 Sebastien Audet1,2*Valerie Triassi1
Sebastien Audet1,2*Valerie Triassi1 Myriam Gelinas3Nab Legault-Cadieux1,2Vincent Ferraro3
Myriam Gelinas3Nab Legault-Cadieux1,2Vincent Ferraro3 Antoine Duquette1,2,4,5
Antoine Duquette1,2,4,5 Martine Tetreault1,2
Martine Tetreault1,2- 1University of Montreal Hospital Research Center (CRCHUM), Montreal, QC, Canada
- 2Department of Neurosciences, University of Montreal, Montreal, QC, Canada
- 3Department of Medicine, University of Montreal Hospital Centre (CHUM), Montreal, QC, Canada
- 4Neurology Service, Department of Medicine, André-Barbeau Movement Disorders Unit, University of Montreal Hospital (CHUM), Montreal, QC, Canada
- 5Genetic Service, Department of Medicine, University of Montreal Hospital (CHUM), Montreal, QC, Canada
Background: Episodic ataxias are rare neurological disorders characterized by recurring episodes of imbalance and coordination difficulties. Obtaining definitive molecular diagnoses poses challenges, as clinical presentation is highly heterogeneous, and literature on the underlying genetics is limited. While the advent of high-throughput sequencing technologies has significantly contributed to Mendelian disorders genetics, interpretation of variants of uncertain significance and other limitations inherent to individual methods still leaves many patients undiagnosed. This study aimed to investigate the utility of multi-omics for the identification and validation of molecular candidates in a cohort of complex cases of ataxia with episodic presentation.
Methods: Eight patients lacking molecular diagnosis despite extensive clinical examination were recruited following standard genetic testing. Whole genome and RNA sequencing were performed on samples isolated from peripheral blood mononuclear cells. Integration of expression and splicing data facilitated genomic variants prioritization. Subsequently, long-read sequencing played a crucial role in the validation of those candidate variants.
Results: Whole genome sequencing uncovered pathogenic variants in four genes (SPG7, ATXN2, ELOVL4, PMPCB). A missense and a nonsense variant, both previously reported as likely pathogenic, configured in trans in individual #1 (SPG7: c.2228T>C/p.I743T, c.1861C>T/p.Q621*). An ATXN2 microsatellite expansion (CAG32) in another late-onset case. In two separate individuals, intronic variants near splice sites (ELOVL4: c.541 + 5G>A; PMPCB: c.1154 + 5G>C) were predicted to induce loss-of-function splicing, but had never been reported as disease-causing. Long-read sequencing confirmed the compound heterozygous variants configuration, repeat expansion length, as well as splicing landscape for those pathogenic variants. A potential genetic modifier of the ATXN2 expansion was discovered in ZFYVE26 (c.3022C>T/p.R1008*).
Conclusion: Despite failure to identify pathogenic variants through clinical genetic testing, the multi-omics approach enabled the molecular diagnosis in 50% of patients, also giving valuable insights for variant prioritization in remaining cases. The findings demonstrate the value of long-read sequencing for the validation of candidate variants in various scenarios. Our study demonstrates the effectiveness of leveraging complementary omics technologies to unravel the underlying genetics in patients with unresolved rare diseases such as ataxia. Molecular diagnoses not only hold significant promise in improving patient care management, but also alleviates the burden of diagnostic odysseys, more broadly enhancing quality of life.
1 Introduction
Ataxias encompass neurodegenerative disorders characterized by impaired coordination and balance, but also a remarkable clinical and genetic heterogeneity. With over 160 genes known to affect cerebellar functions, ataxia-related manifestations are found in a vast array of conditions such as spinocerebellar ataxias (SCAs), autosomal recessive cerebellar ataxias, episodic ataxias (EAs), spastic paraplegias, and mitochondrial disorders (Perlman et al., 1993; Synofzik et al., 2023). Furthermore, acquired ataxia can stem from non-genetic cerebellar damage, which can arise from autoimmunity, metabolic toxicity, vitamin deficiency, and infections (Nachbauer et al., 2015). These disorders exhibit a broad range of clinical features, most commonly unstable gait, dysarthria, visual disturbances, cerebellar atrophy, and muscle weakness. In addition to the age of onset and severity of clinical phenotypes being highly variable, disease presentation can extend beyond cerebellar functions, resulting in multi-systemic manifestations (Traschutz et al., 2021; Roberts et al., 2022). Part of the clinical heterogeneity and variable penetrance observed between ataxic individuals can be attributed to the diversity of potential pathogenic genomic alteration (Hersheson et al., 2012): single nucleotide polymorphisms, structural variants, copy number variants, repeat expansions, and splicing defects have all previously been reported throughout the extensive causative gene list. Genetic modifiers, while not fully understood, are also believed to play a role in the phenotypic spectrum of ataxia conditions (Rahit and Tarailo-Graovac, 2020). These factors underscore the need for a better understanding and classification of molecular causes, but also highlight the need for personalized clinical genetic testing methods.
EA is an intriguing category of ataxias that shares a lot of similarities to autosomal dominant SCAs, but distinguishes itself through the episodic nature of clinical ataxic manifestations, which can be triggered by various factors such as emotional stress or physical exertion (Choi and Choi, 2016). This atypical intermittence of symptoms, along with a frequent late-onset presentation, introduces additional layers of complexity to the diagnosis. Cases of progressive EA developing into chronic cerebellar ataxia also complicate the differential diagnosis (Giunti et al., 2020). There are officially eight subtypes of EA, although recent case reports suggest there could be up to eleven forms observed to this day (Hassan, 2023). In fact, pathogenic variants in KCNA1 (EA1) and CACNA1A (EA2) are almost the only ones to be observed in multiple families, and represent the majority of EA cases (Sintas et al., 2017; Paulhus et al., 2020). In contrast, the other six subtypes were characterized mostly in single families (Escayg et al., 2000; Jen et al., 2005; Conroy et al., 2014), locating only three causative genes in CACNB4 (EA5), SLC1A3 (EA6), and UBR4 (EA8). This is consistent with the notion that around 40% of patients who present late-onset phenotypes without an extensive family history do not obtain a molecular diagnosis despite a comprehensive evaluation (Perlman et al., 1993). Recent reports also suggest EA phenotypes can be caused by variants in genes traditionally associated with chronic disorders, and may often be overlooked or misdiagnosed in favor of more prevalent conditions (Hassan, 2023). Although recent advances have yielded new causative genes in FGF14 (Schesny et al., 2019), SCN2A (Schwarz et al., 2016), and CACNA1G (Gazulla et al., 2021), there is a clear need to investigate ataxia patients with episodic features to better define genetic etiology and improve clinical genetic screenings.
Whole genome sequencing (WGS) enables a complete overview of an individual’s genomic alterations, maximizing the chances of identifying novel pathogenic variants. The major limitation of WGS is that each individual carries millions of variants, with the majority of them still classified as variants of uncertain significance (VUS), comprising the majority of non-coding alterations as research on the matter is still sparse (Scacheri and Scacheri, 2015). This complicates variant prioritization, limiting the technology mostly to the exome regions, but still enabling a lower rate of inconclusive testing due to VUS than gene panels (Rehm et al., 2023). RNA sequencing (RNA-seq) on the other hand, offers valuable insights on the potential molecular effects of variants through expression and splicing data (Yepez et al., 2022). A multi-omics approach integrating genomic and transcriptomic sequencing provides a more comprehensive depiction of a patient’s genetic profile, thereby enhancing the potential for identifying previously unreported pathogenic variants in known or novel genes (Lunke et al., 2023). The latter does require further functional validation, which is where the versatility of long-read sequencing (LRS) is highly interesting. It is been shown that the technology allows for precise characterization of transcriptomic landscapes (Mezreani et al., 2022), large structural variants (Miller et al., 2022), and even repeat expansion (Pellerin et al., 2023). Improvements in the capacity to quickly identify underlying causes of genetic disorders offers many benefits to patients, including better care management and treatment options following the molecular diagnosis. Furthermore, with the continuous advancements in personalized genetic therapy tools, such as antisense oligonucleotides, gaining a comprehensive understanding of the genetic variations at play becomes increasingly crucial. Molecular diagnoses now present a promising perspective for developing effective treatments tailored to these individuals (Kulkarni et al., 2021).
In this study, we describe a multi-omics approach to uncover potential disease-causing variants in late-onset ataxia patients with episodic features lacking molecular diagnoses despite exhaustive clinical and genetic evaluation. We combined WGS, RNA-seq as well as LRS to identify variants and validate segregation of alleles, splicing defects and repeat expansions. Our results highlight the need to broaden genetic testing in unsolved patients with primary EA or EA-like phenotypes.
2 Materials and methods
2.1 Participants and clinical investigation
The cohort is comprised of 7 probands from different families, with a single affected family member in patient #6 (related to patient #5). All participants were referred for episodes of ataxia and had a comprehensive clinical evaluation in July 2019 at the time of specimen collection. Neurological and general examination were performed and the Inventory of Non-Ataxia Signs (INAS) as well as the Scale for the Assessment and Rating of Ataxia (SARA) were recorded (Schmitz-Hubsch et al., 2006; Jacobi et al., 2013). Medical files were reviewed to document previous genetic panels and imaging studies. Acquired causes of ataxia were investigated through systematic questioning as well as laboratory tests including vitamin levels, and autoimmune and paraneoplastic markers. The Movement Disorders Society criteria were used to rule out multi-system atrophy (Wenning et al., 2022). All data was recorded in the REDCap database software developed at Vanderbilt University (Harris et al., 2019). Participants were recruited for further sequencing in light of a suspected complex ataxia without a molecular diagnosis. The study was approved by the hospital’s ethics committee, and recruited participants signed an informed consent form authorizing genetic analysis for research purposes.
2.2 Sample preparation
Participants had blood samples drawn as part of their clinical work-up. Peripheral blood mononuclear cells were isolated using a standard Ficoll-paque centrifugation protocol, and 2.5 million cells pellets were frozen at −80 °C prior to their extraction with a column purification kit (Norgen Biotek™). DNA and RNA were quantified using the QuBit system (Thermo Fisher Scientific™) with high-sensitivity kits prior to storage at recommended temperatures for sequencing and validation experiments.
