 Abrar Bawatneh
Abrar Bawatneh Alaa Darwish2
Alaa Darwish2 Hisham M. Darwish
Hisham M. Darwish
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 11 January 2024
Sec. Genetics of Common and Rare Diseases
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1292073
This article is part of the Research TopicInborn errors of Carbohydrate MetabolismView all 15 articles
Introduction: Diabetes Mellitus is a group of metabolic disorders characterized by hyperglycemia secondary to insulin resistance or deficiency. It is considered a major health problem worldwide. T1DM is a result of a combination of genetics, epigenetics, and environmental factors. Several genes have been associated with T1DM, including HLA, INS, CTLA4, and PTPN22. However, none of these findings have been based on linkage analysis because it is rare to find families with several diabetic individuals. Two Palestinian families with several afflicted members with variations in the mode of inheritance were identified and selected for this study. This study aimed to identify the putative causative gene(s) responsible for T1DM development in these families to improve our understanding of the molecular genetics of the disease.
Methods: One afflicted member from each family was selected for Whole-Exome Sequencing. Data were mapped to the reference of the human genome, and the resulting VCF file data were filtered. The variants with the highest phenotype correlation score were checked by Sanger sequencing for all family members. The confirmed variants were analyzed in silico by bioinformatics tools.
Results: In one family, the IGF1R p.V579F variant, which follows autosomal dominant inheritance, was confirmed and segregated in the family. In another family, the NEUROD1 p.P197H variant, which follows autosomal recessive inheritance, was positively confirmed and segregated.
Conclusion: IGF1R p.V579F and NEUROD1 p.P197H variants were associated with T1DM development in the two inflicted families. Further analysis and functional assays will be performed, including the generation of mutant model cell systems, to unravel their specific molecular mechanism in the disease development.
Diabetes mellitus is a group of metabolic disorders characterized by the presence of hyperglycemia secondary to insulin resistance or deficiency, and it is associated with abnormalities in lipid and protein metabolism and electrolyte disturbances (Kharroubi and Darwish, 2015). Diabetes is considered a great health problem worldwide, with about 425 million adults having been recorded as affected in 2017 (Tremblay and Hamet, 2019). Diabetes mellitus can be classified into type 1 diabetes (T1D), which is characterized by insulin deficiency, type 2 diabetes (T2D), which is characterized by resistance to the action of insulin, gestational diabetes, which develops in women during pregnancy, and Maturity-Onset Diabetes of the Young (MODY), which is characterized by autosomal dominant inheritance and non-dependence on insulin (Anik et al., 2015). The annual health report in Palestine in 2018 reported diabetes health complications as the fifth-ranked cause of death (about 7.5% of all deaths). In 2018, the incidence rate of diabetes mellitus in the West Bank region was reported as 210.7 cases per 100,000 people. There were 2,420 cases among males and 3,135 among females. The mortality rate was 20.4 per 100,000 people (Health annual Palestine report, 2018).
T1D is an autoimmune disease in which insulin is deficient due to the destruction of pancreatic beta cells. There are two forms of T1DM: type 1A, which results from cell-mediated attack and is characterized by the presence of autoantibodies to beta cells, and type 1B, which results from unknown causes and includes unmeasurable autoantibodies or monogenic diabetes (Nyaga et al., 2018; Giwa et al., 2020). Patients are diagnosed in the first few months after birth based on the absence of insulin, detection of C-peptide, and presence of antibodies in the serum. These patients will be treated for life without interruption by insulin and are monitored for several metabolic indicators. T1D is the most common form in children and white persons, accounting for 80% of childhood diabetes in the United States (Redondo et al., 2017). The risk of the disease increases in relatives and siblings and exceeds 70% in identical twins (Bradfield et al., 2011). This clearly implies that T1D is associated with genetic factors. Overall, T1D represents a multifactorial disease that develops from the interplay of genetic, epigenetic, and environmental factors, and it can be affected by age, ethnicity, race, geography, and socioeconomic status (Tremblay and Hamet, 2019).
Genetics are responsible for 80% of hereditary T1D; the HLA region on chromosome 6p21 accounts for 40%–50% of familial T1D. Variation in this region is correlated with the role of autoimmunity in T1D development and can increase its risk or be protective. HLA DR and DQ (part of MCH class II) form alpha-beta heterodimer cell surface antigens and have the strongest association with T1D. The Type 1 Diabetes Genetics Consortium showed that DRB1*0301-DQA1*0501-DQB1*0201, DRB1*0405-DQA1*0301-DQB1*0302, and DRB1*0401-DQA1*0301-DQB*0302 are the most susceptible haplotypes. Whereas, DRB1*1501-DQA1*0102-DQB1*0602, DRB1*1401-DQA1*0101-DQB1*0503, and DRB1*0701-DQA1*0201-DQB1*0303 are the most protective haplotypes (Erlich et al., 2013).
The insulin gene region on chromosome 11 (locus 11p15) constituted the next strongest genetic factor in T1D, and the third associated locus was the cytotoxic T lymphocyte-associated protein 4 (CTLA4) gene (Cronin et al., 2017). The protein tyrosine phosphatase non-receptor type 22 (PTPN22) gene was reported to be the fourth identified locus in 2004 (Bottini et al., 2004). Later, the interleukin 2 receptor alpha (IL2RA) gene (Vella et al., 2005) and the interferon-induced helicase C domain 1 (IFIH1) gene on chromosome 2q24.3 (Smyth et al., 2006) were reported as the fifth and sixth associated loci, respectively. In a Japanese study, the FOX3P gene, which plays a role in the regulation of T-cell differentiation, was also found to be associated with T1D (Bassuny et al., 2003). Genome-wide association studies were later started to search for further loci associated with T1D (Bakay et al., 2019). They confirmed previous loci and showed evidence of new loci on chromosomes 16p13, 12q13, 12q24, and 18p11. The 16p13 region contains the KIAA0350 gene, which lies within a 233 kb LD block and is suspected to produce a sugar-binding C-type lectin (Hakonarson et al., 2007; Todd et al., 2007; Concannon et al., 2008). Regions like 12q13 and 12q24 contain many genes in which some of these loci play a role in immune signaling and insulin production, and they are evidenced to be associated with T1D genes such as ERBB3, CYP27B1, and SH2B3 (Cooper et al., 2012; Groop and Pociot, 2014). The 18p11 region contains the PTPN2 gene, which has a function in the immune system and cytokine-induced apoptosis (Groop and Pociot, 2014; Pociot, 2017). Other groups conducted their investigation on 2,496 families inflicted with T1D genotyped with a panel of 6,090 SNPs and reported additional T1D-dependent loci on 21q22.3 in the UBASH3A locus (Concannon et al., 2008).
Arab countries have one of the highest incidence and prevalence rates of T1D, which varies from low prevalence (2.54/100,000) in Oman to significantly higher values in Saudi Arabia (29/100,000) (Zayed, 2016). The DRB1*030101-DQB1*0201 haplotypes increased the risk of T1D in Tunisia, Lebanon, and Bahrain, whereas the DRB1*040101-DQB1*0302 haplotype was highly associated with Tunisia and Bahrain but is protective among the Lebanese population (Al-Jenaidi et al., 2005; Stayoussef et al., 2009). The most common HLA haplotypes in Egypt were DRB1*0301-DRB3*0201- DQA1*0501-DQB1*0201 and DRB1*04:02-DQA1*03- DQB1*03:02, while the protective one was DRB1*04:03-DQA1*03DQB1*03:02 (El-Amir et al., 2015). Studies from Morocco showed that Moroccans share some susceptible and protective haplotypes with other populations, such as those from Tunisia, Algeria, Spain, and France, including DRB1*08-DQA1*0401-DQB1*0402 and DRB1*07-DQA1*0201-DQB1*020 haplotypes, respectively (Nadia et al., 2012). Concerning non-HLA genes, several studies reported T1D in Arabs was associated with PTPN22, CTLA4, IL15, ZAP70, CD3z, CD28, TCRβ, and BANK1 (Zayed, 2016). However, no previous studies on the molecular genetics of T1D in Palestine have been reported. We have identified two Palestinian families with several affected members with clear variations in the inheritance pattern of the disease. We hypothesized the presence of putative gene mutations that have a direct role in developing T1DM in these families. Therefore, the objective of this study was to investigate the molecular genetics of T1D development in these families using WES technology. Such genes will provide valuable information to increase our understanding of the molecular genetics basis of Type 1 diabetes and provide tools for early diagnosis and clinical intervention to improve the survival and life quality of these patients before severe complications develop in these individuals.
