson et al., 1990; Maldonado et al., 1996). DNA-PKcs phosphorylates many transcriptional regulators, including RNAPII and RNAPI, TBP, TFIIB, TRIM28, Sp1, Oct1 and Oct2, AR, NRE, c-Myc, and c-Jun (Dvir et al., 1992; Lees-Miller, 1996). The availability of DNA-PKcs at the active transcription sites modulates the function of many transcription factors like autoimmune regulator (AIRE), p53, and erythroblast transformation-specific related gene (ERG) by direct interactions (Goodwin and Knudsen, 2014). DNA-PKcs may mediate transcription initiation by interacting with TopoIIβ and PARP1 in the presence of DSB (Ju et al., 2006; Ju and Rosenfeld, 2006). The effects of DNA-PKcs on global gene expression are not well-characterized. In this study, we analyzed global gene expression with and without DNA damage in cell lines expressing wild-type or kinase-inactive DNA-PKcs to elucidate the roles DNA-PKcs has in regulating gene expression in kinase-dependent and -independent manners, alone or following damage.
Materials and methods
Cell culture
V3-derived Chinese hamster ovary (CHO) cell lines were kindly provided by Dr. Katherine Meek, complemented with either human wild-type (WT) or kinase-inactive (K3753R (KR)) DNA-PKcs (Neal et al., 2014). Cells were cultured in α-MEM (Life Technologies, Waltham, MA) supplemented with 10% FBS (Millipore Sigma, St. Louis, MO), 1% penicillin/streptomycin (Life Technologies), 200 μg/mL G418 (Life Technologies), and 10 μg/mL puromycin (Santa Cruz Biotechnology, Santa Cruz, CA) at 37°C with 5% CO2 and 100% humidity. All chemicals were purchased from Millipore Sigma unless otherwise indicated.
Immunoblotting
Whole-cell lysates were prepared using RIPA buffer [50 mM Tris (pH 7.4), 2 mM EDTA, 150 mM NaCl, 0.1% SDS, and 1.0% Triton X-100] supplemented with Halt phosphatase and protease inhibitor cocktails (Fisher Scientific, Waltham, MA). The protein was quantified using the Pierce BCA Protein Assay (Fisher Scientific) as per the manufacturer’s instructions. The protein (25 µg) was subjected to SDS-PAGE, transferred to PVDF membranes, and blocked with 5% non-fat dried milk in 1X TBS-T (Tris-buffered saline with 0.1% Tween-20) for 1 h at room temperature (RT), and then a primary antibody recognizing DNA-PKcs (Abcam) was added overnight at 4°C in blocking buffer. Membranes were washed, and then an HRP-conjugated secondary antibody (Jackson ImmunoResearch, West Grove, Pennsylvania) was added for 1 h at RT. The DNA-PKcs protein was assessed using the Clarity Western ECL Substrate (Bio-Rad, Hercules, CA) and imaged using a ChemiDoc MP Imaging System (Bio-Rad). Total vinculin levels (Santa Cruz) were assessed as a loading control. To validate the expression of selected proteins, cells were treated with DMSO (0.1% v/v) or etoposide for 24 h. Whole-cell lysates were prepared as mentioned above. To detect ATAD2 and ARRB1, the following primary antibodies were used at a 1:1000 dilution: anti-ATAD2 (Cell Signaling) and anti-ARRB1 (Cell Signaling) in recommended blocking buffer. The concentration of the secondary antibody was applied as described above.
Clonogenic survival assay
We assessed clonogenic survival as previously described (Joshi et al., 2019). Plating efficiency (PE) and surviving fraction (SF) were calculated: PE = (number of colonies formed divided by the number of cells seeded) ×100; SF = PE of treated cells divided by PE of control cells. Each data point is the average of three independent biological replicates.
Cell viability
Cells were cultured in complete media in white-walled 96-well plates (CoStar, Corning, NY) for 24 h, treated with increasing concentrations of etoposide or DMSO, and then incubated for 72 h. Cell viability was quantified using the CellTiter-Glo Assay (Promega, Madison, WI) as per the manufacturer’s instructions.
RNA isolation
Cells were treated with DMSO (0.1% v/v) or 20 µM etoposide for 24 h. Total RNA from three biological replicates of genotypes or treatments were isolated using the TRIzol reagent (Millipore Sigma). RNA was dissolved in nuclease-free water followed by spectrophotometric analysis of the RNA quantity; RNA was subjected to electrophoresis on an agarose gel to assess RNA quality and purity.
Transcriptome library preparation and sequencing
RNA libraries were constructed using the NEBNext Ultra RNA Library Prep Kit for Illumina following the manufacturer’s instructions (NEB, Ipswich, MA, USA). Briefly, mRNAs were first enriched with Oligo (dT) beads. Enriched mRNAs were fragmented for 15 min at 94°C. First-strand and second-strand cDNAs were subsequently synthesized. cDNA fragments were end-repaired and adenylated at 3′ ends, and universal adapters were ligated to cDNA fragments, followed by index addition and library enrichment by limited-cycle PCR. The sequencing libraries were validated on the Agilent TapeStation (Agilent Technologies, Palo Alto, CA, USA) and quantified by using the Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA) as well as by quantitative PCR (KAPA Biosystems, Wilmington, MA, USA). The sequencing libraries were then clustered on a single lane of a flow cell. After clustering, the flow cell was loaded on the Illumina HiSeq instrument (4000 or equivalent) according to the manufacturer’s instructions. The samples were sequenced using a 2 × 150 bp paired-end (PE) configuration. Image analysis and base calling were conducted by HiSeq Control Software (HCS). Raw sequence data (.bcl files) generated from Illumina HiSeq were converted into fastq files and de-multiplexed using Illumina’s bcl2fastq 2.17 software. One mismatch was allowed for index sequence identification.
Differentially expressed gene analysis
The low-quality raw reads (fastq format) were filtered based on the Q30 and GC content, and then the Illumina adapters were trimmed of reads for a minimum read length of 36 bases using Trimmomatic v0.34 (Bolger et al., 2014). The index of the Chinese hamster reference genome (CHOK1GS_HDv1) was built using HISAT2 v2.1.0 (Kim et al., 2015). The Hisat2 v2.2.1 tool was used to align the reads to the genome sequences in the FASTA format, and the output aligned reads in the binary alignment map (BAM) format were translated into the transcriptomes of each sample using the StringTie v2.0 tool, which uses a novel network flow algorithm as well as an optional de novo assembly step to assemble and quantitate full-length transcripts representing multiple splice variants for each gene locus (Pertea et al., 2015). The StringTie outputs (GTF files) were merged to create a single master transcriptome GTF with the exact same naming and numbering scheme across all transcripts. The feature count tool implemented under Subread v2.0 was used to quantify transcripts assembled by StringTie mapped to each gene (Liao et al., 2014). Eventually, the differentially expressed (DE) gene profiles were statistically analyzed through edgeR and limma R libraries (Robinson et al., 2010; Ritchie et al., 2015).
Biological pathway analysis
The ClusterProfiler R tool was applied on the DEGs to demonstrate the functional pathways and gene network enrichment analysis (Yu et al., 2012). This analysis provides the information related to the biological pathways significantly enriched in up or downregulated genes through mediating the Kyoto Encyclopedia of Genes and Genomes (KEGG) database and Gene Ontology (GO) terms (using a standard false discovery rate (FDR) <0.05 and p-value cutoff 0.05).
Regulatory network analysis
The upstream regulator network analysis was performed through the ingenuity pathway analysis (IPA, QIAGEN) platform. The IPA casual network approach was applied on contrast matrices, by having gene symbol, log fold change, and p-value <0.05 of each gene, to characterize the upstream regulators as well as master regulators (the root of network) responsible for driving a set of target genes (Krämer et al., 2014).
qPCR
RNA-Seq data were validated by qPCR from a new set of RNAs isolated from the CHO cells treated with the same treatment or control. We selected 14 master regulators based on their function in inflammatory regulation, neuronal development, and chromatin modification, including CXCR3, HIF1, IGFBP2, IL17RA, FLT1, IL1B, IL20, SPP1, STAT5, KDR, PDGF-BB, TNF, VEGF, and VEGFA. We selected 17 differentially expressed genes regulated by those master regulators and are involved in the functions we were interrogating. Primers were designed from the selected mRNA sequences using the Pickprimers tool of NCBI. Sequences of all primers are listed in Table 1. All primers were validated for qPCR using iTaq Universal SYBR Green Supermix (Bio-Rad) and the CFX Connect Real-Time PCR Detection System (Bio-Rad). RNA from three biological replicates was isolated using the TRIzol reagent (Millipore Sigma) and DNA removed with the TURBO DNA-free™ Kit (Ambion). cDNA was synthesized from 1 μg mRNA using an iScript cDNA Synthesis Kit (Bio-Rad) according to the manufacturer’s instructions. Twenty nanograms of cDNA was subjected to qPCR according to the manufacturer’s protocol. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a housekeeping gene, and the relative expression was analyzed by Cq values of the target and control genes using the 2-delta delta Ct method (Livak and Schmittgen, 2001).