2.3 Whole genome sequencing
DNA samples were diluted in TE buffer and loaded onto Twin-Tec sequencing plate (Eppendorf™) for quality assessment, library preparation, and sequencing from Centre d’expertise et de services Génome Québec (CESGQ). The shotgun PCR-free genomic library was prepared using dual-index adapters from Integrative DNA technologies™. Illumina™ NovaSeq 6000 S4 sequencers provided at least 70 million paired-end reads (150bp) coverage for each sample.
2.4 RNA sequencing
RNA samples were diluted with molecular grade water in 1.5 mL tubes for transport on dry ice to CESGQ. Quality control was performed with Agilent™ Bioanalyzer 2100 to ensure an integrity value (RIN) greater than 8.0. Library preparation was performed by PolyA capture method with next-dual adapters from New England Biolabs™. Illumina™ NovaSeq 6000 S4 technology generated a minimum of 50 million paired-end reads (100bp) coverage for each sample.
2.5 Multi-omics variant analysis
For both sequencing data, quality was assessed with MultiQC and FastQ files were processed with trimmomatic prior to their alignment on the hg19 reference genome (Bolger et al., 2014; Ewels et al., 2016). Whole genome data was aligned using BWA, while STAR was used for RNA-seq (Li and Durbin, 2009; Dobin et al., 2013). Variant calling was performed using GATK best practices recommendations for both datasets (McKenna et al., 2010), and annotations were added using ANNOVAR (Wang et al., 2010). Variants with a minor allele frequency greater than 1% in gnomAD were excluded. Multiple tools supplemented the genomic investigation: CNVkit and LUMPY were used to cover calling of structural variants of all sizes (Layer et al., 2014; Talevich et al., 2016), ExpansionHunter and STRetch predicted expansions of microsatellites in known pathogenic regions as well as genome-wide (Dashnow et al., 2018; Dolzhenko et al., 2020), and SpliceAI predicted variants impacting splicing in coding and non-coding regions (Jaganathan et al., 2019). Quantification of RNA expression utilizing both Salmon and featureCount (Liao et al., 2014; Patro et al., 2017), as well as differential expression analysis using DESeq2 (Love et al., 2014), served for gene expression examination. Non-canonical splicing junction calling with rMATS also helped with variant interpretation (Shen et al., 2014). Custom scripts were used to further filter and process the data, in respect to standardized best practices for clinical variant prioritization, and utilized in silico predictions such as the Combined Annotation-Dependent Depletion (CADD ≥10) scores (Rentzsch et al., 2019). Relevance of the variants to phenotypes was assessed with literature reports from GeneCards (Stelzer et al., 2016), The Human Protein Atlas (Uhlen et al., 2019), Uniprot (UniProt, 2023), gnomAD (Chen et al., 2022), OMIM (Hamosh et al., 2005), Mutation Taster (Steinhaus et al., 2021) and ClinVar (Landrum et al., 2018). Overall, non-synonymous low-frequency variants predicted to be damaging, putative splicing variants, coding indels, CNVs and repeat expansion in genes directly or indirectly associated with ataxias were prioritized.
2.6 Quantitative polymerase chain reaction
RNA from cohort samples was obtained as described in the sample preparation section, while healthy controls had previously been extracted with a TRIzol (Thermo Fisher Scientific™) protocol. Hence, both were used separately to control for batch effect. RNA reverse transcription was carried out using SuperScript Vilo mix (Thermo Fisher Scientific™). 10ng of cDNA was loaded into a 384-well plate (Thermo Fisher Scientific™) along with a FAM-MGB Taqman probe for a gene of interest and an optimized fluorescence master mix (Thermo Fisher Scientific™). Probes for ELOVL4, PMPCB, and ZFYVE26 (Hs00224122_m1; Hs01012489_m1; Hs00188704_m1) were normalized using the housekeeping gene ACTB (Hs01060665_g1). Expression levels were assessed through relative ΔΔCT quantifications obtained from Applied Biosystems™ QuantStudio 6/7 systems.
2.7 Long-read sequencing
Libraries were prepared from either genomic DNA or reverse transcribed cDNA, and were PCR amplified (30 cycles) using the LongAmp 2X Taq Polymerase (New England Biolabs™). Primers designed with primer3 (Untergasser et al., 2012) targeted one of SPG7, ELOVL4, PMPCB, or ATXN2 (Supplementary Table S1). Libraries end prepping utilized NEBNext Ultra II End Repair/dA-Tailing Module (New England Biolabs™; Cat. E7546) to allow barcode ligation (kit SQK-NBD112.24) using the Blunt/TA ligase (New England Biolabs™; Cat. M0367) prior to Nanopore™ sequencing adapters ligation with the NEBNext Quick Ligation Module (New England Biolabs™; Cat. E6056). Purification with included AMPure XP beads is performed between each step for optimal results, with the last clean-up utilizing a size-specific retention buffer from Oxford Nanopore™. Multiplexed libraries were quantified using the QuBit4 fluorometric system for optimal flow cell loading and maximized sequencing over a 72-h period. Generated data were processed with Guppy for high-accuracy base calling and demultiplexing (Wick et al., 2019), minimap2 for alignment to the hg19 reference genome (Li, 2018), and PycoQC for quality assessment (Leger and Leonardi, 2019). Transcriptomic results were then generated using FLAIR as recommended by the authors (Tang et al., 2020). The “align” and “correct” functions used hg19 reference genome and Gencode v19 annotations (Frankish et al., 2021), “collapse” was set in best-only mode with a threshold of 500 supporting reads, and “quantify” utilized the–trust-end parameters since adapters were removed during demultiplexing. Targeted repeat expansion was quantified using tandem-genotypes tool with standard parameters (Mitsuhashi et al., 2019). The resulting data were used to construct the customized plots presented in the article.
3 Results
3.1 Individual #1 SPG7
Individual #1 is a male proband that was diagnosed with a progressive ataxia in his mid-fifties. However, there was noticeable but stable signs of speech and coordination difficulties since childhood. He is the only patient in which a potential acquired form of ataxia was not initially ruled out, as he recovered from a subdural hematoma after a car accident in his mid-twenties. Family history is limited, but two family members exhibited balance and speech issues, similar to those of individual #1, in the late stage of their lives. Clinical manifestations include strong episodes of imbalance with vertigo, ataxic gait, and nystagmus. Ophthalmoparesis as well as multiple auditory phenotypes such as progressive sensorineural hearing loss underscored the possibility of a mitochondrial disorder. Cerebellar atrophy was also observed by MRI. Genetic testing for both ataxia and mitochondrial relevant genes yielded mostly negative results (Table 1). Whole genome sequencing on the other hand revealed two distinct variants in the SPG7 coding region (Supplementary Figure S1), a well-studied gene that causes an autosomal recessive spastic paraplegia as well as recessive cerebellar ataxias (Choquet et al., 2016a). The gene was prioritized due to the clear genotype-phenotype association, and the pathogenic potential of the identified variants (Supplementary Table S1). The missense (NM_003119: c.2228T>C/p.I743T) had previously been reported as likely pathogenic multiple times in ClinVar, while the nonsense variant (NM_003119: c.1861C>T/p.Q621*) has a near-zero allele frequency and has only recently been submitted as likely pathogenic. Both variants were also seen in the RNA-seq data, and were confirmed by Sanger sequencing for both the DNA and RNA (Supplementary Figure S1). In order to assess if both alleles were functionally affected by those variants, targeted LRS of complete SPG7 mRNA amplicons confirmed that the two variants were in trans configuration (Figures 1A,B). Indeed, while reads from the control sample shows indistinguishable alleles containing no variants, reads from the patient, which can be confirmed by the homozygous synonymous variant (NM_003119: c.2064G>A/p.R688R), exhibit two clearly distinct alleles. Reads from individual #1 carry either the missense or the stop-gain variants, but never both. The identification of compound heterozygosity of two SPG7 variants, previously reported as likely pathogenic according to ACMG guidelines (Richards et al., 2015), offers strong evidence of pathogenicity and sufficed to obtain a clinical diagnosis.
