
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet., 18 December 2023
Sec. Genetics of Common and Rare Diseases
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1282790
This article is part of the Research TopicInborn errors of Carbohydrate MetabolismView all 15 articles
 Matthew M. Gayed1
Matthew M. Gayed1 Paulo Sgobbi2
Paulo Sgobbi2 Wladimir Bocca Viera De Rezende Pinto2
Wladimir Bocca Viera De Rezende Pinto2 Priya S. Kishnani1*†
Priya S. Kishnani1*† Rebecca L. Koch1†
Rebecca L. Koch1†Introduction: Adult polyglucosan body disease (APBD) has long been regarded as the adult-onset form of glycogen storage disease type IV (GSD IV) and is caused by biallelic pathogenic variants in GBE1. Advances in the understanding of the natural history of APBD published in recent years have led to the use of discrete descriptors (“typical” versus “atypical”) based on adherence to traditional symptomatology and homozygosity for the p.Y329S variant. Although these general descriptors are helpful in summarizing common findings and symptoms in APBD, they are inherently limited and may affect disease recognition in diverse populations.
Methods: This case series includes three American patients (cases 1–3) and four Brazilian patients (cases 4–7) diagnosed with APBD. Patient-reported outcome (PRO) measures were employed to evaluate pain, fatigue, and quality of life in cases 1–3.
Results: We describe the clinical course and diagnostic odyssey of seven cases of APBD that challenge the utility and efficacy of discrete descriptors. Cases 1–3 are compound heterozygotes that harbor the previously identified deep intronic variant in GBE1 and presented with “typical” APBD phenotypically, despite lacking two copies of the pathogenic p.Y329S variant. Patient-reported outcome measures in these three cases revealed the moderate levels of pain and fatigue as well as an impacted quality of life. Cases 4–7 have unique genotypic profiles and emphasize the growing recognition of presentations of APBD in diverse populations with broad neurological manifestations.
Conclusion: Collectively, these cases underscore the understanding of APBD as a spectrum disorder existing on the GSD IV phenotypic continuum. We draw attention to the pitfalls of commonly used genetic testing methods when diagnosing APBD and highlight the utility of patient-reported outcome questionnaires in managing this disease.
Adult polyglucosan body disease (APBD) is the adult-onset form of glycogen storage disease type IV (GSD IV) and is caused by biallelic pathogenic variants in GBE1 which encodes the glycogen-branching enzyme (GBE) (Robitaille et al., 1980; Mochel et al., 2012; Souza et al., 2021). Reduced or deficient GBE activity disrupts normal glycogen synthesis and leads to the formation of glycogen with elongated outer chains resembling plant amylopectin (polyglucosan) (Koch et al., 2023). The polyglucosan aggregates and accumulates in various tissues, including the central nervous system, peripheral nervous system, skeletal muscle, heart, and lungs (Peress et al., 1979; Robitaille et al., 1980; Postler et al., 2002; Sindern et al., 2003; Dainese et al., 2013; Sullivan et al., 2019). Characteristic symptoms of APBD include progressive neurogenic bladder, spastic paraparesis, and sensorimotor axonal peripheral neuropathy, with the typical onset between the fourth and sixth decades of life (Robitaille et al., 1980; Mochel et al., 2012; Koch et al., 2023). While formal epidemiological studies categorizing the number of patients affected globally are yet to be conducted, it has been established that the pathogenic missense variant, NM_000158.4(GBE1):c.986A>C (p.Tyr329Ser; p.Y329S) (National Center for Biotechnology Information, 2023a), occurs with greater frequency in patients with Ashkenazi Jewish ancestry (estimated carrier rate 1:35–1:48) (Bao et al., 1996; Hussain et al., 2012; Schwartz et al., 2020).
APBD was first described by Robitaille et al. (1980) and Mochel et al. (2012) published the first natural history study in a cohort of 50 patients with APBD. This foundational work detailed APBD symptomatology and common misdiagnoses, as well as described the characteristic findings on magnetic resonance imaging (MRI): hyperintense white matter abnormalities predominant in the periventricular region, as well as medullary and spinal atrophy, on T2-weighted/fluid-attenuated inversion recovery (FLAIR) imaging (Mochel et al., 2012). The investigation was conducted primarily in patients homozygous for the p.Y329S variant but also included a subset of heterozygote patients without a known second variant (Mochel et al., 2012). Shortly thereafter, Akman et al. (2015) identified a novel pathogenic deep intronic variant, NM_000158.4(GBE1):c.2053-3358_2053-3350delinsTGTTTTTTACATGACAGGT (National Center for Biotechnology Information, 2023b) (herein referred to as the identified deep intronic variant), in symptomatic patients heterozygous for the p.Y329S variant without a previously known second mutation. Since the discovery of this identified deep intronic variant, there have not been extensive, longitudinal phenotypic descriptions of patients confirmed to have this intronic variant (Akman et al., 2015; Grunseich et al., 2021). Although clinical heterogeneity has been acknowledged, more recent work in 2021 sought to delineate APBD into categories of “typical” versus “atypical” on the basis of adherence to the aforementioned traditional symptomatology and homozygosity for the p.Y329S variant (Souza et al., 2021). Yet, APBD has been reported in a variety of patients of various ages, with or without Ashkenazi Jewish ancestry, and there continues to be additional phenotypes reported through multiple case series since the initial characterizations of the disorder (Ferguson et al., 1983; Bruno et al., 1993; Ziemssen et al., 2000; Klein et al., 2004; Billot et al., 2013; Souza et al., 2021).