Two Palestinian families inflicted with Type 1 diabetes mellitus were included in this study and referred to us by a specialized local Medical Center (Layan Center) in Ramallah, Palestine. They were confirmed to have T1DM based on specific parameters including high serum glucose (above 350 g/dL), high levels of HbA1C (>7%), Low levels of C peptide, the presence of autoantibodies, and insulin dependence. Family I was composed of diabetic parents, six diabetic offspring, and one non-diabetic daughter, indicating the presence of a potentially dominant gene factor. Family II was composed of non-diabetic parents, three diabetic offspring, and a non-diabetic daughter, indicating the presence of a potentially recessive gene mutation. Figure 1 shows the pedigree of both families. Written informed consent was obtained from all participants.
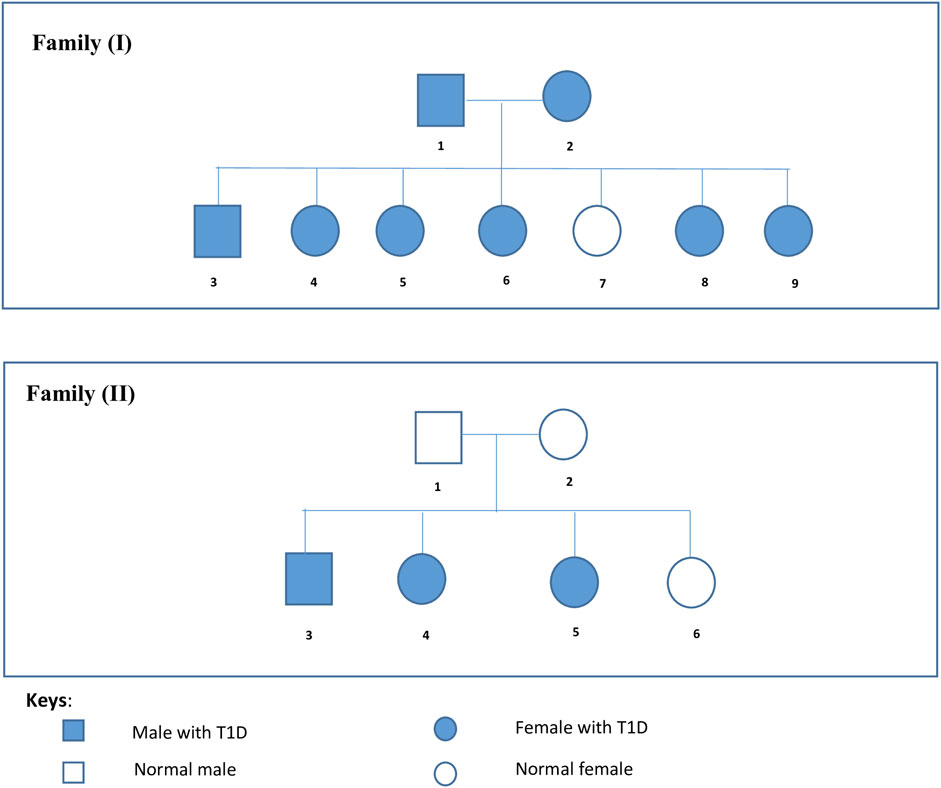
FIGURE 1. Families’ Pedigrees. Family I is composed of diabetic parents, six diabetic offspring, and one non-diabetic daughter. Family II is composed of non diabetic parents, three diabetic offspring, and a non-diabetic daughter.
Genomic DNA was extracted from peripheral blood samples according to a commercially available kit (Master PureTM Genomic DNA Purification Kit, Epicenter Technology Co. Cat.No.MG71100) (Mcd, 2012). Extracted DNA was checked for purity, concentration size, and integrity using Nanodrop spectrophotometer and Agarose Gel Electrophoresis and stored at −30°C.
Samples were subjected to whole-exome sequencing using Illumina Nextseq550 according to the manufacturer’s instructions (Illumina, 2021).
Fastq paired-end reads were mapped to the reference human genome version GRCh38 using the Burrows-Wheeler Alignment Tool with the BWA-MEM algorithm. The mapped reads in the bam file were filtered for the following two criteria; first, we retained only paired reads for which both the forward and the reverse read have been mapped to the reference successfully using Samtools. Second, PCR duplicates were removed using the RmDup tool. Filtered mapped reads were then used to call the variants using the FreeBayes variant detector to identify SNPs (single-nucleotide polymorphisms) and indels (insertions and deletions). The list of variants produced in a VCF format were filtered and prioritized with respect to their potential relevance for features that have been used, and a VCF file containing annotations of variant effects was produced. Finally, we filtered out variants that are unlikely to have a pathogenic effect like variants in UTRs, those upstream/downstream of the gene, deep intronic variants (but we leave variants that affect splice sites), and other non-coding variants. In addition, we filtered out variants that are common in population databases such as Gnomad. Furthermore, we prioritized variants that have been reported as pathogenic in databases like Clinvar. The aligned data were deposited in the SRA database under the accession number SRR26632668.
We focused on non-synonymous exonic variants with high correlation scores present in the affected family members and not present in the unaffected family members.
Primer 3 web (https://primer3.ut.ee/) was used to design the primers listed in Table 1 below for PCR amplification and direct sanger sequencing of selected regions in the indicated genes with the identified potential mutant variants.

TABLE 1. The sequences and characteristics of the variants’ primers.
Samples were sequenced using the BigDye™ Terminator Cycle Sequencing Kit and Applied Biosystems Genetic Analyzer according to the manufacturer’s instructions (Thermo Fisher Scientific, 2015).
The Mutation Mapper tool of cBioPortal was used to map mutations on proteins and their domains. The conservation of the variant locus was detected using the COBALT Alignment tool. Several prediction tools were used to predict the impact of amino acid substitution on protein structure and function, including PolyPhen-2, PROVEAN, FATHMM, SIFT, Mutation Taster, GVGD, and LIST-S2. The HaploR tool from R package was used to analyze motif changing.
Ethical approval for this study was provided. A written informed consent form was obtained from all participating subjects in the study. Written informed consent was obtained from the individuals for the publication of any potentially identifiable images or data included in this article.
Variants of the INS, HNF1A, KCNJ11, and IGF1R genes were found to have high correlation b based on data analysis. The first three variants were tested by Sanger sequencing but showed no segregation in the family because both diabetic patients and non-diabetic individuals have similar genotypes.
Variants of the IGF1R gene were identified in T1DM patients but not in non-diabetic ones. The IGF1R gene is located on chromosome 15, and the identified variant was a missense mutation where Guanine is replaced by Thymidine at position 1735 (c.1735G>T), leading to replacing Valine with Phenylalanine at codon number 579. This variant was confirmed by Sanger sequencing and segregated in the family (genotypes match the phenotype). Members I-1, I-2, I-4, and I-9 are heterozygous GT, non-diabetic member I-7 is wild-type GG, and members I-5, I-6, I-8 are homozygous TT, as shown in Figure 2.