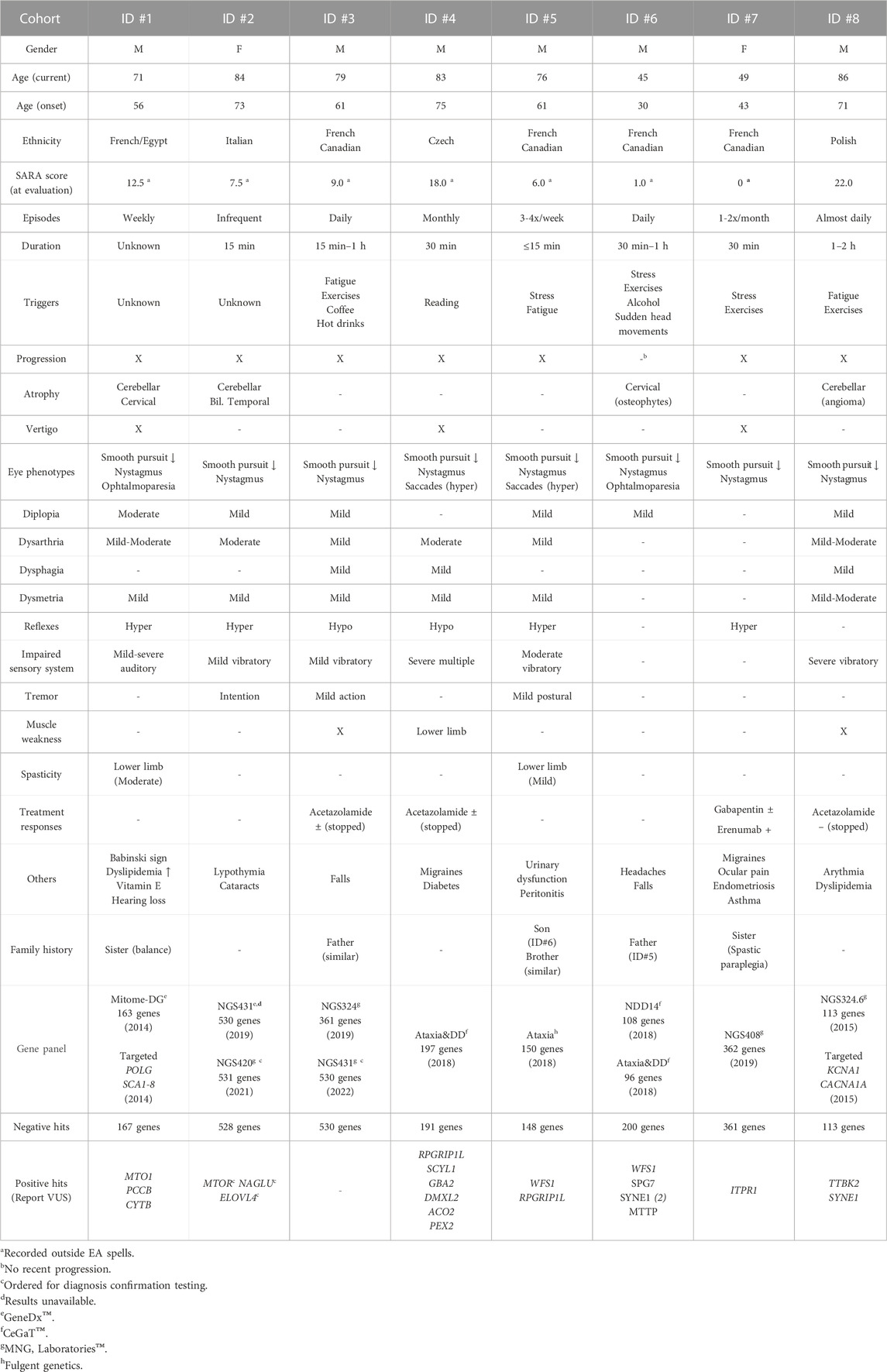
TABLE 1. Clinical presentation summary of ataxia cohort.
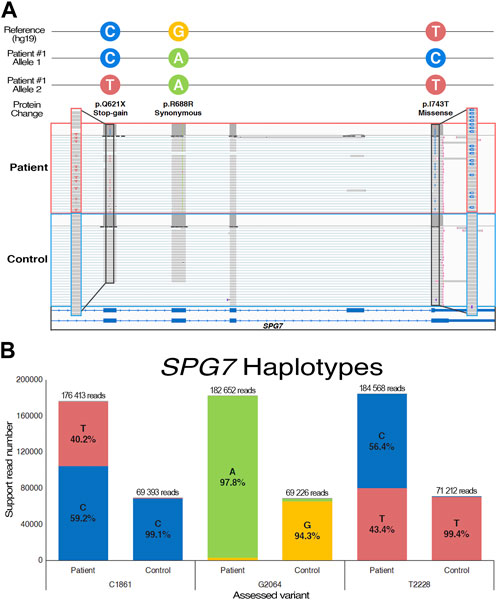
FIGURE 1. Long-read sequencing of the SPG7 cDNA from individual #1. (A) Visual representation of the trans-configured variants on transcriptomic reads using the Integrative Genome Viewer. (B) Allele quantification utilizing a patient-specific synonymous variant (c.2064G>A) to confirm sample identity and base-calling accuracy. Control data was produced using comparable cDNA from a healthy sample.
3.2 Individual #2 ELOVL4
First signs of disease in individual #2, a female proband without family history, manifested around age 73. Clinical presentation is predominantly characterized by episodes of dysarthria, but disease progression also led to ataxic gait as well as important episodes of nystagmus and diplopia. MRI revealed progressive atrophy of the cerebellum, but also a bilateral atrophy of temporal lobes. The latter prompted additional assessment of cognitive functions, which revealed that a mild cognitive impairment was in development (Hobson, 2015). Clinical genetic testing identified no pathogenic candidates (Table 1), but whole genome sequencing was able to detect an intronic variant in ELOVL4 (NM_022726: c.541 + 5G>A), a gene known to cause autosomal dominant SCA34 (Supplementary Figure S1). Given the variant’s high pathogenicity score, null allele frequency in public databases, and its clear potential effect on the gene, it was prioritized for validation. While the variant has never been reported before, in silico predictions suggest it to be disease-causing through loss of exon 4 splice donor site (Supplementary Table S1). Coverage of ELOVL4 in the RNA-seq was too low for variant calling, but it does appear in a few reads, and splice junctions suggested skipping of exon 4. After the variant was confirmed through Sanger sequencing (Supplementary Figure S1), PCR amplification of ELOVL4 complete coding sequence enabled the analysis of its transcriptomic landscape through LRS (Figure 2A). The sequencing data confirmed that the intronic variant significantly caused skipping of exon 4, with an additional event that skipped both exon 4 and 5 (Figure 2B). While LRS isoform ratio analysis is relative, quantification of ELOVL4 resulting mRNA levels was performed by qPCR (Figure 2C). The expression level is decreased by about 50% compared to healthy and diseased controls, suggesting nonsense-mediated decay of the alternatively spliced transcripts. The splicing alteration and subsequent decreased expression of ELOVL4 observed in our patient demonstrate the deleterious effect of c.541 + 5G>A and is sufficient to classify this variant as pathogenic according to ACMG guidelines.
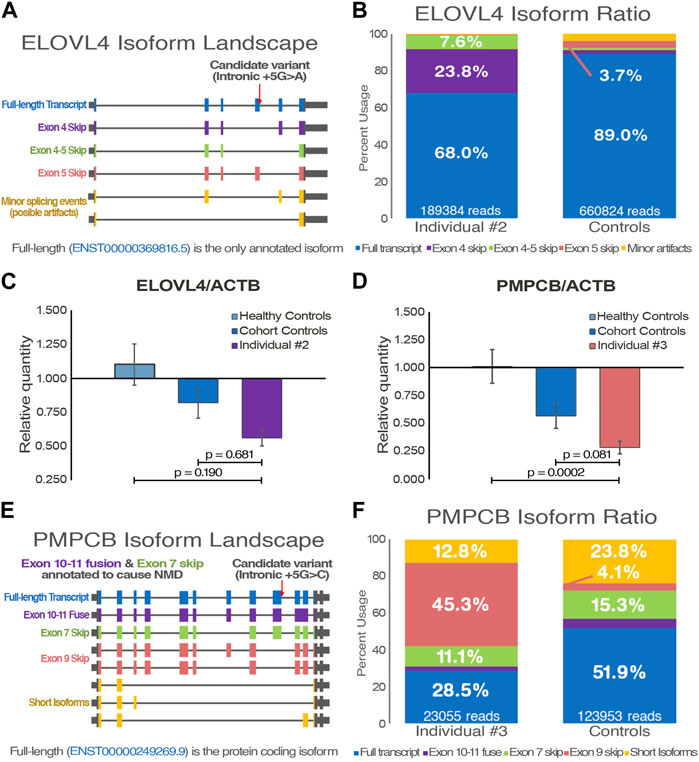
FIGURE 2. Functional transcriptomic validation of genes with suspected alternative splicing variants. (A) Visual representation of the ELOVL4 isoform landscape following alignment on the hg19 reference genome. (B) FLAIR quantification of annotated and de novo ELOVL4 transcripts from individual #2 and control samples (n = 3). (C, D) Gene expression quantification of qPCR data with an ELOVL4 probe targeting exon 1–2 junction (NM_022726) and a PMPCB probe targeting exon 4–5 junction (NM_004279). ACTB is used for data normalization; Ataxia individuals are compared to both healthy (n = 3) and cohort (n = 2) controls. One-way ANOVA and Tukey’s post hoc multiple comparisons test were applied to calculate adjusted p-values and assess statistical significance. (E) Visual representation of the PMPCB isoform landscape following hg19 alignment. (F) FLAIR quantification of annotated and de novo PMPCB transcripts from individual #3 and control samples (n = 3).
3.3 Individual #3 PMPCB
Individual #3 is a male proband who was diagnosed with progressive EA at age 61. Acute episodes are characterized mainly by gait ataxia, dysarthria, and nystagmus. Progressive ataxia baseline is accompanied by worsening of the upper and lower limbs muscle weakness after episodes. Mild action tremor was also noted, and inconsistent dysphagia has been observed (Table 1). While family history is incomplete, the father did exhibit signs of late-onset ataxia a few years prior to his death. Individual #3 also mentioned he has a few brothers with potential imbalance problems, which could suggest an autosomal dominant transmission, but clinical follow-up could not be achieved. Both WGS and RNA-seq identified an intronic variant in PMPCB (NM_004279: c.1154 + 5G>C) that is predicted to induce skipping of exon 9 (Supplementary Figure S1). Once more, analysis led to prioritization of the variant due to high scores from pathogenicity prediction tools, low allele frequency, and literature supporting a role in cerebellar functions (Supplementary Table S1). Transcriptomic data, despite a relatively low coverage, does support this potential alternative transcript. DNA Sanger sequencing confirmed the variant before further assessment by qPCR and LRS (Supplementary Figure S1). A significant decrease of over 50% of the gene expression in qPCR data suggests that the affected allele is eliminated through nonsense-mediated decay (Figure 2D). Amplification of the most commonly expressed annotated transcript by targeting shared exons yielded a near complete transcriptomic landscape of PMPCB (Figure 2E). LRS data once again strongly support the loss of exon 9 in multiple transcripts (Figure 2F), and provides a probable explanation for the nonsense-mediated decay. While they are not regarded as the main disease-causing variants, it is important to note that the WGS also uncovered two variants in CACNA1G, albeit neither are considered likely pathogenic due to their considerable allele frequencies.