Due to symptoms overlapping with other neurological disorders, APBD is frequently misdiagnosed; the most common misdiagnoses include multiple sclerosis, cerebral small vessel disease, peripheral neuropathy, amyotrophic lateral sclerosis, and benign prostatic hypertrophy in men (Hellmann et al., 2015). As a result, APBD patients often face prolonged diagnostic delays and may undergo unnecessary and potentially invasive procedures (Hellmann et al., 2015). Delay in diagnosing APBD is likely complicated by a number of factors including lack of awareness regarding the disorder and its associated presentations (Mochel et al., 2012; Hellmann et al., 2015). Collective understanding also continues to be hindered by a lack of longitudinal, comprehensive clinical history data in the medical literature, particularly for those with uncommon genotypes or presenting in earlier life. To address this, we present the clinical history and diagnostic odyssey for multiple patients with APBD receiving care in the United States and Brazil.
We describe three patients from the United States who are compound heterozygotes for the identified deep intronic variant in GBE1 and utilize patient-reported outcome (PRO) measures to shed light on quality of life as well as other functional limitations: fatigue and pain. In addition, we detail four cases from Brazil with novel GBE1 genotypes not previously described in patients with APBD and with broad neurological presentations.
As part of an international collaboration, patients with a confirmed diagnosis of APBD were included for participation in a longitudinal, retrospective natural history study (Duke University Institutional Review Board Pro00060753, ClinicalTrials.gov Identifier: NCT02683512). All available paper and electronic medical records were reviewed for participants who consented to the study. Collaborating institutions managing patients with APBD for whom direct consent was not possible submitted de-identified participant data using a targeted, study-specific spreadsheet. Participant data were entered into REDCap electronic data capture tools hosted at Duke University. Ages reported in this study represent available information in patient records and an estimation when exact ages were not available.
To evaluate symptoms of pain and fatigue as well as health-related quality of life, patients were asked to complete a series of validated PRO surveys. Due to institution research ethics restrictions, only United States-based participants were eligible to complete the survey (patients 1–3). The details of each validated survey and scoring are provided in Supplementary Material. Surveys were distributed and returned in April 2023. The results represent one time point (Supplementary Table S1). Survey scoring and selection details are provided in Supplementary Material.
Aspects of pain and its interference with daily life were measured using the Brief Pain Inventory (BPI) (Cleeland and Ryan, 1994; Tan et al., 2004; Güngör et al., 2013) and the Patient-Reported Outcomes Measurement Information System (PROMIS) Short Form v1.1—Pain Interference 8a (PI-8a) (Amtmann et al., 2010; Cella et al., 2010; Rothrock et al., 2010). Fatigue and its impact on quality of life were measured using the Brief Fatigue Inventory (BFI) (Mendoza et al., 1999; Shahid et al., 2012; Liu et al., 2022; Ritchie et al., 2023) and the PROMIS Short Form v1.0—Fatigue 13a (FACIT-Fatigue) (Lai et al., 2011; Cella et al., 2016). Here, pain and fatigue as measured by the BPI and BFI, respectively, were defined as no interference (0), mild (1–3), moderate (4–6), and severe (7–10) (Serlin et al., 1995; Mendoza et al., 1999; Li et al., 2007; Deandrea et al., 2008; Shahid et al., 2012; Güngör et al., 2013; Liu et al., 2022; Ritchie et al., 2023). PI-8a and FACIT-Fatigue T-scores were calculated using HealthMeasures Scoring Service, powered by Assessment Center (https://www.assessmentcenter.net/ac_scoringservice), and cut-offs for normal, mild, moderate, and severe pain interference were defined as >55, 55 to >60, 60 to >70, and >70, respectively (Cella et al., 2010; Rothrock et al., 2010; HealthMeasures, 2023).
Quality of life was measured using the EuroQol Research Foundation EQ-5D-5L (Janssen et al., 2013; Devlin et al., 2018; Janssen et al., 2018; Pickard et al., 2019) survey and the RAND Corporation 36-Item Health Survey 1.0 (SF-36) (Ware and Sherbourne, 1992; RAND Corporation, 2023). The EQ-5D-5L measures the quality of life across five dimensions: mobility, self-care, usual activities, pain/discomfort, and anxiety/depression; health states were then converted into a utility index value for each participant (Janssen et al., 2013; Devlin et al., 2018; Janssen et al., 2018; EuroQol Research Foundation, 2019; Pickard et al., 2019). The SF-36 assesses quality of life in eight domains: physical functioning, role limitations due to physical health, role limitations due to emotional problems, energy/fatigue, emotional wellbeing, social functioning, pain, and general health (Ware and Sherbourne, 1992; RAND Corporation, 2023). Individual patient utility index values (EQ-5D-5L) and health scores (SF-36) were then compared to published norm values (Jiang et al., 2021; RAND Corporation, 2023).