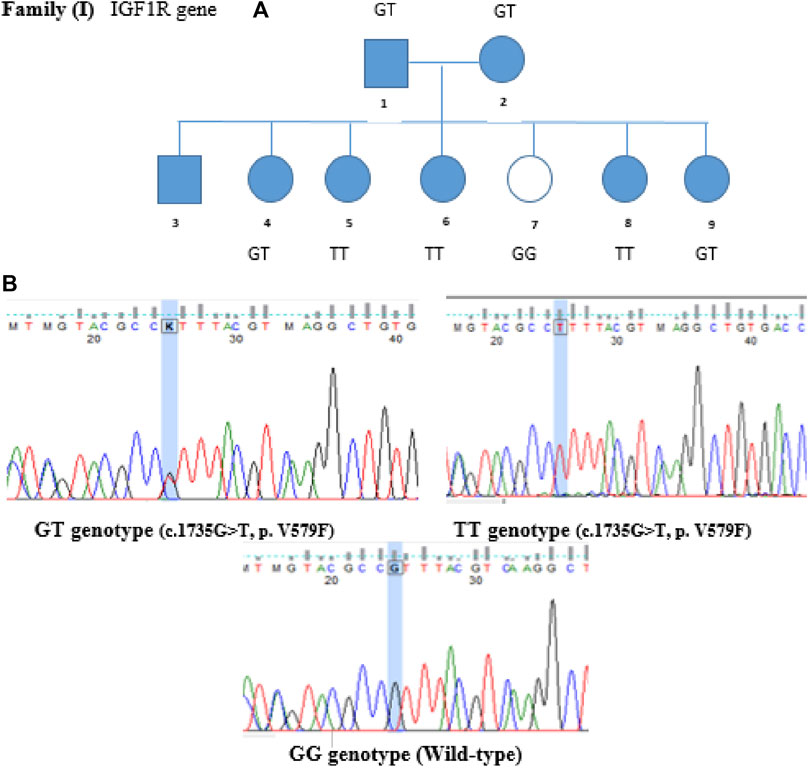
FIGURE 2. Family segregation of IGF1R variant. (A) Genotype results of each family member shown in the family pedigree. (B) Sanger Sequencing results of the detected variant in Family I.
Mutation conservation, functional location, and pathogenicity were further investigated. The IGF1R protein is composed of five domains: two Receptor-L domains, a Furin-like cysteine-rich region, a Fibronectin type III domain, and a Protein tyrosine kinase domain. IGF1R p.V579F is located between the Receptor-L domain and Fibronectin type III domain, as shown in Figure 3. This region is highly conserved, as shown in Figure 4. Several prediction tools were used to investigate whether the IGF1R p.V579F variant has an impact on the biological function and structure of the protein. The variant is predicted to be neutral and tolerated according to PROVEAN and SIFT tools. It is also predicted to be benign by the PolyPhen-2 tool. Based on the FATHMM prediction tool, IGF1R p.V579F will be tolerated. However, according to the FATHMM-MKL and LIST-S2 tools, it is predicted to be damaging. The IGF1R p.V579F variant is disease-causing based on the Mutation Taster tool and is classified in the C45 class of Align-GVGD classes, which means it is pathogenic.

FIGURE 3. Location of IGF1R p.V579F variant on IGF1R protein.
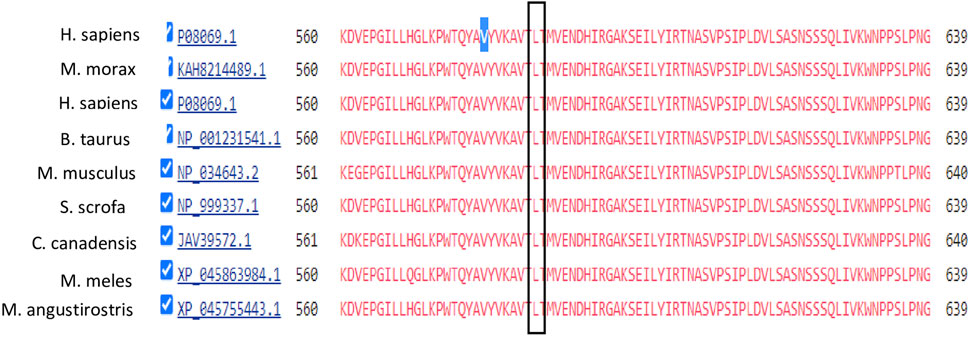
FIGURE 4. Conservation analysis of IGF1R V579 locus using COBALT alignment tool.
In the second family, we identified variants in the ABCC8, RYR1, CTRC, and NEUROD1 genes. The first three variants were tested by Sanger sequencing but showed no segregation in the family. However, a variant in the NEUROD1 gene that is located on chromosome 2 was segregated. The identified variant c.590G>T is a missense mutation where Guanine is replaced by Thymidine at position 590 replacing Proline with Histidine. This variant was validated by Sanger sequencing and segregated in Family II, as shown in Figure 5. Members II-1 and II-2 (Parents) have heterozygous GT genotypes, whereas members II-3, II-4, and II-5 (diabetic offspring) have homozygous TT genotypes. The non-diabetic daughter (member II-6) has a wild-type GG genotype. Therefore, the segregation matches the observed expression of the disease.
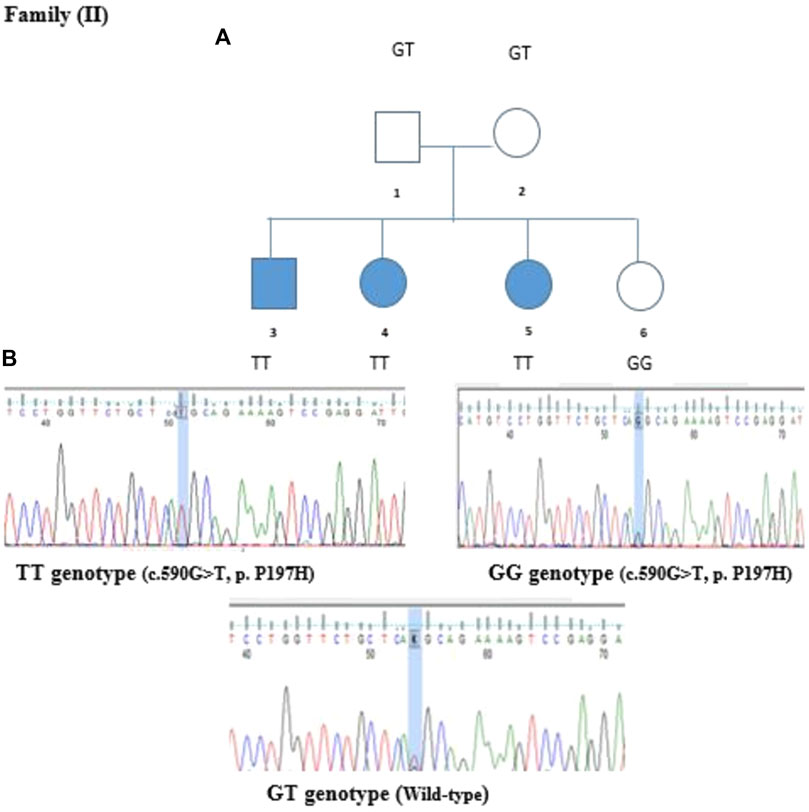
FIGURE 5. Family segregation of NEUROD1 variant. (A) Genotype results of each family member shown in the family pedigree. (B) Sanger Sequencing results of the detected variant in Family II.