3.4 Individual #4 ATXN2
Acute episodes of dysarthria, vertigo and ataxia first happened when individual #4 was 75 years old. Disease progression was observed despite a positive response to acetazolamide, as episodes are more frequent and now include nystagmus, migraines, as well as muscle weakness and paresthesia of the lower limbs. The condition’s progression into a chronic state remains uncertain but plausible. Clinical genetic testing, which did not cover repeat expansions, included 197 ataxia-related genes and identified no variants that could explain individual #4 phenotypes (Table 1). Using WGS data (Supplementary Figure S1), the Expansion Hunter tool predicted a repeat expansion of ATXN2 exon 1 CAG microsatellite, which was prioritized due to the clear association to cerebellar ataxia, and a lack of other clear causative variants. Using LRS on ATXN2 DNA amplicons, alleles of 22 and 32 repeats were detected in individual #4 (Figures 3A,B), positioning him directly on the SCA2 threshold of variable penetrance and delayed onset (Almaguer-Mederos et al., 2010). Interestingly, both sequencing datasets also identified a heterozygous nonsense variant in ZFYVE26 (NM_015346: c.3022C>T/p.R1008*), which has been reported as pathogenic (Supplementary Table S1). For this reason, we hypothesized that the variant could potentially act as a disease modifier of the patient’s phenotype. The variant was confirmed by Sanger sequencing (Supplementary Figure S1), and a strong decrease in the gene expression was observed by qPCR (Figure 3C), suggesting nonsense-mediated decay of the affected transcript once more.
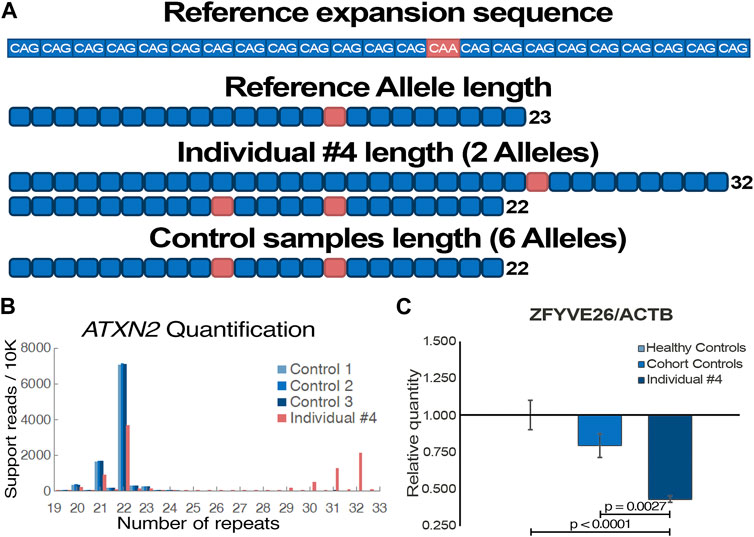
FIGURE 3. Functional validation of individual #4 pathogenic genomic alterations. (A) Visual representation of ATXN2 trinucleotide repeat expansion sequence and length from individual #4 and control samples (n = 3) following targeted long-read sequencing. (B) Tandem-genotypes quantification of the samples’ ATXN2 trinucleotide repeat lengths. Supporting read numbers are scaled per 10,000 reads to facilitate visualization of samples alleles. (C) Gene expression quantification of qPCR data with a ZFYVE26 probe targeting exon 16–17 junction (NM_015346). ACTB is used for data normalization; Individual #4 is compared to both healthy (n = 3) and cohort (n = 2) controls. One-way ANOVA and Tukey’s post hoc multiple comparisons test were applied to calculate adjusted p-values and assess statistical significance.
3.5 Individual #5 and #6
Individual #5 is a male proband who first consulted for episodes of gait instability around age 61, but had short transitory episodes of diplopia in his mid-thirties. Additional clinical manifestations following disease progression include dysarthria, nystagmus, hyperreflexia and mild postural tremor. Family history revealed that one of five siblings also exhibits gait imbalance and mild tremors akin to those of the patient. He is also the father of individual #6, who exhibits a similar early ocular phenotype, as well as an unaffected daughter. Individual #6 disease onset was around age 30, which could be due to a mild traumatic brain injury that occurred 10 years prior, but an anticipation effect was considered. As expected from his father’s medical history, there has been no disease progression yet, and the main feature of episodes is diplopia. Neither patient exhibit cerebellar or cervical atrophy. Genetic testing, which covered over 200 ataxia genes, identified only one common VUS in WFS1, and no clear disease-causing de novo variants (Table 1). WGS and RNA-seq did not detect pathogenic variants in known ataxia genes, but did identify shared VUS in two genes that we are currently investigating as potentially relevant to cerebellar functions. VUS unique to a single patient are also being considered in case of distinct pathologies.
3.6 Individual #7 and #8
Individual #7 is a female proband with a disease onset at age 43. Acute episodes of vertigo, nystagmus, and migraines have progressed in intensity over time, but few new clinical manifestations have been observed. Ataxia is mainly observed in the lower limbs, and there is no cerebellar atrophy at this time. Lack of family history suggests a de novo pathology, albeit her mother has history of epilepsy, but clinical genetic testing yielded no candidate variants, as a single ITPR1 VUS was found (Table 1). Individual #8 is a male proband who first exhibited episodic signs of cerebellar dysfunction around the age of 71. Symptoms were typical: ataxic gait, nystagmus, dysarthria, as well as cerebellar atrophy which was revealed by CT-scan. Although current clinical presentation appears to fit EA, the increasing frequency and duration of episodes implies that progression into a chronic state cannot be ruled out. Gene panels for 115 genes failed to identify conclusive pathogenic candidates, revealing VUS only in TTBK2 and SYNE1 (Table 1). WGS and RNA-seq variant analysis also did not identify obvious candidates, but variants in genes with brain-restricted expression are currently being investigated as potential novel causes of the individual’s phenotypes.
4 Discussion
The results of this study highlight the relevance of multi-omics approaches in genetic investigations deemed complex following a failure from first-line clinical testing, or even in contexts where the suspected pathology is not clear. Although phenotype-driven genomic investigations are generally efficient diagnostic strategies, certain Mendelian disorders pose additional challenges that render the option less viable, such as a late onset of phenotypes, high clinical heterogeneity, variable penetrance, and unpredictable disease progression (Pengelly et al., 2017). Our cohort of individuals with late-onset progressive EA-like disorders presented several of these challenges, as most had limited family history and showed no identifiable causative variants in standard gene panels testing (Table 1). In this study, the rationale behind the combination of WGS and RNA-seq lies in the knowledge that known pathogenic variants are no longer expected, albeit some could have been missed during clinical probing. In an exploratory context, the WGS technology offers the most comprehensive overview of a patient’s genomic state (Koboldt, 2020). While there is currently limited information available regarding the biological relevance of non-coding regions, which represent 98% of the genome, it is thought that up to 82% of it could have regulatory functions (Gloss and Dinger, 2018). Although most non-coding variants might have less direct pathogenic impacts, they undoubtedly contribute to the relatively modest clinical diagnostic success rates, around 25%–50%, obtained from clinical methods focusing primarily on coding regions (Scacheri and Scacheri, 2015; Ellingford et al., 2022). Despite the advancements in algorithms that predict functional impacts of whole genome variants, such as SpliceAI which demonstrates impressive performances and will likely improve further along with new literature (Jaganathan et al., 2019), the interpretation of non-coding variants as well as VUS remains the significant limitation of WGS. As such, RNA-seq offers a complementary functional insight on the transcriptomic states of genes carrying those variants (Yepez et al., 2022; Lunke et al., 2023). This dynamic view of gene expression and splicing in relation to genomic alterations is highly valuable to variant classification and prioritization. Therefore, we believe the approach offers the best odds at reaching a molecular diagnostic in patients with Mendelian disorders deemed complex.
Identification of non-synonymous mutations in the SPG7 gene of individual #1 was a surprising finding, as they technically should have been caught by gene panel testing. Unsuccessful identification of causal variants by first-line clinical testing can stem from technical or bioinformatics issues (Murray et al., 2022), but in this case was simply due to the fact that SPG7 was not included on the selected ataxia and mitochondrial genes panel at the time of testing (Table 1). This underscores the dynamic nature of VUS literature, but also that leveraging more complete sequencing approaches enables retrospective analysis of emerging pathogenic gene associations. The SPG7 gene encodes a mitochondrial metalloprotease, and its dysfunction is a common cause of an adult form of recessive spastic paraplegia and cerebellar ataxias (Pfeffer et al., 2015). The patient’s observed phenotypes are consistent with SPG7-related pathologies, which encompass both complicated and pure forms, with some carriers even receiving a diagnosis of spastic ataxia. It is important to note that while the individual made a near complete recovery from the subdural hematoma he suffered in his early adulthood, it is not possible to conclude on whether it influenced the clinical phenotypic presentation or not. However, both variants of individual #1 have been reported as likely pathogenic at least once: the missense variant (NM_003119: c.2228T>C/p.I743T) causes a change in amino acid within the peptidase domain, which is predicted to have deleterious effects in silico (Supplementary Table S1), while the stop-gain variant (NM_003119: c.1861C>T/p.Q621*) almost certainly causes loss-of-function through protein truncation and nonsense-mediated decay (Choquet et al., 2016a). The usage of LRS to clearly demonstrate the trans configuration of the two pathogenic variants is sufficient evidence to obtain a clear diagnosis of recessive spastic ataxia with episodic manifestations. Although persistence of symptoms is suspected to arise from future disease progression, the episodic nature of phenotypes was a prominent feature of clinical presentation. The results also highlighted a strength of the technology, which is the ability to perform accurate genome-wide allele phasing prior to variant calling. This feature is valuable to the identification of compound heterozygosity, especially in late-onset cases where parents are unavailable for segregation analysis (Akbari et al., 2021).