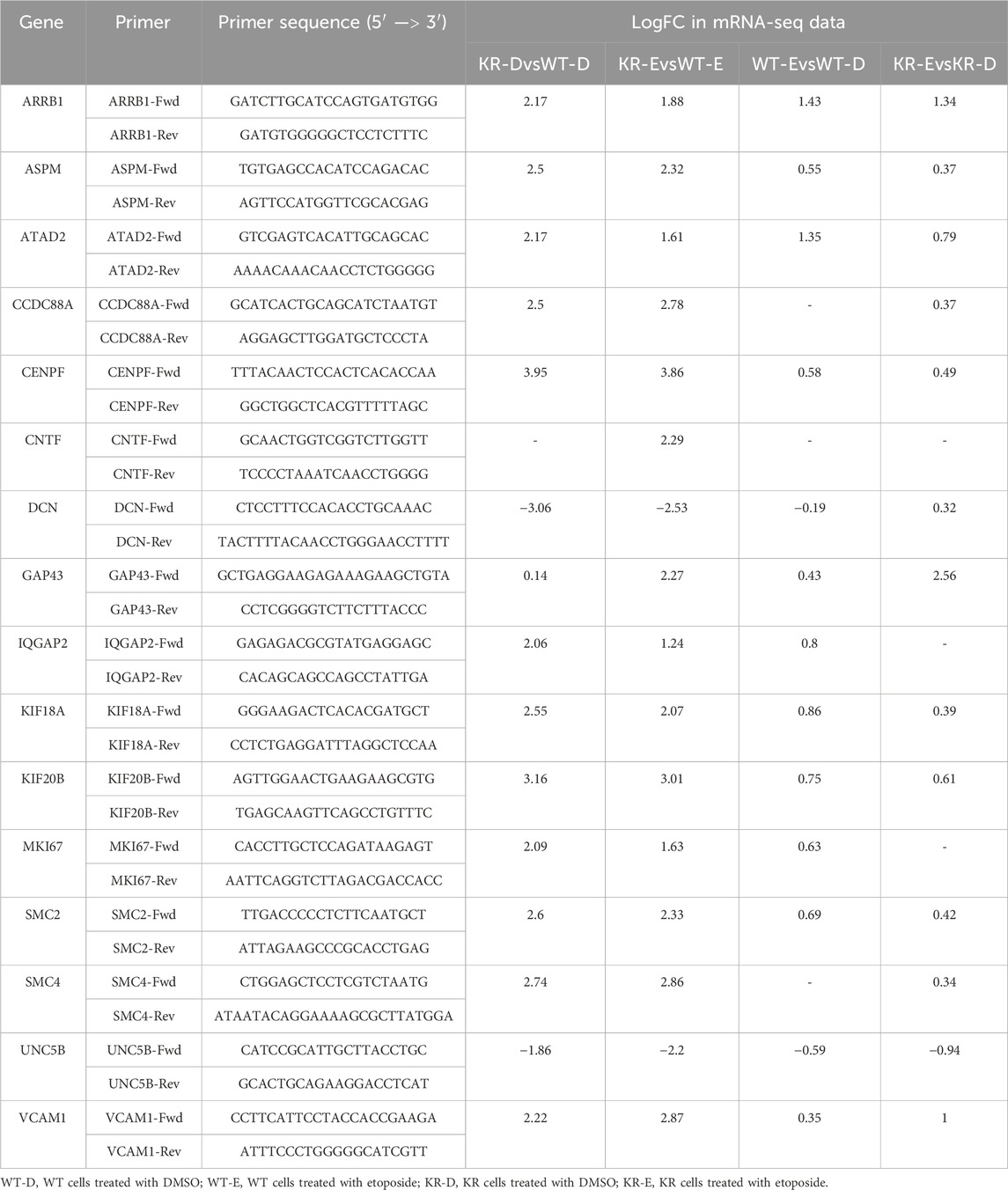
Table 1. Primer sequences of select genes with fold changes observed during mRNA sequencing analysis.
Statistical analysis
Cell viability was analyzed using non-linear regression function in GraphPad Prism (version 8; San Diego, CA) comparing viability as a function of the log etoposide dose. Data points represent an average of the three biological replicates. The area under the curve (AUC) for viability and colony-forming assays was assessed using GraphPad Prism, and the resultant total peak AUC and standard error per sample were compared using one-way ANOVA with an ad hoc Tukey’s multiple comparisons test. qPCR results were assessed using two-way ANOVA with Fisher’s LSD test to detect the significant differences among the mean relative expression levels.
Results
DNA-PKcs promotes survival following exposure to etoposide
We confirmed DNA-PKcs production by Western blotting on WT and KR CHO cells. DNA-PKcs is produced equally in both WT and KR cells (Figure 1A). Then, we assessed the effect of DNA-PKcs on cell survivability after exposure to the topoisomerase II inhibitor, etoposide. Consistent with previous reports, cells lacking DNA-PKcs kinase activity (KR cells) are more sensitive to etoposide (Figures 1B–E) (Convery et al., 2005).
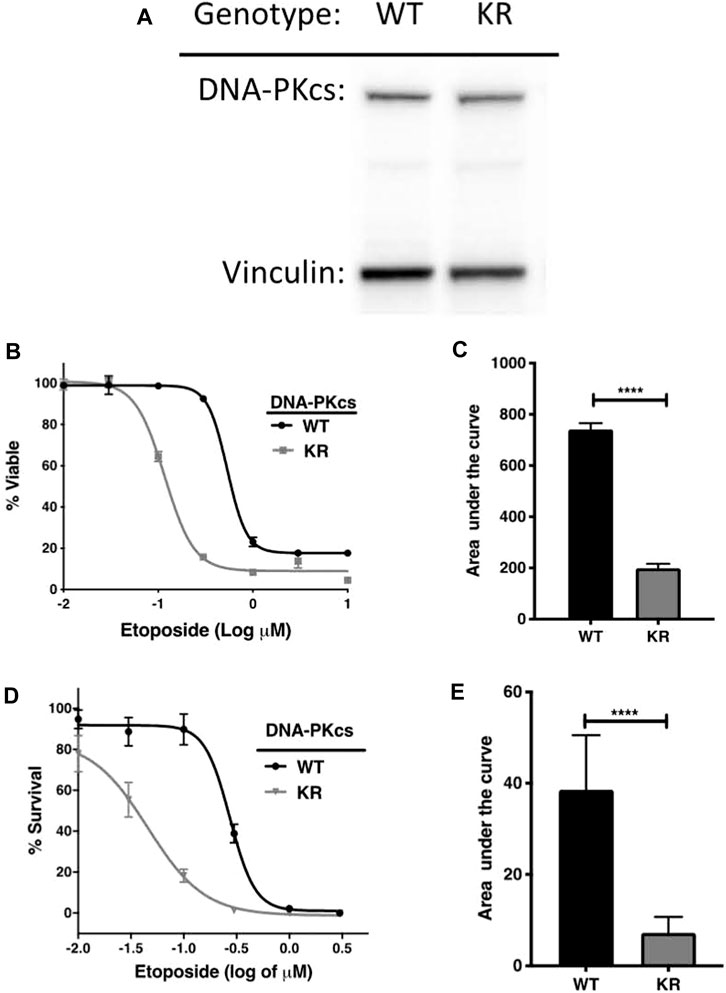
Figure 1. DNA-PKcs protects cells from etoposide toxicity via its kinase domain. (A) Evaluation of DNA-PKcs proteins in CHO variants by Western blot. (B, C) Cell survival profiles of CHO cells assessed using the CellTiter-Glo (CTG) assay with increasing doses (0.003–1 μM) of etoposide at 72 h post-exposure with the associated area under the curve analysis. (D, E) Clonogenic survival of DNA-PKcs WT or KR cells following exposure to etoposide (0.003–1 μM) with the associated area under the curve analysis, ****p < 0.0001.
Differential expression mediated by DNA-PKcs
We assessed the replicates of two isogenic CHO genotypes, WT and KR, treated with either DMSO or etoposide by principal component analysis (PCA; Supplementary Figure S1). All replicates within the genotype and treatment combinations (such as WT–DMSO, WT–etoposide, KR–DMSO, or KR–etoposide) clustered closely, indicating that the replicates strongly correlated with one another. A total of 12,759 genes were of sufficient quality to allow assessment of potential differential regulation in our samples (Table 2). In each of our comparisons, approximately 7,000 genes were differentially expressed, either up or downregulated. We assessed genotype-specific differentially expressed genes (DEGs) by comparing DMSO-treated WT versus KR gene expression. Etoposide-induced alterations within each genotype were compared to those induced by DMSO alone, and finally, differential expression due to drug treatment in WT was compared to that in KR cells. Each red dot represents a single gene with a significant (p < 0.05) positive log-fold change, and the blue dot denotes a gene with a significant negative log-fold change (Supplementary Figures S2A–D). Upon assessing DEGs in KR following etoposide exposure to those in WT, we found most genes are induced independent of DNA-PKcs kinase activity, as these were upregulated in both KR and WT (Figure 2A), where 571 of the upregulated genes were discretely induced in WT and 1,251 were observed only in KR cells. In comparing the diminished expression, 2,338 of 4,466 genes were downregulated independently of DNA-PKcs kinase in both KR and WT in response to etoposide exposure (Figure 2B). The number of privately downregulated DEGs is similar in WT and KR cells (Figure 2B).
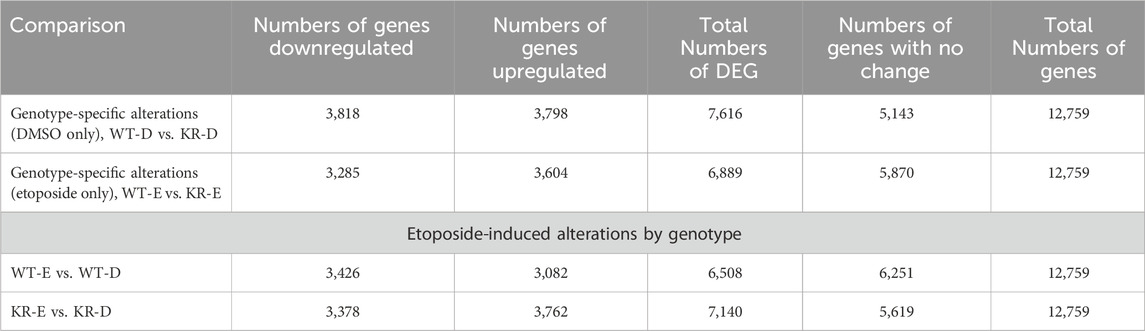
Table 2. Number of genes identified for assessment of potential differential regulation based on genotype.
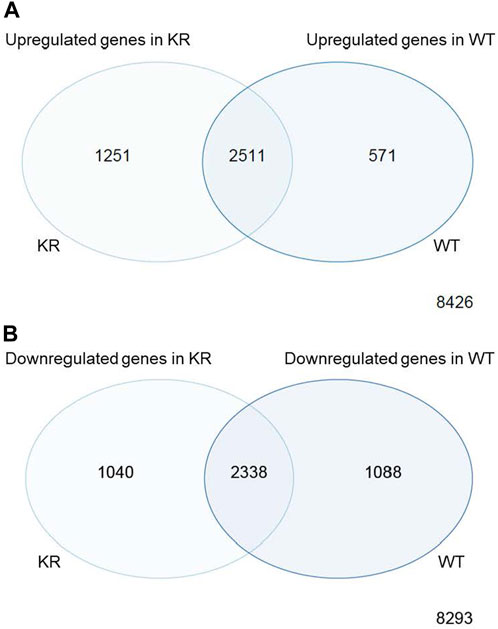
Figure 2. Comparison of shared versus discreet differentially regulated genes in DNA-PK wild-type (WT) versus kinase-inactive (KR) cells. (A) Shared or exclusively upregulated genes in KR and WT cells after etoposide exposure. (B) Shared or exclusively downregulated genes in KR and WT cells after etoposide exposure.