The NEUROD1 protein is composed of two domains: the helix loop helix domain and the transactivation domain. The NEUROD1 p.P197H variant is located at the transactivation domain of the NEUROD1 protein (Figure 6) in a highly conserved region, as shown in Figure 7. The NEUROD1 p.P197H variant is predicted to be damaging according to the PROVEAN, LIST-S2, and FATHMM-XF tools. It is also predicted to be possibly damaging according to the PolyPhen-2 tool. The variant was predicted to be disease-causing by the GVGD tool and pathogenic by the Mutation Taster tool. On the other hand, the NEUROD1 p.P197H variant is predicted to be tolerated according to the FATHMM and FATHMM-MKL tools.
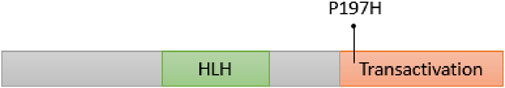
FIGURE 6. Location of NEUROD1 p.P197H variant on NEUROD1 protein.
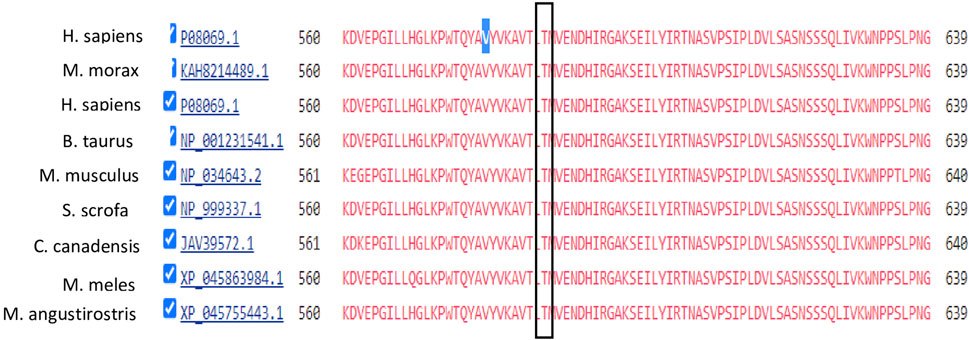
FIGURE 7. Conservation analysis of IGF1R V579 locus using COBALT alignment tool.
To detect whether the NEUROD1 p.P197H variant affects the binding motif of any transcription factor, the HaploR tool was used. Figure 8 shows the variant is located within the PAX motif.
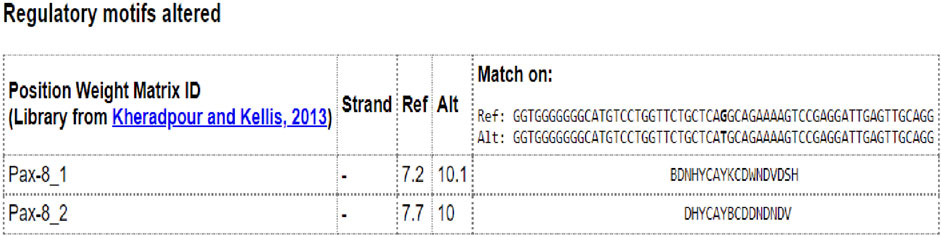
FIGURE 8. In silico analysis of NEUROD1 p.P197H variant by HaploR tool (motifs alteration).
The indicated variants in the IGF1R and NEUROD1 genes were tested in genomic DNA samples from 80 non-familial T1D patients available in our laboratory. The results showed these variants are unique mutations found in the study’s two families.
Despite several gene mutations having been linked with type 1 diabetes mellitus, the molecular mechanisms of the disease are not fully understood. This hampers the presence of specific genetic factors that require increased efforts to unravel the transmittance of the disease within generations. This work was initiated to study two families with T1DM inherited between consecutive generations. Two putative candidate variants were prioritized and were checked for confirmation by Sanger sequencing and family segregation.
In the first family, T1D follows autosomal dominant inheritance; the IGF1R p.V579F variant was associated with T1DM in the family confirmed by Sanger sequencing and segregated in the family with the phenotype. This variant showed uncertain significance according to ACMG classification (The American College of Medical Genetics). No previous studies reported the association between this variant and T1DM or other diseases. However, the Val579 residue is highly conserved, indicating its importance in the protein with potential pathogenicity. This was confirmed with several in silico prediction tools, including FATHMM-MKL, LIST-S2, GVGD, and Mutation Taster, which predicted the variant to be damaging and disease-causing. Other tools predicted the variant to be benign, including PolyPhen-2, SIFT, and PROVEAN. This shows the necessity for further analysis and studies, including studies on the effects of the IGF1R p.V579F variant on the 3D structure of the protein and its functional implications. The IGF1R gene is located in chromosome 15, which encodes the insulin growth factor 1 receptor that binds with high affinity to insulin growth factor (IGF1) and with lower affinity to insulin. The structure of IGF1R and insulin receptors are very similar. Both receptors consist of a tetramer of two α subunits representing the extracellular ligand-binding domain and two β subunits representing the transmembrane domain with tyrosine kinase activity. The two subunits are linked with disulfide bonds (Hakuno and Takahashi, 2018). The IGF1 binds to the α subunits of IGF1R, causing conformational changes and stimulation of β subunit activity. This leads to autophosphorylation and transphosphorylation of tyrosine (especially tyrosine 1131, 1135, 1136, and 1221), increasing its kinase activity and thus activating and recruiting the insulin receptor substrate (IRS), CT10 regulator of kinase (CRK), and Src homology and Collagen (SHC) adaptor proteins. The latter will transduce and activate downstream signaling pathways, including the MAPK/RasRaf-Erk pathway, phosphatidylinositol-3-kinase/AKT/mTOR (PI3K/AKT) pathway, Janus kinase/signal transducer, and activator of transcription (JAK/STAT) pathway, resulting in stimulation cell proliferation, survival, and differentiation (Arnaldez and Helman, 2012). Moreover, activated AKT can activate mTOR and inhibit FOXO1 and GSK3 downstream molecules involved in cellular processes and glucose homeostasis, resulting in gluconeogenesis inhibition and increasing glycogen and protein synthesis (Jung and Suh, 2015).
The IGF1R gene is highly expressed in 9% of cancer cells, including lung adenocarcinoma, cutaneous melanoma, breast cancer, and colon adenocarcinoma (Farabaugh et al., 2015). It plays an important role in phenotypic and oncogenic transformation by activating signaling pathways downstream. IGF1R can then interact with oncogenes such as RAS, c-MYC, and epidermal growth factor receptor (EGFR), causing an increase in cell migration, invasion, poor prognosis, and shorter survival rate (Sun et al., 2017).