Case 1 is a White American male with known Ashkenazi Jewish ancestry who initially presented at age 51.4 years due to fecal incontinence and urinary retention. He reported to the emergency department due to an episode of sudden, forceful loss of fecal contents. Preliminary abdominal X-ray imaging returned benign, and he was recommended to begin fiber supplementation. Of note, he was also found to be retaining urine at this visit. At age 52.6 years following multiple subsequent episodes of fecal incontinence, he was evaluated by the gastroenterology department and anorectal manometry was performed, revealing sphincter dysfunction. MRI of the lumbar spine at 52.7 years did not reveal a cause for the fecal incontinence or sphincter dysfunction. MRI of the brain without contrast, followed by MRA (magnetic resonance angiography) of the head and, subsequently, MRI of the brain with and without contrast and of the cervical, thoracic, and lumbar spine were performed together, revealing evidence of leukodystrophy with findings of white matter abnormalities, cerebral and cerebellar volume loss, spinal cord atrophy, and vascular anomalies (Table 1). Evaluation by the neurology department at age 53.3 years disclosed a history of bladder dysfunction and erectile dysfunction from age 44.5 years, as well as gait abnormalities starting at age 49.6 years. Bladder dysfunction was diagnosed as benign prostatic hypertrophy, although a review of records showed resistance to multiple medications. He was noted to have bilateral pes cavus, ataxic gait, and abnormal reflexes (increased knee jerk, decreased ankle jerk, and upgoing plantar reflex bilaterally). A leukodystrophy gene panel returned without notable findings. At age 53.5 years, he underwent cystoscopy, showing urethral obstruction (Table 2), and had a prostatic urethral lift procedure at age 54.0 years. Despite this procedure, he continued to develop symptoms of urinary retention and elevated post void residuals. To further evaluate gait disturbances, he underwent the electromyography and nerve conduction velocity test (EMG/NCS) at age 54.4 years, showing a length-dependent sensorimotor axonal neuropathy in the right lower extremity (Table 3).
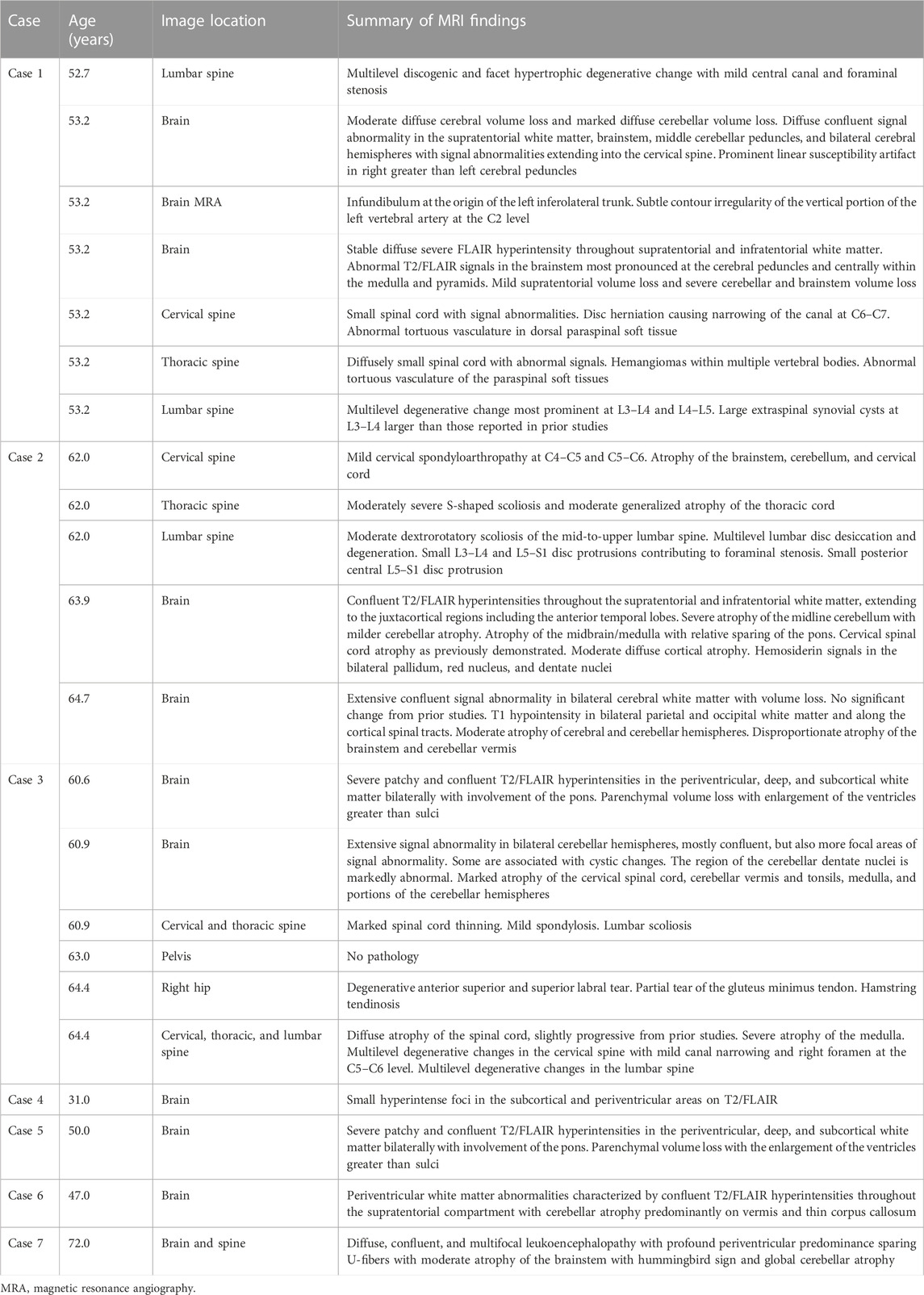
TABLE 1. Results of MRI in cases with APBD.