Intronic variants offer compelling illustrations of the benefits of combining WGS and RNA-seq. The ELOVL4 variant (NM_022726: c.541 + 5G>A) was called by GATK in the genomic dataset, but the coverage was seemingly too low to be detected in the RNA-seq data. However, a manual examination of the transcriptomic landscape of ELOVL4 using Integrated Genome Viewer (IGV) revealed that a few reads did support the in silico prediction of exon 4 splice donor loss resulting in skipping (Supplementary Table S1). The observation greatly assisted in prioritizing this variant, as potential splicing events frequently have otherwise unremarkable pathogenic scores compared to non-synonymous variants called in WGS (Rentzsch et al., 2021). Once more, leveraging LRS as a functional validation tool, we performed targeted sequencing of ELOVL4 transcripts in individual #2, which confirmed a clear shift in isoform ratio (Figure 2B). Interestingly, our findings revealed that exon 5 exhibited a natural predisposition to splice defects, resulting in approximately 25% of the patient’s alternatively spliced transcript skipping both exon 4 and 5 instead of only the predicted exon 4. Moreover, individual #2 presents a near 10-fold increase of exon 4 skipping, which represents around 30% of all reads and supports the hypothesis that alternatively spliced transcripts are subject to nonsense-mediated decay. The decrease in mRNA expression also corroborates this hypothesis (Figure 2C). ELOVL4 encodes an enzyme that is involved in fatty acids biosynthesis, and is already associated to the dominant form of SCA34 (Gyening et al., 2023). Both abnormal exon-skipping events observed in individual #2 also induce a frameshift, leading to major structural changes at the protein level which ultimately results in a complete loss of native catalytic functions. The rationale we posit for the prominent episodic nature of the patient’s phenotypes is that the biological role of ELOVL4 is dosage-dependent: in baseline circumstances, cells compensate the enzyme function with their healthy allele, effectively distinguishing and degrading the alternatively spliced transcripts in parallel (Fahrner and Bjornsson, 2014). However, events that lead to increased enzymatic processing of fatty acids would act as EA triggers. This hypothesis could also provide an explanation for the absence of erythrokeratodermia, a frequently observed but not obligatory persistent phenotype of SCA34 (Nishide et al., 2023).
Individual #3 presents a very similar case, except the PMPCB variant (c.1154 + 5G>C) was called in both datasets, albeit with lower pathogenicity prediction scores. Like the previous case, IGV examination supported the in silico prediction of splice donor loss and exon 9 skipping. Intriguingly, LRS revealed that while only the full-length transcript (ENST00000249269.9) of PMPCB is expected to be functional (Vogtle et al., 2018), minor isoforms carrying alternative splicing such as exon 7 skipping or a fuse of exon 10 and 11 accounted for nearly 50% of all reads in control samples (Figure 2F). Nevertheless, the isoform ratio analysis still reveals a clear shift for individual #3, whose data suggest that all transcripts from the variant-carrying allele are predictably missing exon 9, even as other alternative splicing events also occur. While transcripts missing exon 9 account for 45% of reads in the LRS data, the 2-fold decrease observed in mRNA expression data (Figure 2D), as well as in the RNA-seq (Supplementary Table S1), suggests nonsense-mediated decay of affected allele may still be happening. The PMPCB gene encodes for the catalytic subunit of the essential mitochondrial processing protease (MPP), and the bi-allelic loss-of-function has been shown to cause multiple mitochondrial dysfunctions syndrome-6 (MMDS6), a severe and early-onset neurodegenerative disorder (Vogtle et al., 2018). While individual #3 phenotypes are less severe and more tardive, there is a strong overlap in the type of clinical manifestation, and a milder autosomal dominant presentation of a known harsh recessive disorder has been observed numerous times in rare disorders literature (Tetreault et al., 2015). As loss of exon 9 results in the loss of a significant part of the MPP-β, specifically two α-helix and two β-sheets towards the inside of the protein (UniProt, 2023), in silico tools unanimously predict misfolding and complete loss-of-function of the resulting peptidase (Supplementary Table S1). As MPP-β is essential for the processing of proteins associated with neurodegeneration, namely FXN and PINK1 (Schmucker et al., 2008; Greene et al., 2012), it is highly probable its alteration negatively impacts cerebellar functions, and could even explain the mild action tremors observed in individual #3. Therefore, we propose a similar dosage-dependent pathogenic effect of the variant, where a single healthy PMPCB allele is enough for baseline functions, but increased mitochondrial activity causes dysfunctions that are similar to those seen in MMDS6, albeit on a smaller scale. While further functional investigation and additional patients will help understand the role of PMPCB in ataxic phenotype, we posit that ataxia gene panels would benefit from the inclusion of the gene in a similar fashion to its partner, PMPCA (Choquet et al., 2016b). Given that the two are functional partners, it is reasonable to hypothesize disruptions of PMPCB functions, thereby hindering MPP mitochondrial role, could cause a very similar condition (Kunova et al., 2022).
While RNA-seq provided valuable insights into potential pathogenic alternative splicing and changes in expression associated with candidate variants, we firmly believe that the additional information offered by LRS makes it a formidable asset for the functional validation of VUS. As long-read technology providers approach the capability of offering full-length whole-transcriptome data with unique molecular identifiers, accurate assessment of differential expression at the isoform level becomes feasible. This extent of data resolution holds immense potential, especially as pathogenic effects of variants can be driven in isoform-specific manners (Rudell et al., 2019), and compensatory mechanisms underlying discrepancies between gene expression and functional impact could be more readily identified. Although the RNA-seq and qPCR data did not demonstrate statistically significant, albeit substantial, decreases in ELOVL4 levels for the patient, examination of the LRS results clearly revealed a significant decrease in functional transcripts (Figure 2B). Hence, it enables the hypothesis that there is compensatory expression from the healthy allele to partially rescue the function, which could help elucidate the episodic nature of the phenotypes. The implementation of phased calling features, currently being optimized for long-read platforms, is poised to reveal numerous additional instances of this phenomenon (Akbari et al., 2021). Moreover, LRS is currently being utilized to construct a highly accurate and cell-specific human transcriptome that does not rely on isoform prediction, directly enhancing the ability to detect abnormal splicing and isoform-specific differential expression (Reese et al., 2023). The decreasing cost per sample and the steady improvement in LRS base calling accuracy have been effectively mitigating current limitations of the technology, underscoring its growing potential for clinical genetics.
In a similar manner, increased length of native genomic reads, as well as the ability to detect nucleotide modifications such as methylation, give a slight edge to the potential of LRS (Akbari et al., 2021). One of the shortcomings of traditional WGS is the difficulty of detecting structural variants larger than a few hundred base pairs. As standard sequencing read length is too short to confidently anchor the alternative sequences to clear break points in the genome, most structural variant calling tools for short reads derive information from atypical read-splitting, inconsistent read pairs, and abnormally low read depth for the region (Kosugi et al., 2019). Assembly approaches, which aim to make contigs spanning as far as possible from the break points, along with combinatory tools, generally perform best as they more accurately detect abnormal events. LRS approaches provide a significant advantage in identifying large structural variants, as every read generated by this technology contains information comparable to a contig. Similarly, repetitive regions have long represented a fundamental challenge for variant calling of sequencing. As such, Expansion Hunter is one of the few tools that can be used on short-read data to call repeat expansions, a common cause of neurodegenerative disorders (Dolzhenko et al., 2020). Moreover, it still just offers a broad prediction of the microsatellite expansion length. Therefore, LRS offers a unique opportunity to observe full-length pathogenic repeat expansions anchored to the reference genome. As tools are being developed and optimized, it is feasible to detect and quantify the number of short-tandem repeats in each sample, which will likely lead to the detection of numerous new repeat expansion disorders (Pellerin et al., 2023). Individual #4 offered a neat example of the clinical potential of targeted repeat expansion amplification, as precise repeat quantification could be used in multiple ataxia-related disorder testing. Combined with phased-alignment, it is possible to obtain a more accurate quantification of the repeat number on each allele, revealing here that the patient is directly on the threshold of variable penetrance (Laffita-Mesa et al., 2012). It is interesting to observe the characteristic loss of one of the CAA interruptions, which is believed to influence functional transcripts by disrupting the formation of CAG hairpins. While mechanisms of clinical variability in ATXN2 expansions remains unclear, it is hypothesized that these CAG hairpins contribute to pathogenicity by altering protein interactions, as well as mRNA localization and processing in a manner that may be tissue-specific (Marti, 2016; Fournier et al., 2018). Alleles of ≥31 CAG repeats have also been shown to be risk alleles for amyotrophic lateral sclerosis with or without frontotemporal dementia, suggesting ATXN2 pathogenic mechanisms are complex and dynamic (Glass et al., 2022), and that other variants or genes may be implicated in the variable pathogenesis. Although rare, alleles of 32 and 33 repeats have been described as significantly delaying the age of onset (Daoud et al., 2011; Neuenschwander et al., 2014). This finding is consistent with the episodic and late-onset nature of the phenotypes compared to typical SCA2 patients with large ATXN2 expansions (Figueroa et al., 2017).