Several studies suggest a close link between IGF1R function and glucose metabolism. It plays several physiological roles in glucose metabolism and tissue development including growth, heart ventricular development, neutrophils differentiation, and brain development (Wit and Walenkamp, 2013; Moody et al., 2014; Yuan et al., 2018; Jin et al., 2019). Mouse models with IGF1R knockout showed a significant decrease in their weight after birth. These animals suffer from reduced glucose tolerance in addition to several organ failures, leading to earlier death (Hakuno and Takahashi, 2018). The significant association between IGF1R expression and diabetes is evident (Fowlkes et al., 2012). One previous study found that the absence of IGF1R in beta cells in mice caused glucose intolerance and deficiency in insulin secretion (Xuan et al., 2002). Another study showed that the loss of one allele of IGF1R is associated with glucose intolerance and later insulin resistance, resulting in low birth weight and growth (Garg et al., 2011). Additionally, osteopathy defects, which are serious complications of T1DM and T2DM, result from interruption of insulin and IGF1R (Fowlkes et al., 2012). Noncoding RNAs (LncRNA and Micro RNAs) have a major role in disease development, including diabetes and cancer, through the regulation of IGF1R or other IGF signaling molecules (Chen et al., 2019). Interestingly, knockout of miR-375 in mice model revealed insulin resistance and decreased glucose homeostasis, leading to diabetes, an effect that was associated with upregulation of IGF1R (Kumar et al., 2021). The IGF1R p.V579F is predicted to be a loss-of-function mutation affecting insulin release from beta cells. The significant association between IGF1R and cancer results from IGF1R gain-of-function mutations. Therefore, diabetic patients from Family I are not expected to develop cancer due to this variant. In contrast, we cannot exclude the possibility that some IGF1R gain-of-function mutations are associated with diabetes as previous studies showed (Engberding et al., 2009; Kumar et al., 2021). One possible explanation is that upregulated IGF1R may suppress insulin signaling through a direct effect on the insulin receptor itself or its signaling mechanisms. Therefore, the collective indication of these studies and the present study indicate a significant correlation between the IGF1R signaling pathway and disruption in glucose metabolism leading to diabetes.
In the family with autosomal recessive inheritance, the NEUROD1 p.P197H variant was found to be associated with T1DM. This was a missense mutation where Guanine is replaced by Thymidine at position 590 resulting in Histidine replacing Proline at codon 197. This exchange can lead to a major change in the protein structure since the variant is located in the Neuronal helix-loop-helix transactivation domain and its locus is well conserved, indicating this residue has functional significance. It is predicted to have deleterious functional consequences by several prediction tools, including PolyPhen-2, PROVEAN, SIFT, GVGD, LIST-S2, and FATHMM-XF.
The neurogenic differentiation 1 (NEUROD1) gene is located in chromosome 2 and encodes the basic helix-loop-helix (bHLH) transcription factor. These factors regulate the expression of genes through binding to a consensus sequence CANNTG, known as E-box. NEUROD1 combines with E2A-encoded proteins and HEB-encoded proteins. The HLH region of NEUROD1 is responsible for the dimerization between the bHLH protein and the basic region, mediating protein–DNA interactions. The NEUROD1 gene is expressed in the intestine, pituitary gland, some central and peripheral nerves, and pancreatic islet cells. In the intestine, it stimulates the release of secretin hormone (Ray and Leiter, 2007). In the pituitary gland, it regulates the expression of proopiomelanocortin (POMC), which is a precursor of some important hormones (Parvin et al., 2017). In the nervous system, it motivates the formation of neurites, and its name is derived from this activity. This gene is also known as BETA2 due to its function in activating the insulin gene in beta cells. Moreover, it has an important role in the development of the pancreas (Sharma et al., 1999). A previous study showed that mice lacking NEUROD1 −/− exhibited serious complications of the pancreas, which causes a decline in beta cells, leading to severe diabetes (Naya et al., 1997).
NEUROD1 forms a heterodimer with a bHLH transcription factor called E4, which binds to the E-box binding site in the promoter region of insulin (INS), glucokinase (GCK), sulfonylurea receptor 1 (SUR1), Paired box (PAX), and islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP) genes and activates their expression (Rubio-Cabezas et al., 2010). These genes are known to play important roles in glucose homeostasis besides differentiation and development of pancreatic cells (Panneerselvam et al., 2019). This explains the connection between the NEUROD1 gene and diabetes. NEUROD1 is also involved in beta cell dysfunction during chronic hyperglycemia through two mechanisms. First, high glucose concentration will activate the expression of a small heterodimer partner (SHP) gene, which in turn inhibits p300-mediated pancreatic duodenal homeobox factor 1 (PDX1) and NEUROD1, resulting in insulin gene downregulation. Second, a decreased protein phosphatase 2 (PP2A) level will activate cyclic AMP-responsive element-binding protein (CREB), leading to NEUROD1 and INSULIN repression (Horikawa and Enya, 2019; Bohuslavova et al., 2021). The detected NEUROD1 P197H variant is found within a motif sequence specific to the PAX transcription factor. This can explain its role in developing diabetes. Further validation can be tested using CHIP-sequencing.
The NEUROD1 gene has two exons; the first one is not translated while the second encodes for a protein with several motifs. Four variants have been identified in exon 2. One variant is p.Ala45Thr, the second one is p. Pro197His, and the third one is p.Arg111Leucine, which is located in the proximal basic portion of the basic HLH domain. The fourth variant is an insertion of cytosine in codon 206, defined as c.206 + C, leading to nonsense mutation and a premature stop codon. Malecki M. et al. studied these variants in autosomal dominant families inflicted with T2DM. The first two variants, p.Ala45Thr and p.Pro197His, were not associated with T2DM, whereas the latter two variants, p.Arg111Leu and c.206 + C, were associated with T2DM development by affecting the ability of NEUROD1 to bind E-box and active CBP/p300, respectively (Malecki et al., 1999; Malecki et al., 2003).
Previous studies reported that the NEUROD1 p.Ala45Thr variant was associated with T1DM but not T2DM (Iwata et al., 1999; Hansen et al., 2000). In another Japanese case-control study, the researchers hypothesized that the NEUROD1 gene can affect the development and onset of T1DM since it acts on initial islet precursors (Yamada et al., 2001). They studied NEUROD1 polymorphisms in 105 T1DM patients and 122 nondiabetic controls, and the diabetic patients were classified into groups according to their onset pattern. Interestingly, a significant difference in NEUROD1 polymorphisms between cases and the control was detected in the acute-onset group (Yamada et al., 2001). This revealed the clear association of the NEUROD1 gene with T1DM development. A study in 2010, aimed to examine the effect of NEUROD1 mutations on patients with permanent monogenic neonatal diabetes (PNDM), found two rare homozygous mutations. The first was a duplication of a single base pair (c.364dupG), while the second was a deletion of two base pairs CT (c.427_428del)X. Both mutations lead to the absence of the transactivation domain from the protein due to premature truncation of the C terminus (p.Asp122Glyfs*12 and p.Leu143Alafs*55, respectively) as shown in Figure 9. These two indicated patients had some neurological abnormalities, including weakening of vision and hearing, cerebellar hypoplasia, and development delay. This leads to the conclusion that NEUROD1 plays an important role in the pancreas as well as in the nervous system (Rubio-Cabezas et al., 2010).
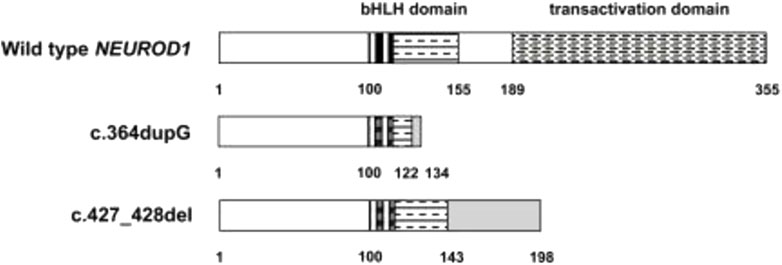
FIGURE 9. The effects of c.364dupG and c.427_428del mutations on NEUROD1 protein structure.