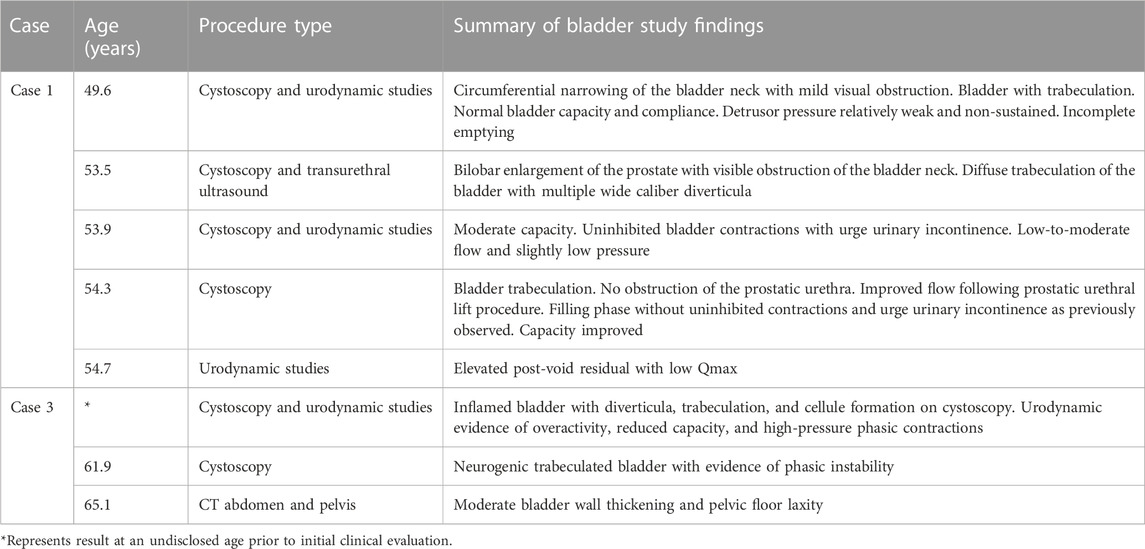
TABLE 2. Results of bladder studies in cases with APBD.

TABLE 3. Results of EMG/NCS in cases with APBD.
At age 54.6 years, he was formally diagnosed with APBD after research reanalysis of the prior leukodystrophy panel identified NM_000158.4(GBE1):c.760A>G (p.Thr254Ala; p.T254A)—a variant with conflicting interpretations of pathogenicity (National Center for Biotechnology Information, 2023d)—and the identified deep intronic variant. At age 54.7 years, he underwent repeat EMG, confirming length-dependent, axonal, sensorimotor peripheral neuropathy (Table 3) and, subsequently, began clean intermittent catheterizations due to urinary retention. He was evaluated at the Duke Metabolic Genetics clinic at age 55.5 years and reported ongoing symptoms of bladder dysfunction and gait instability, as well as intermittent episodes of fecal incontinence at least once every 6 months. During that visit, he reported an overall improvement of bladder control with clean intermittent catheterizations and has not had any resultant infections. However, he disclosed experiencing increased fatigue, noting that he was no longer able to walk distances greater than a mile due to sensation of muscle weakness as well as profound tiredness following meals. Muscle weakness was further evaluated using muscle ultrasound, with findings suggestive of myopathy in conjunction with an elevated creatine kinase level (516 U/L). Case 1 has no known liver or cardiac manifestations of GSD IV; routine echocardiograms and electrocardiograms performed between ages 47.6 and 54.7 years were normal, and alanine transaminase (ALT) levels have always been within normal limits, most recently measured at age 56.0 years.
Case 2 is a White American female with Ashkenazi Jewish ancestry, who presented at age 61.9 years to an orthopedist with symptoms of worsening gait, frequent falls, poor hand dexterity, and paresthesia and pain in the proximal left lower extremity initially thought to be due to worsening of longstanding kyphoscoliosis. On further review, postural changes and gait instability began as early as ages 45 and 55 years, respectively. MRI of the spine at age 62.0 years revealed atrophy of the cerebellum, cervical, and thoracic spinal cord (Table 1). EMG/NCS performed at age 62.2 years also displayed evidence of polyneuropathy (Table 3).