The concept of genetic modifiers, although predominantly explored in the context of genome-wide association studies, holds substantial relevance to the field of clinical genetics, offering potential explanations for the high phenotypic heterogeneity observed in complex genetic disorders (Rahit and Tarailo-Graovac, 2020). The suggested impact of disease modifiers encompasses not only the diversity and severity of observed symptoms, but could potentially influence the age of onset as well as the rate of disease progression (Li et al., 2021). Individual #4 presents an interesting example of the potential of a genetic modifier impacting disease phenotype. The very significant decrease in gene expression (Figure 3C), which supports in silico predictions of a non-functional protein resulting from the truncating variant (Supplementary Table S1), is partly compelling evidence that ZFYVE26 is implicated in the patient’s phenotype. Furthermore, the patient clearly present clinical features associated with both ATXN2 and ZFYVE26 related disorders. Nevertheless, additional validation is required to confirm a genetic modifier role, as related spastic paraplegias are generally recessive (Saffari et al., 2023). Additional SCA2 patients carrying heterozygous pathogenic variants in ZFYVE26 or modeling of both variants could help confirm this hypothesis. In addition, having access to isoform level quantification as well as epigenetic data directly from the combined sequencing results may have provided the additional information required to confidently make the assessment on the role of ZFYVE26. Indeed, the growing capability to evaluate epigenetic markers using LRS data is highly interesting in the context of predicting the functional effects of VUS, and particularly non-coding variants (Frydas et al., 2022). There are several examples in the literature describing genetic modifiers acting in synergy with the main genetic variant to influence phenotypes. Illustratively, a SQSTM1 variant known to cause a multisystem proteinopathy through stress granule impairment was enhanced synergistically by a variant in TIA1, gene annotated to cause a distinct myopathy (Lee et al., 2018). This concept has consequently been explored to elucidate the marked clinical diversity observed in SCAs (Wang et al., 2019). As clinical genetic testing decisions mostly rely on the standard phenotype-driven approach, improving the classification of genotype-phenotype relationships can aid healthcare providers in obtaining accurate diagnoses through first-line testing. Additionally, it has been proposed that modifier variants can influence, whether positively or negatively, the efficacy of therapeutic interventions, akin to haplotype-specific drug responses (Ahsan et al., 2020). This emphasizes the importance of identifying and understanding disease modifiers, as it can significantly impact multiple facets of clinical genetics and pharmacogenomics, aligning with the goal of achieving personalized medicine.
Despite being more time and resource consuming, extensive exploratory research on patients with suspected complex genetic causes are essential to the broader improvement of clinical genetics. Reclassification of VUS following functional assessments, as well as identification of novel causative genes and molecular mechanisms of pathogenesis greatly assist future variant prioritization during clinical genetic testing. Despite having a relatively small cohort, since atypical EA are highly rare and complex disorders, the combination of WGS and RNA-seq enabled the identification of pathogenic variants in four individuals (50%). Three of them should improve genotype-phenotype classification of known ataxia genes, as the variants seemingly cause atypical episodic clinical manifestations that otherwise resemble their classic disorder counterpart. Although PMPCB was previously only associated to a severe recessive mitochondrial disorder with ataxia phenotypes, we propose that loss-of-function of a single allele can lead to a milder late-onset dominant neurodegenerative phenotype. Our findings provide further evidence to broaden the genetic analysis to other genes, such as those directly or indirectly affecting ion channels or mitochondrial function, in patients presenting EA-like phenotypes or typical EA who do not carry disease variants in the respective genes (Giunti et al., 2020). Additionally, we are optimistic that the candidate variants identified in genes that are not yet associated to neurodegenerative disorders for the four remaining participants may lead to an improved diagnostic yield. While related literature is limited, elements such as suspected function, expression patterns, interaction partners, and preliminary results all support a potential role in cerebellar functions. Novel associations that may result from the on-going extensive tissue-appropriate modeling and functional validation would further support the efficiency of the approach in clinical contexts. While satisfied with the results obtained through the presented multi-omics approach, we believe an LRS-centered adaptation could perform even better, as part of the functional validation could be accomplished with the initial sequencing data. The proposed approach would offer genomic, transcriptomic, and epigenetic information in the most optimal resolution currently available, increasing the likelihood of attaining a molecular diagnosis. Consequently, we hope its application will benefit a broader spectrum of Mendelian disorders. Ultimately, the goal is to help define an optimal workflow of genetic testing, leveraging the benefits of continually evolving molecular technologies. Despite the perceived high cost of extensive second-line genetic testing, its value lies in the profound knowledge it offers. Moreover, it has the potential to reduce future care management costs through the development of personalized therapeutic strategies, thus maximizing the overall benefits for the patient.
Data availability statement
The data presented in this study are deposited in the NCBI BioProject repository, accession number PRJNA1014106, https://www.ncbi.nlm.nih.gov/bioproject/1014106. Variant reports have also been deposited in NCBI ClinVar repositories. The names of the repositories and accession numbers can be found below: https://www.ncbi.nlm.nih.gov/clinvar/variation/215218/, VCV000215218.46 (SPG7-missense). https://www.ncbi.nlm.nih.gov/clinvar/variation/1526041/, VCV001526041.4 (SPG7-nonsense). https://www.ncbi.nlm.nih.gov/clinvar/variation/2580357/, VCV002580357.1 (ELOVL4). https://www.ncbi.nlm.nih.gov/clinvar/variation/2580358/, VCV002580358.1 (PMPCB). https://www.ncbi.nlm.nih.gov/clinvar/variation/948359/, VCV000948359.5 (ZFYVE26).
Ethics statement
The studies involving humans were approved by the appropriate institutional review board of the University of Montreal Hospital (NO. 15.221). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
SA: Data curation, Formal Analysis, Investigation, Methodology, Software, Validation, Visualization, Writing–original draft, Writing–review and editing. VT: Writing–review and editing, Software. MG: Writing–review and editing, Data curation. NL-C: Writing–review and editing, Validation. VF: Resources, Writing–review and editing. AD: Conceptualization, Writing–review and editing. MT: Conceptualization, Funding acquisition, Methodology, Project administration, Supervision, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This project was supported by the Courtois Foundation and the National Ataxia Foundation (#618428). SA received a master’s degree fellowship from the Canadian Institute of Health Research and NLC from the Neuroscience department of Université de Montréal. MT received a Junior 1 salary award from the Fond de recherche du Québec—Santé.
Acknowledgments
The authors are grateful to the patients and their families for their willing participation to this study. We would also like to thank the André-Barbeau movement disorders unit for blood sampling within the framework of a biobank, as well as Génome Québec for performing the short-read sequencing with the Illumina NovaSeq sequencer. The McGill Trempe lab’s expert insights on the effect of the PMPCB variant on its protein misfolding are also greatly appreciated. This research was enabled in part by the bioinformatic resources access provided by Calcul Québec (beluga.calculcanada.ca) and the Digital Research Alliance of Canada (alliancecan.ca).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1304711/full#supplementary-material
References
Ahsan, T., Urmi, N. J., and Sajib, A. A. (2020). Heterogeneity in the distribution of 159 drug-response related SNPs in world populations and their genetic relatedness. PLoS One 15 (1), e0228000. doi:10.1371/journal.pone.0228000
Akbari, V., Garant, J. M., O'Neill, K., Pandoh, P., Moore, R., Marra, M. A., et al. (2021). Megabase-scale methylation phasing using nanopore long reads and NanoMethPhase. Genome Biol. 22 (1), 68. doi:10.1186/s13059-021-02283-5
Almaguer-Mederos, L. E., Falcon, N. S., Almira, Y. R., Zaldivar, Y. G., Almarales, D. C., Gongora, E. M., et al. (2010). Estimation of the age at onset in spinocerebellar ataxia type 2 Cuban patients by survival analysis. Clin. Genet. 78 (2), 169–174. doi:10.1111/j.1399-0004.2009.01358.x
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi:10.1093/bioinformatics/btu170
Chen, S., Francioli, L. C., Goodrich, J. K., Collins, R. L., Kanai, M., Wang, Q., et al. (2022). A genome-wide mutational constraint map quantified from variation in 76,156 human genomes. bioRxiv. doi:10.1101/2022.03.20.485034
Choi, K. D., and Choi, J. H. (2016). Episodic ataxias: clinical and genetic features. J. Mov. Disord. 9 (3), 129–135. doi:10.14802/jmd.16028
Choquet, K., Tetreault, M., Yang, S., La Piana, R., Dicaire, M. J., Vanstone, M. R., et al. (2016a). SPG7 mutations explain a significant proportion of French Canadian spastic ataxia cases. Eur. J. Hum. Genet. 24 (7), 1016–1021. doi:10.1038/ejhg.2015.240
Choquet, K., Zurita-Rendon, O., La Piana, R., Yang, S., Dicaire, M. J., Rare, C., et al. (2016b). Autosomal recessive cerebellar ataxia caused by a homozygous mutation in PMPCA. Brain 139 (Pt 3), e19. doi:10.1093/brain/awv362
Conroy, J., McGettigan, P., Murphy, R., Webb, D., Murphy, S. M., McCoy, B., et al. (2014). A novel locus for episodic ataxia:UBR4 the likely candidate. Eur. J. Hum. Genet. 22 (4), 505–510. doi:10.1038/ejhg.2013.173
Daoud, H., Belzil, V., Martins, S., Sabbagh, M., Provencher, P., Lacomblez, L., et al. (2011). Association of long ATXN2 CAG repeat sizes with increased risk of amyotrophic lateral sclerosis. Arch. Neurol. 68 (6), 739–742. doi:10.1001/archneurol.2011.111