In maturity-onset diabetes of the youth (MODY), several studies demonstrated some NEUROD1 candidate mutations, including NEUROD1 p.Pro197His, p.Asp202Glu, p.Leu157Arg, and p.Arg103Pro (Ang et al., 2016; Aǧladioǧlu et al., 2016; Szopa et al., 2016; Horikawa et al., 2018; Demirci et al., 2021). Recently, another heterozygous mutation, NEUROD1 p.Met114Leu (c.340A > C), was reported in an Italian patient with MODY6 (Brodosi et al., 2021). This variant is predicted to be pathogenic based on many prediction tools and was later confirmed in a French family (Bouillet et al., 2020). In Latin America, a novel frameshift deletion (p.Phe256Leufs*2) in NEUROD1 was reported in the MODY6 family (Abreu et al., 2019). Horikawa Y. and Enya H. concluded that heterozygous mutations in the NEUROD1 gene are associated with MODY6 whereas homozygous mutations cause neonatal diabetes (Horikawa and Enya, 2019; Oliveira et al., 2020). The Diabetic members of Family II (II-2, II-3, and II-4) did not have neurological disorders. Similarly, some patients in previous studies have homozygous mutations in the NEUROD1 gene with no neurological abnormalities (Wang et al., 2015). The reasons for this are not clear; however, one possibility suggests that the missense mutations P197H do not affect the domain needed for neurological functions.
A previous study aimed to determine if genetic variation in MODY genes can affect the response to insulin-sensitizing interventions. The study included individuals with a high risk of developing diabetes (high fasting glucose, being overweight, etc.). These subjects were divided into three groups; the first group took a placebo, the second group took a metformin drug twice a day, and the third group experienced lifestyle intervention. Different variants in several MODY genes, including HNF4A, GCK, HNF1A, PDX1, HNF1B, and NEUROD1, were genotyped in all participants. There was a significant association between HNF4A, HNF1B, and NEUROD1 variants and treatment response to metformin and lifestyle intervention. One minor allele (rs6719578) in the NEUROD1 gene showed an increase in insulin secretion in the metformin group (Billings et al., 2017). Therefore, NEUROD1 variants can also be used in treatment programming. Its genotypes can predict the treatment response, thus it affects the selection of the drug and the doses given.
In conclusion, NEUROD1 p.P197H and IGF1R p.V579F constitute high susceptibility mutations for T1DM development. Further studies should be done to confirm their causative relationship in T1DM and its mechanisms. One approach is to develop human in vitro modeling system(s) using selected cells in culture transformed with the mutation by CRISPR-CAS9 technology and study various responses, including glucose-stimulated insulin secretion, and the expression and activity of several genes involved in glucose metabolism. The data generated from these studies can be extended to the development of mouse models for long-term studies. These investigations will help to extend our understanding of the pathogenesis of T1D for early diagnoses and more effective clinical intervention.
The original contributions presented in the study are publicly available. This data can be found here: https://www.ncbi.nlm.nih.gov/sra/SRR26632668.
Written informed consent was obtained from the individuals for the publication of any potentially identifiable images or data included in this article.
AB: Data curation, Formal Analysis, Validation, Writing–original draft. AD: Formal Analysis, Software, Conceptualization, Visualization, Writing–review and editing. HE: Conceptualization, Data curation, Resources, Writing–review and editing. HD: Conceptualization, Formal Analysis, Funding acquisition, Project administration, Resources, Supervision, Writing–review and editing, Data curation, Investigation, Visualization.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors are grateful to the faculty of graduate studies at the Arab American University for providing support for this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abreu, G. de M., Tarantino, R. M., Cabello, P. H., Zembrzuski, V. M., da Fonseca, A. C. P., Rodacki, M., et al. (2019). The first case of NEUROD1-MODY reported in Latin America. Mol. Genet. Genomic Med. 7, e989. doi:10.1002/mgg3.989
Aǧladioǧlu, S. Y., Aycan, Z., Çetinkaya, S., Baş, V. N., Önder, A., Peltek Kendirci, H. N., et al. (2016). Maturity onset diabetes of youth (MODY) in Turkish children: sequence analysis of 11 causative genes by next generation sequencing. J. Pediatr. Endocrinol. Metab. 29, 487–496. doi:10.1515/jpem-2015-0039
Al-Jenaidi, F. A., Wakim-Ghorayeb, S. F., Al-Abbasi, A., Arekat, M. R., Irani-Hakime, N., Najm, P., et al. (2005). Contribution of selective HLA-DRB1/DQB1 alleles and haplotypes to the genetic susceptibility of type 1 diabetes among Lebanese and Bahraini Arabs. J. Clin. Endocrinol. Metab. 90, 5104–5109. doi:10.1210/jc.2005-1166
Ang, S. F., Lim, S. C., Tan, C. S., Fong, J. C., Kon, W. Y., Lian, J. X., et al. (2016). A preliminary study to evaluate the strategy of combining clinical criteria and next generation sequencing (NGS) for the identification of monogenic diabetes among multi-ethnic Asians. Diabetes Res. Clin. Pract. 119, 13–22. doi:10.1016/j.diabres.2016.06.008
Anik, A., Çatli, G., Abaci, A., and Böber, E. (2015). Maturity-onset diabetes of the young (MODY): an update. J. Pediatr. Endocrinol. Metabolism 28, 251–263. doi:10.1515/jpem-2014-0384
Arnaldez, F. I., and Helman, L. J. (2012). “Targeting the insulin growth factor receptor 1,” in Hematology/oncology clinics of north America (Berlin, Germany: Springer). doi:10.1016/j.hoc.2012.01.004
Bakay, M., Pandey, R., Grant, S. F. A., and Hakonarson, H. (2019). The genetic contribution to type 1 diabetes. Curr. Diab. Rep. 19, 116. doi:10.1007/s11892-019-1235-1
Bassuny, W. M., Ihara, K., Sasaki, Y., Kuromaru, R., Kohno, H., Matsuura, N., et al. (2003). A functional polymorphism in the promoter/enhancer region of the FOXP3/Scurfin gene associated with type 1 diabetes. Immunogenetics 55, 149–156. doi:10.1007/s00251-003-0559-8
Billings, L. K., Jablonski, K. A., Warner, A. S., Cheng, Y. C., McAteer, J. B., Tipton, L., et al. (2017). Variation in maturity-onset diabetes of the young genes influence response to interventions for diabetes prevention. J. Clin. Endocrinol. Metab. 102, 2678–2689. doi:10.1210/jc.2016-3429
Bohuslavova, R., Smolik, O., Malfatti, J., Berkova, Z., Novakova, Z., Saudek, F., et al. (2021). Neurod1 is required for the early α and β endocrine differentiation in the pancreas. Int. J. Mol. Sci. 22, 6713. doi:10.3390/ijms22136713
Bottini, N., Musumeci, L., Alonso, A., Rahmouni, S., Nika, K., Rostamkhani, M., et al. (2004). A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat. Genet. 36, 337–338. doi:10.1038/ng1323
Bouillet, B., Crevisy, E., Baillot-Rudoni, S., Gallegarine, D., Jouan, T., Duffourd, Y., et al. (2020). Whole-exome sequencing identifies the first French MODY 6 family with a new mutation in the NEUROD1 gene. Diabetes Metab. 46, 400–402. doi:10.1016/j.diabet.2020.03.001
Bradfield, J. P., Qu, H. Q., Wang, K., Zhang, H., Sleiman, P. M., Kim, C. E., et al. (2011). A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genet. 7, e1002293. doi:10.1371/journal.pgen.1002293
Brodosi, L., Baracco, B., Mantovani, V., and Pironi, L. (2021). NEUROD1 mutation in an Italian patient with maturity onset diabetes of the young 6: a case report. BMC Endocr. Disord. 21, 202. doi:10.1186/s12902-021-00864-w
Chen, B., Li, J., Chi, D., Sahnoune, I., Calin, S., Girnita, L., et al. (2019). Non-coding RNAs in IGF-1R signaling regulation: the underlying pathophysiological link between diabetes and cancer. Cells 8, 1638. doi:10.3390/cells8121638
Concannon, P., Onengut-Gumuscu, S., Todd, J. A., Smyth, D. J., Pociot, F., Bergholdt, R., et al. (2008). A human type 1 diabetes susceptibility locus maps to chromosome 21q22.3. Diabetes 57, 2858–2861. doi:10.2337/db08-0753
Cooper, J. D., Howson, J. M. M., Smyth, D., Walker, N. M., Stevens, H., Yang, J. H. M., et al. (2012). Confirmation of novel type 1 diabetes risk loci in families. Diabetologia 55, 996–1000. doi:10.1007/s00125-012-2450-3
Cronin, R., Dias, N., and Yung Peng, R. K. (2017). 乳鼠心肌提取 HHS public access. Physiol. Behav. 176, 139–148.