DNA-PKcs alters the transcription of multiple genes involved in various cellular processes
DEGs were analyzed with the KEGG pathway database and were involved in diverse cellular processes (Figures 3A–F). We analyzed these pathways regulated by DEGs to ascertain significantly altered biological processes within WT and KR cells in control (DMSO) or DNA-damaging conditions. We compared the biological pathways populated by the DEGs in WT and KR cells without etoposide treatment. A total of 14 biological pathways were enriched in upregulated DEGs (Supplementary Figure S3A), and seven pathways were associated with downregulation (Supplementary Figure S3B) in KR cells compared to WT. We observed that several genes involved in DNA damage repair pathways were upregulated in KR cells (Figure 3A and Supplementary Figure S3A). Genes upregulated in KR cells compared to WT cells were predominantly involved in cell cycle, cytokine signaling and associated anomalies, and cytoskeletal regulation (Supplementary Figure S3B). Genes upregulated in KR compared to WT were involved with arrhythmogenic right ventricular cardiomyopathy (ARVC), hypertrophic cardiomyopathy (HCM), focal adhesion, axon guidance, and cytokine signaling pathways, indicating DNA-PKcs kinase-dependent regulation of these pathways. In contrast, genes involved in protein processing and degradation, autophagy, and protein and sugar metabolism were less abundant in KR cells than in WT cells (Figure 5B; Supplementary Figure S3B).

Figure 3. Diverse biological pathways regulated by the DEGs from the mRNA seq data found from KEGG pathway analysis. Upregulated genes are represented with red (highest positive fold change) and blue (lowest positive fold change) dots, and downregulated genes are represented with blue (lowest negative fold change) and green (highest negative fold change) dots. Corresponding pathways regulated by DEGs are represented with a solid tangerine circle. The size of the correlates with the number of genes. (A, B) Upregulated and downregulated, respectively, genes in KR cells compared to WT; (C, D) upregulated and downregulated, respectively, genes in WT cells after etoposide exposure; (E, F) upregulated and downregulated, respectively, genes in KR cells after etoposide exposure.
Etoposide-induced differentially regulated biological pathways within WT and KR cells
We analyzed the role of DNA damage in differentially regulated biological pathways between cell lines. A total of 34 pathways were upregulated following etoposide treatment in WT cells (Supplementary Figure S4A), whereas only two pathways were suppressed in WT cells (Supplementary Figure S4B). In KR cells, 37 pathways were upregulated (Supplementary Figure S4C) and three pathways were downregulated (Supplementary Figure S4D) after etoposide treatment. Comparing KR to WT following drug treatment, five pathways were uniquely upregulated in WT, and eight were observed only in KR following drug treatment. Out of 37 pathways, 29 were shared between genotypes, (Figure 4; Supplementary Figures S4A and C) indicating that genes represented here are upregulated independent of DNA-PKcs kinase activity. Three pathways decreased in response to etoposide treatment, in which two were observed in both cell lines, again indicating they are regulated in a DNA-PKcs kinase-independent manner (Figure 4; Supplementary Figures S4B and D). Multiple DNA damage repair processes were commonly upregulated in both cell types after etoposide treatment, whereas NHEJ was exclusively induced in WT cells (Figure 4). Other commonly upregulated pathways including cell cycle, DNA replication, cytokine signaling, calcium signaling, and extracellular matrix (ECM)–receptor interactions are observed in both WT and KR cells. The two common pathways that were downregulated in both WT and KR cells were related to ribosome biogenesis and protein processing (Figure 4).
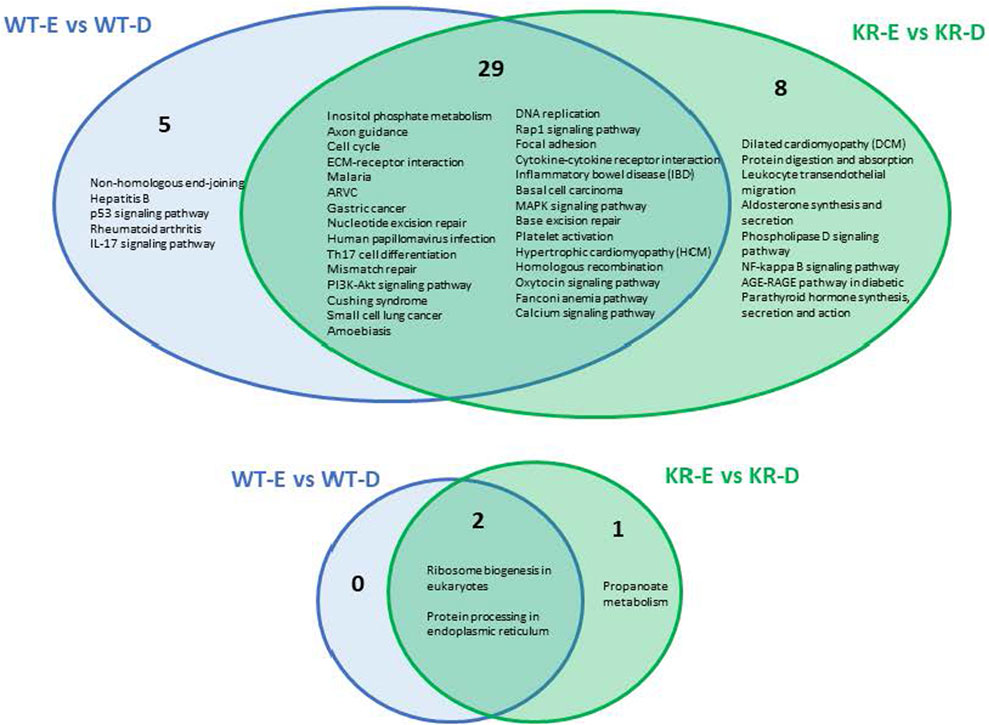
Figure 4. Comparison of shared versus divergent etoposide-induced biological pathways altered by differential gene regulation in DNA-PKcs wild-type (WT) compared to kinase-inactive (KR) cells. Etoposide-induced upregulated biological pathways in WT and KR cells. Etoposide-induced downregulated biological pathways in WT and KR cells.
Differentially regulated biological processes between etoposide-exposed WT and KR cells
We assessed the biological pathways impacted by differential gene regulation between each genotype after exposure to etoposide to discern how DNA-PKcs regulates expression changes following topoisomerase II inhibition (Figures 5A, B). When comparing the effects of etoposide on gene expression between genotypes, we found several genes upregulated in KR cells compared to WT cells in a kinase-dependent and etoposide-independent fashion. These upregulated genes were involved in multiple kinase-driven signaling pathways, including many involved in cancer biology, such as TNF signaling, cell–cell and cell–matrix interactions, and DNA damage response pathways. Genes involved in pathways related to human papilloma virus infection were also upregulated in etoposide-treated KR cells compared to etoposide-treated WT cells (Figure 6A). Downregulated genes in KR cells compared to WT after etoposide exposure are involved in mainly protein processing and carbon metabolism (Figure 6B).
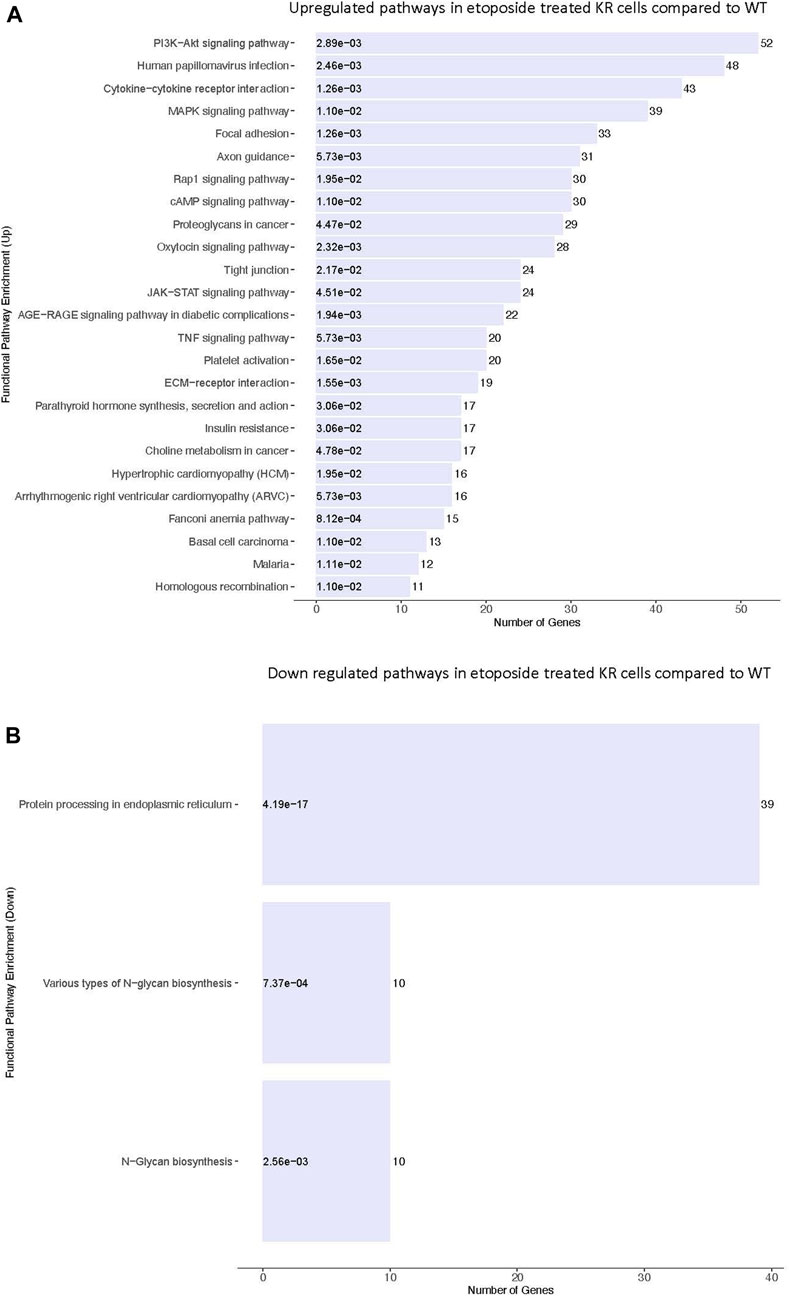
Figure 5. Comparative assessment of etoposide-induced biological pathways altered by differential gene regulation in DNA-PKcs wild-type (WT) and kinase-inactive (KR) cells. (A) Upregulated biological pathways in etoposide-treated KR cells compared to WT genotype-specific upregulated biological processes. (B) Downregulated biological pathways in etoposide-treated KR cells compared to WT.