Dashnow, H., Lek, M., Phipson, B., Halman, A., Sadedin, S., Lonsdale, A., et al. (2018). STRetch: detecting and discovering pathogenic short tandem repeat expansions. Genome Biol. 19 (1), 121. doi:10.1186/s13059-018-1505-2
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29 (1), 15–21. doi:10.1093/bioinformatics/bts635
Dolzhenko, E., Bennett, M. F., Richmond, P. A., Trost, B., Chen, S., van Vugt, J., et al. (2020). ExpansionHunter Denovo: a computational method for locating known and novel repeat expansions in short-read sequencing data. Genome Biol. 21 (1), 102. doi:10.1186/s13059-020-02017-z
Ellingford, J. M., Ahn, J. W., Bagnall, R. D., Baralle, D., Barton, S., Campbell, C., et al. (2022). Recommendations for clinical interpretation of variants found in non-coding regions of the genome. Genome Med. 14 (1), 73. doi:10.1186/s13073-022-01073-3
Escayg, A., De Waard, M., Lee, D. D., Bichet, D., Wolf, P., Mayer, T., et al. (2000). Coding and noncoding variation of the human calcium-channel beta4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am. J. Hum. Genet. 66 (5), 1531–1539. doi:10.1086/302909
Ewels, P., Magnusson, M., Lundin, S., and Kaller, M. (2016). MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32 (19), 3047–3048. doi:10.1093/bioinformatics/btw354
Fahrner, J. A., and Bjornsson, H. T. (2014). Mendelian disorders of the epigenetic machinery: tipping the balance of chromatin states. Annu. Rev. Genomics Hum. Genet. 15, 269–293. doi:10.1146/annurev-genom-090613-094245
Figueroa, K. P., Coon, H., Santos, N., Velazquez, L., Mederos, L. A., and Pulst, S. M. (2017). Genetic analysis of age at onset variation in spinocerebellar ataxia type 2. Neurol. Genet. 3 (3), e155. doi:10.1212/NXG.0000000000000155
Fournier, C., Anquetil, V., Camuzat, A., Stirati-Buron, S., Sazdovitch, V., Molina-Porcel, L., et al. (2018). Interrupted CAG expansions in ATXN2 gene expand the genetic spectrum of frontotemporal dementias. Acta Neuropathol. Commun. 6 (1), 41. doi:10.1186/s40478-018-0547-8
Frankish, A., Diekhans, M., Jungreis, I., Lagarde, J., Loveland, J. E., Mudge, J. M., et al. (2021). Gencode 2021. Nucleic Acids Res. 49 (D1), D916–D923. doi:10.1093/nar/gkaa1087
Frydas, A., Wauters, E., van der Zee, J., and Van Broeckhoven, C. (2022). Uncovering the impact of noncoding variants in neurodegenerative brain diseases. Trends Genet. 38 (3), 258–272. doi:10.1016/j.tig.2021.08.010
Gazulla, J., Izquierdo-Alvarez, S., Ruiz-Fernandez, E., Lazaro-Romero, A., and Berciano, J. (2021). Episodic vestibulocerebellar ataxia associated with a CACNA1G missense variant. Case Rep. Neurol. 13 (2), 347–354. doi:10.1159/000515974
Giunti, P., Mantuano, E., and Frontali, M. (2020). Episodic ataxias: faux or real? Int. J. Mol. Sci. 21 (18), 6472. doi:10.3390/ijms21186472
Glass, J. D., Dewan, R., Ding, J., Gibbs, J. R., Dalgard, C., Keagle, P. J., et al. (2022). ATXN2 intermediate expansions in amyotrophic lateral sclerosis. Brain 145 (8), 2671–2676. doi:10.1093/brain/awac167
Gloss, B. S., and Dinger, M. E. (2018). Realizing the significance of noncoding functionality in clinical genomics. Exp. Mol. Med. 50 (8), 97–98. doi:10.1038/s12276-018-0087-0
Greene, A. W., Grenier, K., Aguileta, M. A., Muise, S., Farazifard, R., Haque, M. E., et al. (2012). Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 13 (4), 378–385. doi:10.1038/embor.2012.14
Gyening, Y. K., Chauhan, N. K., Tytanic, M., Ea, V., Brush, R. S., and Agbaga, M. P. (2023). ELOVL4 mutations that cause spinocerebellar ataxia-34 differentially alter very long chain fatty acid biosynthesis. J. Lipid Res. 64 (1), 100317. doi:10.1016/j.jlr.2022.100317
Hamosh, A., Scott, A. F., Amberger, J. S., Bocchini, C. A., and McKusick, V. A. (2005). Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 33, D514–D517. doi:10.1093/nar/gki033
Harris, P. A., Taylor, R., Minor, B. L., Elliott, V., Fernandez, M., O'Neal, L., et al. (2019). The REDCap consortium: building an international community of software platform partners. J. Biomed. Inf. 95, 103208. doi:10.1016/j.jbi.2019.103208
Hassan, A. (2023). Episodic ataxias: primary and secondary etiologies, treatment, and classification approaches. Tremor Other Hyperkinet Mov. (N Y) 13, 9. doi:10.5334/tohm.747
Hersheson, J., Haworth, A., and Houlden, H. (2012). The inherited ataxias: genetic heterogeneity, mutation databases, and future directions in research and clinical diagnostics. Hum. Mutat. 33 (9), 1324–1332. doi:10.1002/humu.22132
Hobson, J. (2015). The Montreal Cognitive assessment (MoCA). Occup. Med. (Lond) 65 (9), 764–765. doi:10.1093/occmed/kqv078
Jacobi, H., Rakowicz, M., Rola, R., Fancellu, R., Mariotti, C., Charles, P., et al. (2013). Inventory of Non-Ataxia Signs (INAS): validation of a new clinical assessment instrument. Cerebellum 12 (3), 418–428. doi:10.1007/s12311-012-0421-3
Jaganathan, K., Kyriazopoulou Panagiotopoulou, S., McRae, J. F., Darbandi, S. F., Knowles, D., Li, Y. I., et al. (2019). Predicting splicing from primary sequence with deep learning. Cell 176 (3), 535–548. doi:10.1016/j.cell.2018.12.015
Jen, J. C., Wan, J., Palos, T. P., Howard, B. D., and Baloh, R. W. (2005). Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology 65 (4), 529–534. doi:10.1212/01.wnl.0000172638.58172.5a
Koboldt, D. C. (2020). Best practices for variant calling in clinical sequencing. Genome Med. 12 (1), 91. doi:10.1186/s13073-020-00791-w
Kosugi, S., Momozawa, Y., Liu, X., Terao, C., Kubo, M., and Kamatani, Y. (2019). Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 20 (1), 117. doi:10.1186/s13059-019-1720-5
Kulkarni, J. A., Witzigmann, D., Thomson, S. B., Chen, S., Leavitt, B. R., Cullis, P. R., et al. (2021). The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 16 (6), 630–643. doi:10.1038/s41565-021-00898-0
Kunova, N., Havalova, H., Ondrovicova, G., Stojkovicova, B., Bauer, J. A., Bauerova-Hlinkova, V., et al. (2022). Mitochondrial processing peptidases-structure, function and the role in human diseases. Int. J. Mol. Sci. 23 (3), 1297. doi:10.3390/ijms23031297
Laffita-Mesa, J. M., Velazquez-Perez, L. C., Santos Falcon, N., Cruz-Marino, T., Gonzalez Zaldivar, Y., Vazquez Mojena, Y., et al. (2012). Unexpanded and intermediate CAG polymorphisms at the SCA2 locus (ATXN2) in the Cuban population: evidence about the origin of expanded SCA2 alleles. Eur. J. Hum. Genet. 20 (1), 41–49. doi:10.1038/ejhg.2011.154
Landrum, M. J., Lee, J. M., Benson, M., Brown, G. R., Chao, C., Chitipiralla, S., et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46 (D1), D1062–D1067. doi:10.1093/nar/gkx1153
Layer, R. M., Chiang, C., Quinlan, A. R., and Hall, I. M. (2014). LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 15 (6), R84. doi:10.1186/gb-2014-15-6-r84
Lee, Y., Jonson, P. H., Sarparanta, J., Palmio, J., Sarkar, M., Vihola, A., et al. (2018). TIA1 variant drives myodegeneration in multisystem proteinopathy with SQSTM1 mutations. J. Clin. Invest. 128 (3), 1164–1177. doi:10.1172/JCI97103
Leger, A., and Leonardi, T. (2019). pycoQC, interactive quality control for Oxford Nanopore Sequencing. J. Open Source Softw. 4 (34), 1236. doi:10.21105/joss.01236
Li, C., Ou, R., Chen, Y., Gu, X., Wei, Q., Cao, B., et al. (2021). Genetic modifiers of age at onset for Parkinson's disease in asians: a genome-wide association study. Mov. Disord. 36 (9), 2077–2084. doi:10.1002/mds.28621
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34 (18), 3094–3100. doi:10.1093/bioinformatics/bty191
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Liao, Y., Smyth, G. K., and Shi, W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30 (7), 923–930. doi:10.1093/bioinformatics/btt656
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 550. doi:10.1186/s13059-014-0550-8
Lunke, S., Bouffler, S. E., Patel, C. V., Sandaradura, S. A., Wilson, M., Pinner, J., et al. (2023). Integrated multi-omics for rapid rare disease diagnosis on a national scale. Nat. Med. 29 (7), 1681–1691. doi:10.1038/s41591-023-02401-9
Marti, E. (2016). RNA toxicity induced by expanded CAG repeats in Huntington's disease. Brain Pathol. 26 (6), 779–786. doi:10.1111/bpa.12427
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20 (9), 1297–1303. doi:10.1101/gr.107524.110
Mezreani, J., Audet, S., Martin, F., Charbonneau, J., Triassi, V., Bareke, E., et al. (2022). Novel homozygous nonsense mutation of MLIP and compensatory alternative splicing. NPJ Genom Med. 7 (1), 36. doi:10.1038/s41525-022-00307-y
Miller, D. E., Lee, L., Galey, M., Kandhaya-Pillai, R., Tischkowitz, M., Amalnath, D., et al. (2022). Targeted long-read sequencing identifies missing pathogenic variants in unsolved Werner syndrome cases. J. Med. Genet. 59 (11), 1087–1094. doi:10.1136/jmedgenet-2022-108485
Mitsuhashi, S., Frith, M. C., Mizuguchi, T., Miyatake, S., Toyota, T., Adachi, H., et al. (2019). Tandem-genotypes: robust detection of tandem repeat expansions from long DNA reads. Genome Biol. 20 (1), 58. doi:10.1186/s13059-019-1667-6
Murray, M. F., Khoury, M. J., and Abul-Husn, N. S. (2022). Addressing the routine failure to clinically identify monogenic cases of common disease. Genome Med. 14 (1), 60. doi:10.1186/s13073-022-01062-6
Nachbauer, W., Eigentler, A., and Boesch, S. (2015). Acquired ataxias: the clinical spectrum, diagnosis and management. J. Neurol. 262 (5), 1385–1393. doi:10.1007/s00415-015-7685-8
Neuenschwander, A. G., Thai, K. K., Figueroa, K. P., and Pulst, S. M. (2014). Amyotrophic lateral sclerosis risk for spinocerebellar ataxia type 2 ATXN2 CAG repeat alleles: a meta-analysis. JAMA Neurol. 71 (12), 1529–1534. doi:10.1001/jamaneurol.2014.2082
Nishide, M., Le Marquand, K., Davis, M. R., Halmagyi, G. M., Fellner, A., Narayanan, R. K., et al. (2023). Two new families and a literature review of ELOVL4-associated spinocerebellar ataxia type 34. Cerebellum. 23 (1). doi:10.1007/s12311-023-01522-8
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., and Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14 (4), 417–419. doi:10.1038/nmeth.4197
Paulhus, K., Ammerman, L., and Glasscock, E. (2020). Clinical spectrum of KCNA1 mutations: new insights into episodic ataxia and epilepsy comorbidity. Int. J. Mol. Sci. 21 (8), 2802. doi:10.3390/ijms21082802
Pellerin, D., Danzi, M. C., Wilke, C., Renaud, M., Fazal, S., Dicaire, M. J., et al. (2023). Deep intronic FGF14 GAA repeat expansion in late-onset cerebellar ataxia. N. Engl. J. Med. 388 (2), 128–141. doi:10.1056/NEJMoa2207406
Pengelly, R. J., Alom, T., Zhang, Z., Hunt, D., Ennis, S., and Collins, A. (2017). Evaluating phenotype-driven approaches for genetic diagnoses from exomes in a clinical setting. Sci. Rep. 7 (1), 13509. doi:10.1038/s41598-017-13841-y
Perlman, S. (1993). “Hereditary ataxia overview,” in GeneReviews (Seattle WA: University of Washington).