Demirci, D. K., Darendeliler, F., Poyrazoglu, S., Al, A. D. K., Gul, N., Tutuncu, Y., et al. (2021). Monogenic childhood diabetes: dissecting clinical heterogeneity by next-generation sequencing in maturity-onset diabetes of the young. Omi. A J. Integr. Biol. 25, 431–449. doi:10.1089/omi.2021.0081
El-Amir, M. I., El-Feky, M. A., Laine, A. P., Härkönen, T., El-Badawy, O., Eltayeb, A. A., et al. (2015). Risk genes and autoantibodies in Egyptian children with type 1 diabetes - low frequency of autoantibodies in carriers of the HLA-DRB1*04:05-DQA1*03-DQB1*02 risk haplotype. Diabetes. Metab. Res. Rev. 31, 287–294. doi:10.1002/dmrr.2609
Engberding, N., San Martín, A., Martin-Garrido, A., Koga, M., Pounkova, L., Lyons, E., et al. (2009). Insulin-like growth factor-1 receptor expression masks the antiinflammatory and glucose uptake capacity of insulin in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 29, 408–415. doi:10.1161/ATVBAHA.108.181727
Erlich, H. A., Valdes, A. M., McDevitt, S. L., Simen, B. B., Blake, L. A., McGowan, K. R., et al. (2013). Next generation sequencing reveals the association of DRB3*02:02 with type 1 diabetes. Diabetes 62, 2618–2622. doi:10.2337/db12-1387
Farabaugh, S. M., Boone, D. N., and Lee, A. V. (2015). Role of IGF1R in breast cancer subtypes, stemness, and lineage differentiation. Front. Endocrinol. 6, 59. doi:10.3389/fendo.2015.00059
Fowlkes, L., Bunn R, C., and Thrailkill, K. M. (2012). Contributions of the insulin/insulin-like growth factor-1 Axis to diabetic osteopathy. J. Diabetes Metab. 1, S1-003. doi:10.4172/2155-6156.s1-003
Garg, N., Thakur, S., Alex McMahan, C., and Adamo, M. L. (2011). High fat diet induced insulin resistance and glucose intolerance are gender-specific in IGF-1R heterozygous mice. Biochem. Biophys. Res. Commun. 413, 476–480. doi:10.1016/j.bbrc.2011.08.123
Giwa, A. M., Ahmed, R., Omidian, Z., Majety, N., Karakus, K. E., Omer, S. M., et al. (2020). Current understandings of the pathogenesis of type 1 diabetes: genetics to environment. World J. Diabetes 11, 13–25. doi:10.4239/wjd.v11.i1.13
Groop, L., and Pociot, F. (2014). Genetics of diabetes - are we missing the genes or the disease? Mol. Cell. Endocrinol. 382, 726–739. doi:10.1016/j.mce.2013.04.002
Hakonarson, H., Grant, S. F. A., Bradfield, J. P., Marchand, L., Kim, C. E., Glessner, J. T., et al. (2007). A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature 448, 591–594. doi:10.1038/nature06010
Hakuno, F., and Takahashi, S. I. (2018). 40 years of IGF1: IGF1 receptor signaling pathways. J. Mol. Endocrinol. 61, T69-T86. doi:10.1530/JME-17-0311
Hansen, L., Jensen, J. N., Urioste, S., Petersen, H. V., Pociot, F., Eiberg, H., et al. (2000). NeuroD/BETA2 gene variability and diabetes: no associations to late-onset type 2 diabetes but an A45 allele may represent a susceptibility marker for type 1 diabetes among Danes. Danish Study Group of Diabetes in Childhood, and the Danish IDDM Epidemiology and Genetics Group. Diabetes 49, 876–878. doi:10.2337/diabetes.49.5.876
Health annual Palestine report, (2018). Health annual Palestine report. https://healthclusteropt.org/admin/file_manager/uploads/files/1/Health%20Annual%20Report%20Palestine%202018.pdf.
Horikawa, Y., and Enya, M. (2019). Genetic dissection and clinical features of MODY6 (NEUROD1-MODY). Curr. Diabetes Rep. 19, 12. doi:10.1007/s11892-019-1130-9
Horikawa, Y., Enya, M., Mabe, H., Fukushima, K., Takubo, N., Ohashi, M., et al. (2018). NEUROD1-deficient diabetes (MODY6): identification of the first cases in Japanese and the clinical features. Pediatr. Diabetes 19, 236–242. doi:10.1111/pedi.12553
Illumina, (2021). Illumina DNA prep with enrichment reference guide. https://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/illumina_prep/illumina-dna-prep-with-enrichment-reference-guide-1000000048041-07.pdf.
Iwata, I., Nagafuchi, S., Nakashima, H., Kondo, S., Koga, T., Yokogawa, Y., et al. (1999). Association of polymorphism in the neuroD/BETA2 gene with type 1 diabetes in the Japanese. Diabetes 48, 416–419. doi:10.2337/diabetes.48.2.416
Jin, J., Ravindran, P., Meo, D.Di, and Püschel, A. W. (2019). Igf1R/InsR function is required for axon extension and corpus callosum formation. PLoS One 14, e0219362. doi:10.1371/journal.pone.0219362
Jung, H. J., and Suh, Y. (2015). Regulation of IGF -1 signaling by microRNAs. Front. Genet. 5, 472. doi:10.3389/fgene.2014.00472
Kharroubi, A. T., and Darwish, H. M. (2015). Diabetes mellitus: the epidemic of the century. World J. Diabetes 6, 850–867. doi:10.4239/wjd.v6.i6.850
Kumar, A., Ren, Y., Sundaram, K., Mu, J., Sriwastva, M. K., Dryden, G. W., et al. (2021). miR-375 prevents high-fat diet-induced insulin resistance and obesity by targeting the aryl hydrocarbon receptor and bacterial tryptophanase (tnaA) gene. Theranostics 11, 4061–4077. doi:10.7150/thno.52558
Malecki, M. T., Cyganek, K., Klupa, T., and Sieradzki, J. (2003). The Ala45Thr polymorphism of BETA2/NeuroD1 gene and susceptibility to type 2 diabetes mellitus in a Polish population. Acta Diabetol. 40, 109–111. doi:10.1007/s005920300015
Malecki, M. T., Jhala, U. S., Antonellis, A., Fields, L., Doria, A., Orban, T., et al. (1999). Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat. Genet. 23, 323–328. doi:10.1038/15500
Mcd, C. N. MasterPure TM DNA purification kit. https://shop.biosearchtech.com/dna-rna-extraction-and-purification-kits/dna-and-rna-purification-kits-and-reagents/masterpure-complete-dna-and-rna-purification-kit. (2012).