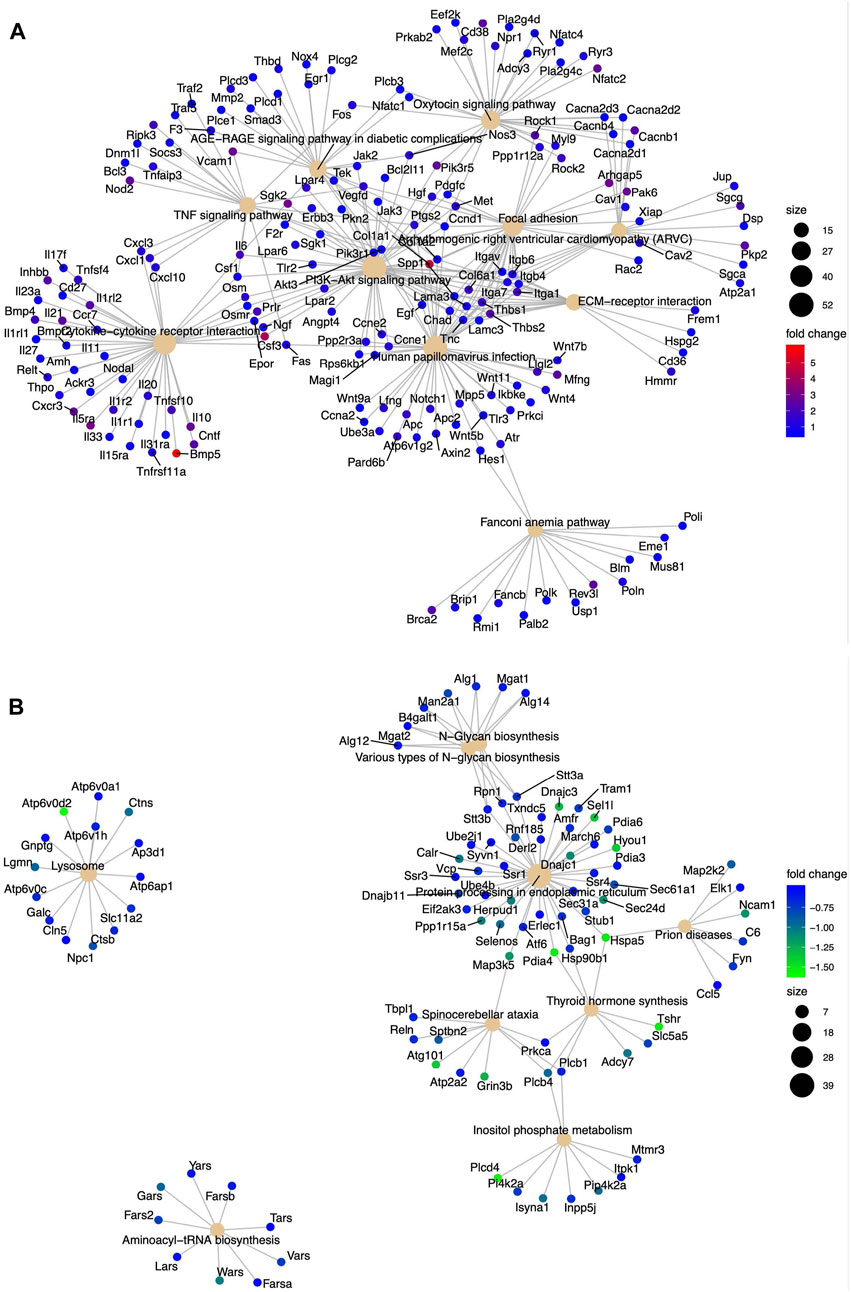
Figure 6. Diverse biological pathways regulated by the DEGs from the mRNA seq data found from KEGG pathway analysis in etoposide-treated WT and KR cells. Upregulated and downregulated genes are represented with blue and red dots and blue and green dots, respectively. Pathways are shown as a solid tangerine circle. (A, B) Upregulated and downregulated, respectively, genes in etoposide-treated KR cells compared to WT.
Analysis of global transcriptional regulators
We evaluated our DEGs using ingenuity pathway analysis (IPA, © 2000–2020, QIAGEN) to determine which master regulators are driving gene expression changes observed in our samples. Master regulators possibly activated or inactivated in response to etoposide exposure to induce or suppress the DEGs are listed in Table 3. A complete summary of IPA-designated master regulators is presented in Supplementary Tables S1–S4. Master regulators controlling genes induced in KR cells without treatment include the estrogen receptor, growth hormone, IGFBP2, and corticotropin-releasing hormone receptor. We also noted an enrichment in proteins involved in proliferation and genes associated with the inflammatory response and angiogenesis. Master regulators diminishing expression include potential tumor suppressors and known modulators of gross or intracellular morphology. IPA reveals that genes regulated by drugs inhibiting VEGF and various pro-growth pathways, including those induced by oncogenes such as Raf, KIT, ALK, and ROS1, are diminished. Overall, this indicates that the cellular milieu induced by expressing DNA-PKcs without functional kinase activity varies substantially compared to that of WT. We analyzed etoposide-induced DEGs in both cell types for detecting the master transcriptional regulators that modify DNA damage-related gene expression changes in WT or KR cells after etoposide exposure (Table 2) Ten master regulators were detected commonly in etoposide-treated cells, whereas most were discreet to each cell type. We contrasted the master regulators between etoposide-treated WT and KR cells. Comparing the upregulated genes in etoposide-treated KR cells to WT cells, we found 14 global regulators, and four upstream global regulators dictated gene repression in the KR.
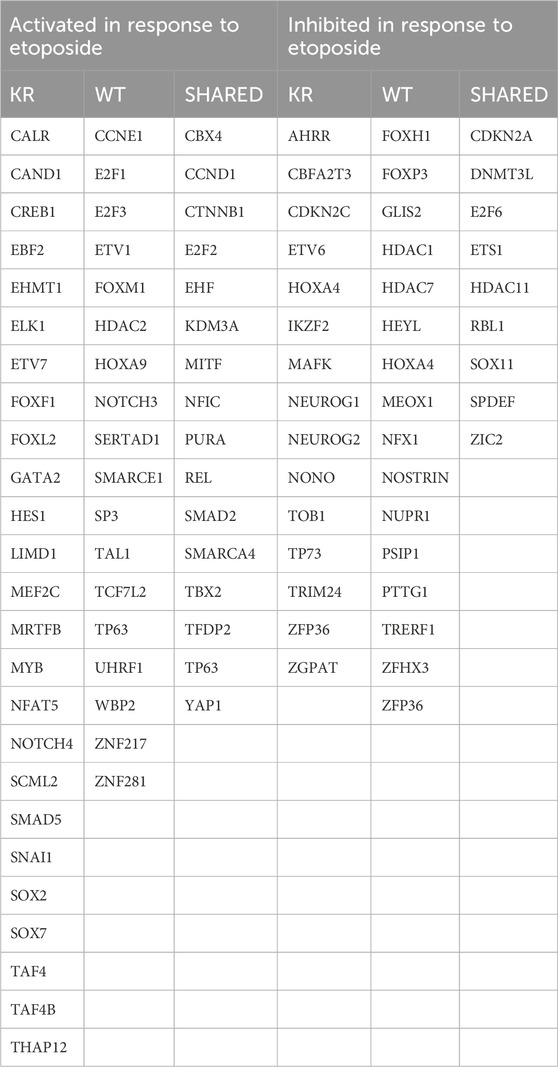
Table 3. Summary of shared or discrete master regulators identified in cells expressing wild-type (WT) or kinase-inactive (KR) DNA-PKcs with or without etoposide exposure.
Relative expression analysis of selected genes by qPCR and immunoblotting
Seventeen DEGs were selected for qPCR based upon the above data. The mean relative expression of DEGs was calculated from the Cq values from the qPCR (Figures 7A–Q). For all genes, the relative expression pattern detected by qPCR followed the same trend observed in RNA-Seq (Table 3). Furthermore, two genes were selected for immunoblotting to validate the protein expression in WT and KR cells (Figures 7R, S). Overall, the trends supported our findings in RNA-Seq and indicated that the kinase activity of DNA-PKcs differentially affects gene regulation with and without exogenous DNA damage.