At age 62.7 years, she was evaluated by the neurology department, at which time she endorsed muscle tightness/stiffness as well as cramping of the right hand, rollator use from approximately age 60 years, as well as long-standing bladder dysfunction from as early as age 49 years beginning initially as urgency and progressing to incontinence requiring the use of pads. An ataxia gene panel and subsequent whole exome sequencing on the same sample were performed at age 63.6 years and returned negative; notably, GBE1 analysis was not performed on the ataxia panel nor was it commented on in the whole exome sequencing report. Following undiagnostic genetic testing results, MRI of the brain was performed at age 63.9 years, revealing white matter hyperintensities as well as cortical, midbrain, and midline cerebellar atrophy (Table 1). At age 64.0 years, mitochondrial gene testing and targeted FMR1 testing were performed and returned negative. She had worsening symptoms including deteriorating balance, weakness of the right leg, and frequent falls. On examination, she was noted to have abnormal reflexes, abnormal gait, and paratonia in the lower extremities. At subsequent evaluations, she reported increased dependency on a rollator for mobility and increasing difficulties with daily life activities.
She was referred for a neurological specialty evaluation at age 64.7 years at which point she reported the continuing progression of symptoms including lower extremity stiffness and weakness as well as bilateral foot drop and progression of bladder dysfunction, with excessive straining to urinate. EMG/NCS indicated severe axonal neuropathy in the lower extremities, in addition to incidental carpal tunnel syndrome in the right arm. Repeat MRI of the brain again showed extensive white matter signal abnormalities as well as cerebral, brainstem, and cerebellar atrophy. Reanalysis of molecular testing was performed at age 65.2 years, detecting the p.Y329S and identified deep intronic variants in GBE1.
Case 2 was reported to have a history of anxiety and was subjectively reported to have declining short-term memory throughout the diagnostic work up period without formal cognition evaluations. She has no known cardiac or hepatic manifestations of GSD IV; ALT levels most recently measured at age 64.7 years were within normal limits.
Case 3 is a White American female with known Ashkenazi Jewish ancestry who initially presented to a specialty multiple sclerosis clinic at age 60.9 years with bladder and bowel dysfunction and increasing difficulty with balance and gait. Bladder dysfunction manifested at approximately age 51 years as increased urgency and later progressed to incontinence and extensive urinary tract infections. Gait instability also began at approximately age 51 years with worsening of coordination and balance over time. She also reported a history of bowel dysfunction marked by constipation and fecal incontinence. Prior to initial evaluation, peripheral neuropathy was noted on EMG/NCS and extensive non-enhancing leukodystrophy on brain MRI (Table 1). Urodynamic studies and cystoscopy revealed reduced bladder capacity and overactivity in addition to bladder wall trabeculation and cellule formation (Table 2).
She was reevaluated at age 61.2 years for adult-onset leukodystrophy. At this visit, she further reported significant fatigue with the onset as early as age 59 years accompanied by frequent headaches and neck pain. She also noted the limited intermittent use of a wheelchair due to gait instability. Physical examination revealed diminished reflexes accompanied by a positive Babinski sign and an unsteady, wide-based gait. Repeat EMG/NCS disclosed prolonged central motor conduction times, indicating abnormal conduction in the descending corticospinal tracts in addition to a motor-predominant axonal polyneuropathy versus polyradiculopathy (Table 3). Mitochondrial DNA testing and whole exome sequencing were performed, and both returned as non-diagnostic. Because of the clinical suspicion for APBD, further evaluation and subsequent gene sequencing of the GBE1 gene led to a diagnosis of APBD at age 62.1 years. She had two pathogenic GBE1 variants (p.Y329S and the identified deep intronic variant) as well as GBE activity less than 10% compared to normal in skin fibroblasts.
Case 3 experienced ongoing deterioration since the time of diagnosis characterized by worsening bladder dysfunction, pain, gait instability, and cognition. Repeat cystoscopy at age 61.9 years again demonstrated neurogenic bladder dysfunction (Table 2). A trial of desmopressin (DDAVP) and pelvic floor physical therapy did not alleviate the symptoms. At age 65.1 years, a CT scan of the abdomen and pelvis revealed moderate bladder wall thickening and pelvic floor laxity/cystocele. She continues to have difficulty with urinary incontinence, voiding, urinary stream strength, recurrent UTIs, and perineal irritation.
Case 3 reports a significant history of pain throughout her disease course. At age 62.1 years, she began experiencing neuropathic, burning pain in the lower extremities up to the level of the thighs. Bladder/pelvic/perineal pain was reported as early as 62.8 years. Due to the unremitting, disabling nature of her pain, case 3 has trialed a number of therapies, such as a vibration table, duloxetine, transcranial magnetic stimulation, spinal procedures (selective nerve root blocks and pulsed radiofrequency ablations), and scrambler therapy—none of which alleviated her pain.
She has noted worsening gait instability and progressed from using a cane to a walker at age 64.3 years and then an increased reliance on a wheelchair by age 64.9 years. Despite regular physical therapy, her condition continues to deteriorate. Case 3 has no known cardiac or hepatic manifestations of GSD IV; she had a normal abdominal CT scan at age 65.1 years, and ALT levels most recently measured at 61.2 years old were within normal limits. Notably, she has a history of acute adjustment disorder with anxiety and insomnia.