Pfeffer, G., Pyle, A., Griffin, H., Miller, J., Wilson, V., Turnbull, L., et al. (2015). SPG7 mutations are a common cause of undiagnosed ataxia. Neurology 84 (11), 1174–1176. doi:10.1212/WNL.0000000000001369
Rahit, K., and Tarailo-Graovac, M. (2020). Genetic modifiers and rare mendelian disease. Genes (Basel) 11 (3), 239. doi:10.3390/genes11030239
Reese, F., Williams, B., Balderrama-Gutierrez, G., Wyman, D., Celik, M. H., Rebboah, E., et al. (2023). The ENCODE4 long-read RNA-seq collection reveals distinct classes of transcript structure diversity. bioRxiv. doi:10.1101/2023.05.15.540865
Rehm, H. L., Alaimo, J. T., Aradhya, S., Bayrak-Toydemir, P., Best, H., Brandon, R., et al. (2023). The landscape of reported VUS in multi-gene panel and genomic testing: time for a change. Genet. Med. 25, 100947. doi:10.1016/j.gim.2023.100947
Rentzsch, P., Schubach, M., Shendure, J., and Kircher, M. (2021). CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 13 (1), 31. doi:10.1186/s13073-021-00835-9
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J., and Kircher, M. (2019). CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47 (D1), D886–D894. doi:10.1093/nar/gky1016
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Roberts, L. J., McVeigh, M., Seiderer, L., Harding, I. H., Corben, L. A., Delatycki, M., et al. (2022). Overview of the clinical approach to individuals with cerebellar ataxia and neuropathy. Neurol. Genet. 8 (5), e200021. doi:10.1212/NXG.0000000000200021
Rudell, J. B., Maselli, R. A., Yarov-Yarovoy, V., and Ferns, M. J. (2019). Pathogenic effects of agrin V1727F mutation are isoform specific and decrease its expression and affinity for HSPGs and LRP4. Hum. Mol. Genet. 28 (16), 2648–2658. doi:10.1093/hmg/ddz081
Saffari, A., Kellner, M., Jordan, C., Rosengarten, H., Mo, A., Zhang, B., et al. (2023). The clinical and molecular spectrum of ZFYVE26-associated hereditary spastic paraplegia: SPG15. Brain 146 (5), 2003–2015. doi:10.1093/brain/awac391
Scacheri, C. A., and Scacheri, P. C. (2015). Mutations in the noncoding genome. Curr. Opin. Pediatr. 27 (6), 659–664. doi:10.1097/MOP.0000000000000283
Schesny, M., Joncourt, F., and Tarnutzer, A. A. (2019). Acetazolamide-responsive episodic ataxia linked to novel splice site variant in FGF14 gene. Cerebellum 18 (3), 649–653. doi:10.1007/s12311-018-0997-3
Schmitz-Hubsch, T., du Montcel, S. T., Baliko, L., Berciano, J., Boesch, S., Depondt, C., et al. (2006). Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 66 (11), 1717–1720. doi:10.1212/01.wnl.0000219042.60538.92
Schmucker, S., Argentini, M., Carelle-Calmels, N., Martelli, A., and Puccio, H. (2008). The in vivo mitochondrial two-step maturation of human frataxin. Hum. Mol. Genet. 17 (22), 3521–3531. doi:10.1093/hmg/ddn244
Schwarz, N., Hahn, A., Bast, T., Muller, S., Loffler, H., Maljevic, S., et al. (2016). Mutations in the sodium channel gene SCN2A cause neonatal epilepsy with late-onset episodic ataxia. J. Neurol. 263 (2), 334–343. doi:10.1007/s00415-015-7984-0
Shen, S., Park, J. W., Lu, Z. X., Lin, L., Henry, M. D., Wu, Y. N., et al. (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. U. S. A. 111 (51), E5593–E5601. doi:10.1073/pnas.1419161111
Sintas, C., Carreno, O., Fernandez-Castillo, N., Corominas, R., Vila-Pueyo, M., Toma, C., et al. (2017). Mutation spectrum in the CACNA1A gene in 49 patients with episodic ataxia. Sci. Rep. 7 (1), 2514. doi:10.1038/s41598-017-02554-x
Steinhaus, R., Proft, S., Schuelke, M., Cooper, D. N., Schwarz, J. M., and Seelow, D. (2021). MutationTaster2021. Nucleic Acids Res. 49 (W1), W446–W451. doi:10.1093/nar/gkab266
Stelzer, G., Rosen, N., Plaschkes, I., Zimmerman, S., Twik, M., Fishilevich, S., et al. (2016). The GeneCards suite: from gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinforma. 54, 1–31. doi:10.1002/cpbi.5
Synofzik, M., Rugarli, E., Reid, E., and Schule, R. (2023). Ataxia and spastic paraplegia in mitochondrial disease. Handb. Clin. Neurol. 194, 79–98. doi:10.1016/B978-0-12-821751-1.00009-9
Talevich, E., Shain, A. H., Botton, T., and Bastian, B. C. (2016). CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 12 (4), e1004873. doi:10.1371/journal.pcbi.1004873
Tang, A. D., Soulette, C. M., van Baren, M. J., Hart, K., Hrabeta-Robinson, E., Wu, C. J., et al. (2020). Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat. Commun. 11 (1), 1438. doi:10.1038/s41467-020-15171-6
Tetreault, M., Gonzalez, M., Dicaire, M. J., Allard, P., Gehring, K., Leblanc, D., et al. (2015). Adult-onset painful axonal polyneuropathy caused by a dominant NAGLU mutation. Brain 138 (Pt 6), 1477–1483. doi:10.1093/brain/awv074
Traschutz, A., Cortese, A., Reich, S., Dominik, N., Faber, J., Jacobi, H., et al. (2021). Natural history, phenotypic spectrum, and discriminative features of multisystemic RFC1 disease. Neurology 96 (9), e1369–e1382. doi:10.1212/WNL.0000000000011528
Uhlen, M., Karlsson, M. J., Zhong, W., Tebani, A., Pou, C., Mikes, J., et al. (2019). A genome-wide transcriptomic analysis of protein-coding genes in human blood cells. Science 366 (6472), eaax9198. doi:10.1126/science.aax9198
UniProt, C. (2023). UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Res. 51 (D1), D523–D531. doi:10.1093/nar/gkac1052
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3--new capabilities and interfaces. Nucleic Acids Res. 40 (15), e115. doi:10.1093/nar/gks596
Vogtle, F. N., Brandl, B., Larson, A., Pendziwiat, M., Friederich, M. W., White, S. M., et al. (2018). Mutations in PMPCB encoding the catalytic subunit of the mitochondrial presequence protease cause neurodegeneration in early childhood. Am. J. Hum. Genet. 102 (4), 557–573. doi:10.1016/j.ajhg.2018.02.014
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38 (16), e164. doi:10.1093/nar/gkq603
Wang, P., Chen, Z., Peng, Y., Cao, L., Li, X., Wang, C., et al. (2019). (CAG)(n) loci as genetic modifiers of age at onset in patients with spinocerebellar ataxia type 1 from mainland China. Eur. J. Neurol. 26 (8), 1130–1136. doi:10.1111/ene.13954
Wenning, G. K., Stankovic, I., Vignatelli, L., Fanciulli, A., Calandra-Buonaura, G., Seppi, K., et al. (2022). The movement disorder society criteria for the diagnosis of multiple system atrophy. Mov. Disord. 37 (6), 1131–1148. doi:10.1002/mds.29005
Wick, R. R., Judd, L. M., and Holt, K. E. (2019). Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 20 (1), 129. doi:10.1186/s13059-019-1727-y
Keywords: genomics, transcriptomics, ataxia, long-read sequencing (LRS), variant of uncertain significance (VUS), whole genome sequencing (WGS), RNA sequencing (RNAseq), multi-omics
Citation: Audet S, Triassi V, Gelinas M, Legault-Cadieux N, Ferraro V, Duquette A and Tetreault M (2024) Integration of multi-omics technologies for molecular diagnosis in ataxia patients. Front. Genet. 14:1304711. doi: 10.3389/fgene.2023.1304711
Received: 29 September 2023; Accepted: 27 November 2023;
Published: 04 January 2024.
Edited by:
Patricia Pelufo Silveira, McGill University, CanadaReviewed by:
Paola Mandich, University of Genoa, ItalyFrancesco Musacchia, Italian Institute of Technology (IIT), Italy
Copyright © 2024 Audet, Triassi, Gelinas, Legault-Cadieux, Ferraro, Duquette and Tetreault. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sebastien Audet, c2ViYXN0aWVuLmF1ZGV0LjRAdW1vbnRyZWFsLmNh