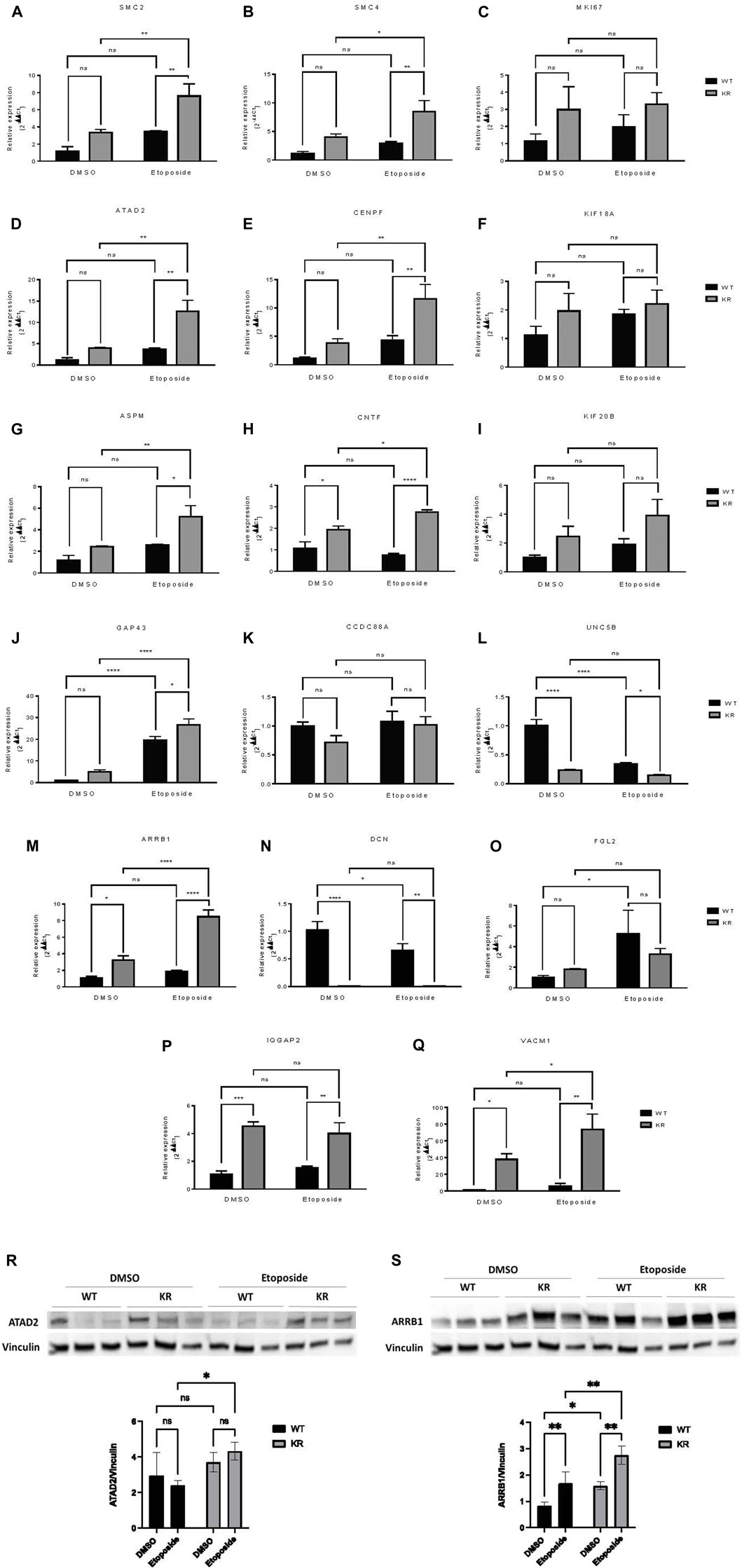
Figure 7. Relative expressions of selected genes detected by qPCR and immunoblotting showed a similar trend with mRNA sequencing data. (A–Q) Seventeen DEGs were selected to perform qPCR to detect their relative expression in WT and KR cells with or without etoposide treatment. The mean relative expression of a DEG was analyzed by Cq values of the target and control genes using the 2−ΔΔCq method after normalizing with the Cq value of GAPDH for any specific sample. (R, S) Two DEGs were selected for immunoblotting to detect their protein expression in WT and KR cells with or without etoposide treatment. Two-way ANOVA with Fisher’s LSD test was used to detect the significant differences among the mean relative expression levels. ns: non-significant, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Discussion and conclusion
DNA-PKcs is a key regulator in two major DNA DSB repair pathways, HR and NHEJ, and is an emerging therapeutic target for multiple cancer subtypes, with most inhibitors preventing its kinase activity (Blackford and Jackson, 2017; Mohiuddin and Kang, 2019; Dylgjeri and Knudsen, 2022). DNA-PKcs is also involved in the phosphorylation of many transcriptional regulators (Collis et al., 2005; Goodwin et al., 2015; Park et al., 2017). We used RNA-Seq to analyze mRNA transcripts isolated from DNA-PKcs WT and kinase-inactivated KR CHO cells after exposure to etoposide. We analyzed our RNA sequencing data and identified the differentially regulated biological pathways that depend on DNA-PKcs kinase activity with or without DNA damage. Previous studies indicated a limited role of DNA-PKcs in transcription after DNA damage; however, data from these studies are not directly comparable to RNA-Seq data (Galloway and Allalunis-Turner, 2000; Bryntesson et al., 2001; An et al., 2005; Goodwin et al., 2015). Novel findings from the current study revealed more than 7,000 DEGs in KR cells compared to WT, while etoposide treatment alone changed the expression of more than 6,500 and 7,100 genes in WT and KR, respectively.
Upregulated genes in DMSO-treated KR cells compared to WT were enriched in three DNA damage repair pathways, including HR. DNA-PKcs is a core component of NHEJ; thus, cells lacking catalytically active DNA-PKcs rely more heavily on HR to repair endogenously formed DSB. We also observed the upregulation of several genes involved in cell cycle regulation and proliferation, in agreement with our previous work, indicating that DNA-PKcs regulates replication in a kinase-dependent fashion in replication stress (Liu et al., 2012; Ashley et al., 2014). Genes upregulated in KR cells compared to WT cells are involved in cell cycle, ARVC, and HCM, indicating that DNA-PKcs regulates gene expression without DNA damage or that these are upregulated in a compensatory manner. Downregulated genes in KR compared to WT cells regulate protein and sugar metabolism (Figure 3B). Goodwin et al. (2015) previously reported that overexpression of various metabolic pathways in DNA-PKcs depleted cancer cells. However, our results found downregulation of genes associated with metabolic pathways in the absence of DNA-PKcs kinase.
We observed that 29 biological pathways were commonly upregulated in both WT and KR cells (Supplementary Figure S4) in a DNA-PKcs kinase-independent manner. Several genes involved in the DNA damage response and inflammatory signaling were upregulated in etoposide-treated KR cells alone, indicating their transcription may be regulated by the kinase activity of DNA-PKcs. Differences in HR-based repair are noted in the WT versus KR cells following exposure to other genotoxic agents, as KR cells promote higher levels of recombination compared to WT cells or cells lacking DNA-PKcs (Ashley et al., 2014).
Etoposide exposure modified the global transcription of several genes in both genotypes. Genes involved in NHEJ are discreetly upregulated in WT cells in the presence of fully functional DNA-PKcs. Genes associated with various DNA repair pathways along with cell cycle and DNA replication were upregulated in both cell lines. Ribosome biogenesis transcripts were downregulated independently of DNA-PKcs, consistent with previous reports which suggest that several DNA repair proteins have a defined role in ribosome biogenesis (Ogawa and Baserga, 2017). Upregulated genes in KR may help the DNA-PKcs kinase-deficient genotypes to survive in the absence of a functional classical NHEJ. Genes involved in inflammation were repressed in WT after etoposide exposure but induced in KR cells. Chronic proinflammatory factors are correlated with persistent DNA damage and induce the expression of IFN-regulated genes (Brzostek-Racine et al., 2011; Karakasilioti et al., 2013). DNA-PKcs kinase may alter the inflammatory milieu via impacting the stability of Egr1, which modulates proinflammatory signaling in T-cells (Waldrip et al., 2021). The robust inflammatory response to DNA damage observed in KR indicates that treatment with a DNA-PKcs inhibitor in combination with chemotherapy or radiotherapy may promote secondary tumors. Further investigations into the clinical ramifications of these data will clarify if DNA-PKcs mitigates the inflammatory response following DNA damage.
In KR, the changes in gene expression were dominantly regulated by the Akt or Wnt/β-catenin signaling pathways, as well as chromatin or developmental modulators (Table 2). Changes in genes dependent on Wnt signaling was more frequently observed in WT cells, indicating DNA-PKcs may regulate this pathway, which is consistent with our previous findings (Gurley et al., 2017). The abundance of regulators associated with development, differentiation, and steroid signaling is likely due to the origin of the cells from ovaries. However, we have previously indicated a role for DNA-PKcs in differentiation and stemness, so these observations warrant additional investigation (Gurley et al., 2017). Master regulators involved in histone modification were found in response to etoposide-mediated DNA damage. DNA-PKcs phosphorylates histone variant H2AX and promotes chromatin decondensation immediately after DSB (Lu et al., 2019). DNA-PKcs-mediated phosphorylation of the histone core after ionizing radiation weakens the DNA-histone binding, which then facilitates the H2A.Z-H2B dimer’s incorporation into reconstituted nucleosomes (Wang et al., 2019). The pattern of changes in histone modifications indicates DNA-PKcs has a role in regulating chromatin after DNA damage.
A subset of genes associated with chromatin modification, neuronal development, and proinflammatory responses was assessed via qPCR. SMC2 and SMC4, components of condensin complex I and II, and CENPF, a component of centromere–kinetochore proteins, all required for mitotic chromosome segregation, were upregulated robustly in KR cells following damage (Figures 7A, B, E) (Varis, Salmela, and Kallio, 2006; Wu and Yu, 2012). Condensin I and II, respectively, participate in DNA single-strand damage and regulate HR-mediated DSB repair (Heale et al., 2006; Wood et al., 2008), whereas overexpression of CENPF is associated with metastasis and co-immunoprecipitates with DNA-PKcs (Du et al., 2010; Yu et al., 2020). KR cells are dependent on HR due to an inability to complete NHEJ, so induction of SMC2, SMC4, or CENPF after etoposide exposure might be expected. On the other hand, ATAD2 is required for histone hyperacetylation, facilitating progression and proliferation in multiple cancers by promoting the expression of some genes from the kinesin family (Liu et al., 2019; Zheng et al., 2015; Zou et al., 2014). Increase in ATAD2 in KR cells (Figures 7D, R) indicates its impact in surviving DNA damage in DNA-PKcs kinase inactivity, consistent with previous findings, where ATAD2 inhibition has shown promising results in inducing apoptosis and autophagy in breast cancer cells (Yao et al., 2020). All of these genes, SMC2, SMC4, CENPF, and ATAD2, could be considered potential cancer therapeutic targets for future studies.