PROs were used to further evaluate the health statuses for cases 1–3 across a series of domains including fatigue, pain, and quality of life (Table 4). On the BPI, averaged scores for pain severity and pain interference were 5.5 (moderate) and 6.1 (moderate), respectively. The mean T-score for the PI-8a was 62.7 (moderate pain interference). On the BFI, the average global fatigue score was 6.5 (moderate); the greatest impacted domain on average was walking ability. The mean T-score for the FACIT-Fatigue was 60.0 (moderate fatigue). Average scores for quality of life domains as measured by the SF-36 were lower than the established national averages, reflecting a significant impact on quality of life. Lastly, EQ-5D-5L assessed quality of life, and the health profiles were 32,233 (case 1), 54,532 (case 2), and 42,442 (case 3); these equate to utility index values of 0.5, 0.048, and 0.044, respectively. When compared to the US national mean utility index value (mean = 0.851, SD = 0.205), case 1 has an index value falling to approximately 1.7 SD from the mean, and cases 2 and 3 have index values falling to 3.9 SD from the mean.
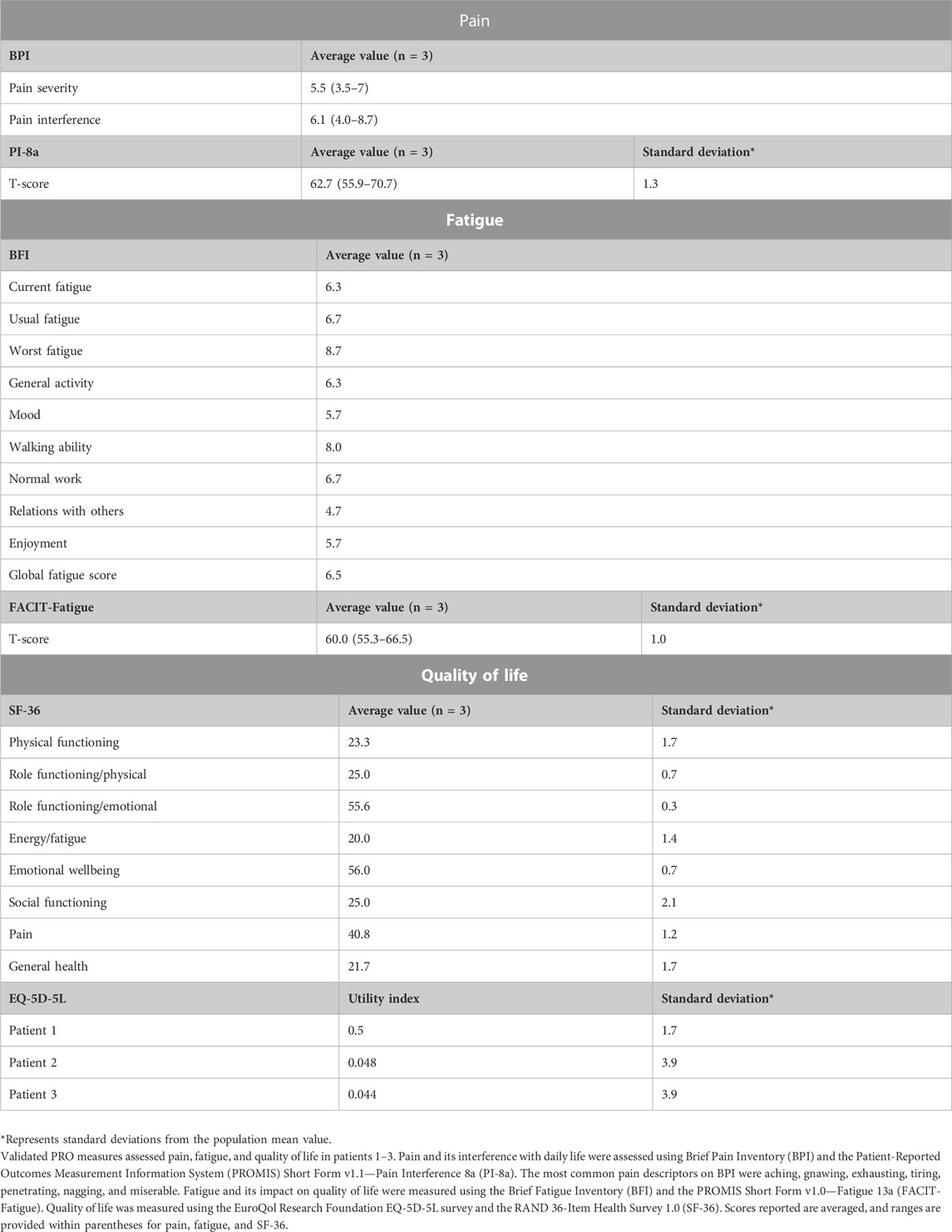
TABLE 4. Results of PRO measures from patients with APBD.