Moody, G., Beltran, P. J., Mitchell, P., Cajulis, E., Chung, Y. A., Hwang, D., et al. (2014). IGF1R blockade with ganitumab results in systemic effects on the GH-IGF axis in mice. J. Endocrinol. 221, 145–155. doi:10.1530/JOE-13-0306
Nadia, B., Chehrazade, B., Ouafa, A., Asmaa, D. B., Ouadghiri, S., and Malika, E. (2012). Human leukocyte antigen (HLA) polymorphism and type 1 diabetes in the Moroccan population. Afr. J. Biotechnol. 11, 16126–16131. doi:10.5897/ajb12.1863
Naya, F. J., Huang, H. P., Qiu, Y., Mutoh, H., DeMayo, F. J., Leiter, A. B., et al. (1997). Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/NeuroD-deficient mice. Genes Dev. 11, 2323–2334. doi:10.1101/gad.11.18.2323
Nyaga, D. M., Vickers, M. H., Jefferies, C., Perry, J. K., and O’Sullivan, J. M. (2018). The genetic architecture of type 1 diabetes mellitus. Mol. Cell. Endocrinol. 477, 70–80. doi:10.1016/j.mce.2018.06.002
Oliveira, S. C., Neves, J. S., Pérez, A., and Carvalho, D. (2020). Maturity-onset diabetes of the young: from a molecular basis perspective toward the clinical phenotype and proper management. Endocrinol. Diabetes Nutr. 67, 137–147. doi:10.1016/j.endinu.2019.07.012
Panneerselvam, A., Kannan, A., Mariajoseph-Antony, L. F., and Prahalathan, C. (2019). PAX proteins and their role in pancreas. Diabetes Res. Clin. Pract. 155, 107792. doi:10.1016/j.diabres.2019.107792
Parvin, R., Saito-Hakoda, A., Shimada, H., Shimizu, K., Noro, E., Iwasaki, Y., et al. (2017). Role of NeuroD1 on the negative regulation of Pomc expression by glucocorticoid. PLoS One 12, e0175435. doi:10.1371/journal.pone.0175435
Pociot, F. (2017). Type 1 diabetes genome-wide association studies: not to be lost in translation. Clin. Transl. Immunol. 6, e162–e167. doi:10.1038/cti.2017.51
Ray, S. K., and Leiter, A. B. (2007). The basic helix-loop-helix transcription factor NeuroD1 facilitates interaction of Sp1 with the secretin gene enhancer. Mol. Cell. Biol. 27, 7839–7847. doi:10.1128/mcb.00438-07
Redondo, M. J., Oram, R. A., and Steck, A. K. (2017). Genetic risk scores for type 1 diabetes prediction and diagnosis. Curr. Diab. Rep. 17, 129. doi:10.1007/s11892-017-0961-5
Rubio-Cabezas, O., Minton, J. A. L., Kantor, I., Williams, D., Ellard, S., and Hattersley, A. T. (2010). Homozygous mutations in NEUROD1 are responsible for a novel syndrome of permanent neonatal diabetes and neurological abnormalities. Diabetes 59, 2326–2331. doi:10.2337/db10-0011
Sharma, A., Moore, M., Marcora, E., Lee, J. E., Qiu, Y., Samaras, S., et al. (1999). The NeuroD1/BETA2 sequences essential for insulin gene transcription colocalize with those necessary for neurogenesis and p300/CREB binding protein binding. Mol. Cell. Biol. 19, 704–713. doi:10.1128/mcb.19.1.704
Smyth, D. J., Cooper, J. D., Bailey, R., Field, S., Burren, O., Smink, L. J., et al. (2006). A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat. Genet. 38, 617–619. doi:10.1038/ng1800
Stayoussef, M., Benmansour, J., Al-Irhayim, A. Q., Said, H. B., Rayana, C. B., Mahjoub, T., et al. (2009). Autoimmune type 1 diabetes genetic susceptibility encoded by human leukocyte antigen DRB1 and DQB1 genes in Tunisia. Clin. Vaccine Immunol. 16, 1146–1150. doi:10.1128/CVI.00105-09
Sun, Y., Sun, X., and Shen, B. (2017). Molecular imaging of IGF-1R in cancer. Mol. Imaging 16, 1536012117736648. doi:10.1177/1536012117736648
Szopa, M., Ludwig-Galezowska, A. H., Radkowski, P., Skupien, J., Machlowska, J., Klupa, T., et al. (2016). A family with the Arg103Pro mutation in the NEUROD1 gene detected by next-generation sequencing - clinical characteristics of mutation carriers. Eur. J. Med. Genet. 59, 75–79. doi:10.1016/j.ejmg.2016.01.002
Thermo Fisher Scientific, (2015). U. BigDye user guide. https://assets.thermofisher.com/TFS-Assets/LSG/manuals/4337035_BDTv31CycSqKt_RUO_UG.pdf.
Todd, J. A., Walker, N. M., Cooper, J. D., Smyth, D. J., Downes, K., Plagnol, V., et al. (2007). Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat. Genet. 39, 857–864. doi:10.1038/ng2068
Tremblay, J., and Hamet, P. (2019). Environmental and genetic contributions to diabetes. Metabolism 100, 153952–153956. doi:10.1016/j.metabol.2019.153952
Vella, A., Cooper, J. D., Lowe, C. E., Walker, N., Nutland, S., Widmer, B., et al. (2005). Localization of a type 1 diabetes locus in the IL2RA/CD25 region by use of tag single-nucleotide polymorphisms. Am. J. Hum. Genet. 76, 773–779. doi:10.1086/429843
Wang, F., Li, H., Xu, M., Li, H., Zhao, L., Yang, L., et al. (2015). A homozygous missense mutation in NEUROD1 is associated with nonsyndromic autosomal recessive retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 56, 150–155. doi:10.1167/iovs.14-15382
Wang, F., Stewart, M., McDermott, S., Kazanjian, A., Vissandjee, B., DesMeules, M., et al. (2012). Migration and diabetes in British Columbia and Quebec: prevalence and health service utilization. Can. J. Public Heal 103, 59–64. doi:10.1007/bf03404070
Wit, J. M., and Walenkamp, M. J. (2013). Role of insulin-like growth factors in growth, development and feeding. World Rev. Nutr. Dietetics 106, 60–65. doi:10.1159/000342546
Xuan, S., Kitamura, T., Nakae, J., Politi, K., Kido, Y., Fisher, P. E., et al. (2002). Defective insulin secretion in pancreatic β cells lacking type 1 IGF receptor. J. Clin. Invest. 110, 1011–1019. doi:10.1172/jci15276
Yamada, S., Motohashi, Y., Yanagawa, T., Maruyama, T., Kasuga, A., Hirose, H., et al. (2001). NeuroD/BETA2 gene G→A polymorphism may affect onset pattern of type 1 diabetes in Japanese. Diabetes Care 24, 1438–1441. doi:10.2337/diacare.24.8.1438
Yuan, J., Yin, Z., Tao, K., Wang, G., and Gao, J. (2018). Function of insulin-like growth factor 1 receptor in cancer resistance to chemotherapy. Oncol. Lett. 15, 41–47. doi:10.3892/ol.2017.7276
Keywords: type diabetes genetics, gene function, molecular genetic, WES, in born errors of metabolism
Citation: Bawatneh A, Darwish A, Eideh H and Darwish HM (2024) Identification of gene mutations associated with type 1 diabetes by next-generation sequencing in affected Palestinian families. Front. Genet. 14:1292073. doi: 10.3389/fgene.2023.1292073
Received: 10 September 2023; Accepted: 04 December 2023;
Published: 11 January 2024.
Edited by:
Ivan Martinez Duncker, Universidad Autónoma del Estado de Morelos, MexicoReviewed by:
Angélica G. Martínez-H, National Institute of Genomic Medicine (INMEGEN), MexicoCopyright © 2024 Bawatneh, Darwish, Eideh and Darwish. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hisham M. Darwish, aGlzaGFtLmRhcndpc2hAYWF1cC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.