Many genes regulating neuronal development (e.g., ASPM, CNTF, and UNC5B) were differentially regulated in a DNA-PKcs kinase-dependent manner with or without damage. ASPM regulates mitotic spindle orientation and astral microtubule dynamics in embryonic neuroblasts (Jiang et al., 2017), and significant overexpression of ASPM was observed in KR cells after etoposide exposure (Figure 7G). However, impairment of DNA DSB repair after ASPM depletion was also reported (Kato et al., 2011). Dual inhibition of DNA-PKcs and ASPM could be an interesting therapeutic strategy. CNTF is a survival factor in neuronal cells and protects cells from inflammatory destruction (Giess et al., 2002; Linker et al., 2002). CNTF was significantly upregulated in KR cells compared to WT and was further induced after etoposide treatment (Figure 7H). UNC5B is involved in NTN1-dependent cell migration and axon guidance (Shao et al., 2017; Hernaiz-Llorens et al., 2021). In breast cancer cells, UNC5B overexpression promotes proliferation and metastasis; however, it also promotes p53-dependent apoptosis (Wu et al., 2020; Arakawa, 2005). We observed a significant downregulation of UNC5B in KR cells without DNA damage (Figure 7L), and in WT cells after DNA damage; hence, DNA-PKcs may regulate UNC5B-NTN1 signaling. To the best of our knowledge, the role of DNA-PKcs in neuronal development is novel and necessitates more detailed investigation as NHEJ is the sole DSB repair pathway for post-mitotic neurons.
Finally, genes involved in proinflammatory responses were also assessed by qPCR. ARRB1 has varying impacts on NF-κB-dependent transcription (Witherow et al., 2004; Cianfrocca et al., 2014). ARRB1 and NF-κB co-expression promotes cancer progression and is correlated with poor prognosis in lung adenocarcinoma (Yu et al., 2015). High expression of ARRB1 was observed in both WT and KR cells after etoposide treatment (Figures 7M, S); thus, DNA-PKcs kinase activity dictates the etoposide-mediated response to ARRB1 gene expression. A similar pattern of expression was observed in SMC2, SMC4, ATAD2, ASPM, CENPF, and CNTF, supporting the idea that DNA-PKcs kinase activity is dictating some gene expression patterns after DNA damage through unknown mechanism(s).
DCN is a leucine-rich extracellular matrix protein which binds to TGF-β, prevents it from binding to its receptor, and may serve as an antagonistic ligand of VEGFR-2 (Khan et al., 2011). DCN induces autophagy and mitophagy in endothelial cells and breast cancer cells, respectively, and inhibits tumor cell proliferation and tumor invasiveness (Goldoni and Iozzo, 2008; Goyal et al., 2014; Neill et al., 2014). As a significant correlation has been found between the disease-free survival of patients with soft tissue sarcoma and reduced DCN expression, it is used as a prognostic marker for metastasis in breast cancer and soft tissue tumors (Matsumine et al., 2007; Goldoni and Iozzo, 2008). Low expression of DCN in KR cells (Figure 7N) indicates that DNA-PKcs’ kinase activity regulates its transcription. In normal non-tumorigenic cells, DCN promotes angiogenesis, while a high DCN level inhibits tumor vascularization (Khan et al., 2011; Mohan et al., 2011). Modified adenovirus co-expressing IL-12 and DCN have already shown potent antitumor effects in an immunogenic tumor model, indicating DCN as a promising cancer immunotherapeutic (Oh et al., 2017).
Expression of IQGAP2, a potential tumor suppressor, is reduced in human primary gastric cancer, ovarian cancer, and hepatocellular carcinoma (Jin et al., 2008; White et al., 2010; Deng et al., 2016). In prostate and ovarian cancer cell lines, IQGAP2 prevents epithelial–mesenchymal transition (EMT), thus reducing the invasiveness of cancer cells (Xie et al., 2012). IQGAP2 may have a role in the IFN-mediated antiviral immune response through NFκB (Brisac et al., 2016). IQGAP2−/− mice exhibit metabolic abnormalities (Vaitheesvaran et al., 2014). We observed a higher expression of IQGAP2 in KR cells compared to WT (Figure 7P) in an etoposide-independent fashion, indicating DNA-PKcs activity may also regulate the metabolic status and inflammation through regulation of IQGAP2 expression. This is also supported by the AGE-RAGE pathway, a central pathway in diabetes, overexpressed in KR cells compared to WT as well as in etoposide-treated KR cells (Figures 3A, 6C, 7A). VCAM1, a member of the Ig superfamily, is involved in trans-endothelial migration of leukocytes during an inflammatory response and promotes tumor cell invasiveness in breast and colon cancer (Cook-Mills, 2002; Zhang et al., 2020). Like IQGAP2, VCAM1 expression is higher in KR cells compared to the WT cells (Figure 7Q). Collectively, our data support the possibility of DNA-PKcs kinase regulation of inflammation (Figure 6A).
Overall, this global transcriptome study suggests a role for DNA-PKcs in transcriptional regulation with or without DNA damage. Our results provide additional knowledge about how DNA-PKcs modifies the transcriptional response to DNA damage, leading to alterations of many essential cellular pathways which ultimately dictate genomic stability. Multiple DNA-PKcs inhibitors are in clinical trials as potential therapeutic interventions in multiple cancer subtypes coupled with traditional chemotherapy and/or radiotherapy; however, we lack a full appreciation of how inhibition of DNA-PKcs kinase will alter the cellular transcriptional response to DNA damage. Our results indicate that impairing DNA-PKcs kinase activity may profoundly impact the transcriptional response to DNA damage, modulating the proclivity for secondary tumor formation via impairing traditional DNA damage signaling and repair, altering transcriptional changes aimed at maintaining genomic stability following DNA damage, and modulating inflammation within the tumor microenvironment.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://doi.org/doi:10.5061/dryad.0zpc8673k. The supplemental information is being hosted by Zenodo and is now published and publicly available here: https://doi.org/10.5281/zenodo.10641696.
Author contributions
SA: data curation, writing–original draft, and writing–review and editing. MN-P: data curation, methodology, validation, visualization, and writing–review and editing. KP: formal analysis, investigation, writing–review and editing. FL: data curation, visualization, and writing–review and editing. JS: data curation, methodology, software, and writing–review and editing. AA: conceptualization, formal analysis, funding acquisition, investigation, project administration, resources, supervision, validation, visualization, writing–original draft, and writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Research reported in this publication was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103451.
Acknowledgments
The authors appreciate the support of Rashmi Joshi in assuring these experiments were complete. The authors would also like to acknowledge all members of AA’s group in supporting the authors’ work.
Conflict of interest
Author JS was employed by the National Center for Genome Resources.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1276365/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Principal component analysis (PCA) of 12 total RNA samples isolated from WT and KR cells treated with DMSO and etoposide. PCA indicates all replicates of each genotype and treatment combinations, WT-DMSO, WT-etoposide, KR-DMSO, or KR-etoposide, were clustered closely within replicates.
SUPPLEMENTARY FIGURE S2 | Differentially expressed genes (DEGs) in DNA-PKcs wild-type (WT) versus kinase-inactive (KR) cells. Each red dot denotes one single gene that has a significant (p < 0.05) positive log-fold change, and the blue dot denotes the gene with a significant negative log-fold change. Each black dot defines a gene with a p-value that was not significantly altered (p > 0.05). (A) DEGs in KR cells compared to WT without etoposide treatment. (B) DEGs in KR cells compared to WT after etoposide exposure. (C) DEGs in etoposide-exposed WT cells compared to its control counterpart. (D) DEGs in etoposide-exposed KR cells compared to its control counterpart.
SUPPLEMENTARY FIGURE S3 | Genotype-specific biological pathways altered by differential gene regulation in DNA-PKcs wild-type (WT) versus kinase-inactive (KR) cells without drug exposure. (A) Genotype-specific over-represented biological processes populated by upregulated genes in KR compared to WT. (B) Genotype-specific under-represented biological processes populated by downregulated genes in KR compared to WT.
SUPPLEMENTARY FIGURE S4 | Etoposide-induced biological pathways altered by differential gene regulation in DNA-PKcs wild-type (WT) and kinase-inactive (KR) cells. (A) Etoposide-induced upregulated biological processes within WT cells. (B) Etoposide-induced downregulated biological processes in WT cells. (C) Etoposide-induced upregulated biological processes within KR cells. (D) Etoposide-induced downregulated biological processes in KR cells.
SUPPLEMENTARY TABLE S1 | Genotypic master regulators controlling the gene expression differences observed in cells producing wild-type (WT) or kinase inactive (KR) DNA-PKcs.
SUPPLEMENTARY TABLE S2 | All master regulators controlling the gene expression differences observed in cells producing wild-type (WT) DNA-PKcs following etoposide treatment.
SUPPLEMENTARY TABLE S3 | Master regulators controlling the gene expression differences observed in cells producing kinase-inactive (KR) DNA-PKcs following etoposide treatment.
SUPPLEMENTARY TABLE S4 | Genotypic master regulators controlling the gene expression differences observed in cells producing wild-type (WT) or kinase inactive (KR) DNA-PKcs after etoposide exposure.