Case 4 is a White Brazilian female with no known Ashkenazi Jewish ancestry. She presented with urinary urgency beginning at age 26 years, followed by frequent urinary tract infections at age 28 years. Bladder dysfunction was notable for urgency and difficulty in emptying. At age 30 years, she experienced muscle cramping in the lower extremities (peroneal muscle group bilaterally) and small-fiber neuropathy symptoms in the distal lower extremities characterized by sensations of burning and tingling. At age 31 years, she exhibited muscle wasting, muscle weakness, easy fatiguability, and pain in the lower limb muscle groups as well as peripheral neuropathy characterized by hyperesthesia and paresthesia. At this time, NCS was performed, which disclosed sensorimotor axonal neuropathy (Table 3). Brain imaging revealed small foci of hyperintensity in the subcortical and periventricular areas on FLAIR and T2-weighted imaging (Table 1). At age 31 years, she underwent gene panel testing for inherited peripheral neuropathies which revealed homozygosity for NM_000158.4(GBE1):c.292G>C (p.Val98Leu; p.V98L), interpreted as a variant of uncertain significance (VUS) in ClinVar (National Center for Biotechnology Information, 2023e). She was not reported to have an unsteady or spastic gait or any cognitive deficits as of age 31 years.
Case 5 is a White Brazilian male with no known Ashkenazi ancestry presenting with numbness and paresthesia in the lower limbs. At age 30, he reported experiencing symptoms of small-fiber neuropathy characterized by burning and tingling in the distal lower extremities. Between age 30 and 50 years, he was misdiagnosed and symptomatically treated for sporadic and idiopathic small-fiber neuropathy with pregabalin without resolution. Evaluation at age 50 years also reported diffuse muscle pain involving the quadriceps femoris muscles and the gastrocnemii bilaterally not associated with exercise. He also had symptoms of peripheral neuropathy, and NCS confirmed moderate axonal sensorimotor polyneuropathy involving the lower and upper limbs (Table 3). He began experiencing bladder dysfunction at age 50 years marked by increased urgency. Physical examination revealed decreased strength in hip extensors, hip flexors, and ankle plantar flexion. Brain MRI revealed periventricular white matter hyperintensities and parenchymal volume loss (Table 1). Genetic testing with a multigene panel for inherited neuropathies at age 50 years revealed homozygosity for NM_000158.4(GBE1):c.534G>C (p.Trp178Cys; p.W178C), interpreted as a VUS (National Center for Biotechnology Information, 2023f).
Case 6 is a White Brazilian female with no known Ashkenazi ancestry who presented with bradykinesia and spastic paraparesis since age 35 years. There was an initial clinical suspicion for juvenile monogenic Parkinson’s disease or neurotransmitter disorders such as Segawa syndrome; a further review of history revealed good clinical improvement with low doses of levodopa. Previous medical history detailed bladder dysfunction beginning at age 32 years, including urinary incontinence and frequent urinary tract infections, as well as constipation beginning at age 44 years. She began using a cane at age 45 years and reported frequent daily falls and continuing unsteady gait on evaluation at age 47 years. NCS performed at 45 years revealed mild axonal sensorimotor polyneuropathy involving the upper and lower limbs (Table 3). At age 47 years, brain MRI identified cerebellar atrophy with periventricular white matter abnormalities (Table 1), and gene panel testing for neurodegenerative disorders identified p.V98L and NM_000158.4(GBE1):c.1909C>T (p.Arg637Ter; p.R637*), the latter interpreted as pathogenic/likely pathogenic (National Center for Biotechnology Information, 2023c). Physical examination from the time of diagnosis revealed preserved muscle strength and normal deep tendon reflexes with bradykinesia, mild spasticity in the lower limbs, and postural instability as well as cognitive impairment [Montreal Cognitive Assessment (MoCA): 22/30] and major depressive disorder.
Case 7 is a Black Brazilian male with no known Ashkenazi Jewish ancestry presenting with constipation, bladder dysfunction, erectile dysfunction, muscle cramping, spasticity, and unsteady gait. Of note, family history is positive for consanguinity as parents are first-degree cousins. The onset of constipation occurred at age 60 years. Bladder dysfunction was characterized by incontinence accompanied by frequent urinary tract infections starting at age 65 years, and EMG/NCS at age 71 years confirmed axonal sensorimotor polyneuropathy (Table 3). At age 72 years, physical examination revealed overall brisk tendon reflexes and reduced strength in hip flexion and extension bilaterally. Brain and spine MRI at age 72 years revealed evidence of leukoencephalopathy with periventricular predominance and atrophy of the brainstem and cerebellum (Table 1). A gene panel for neurodegenerative disorders at age 72 years revealed homozygosity for NM_000158.4(GBE1):c.929A>C (p.Tyr310Ser; p.Y310S). This variant has not been previously reported in the literature in an individual with APBD or entered in ClinVar, nor is it present in the population database gnomAD. Currently, there is limited information on this variant, and thus it is classified as a VUS. As of age 73 years, he began experiencing frequent falls and used a walker for mobility. A review of medical records further revealed history of misdiagnosis with Parkinson’s disease and normal pressure hydrocephalus.
Through our work, we present the detailed, longitudinal clinical history of seven patients with APBD, with the goal of expanding the collective understanding of the phenotypic presentation of APBD.