References
An, J., Xu, Q. Z., Sui, J. L., Bai, B., and Zhou, P. K. (2005). Silencing of DNA-PKcs alters the transcriptional profile of certain signal transduction genes related to proliferation and differentiation in HeLa cells. Int. J. Mol. Med. 16 (3), 455–462. doi:10.3892/ijmm.16.3.455
Arakawa, H. (2005). p53, apoptosis and axon-guidance molecules. Cell Death Differ. 12 (8), 1057–1065. doi:10.1038/sj.cdd.4401601
Ashley, A. K., Shrivastav, M., Nie, J., Amerin, C., Troksa, K., Glanzer, J. G., et al. (2014). DNA-PK phosphorylation of RPA32 Ser4/Ser8 regulates replication stress checkpoint activation, fork restart, homologous recombination and mitotic catastrophe. DNA Repair (Amst) 21, 131–139. doi:10.1016/j.dnarep.2014.04.008
Blackford, A. N., and Jackson, S. P. (2017). ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol. Cell 66 (6), 801–817. doi:10.1016/j.molcel.2017.05.015
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi:10.1093/bioinformatics/btu170
Bouquet, F., Ousset, M., Biard, D., Fallone, F., Dauvillier, S., Frit, P., et al. (2011). A DNA-dependent stress response involving DNA-PK occurs in hypoxic cells and contributes to cellular adaptation to hypoxia. J. Cell Sci. 124 (Pt 11), 1943–1951. doi:10.1242/jcs.078030
Brisac, C., Salloum, S., Yang, V., Schaefer, E. A., Holmes, J. A., Chevaliez, S., et al. (2016). IQGAP2 is a novel interferon-alpha antiviral effector gene acting non-conventionally through the NF-κB pathway. J. Hepatol. 65 (5), 972–979. doi:10.1016/j.jhep.2016.06.028
Bryntesson, F., Regan, J. C., Jeggo, P. A., Taccioli, G. E., and Hubank, M. (2001). Analysis of gene transcription in cells lacking DNA-PK activity. Radiat. Res. 156 (2), 167–176. doi:10.1667/0033-7587(2001)156[0167:aogtic]2.0.co;2
Brzostek-Racine, S., Gordon, C., Van Scoy, S., and Reich, N. C. (2011). The DNA damage response induces IFN. J. Immunol. 187 (10), 5336–5345. doi:10.4049/jimmunol.1100040
Cianfrocca, R., Tocci, P., Semprucci, E., Spinella, F., Di Castro, V., Bagnato, A., et al. (2014). β-Arrestin 1 is required for endothelin-1-induced NF-κB activation in ovarian cancer cells. Life Sci. 118 (2), 179–184. doi:10.1016/j.lfs.2014.01.078
Collis, S. J., DeWeese, T. L., Jeggo, P. A., and Parker, A. R. (2005). The life and death of DNA-PK. Oncogene 24 (6), 949–961. doi:10.1038/sj.onc.1208332
Convery, E., Shin, E. K., Ding, Q., Wang, W., Douglas, P., Davis, L. S., et al. (2005). Inhibition of homologous recombination by variants of the catalytic subunit of the DNA-dependent protein kinase (DNA-PKcs). Proc. Natl. Acad. Sci. U. S. A. 102 (5), 1345–1350. doi:10.1073/pnas.0406466102
Cook-Mills, J. M. (2002). VCAM-1 signals during lymphocyte migration: role of reactive oxygen species. Mol. Immunol. 39 (9), 499–508. doi:10.1016/s0161-5890(02)00206-7
Deng, Z., Wang, L., Hou, H., Zhou, J., and Li, X. (2016). Epigenetic regulation of IQGAP2 promotes ovarian cancer progression via activating Wnt/β-catenin signaling. Int. J. Oncol. 48 (1), 153–160. doi:10.3892/ijo.2015.3228
Du, J., Zhang, Y., Liu, Y., Li, Y., and Zhu, X. (2010). Involvement of Cenp-F in interphase chromatin organization possibly through association with DNA-dependent protein kinase. Acta Biochim. Biophys. Sin. (Shanghai) 42 (12), 839–846. doi:10.1093/abbs/gmq095
Dvir, A., Peterson, S. R., Knuth, M. W., Lu, H., and Dynan, W. S. (1992). Ku autoantigen is the regulatory component of a template-associated protein kinase that phosphorylates RNA polymerase II. Proc. Natl. Acad. Sci. U. S. A. 89 (24), 11920–11924. doi:10.1073/pnas.89.24.11920
Dylgjeri, E., and Knudsen, K. E. (2022). DNA-PKcs: a targetable protumorigenic protein kinase. Cancer Res. 82 (4), 523–533. doi:10.1158/0008-5472.CAN-21-1756
Galloway, A. M., and Allalunis-Turner, J. (2000). cDNA expression array analysis of DNA repair genes in human glioma cells that lack or express DNA-PK. Radiat. Res. 154 (6), 609–615. doi:10.1667/0033-7587(2000)154[0609:ceaaod]2.0.co;2
Giess, R., Mäurer, M., Linker, R., Gold, R., Warmuth-Metz, M., Toyka, K. V., et al. (2002). Association of a null mutation in the CNTF gene with early onset of multiple sclerosis. Arch. Neurol. 59 (3), 407–409. doi:10.1001/archneur.59.3.407
Goldoni, S., and Iozzo, R. V. (2008). Tumor microenvironment: modulation by decorin and related molecules harboring leucine-rich tandem motifs. Int. J. Cancer 123 (11), 2473–2479. doi:10.1002/ijc.23930
Goodwin, J. F., and Knudsen, K. E. (2014). Beyond DNA repair: DNA-PK function in cancer. Cancer Discov. 4 (10), 1126–1139. doi:10.1158/2159-8290.CD-14-0358
Goodwin, J. F., Kothari, V., Drake, J. M., Zhao, S., Dylgjeri, E., Dean, J. L., et al. (2015). DNA-PKcs-Mediated transcriptional regulation drives prostate cancer progression and metastasis. Cancer Cell 28 (1), 97–113. doi:10.1016/j.ccell.2015.06.004
Goyal, A., Neill, T., Owens, R. T., Schaefer, L., and Iozzo, R. V. (2014). Decorin activates AMPK, an energy sensor kinase, to induce autophagy in endothelial cells. Matrix Biol. 34, 46–54. doi:10.1016/j.matbio.2013.12.011
Gurley, K. E., Ashley, A. K., Moser, R. D., and Kemp, C. J. (2017). Synergy between Prkdc and Trp53 regulates stem cell proliferation and GI-ARS after irradiation. Cell Death Differ. 24 (11), 1853–1860. doi:10.1038/cdd.2017.107
Heale, J. T., Ball, A. R., Schmiesing, J. A., Kim, J. S., Kong, X., Zhou, S., et al. (2006). Condensin I interacts with the PARP-1-XRCC1 complex and functions in DNA single-strand break repair. Mol. Cell 21 (6), 837–848. doi:10.1016/j.molcel.2006.01.036
Hernaiz-Llorens, M., Roselló-Busquets, C., Durisic, N., Filip, A., Ulloa, F., Martínez-Mármol, R., et al. (2021). Growth cone repulsion to Netrin-1 depends on lipid raft microdomains enriched in UNC5 receptors. Cell Mol. Life Sci. 78 (6), 2797–2820. doi:10.1007/s00018-020-03663-z
Jackson, S. P., MacDonald, J. J., Lees-Miller, S., and Tjian, R. (1990). GC box binding induces phosphorylation of Sp1 by a DNA-dependent protein kinase. Cell 63 (1), 155–165. doi:10.1016/0092-8674(90)90296-q
Jiang, K., Rezabkova, L., Hua, S., Liu, Q., Capitani, G., Altelaar, A. F. M., et al. (2017). Microtubule minus-end regulation at spindle poles by an ASPM-katanin complex. Nat. Cell Biol. 19 (5), 480–492. doi:10.1038/ncb3511
Jin, S. H., Akiyama, Y., Fukamachi, H., Yanagihara, K., Akashi, T., and Yuasa, Y. (2008). IQGAP2 inactivation through aberrant promoter methylation and promotion of invasion in gastric cancer cells. Int. J. Cancer 122 (5), 1040–1046. doi:10.1002/ijc.23181
Joshi, R. R., Ali, S. I., and Ashley, A. K. (2019). DNA ligase IV prevents replication fork stalling and promotes cellular proliferation in triple negative breast cancer. J. Nucleic Acids 2019, 9170341. doi:10.1155/2019/9170341
Ju, B. G., Lunyak, V. V., Perissi, V., Garcia-Bassets, I., Rose, D. W., Glass, C. K., et al. (2006). A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 312 (5781), 1798–1802. doi:10.1126/science.1127196
Ju, B. G., and Rosenfeld, M. G. (2006). A breaking strategy for topoisomerase IIbeta/PARP-1-dependent regulated transcription. Cell Cycle 5 (22), 2557–2560. doi:10.4161/cc.5.22.3497
Karakasilioti, I., Kamileri, I., Chatzinikolaou, G., Kosteas, T., Vergadi, E., Robinson, A. R., et al. (2013). DNA damage triggers a chronic autoinflammatory response, leading to fat depletion in NER progeria. Cell Metab. 18 (3), 403–415. doi:10.1016/j.cmet.2013.08.011
Karmakar, P., Piotrowski, J., Brosh, R. M., Sommers, J. A., Miller, S. P., Cheng, W. H., et al. (2002). Werner protein is a target of DNA-dependent protein kinase in vivo and in vitro, and its catalytic activities are regulated by phosphorylation. J. Biol. Chem. 277 (21), 18291–18302. doi:10.1074/jbc.M111523200
Kato, T. A., Okayasu, R., Jeggo, P. A., and Fujimori, A. (2011). ASPM influences DNA double-strand break repair and represents a potential target for radiotherapy. Int. J. Radiat. Biol. 87 (12), 1189–1195. doi:10.3109/09553002.2011.624152
Khan, G. A., Girish, G. V., Lala, N., Di Guglielmo, G. M., and Lala, P. K. (2011). Decorin is a novel VEGFR-2-binding antagonist for the human extravillous trophoblast. Mol. Endocrinol. 25 (8), 1431–1443. doi:10.1210/me.2010-0426
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12 (4), 357–360. doi:10.1038/nmeth.3317
Krämer, A., Green, J., Pollard, J., and Tugendreich, S. (2014). Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30 (4), 523–530. doi:10.1093/bioinformatics/btt703
Lees-Miller, S. P. (1996). The DNA-dependent protein kinase, DNA-PK: 10 years and no ends in sight. Biochem. Cell Biol. 74 (4), 503–512. doi:10.1139/o96-054
Liao, Y., Smyth, G. K., and Shi, W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30 (7), 923–930. doi:10.1093/bioinformatics/btt656
Lindahl, T., and Barnes, D. E. (2000). Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 65, 127–133. doi:10.1101/sqb.2000.65.127
Linker, R. A., Mäurer, M., Gaupp, S., Martini, R., Holtmann, B., Giess, R., et al. (2002). CNTF is a major protective factor in demyelinating CNS disease: a neurotrophic cytokine as modulator in neuroinflammation. Nat. Med. 8 (6), 620–624. doi:10.1038/nm0602-620
Liu, N., Funasaka, K., Obayashi, T., Miyahara, R., Hirooka, Y., Goto, H., et al. (2019). ATAD2 is associated with malignant characteristics of pancreatic cancer cells. Oncol. Lett. 17 (3), 3489–3494. doi:10.3892/ol.2019.9960
Liu, S., Opiyo, S. O., Manthey, K., Glanzer, J. G., Ashley, A. K., Amerin, C., et al. (2012). Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res. 40 (21), 10780–10794. doi:10.1093/nar/gks849
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25 (4), 402–408. doi:10.1006/meth.2001.1262
Lu, H., Saha, J., Beckmann, P. J., Hendrickson, E. A., and Davis, A. J. (2019). DNA-PKcs promotes chromatin decondensation to facilitate initiation of the DNA damage response. Nucleic Acids Res. 47 (18), 9467–9479. doi:10.1093/nar/gkz694
Maldonado, E., Shiekhattar, R., Sheldon, M., Cho, H., Drapkin, R., Rickert, P., et al. (1996). A human RNA polymerase II complex associated with SRB and DNA-repair proteins. Nature 381 (6577), 86–89. doi:10.1038/381086a0
Matsumine, A., Shintani, K., Kusuzaki, K., Matsubara, T., Satonaka, H., Wakabayashi, T., et al. (2007). Expression of decorin, a small leucine-rich proteoglycan, as a prognostic factor in soft tissue tumors. J. Surg. Oncol. 96 (5), 411–418. doi:10.1002/jso.20745
Mohan, R. R., Tovey, J. C., Sharma, A., Schultz, G. S., Cowden, J. W., and Tandon, A. (2011). Targeted decorin gene therapy delivered with adeno-associated virus effectively retards corneal neovascularization in vivo. PLoS One 6 (10), e26432. doi:10.1371/journal.pone.0026432
Mohiuddin, I. S., and Kang, M. H. (2019). DNA-PK as an emerging therapeutic target in cancer. Front. Oncol. 9, 635. doi:10.3389/fonc.2019.00635
Neal, J. A., and Meek, K. (2011). Choosing the right path: does DNA-PK help make the decision? Mutat. Res. 711 (1-2), 73–86. doi:10.1016/j.mrfmmm.2011.02.010
Neal, J. A., Sugiman-Marangos, S., VanderVere-Carozza, P., Wagner, M., Turchi, J., Lees-Miller, S. P., et al. (2014). Unraveling the complexities of DNA-dependent protein kinase autophosphorylation. Mol. Cell Biol. 34 (12), 2162–2175. doi:10.1128/MCB.01554-13
Neill, T., Torres, A., Buraschi, S., Owens, R. T., Hoek, J. B., Baffa, R., et al. (2014). Decorin induces mitophagy in breast carcinoma cells via peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) and mitostatin. J. Biol. Chem. 289 (8), 4952–4968. doi:10.1074/jbc.M113.512566
Ogawa, L. M., and Baserga, S. J. (2017). Crosstalk between the nucleolus and the DNA damage response. Mol. Biosyst. 13 (3), 443–455. doi:10.1039/c6mb00740f
Oh, E., Choi, I. K., Hong, J., and Yun, C. O. (2017). Oncolytic adenovirus coexpressing interleukin-12 and decorin overcomes Treg-mediated immunosuppression inducing potent antitumor effects in a weakly immunogenic tumor model. Oncotarget 8 (3), 4730–4746. doi:10.18632/oncotarget.13972
Park, S. J., Gavrilova, O., Brown, A. L., Soto, J. E., Bremner, S., Kim, J., et al. (2017). DNA-PK promotes the mitochondrial, metabolic, and physical decline that occurs during aging. Cell Metab. 26 (2), 447. doi:10.1016/j.cmet.2017.07.005
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33 (3), 290–295. doi:10.1038/nbt.3122
Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W., et al. (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43 (7), e47. doi:10.1093/nar/gkv007
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26 (1), 139–140. doi:10.1093/bioinformatics/btp616
Shao, Q., Yang, T., Huang, H., Alarmanazi, F., and Liu, G. (2017). Uncoupling of UNC5C with polymerized TUBB3 in microtubules mediates netrin-1 repulsion. J. Neurosci. 37 (23), 5620–5633. doi:10.1523/JNEUROSCI.2617-16.2017
Shrivastav, M., De Haro, L. P., and Nickoloff, J. A. (2008). Regulation of DNA double-strand break repair pathway choice. Cell Res. 18 (1), 134–147. doi:10.1038/cr.2007.111
Vaitheesvaran, B., Hartil, K., Navare, A., Zheng, Z., Obroin, P., Golden, A., et al. (2014). Role of the tumor suppressor IQGAP2 in metabolic homeostasis: possible link between diabetes and cancer. Metabolomics 10 (5), 920–937. doi:10.1007/s11306-014-0639-9
Varis, A., Salmela, A. L., and Kallio, M. J. (2006). Cenp-F (mitosin) is more than a mitotic marker. Chromosoma 115 (4), 288–295. doi:10.1007/s00412-005-0046-0
Waldrip, Z. J., Burdine, L., Harrison, D. K., Azevedo-Pouly, A. C., Storey, A. J., Moffett, O. G., et al. (2021). DNA-PKcs kinase activity stabilizes the transcription factor Egr1 in activated immune cells. J. Biol. Chem. 297 (4), 101209. doi:10.1016/j.jbc.2021.101209
Wang, L. Y., He, Y. X., Li, M., Ding, J., Sui, Y., Conaway, J. W., et al. (2019). DNA-dependent protein kinase catalytic subunit (DNA-PKcs) contributes to incorporation of histone variant H2A.Z into nucleosomes. Protein Cell 10 (9), 694–699. doi:10.1007/s13238-019-0632-1
White, C. D., Khurana, H., Gnatenko, D. V., Li, Z., Odze, R. D., Sacks, D. B., et al. (2010). IQGAP1 and IQGAP2 are reciprocally altered in hepatocellular carcinoma. BMC Gastroenterol. 10, 125. doi:10.1186/1471-230X-10-125
Witherow, D. S., Garrison, T. R., Miller, W. E., and Lefkowitz, R. J. (2004). beta-Arrestin inhibits NF-kappaB activity by means of its interaction with the NF-kappaB inhibitor IkappaBalpha. Proc. Natl. Acad. Sci. U. S. A. 101 (23), 8603–8607. doi:10.1073/pnas.0402851101
Wong, R. H., Chang, I., Hudak, C. S., Hyun, S., Kwan, H. Y., and Sul, H. S. (2009). A role of DNA-PK for the metabolic gene regulation in response to insulin. Cell 136 (6), 1056–1072. doi:10.1016/j.cell.2008.12.040
Wood, J. L., Liang, Y., Li, K., and Chen, J. (2008). Microcephalin/MCPH1 associates with the Condensin II complex to function in homologous recombination repair. J. Biol. Chem. 283 (43), 29586–29592. doi:10.1074/jbc.M804080200
Wu, N., and Yu, H. (2012). The Smc complexes in DNA damage response. Cell Biosci. 2, 5. doi:10.1186/2045-3701-2-5
Wu, S., Guo, X., Zhou, J., Zhu, X., Chen, H., Zhang, K., et al. (2020). High expression of UNC5B enhances tumor proliferation, increases metastasis, and worsens prognosis in breast cancer. Aging (Albany NY) 12 (17), 17079–17098. doi:10.18632/aging.103639
Xie, Y., Yan, J., Cutz, J. C., Rybak, A. P., He, L., Wei, F., et al. (2012). IQGAP2, A candidate tumour suppressor of prostate tumorigenesis. Biochim. Biophys. Acta 1822 (6), 875–884. doi:10.1016/j.bbadis.2012.02.019
Xing, M., and Oksenych, V. (2019). Genetic interaction between DNA repair factors PAXX, XLF, XRCC4 and DNA-PKcs in human cells. FEBS Open Bio 9 (7), 1315–1326. doi:10.1002/2211-5463.12681
Yao, D., Zhang, J., Wang, J., Pan, D., and He, Z. (2020). Discovery of novel ATAD2 bromodomain inhibitors that trigger apoptosis and autophagy in breast cells by structure-based virtual screening. J. Enzyme Inhib. Med. Chem. 35 (1), 713–725. doi:10.1080/14756366.2020.1740924
Yoo, S., and Dynan, W. S. (1999). Geometry of a complex formed by double strand break repair proteins at a single DNA end: recruitment of DNA-PKcs induces inward translocation of Ku protein. Nucleic Acids Res. 27 (24), 4679–4686. doi:10.1093/nar/27.24.4679
Yu, B., Chen, L., Zhang, W., Li, Y., Zhang, Y., Gao, Y., et al. (2020). TOP2A and CENPF are synergistic master regulators activated in cervical cancer. BMC Med. Genomics 13 (1), 145. doi:10.1186/s12920-020-00800-2
Yu, G., Wang, L. G., Han, Y., and He, Q. Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16 (5), 284–287. doi:10.1089/omi.2011.0118
Yu, J., Wang, L., Zhang, T., Shen, H., Dong, W., Ni, Y., et al. (2015). Co-expression of β-arrestin1 and NF-кB is associated with cancer progression and poor prognosis in lung adenocarcinoma. Tumour Biol. 36 (8), 6551–6558. doi:10.1007/s13277-015-3349-7
Zhang, D., Bi, J., Liang, Q., Wang, S., Zhang, L., Han, F., et al. (2020). VCAM1 promotes tumor cell invasion and metastasis by inducing EMT and transendothelial migration in colorectal cancer. Front. Oncol. 10, 1066. doi:10.3389/fonc.2020.01066
Zheng, L., Li, T., Zhang, Y., Guo, Y., Yao, J., Dou, L., et al. (2015). Oncogene ATAD2 promotes cell proliferation, invasion and migration in cervical cancer. Oncol. Rep. 33 (5), 2337–2344. doi:10.3892/or.2015.3867
Zou, J. X., Duan, Z., Wang, J., Sokolov, A., Xu, J., Chen, C. Z., et al. (2014). Kinesin family deregulation coordinated by bromodomain protein ANCCA and histone methyltransferase MLL for breast cancer cell growth, survival, and tamoxifen resistance. Mol. Cancer Res. 12 (4), 539–549. doi:10.1158/1541-7786.MCR-13-0459
Keywords: DNA-PKcs, DNA damage repair, non-homologous end-joining, etoposide, gene expression, transcriptome
Citation: Ali SI, Najaf-Panah MJ, Pyper KB, Lujan FE, Sena J and Ashley AK (2024) Comparative analysis of basal and etoposide-induced alterations in gene expression by DNA-PKcs kinase activity. Front. Genet. 15:1276365. doi: 10.3389/fgene.2024.1276365
Received: 11 August 2023; Accepted: 29 January 2024;
Published: 21 March 2024.
Edited by:
Ata Abbas, Case Western Reserve University, United StatesReviewed by:
Atrayee Bhattacharya, Dana–Farber Cancer Institute, United StatesWaldemar Wagner, Polish Academy of Sciences, Poland
Anthony J. Davis, University of Texas Southwestern Medical Center, United States
Copyright © 2024 Ali, Najaf-Panah, Pyper, Lujan, Sena and Ashley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amanda K. Ashley, YXNobGV5YWtAbm1zdS5lZHU=