We began by describing in detail the clinical course of three cases confirmed to have the identified deep intronic variant in GBE1, for which longitudinal descriptions in the medical literature have thus far been limited (Akman et al., 2015; Grunseich et al., 2021). The clinical course and phenotypic presentation of these three cases followed the well-documented pattern of APBD progression previously established largely in populations homozygous for the GBE1 p.Y329S variant (Mochel et al., 2012; Hellmann et al., 2015). Case 1 is the first known case to be reported harboring the identified deep intronic variant without concomitantly harboring the p.Y329S variant in GBE1. This patient presented with bladder dysfunction at age 44 years, followed by gait abnormalities at 49 years, fecal incontinence at 54 years, and progressive fatigue at 55 years. Case 2 developed postural changes at age 45 years, followed by bladder dysfunction at age 49 years, gait instability at 55 years, and peripheral neuropathy at 62 years. Case 3 presented with bladder and bowel dysfunction as well as gait instability at age 51 years; these symptoms were followed by pain resistant to treatment, affecting various parts of the body including the neck, back, legs, and perineal region in the groin. Previous work has suggested defining cases of APBD as “typical” versus “atypical,” largely based on genotype by grouping “typical” cases with homozygosity for the p.Y329S variant in GBE1 (Souza et al., 2021). Cases 1–3 described here displayed features associated with “typical” APBD, despite not harboring homozygosity for the p.Y329S variant. Based on this and other cases in the literature, we suggest avoiding the use of these dichotomous descriptors and instead recommend using the term “APBD” to describe all patients with this diagnosis regardless of genotypic/phenotypic presentation.
We then presented three patients that expanded our understanding of the phenotypical presentation of APBD. Clinically, cases 4–6 represent individuals with APBD in Brazil who are not of Ashkenazi Jewish descent and presented with “traditional” symptoms in early adulthood. Case 4 presented with bladder dysfunction starting at age 26 years, followed by neuropathy and muscle cramping at age 30 years. Case 5 presented with symptoms of small-fiber neuropathy at age 30 years and was diagnosed with APBD at age 50 years, by which time he was also experiencing muscle pain and bladder dysfunction. Case 6 reported bladder dysfunction at age 32 years, gait difficulties marked by bradykinesia and spastic paraparesis at age 35 years, and bowel dysfunction at age 44 years. These cases add to the growing body of evidence of APBD in younger, diverse populations, suggesting that greater attention should be paid to patients of all ages and demographics (Ferguson et al., 1983; Bruno et al., 1993; Ziemssen et al., 2000; Klein et al., 2004; Billot et al., 2013; Souza et al., 2021). We encourage clinicians to maintain high clinical suspicion and to not rule out an APBD diagnosis solely based on ancestry or age of onset. With continued use and improvements in whole exome sequencing and whole genome sequencing technologies, we expect that the diagnosis of GSD IV, including APBD, and our understanding of its natural history will improve.
All phenotypes on the GSD IV continuum, including APBD, are caused by deficiency in functional GBE activity. The genotypic profiles of cases 4–7, when evaluated in combination with their clinical presentation, extend the insight that APBD exists on the GSD IV clinical spectrum. Cases 4 and 6 harbored p.R637* and p.V98L in GBE1, which have previously been reported in patients with pediatric-onset GSD IV presenting with hepatic and/or neuromuscular involvement (Bruno et al., 2004; Janecke et al., 2004; Bruno et al., 2007; Li et al., 2010; Beyzaei et al., 2021). The p.V98L variant was previously reported in ClinVar in a patient with APBD, but no phenotypic information was available. Case 5 presented as homozygous for c.534G>C (p.W178C), a variant that has been reported in ClinVar in a patient with GSD IV but for which no phenotypic presentations have been published. The c.929A>C (p.Y310S) variant detected in case 7 was not previously reported in ClinVar or in the published literature and, therefore, was considered to be novel. Recent work has sought to establish that GSD IV symptomatology and presentation exist on a continuum with overlapping and multisystem involvement, rather than previously defined discrete hepatic or neuromuscular categories (Kiely et al., 2022). Our cases show that recognizing the presence of these variants in combination with the reported symptoms is important for understanding how APBD exists on the GSD IV continuum and for future phenotype–genotype correlations to improve genetic and prognostic counseling. Future work should focus on elucidating the multifaceted symptomatology of GBE deficiency, which has implications on management plans including long-term follow up and counseling.
In presenting a series of patients with APBD representing diverse phenotypic and genotypic presentations, we further suggest the broader inclusion of GBE1 analysis on relevant gene testing panels. The diagnostic pathways for cases 1–3 provide particularly unique insights into the pitfalls of common genetic testing methods. Case 1 underwent genetic testing with a targeted leukodystrophy gene panel which initially returned negative. After strong clinical suspicion, subsequent research re-evaluation of the data from the test nearly 1.3 years later confirmed the diagnosis of APBD. Similarly, case 2 faced a prolonged time for diagnosis after initially receiving negative results on an ataxia gene panel and whole exome sequencing. Moreover, whole exome sequencing also proved undiagnostic for case 3; yet, because of strong clinical suspicion, her providers pursued GBE activity testing and targeted gene re-evaluation that led to her diagnosis. These patients frequently underwent numerous diagnostic studies and evaluations before diagnosis, resulting in increased patient burden and overall costs to healthcare systems. Although we continue to believe that genetic testing is the gold standard, first-line option for diagnosing patients with APBD, our cases demonstrate the common limitations and pitfalls of these methods. To this regard, with the broad phenotypic, genotypic, and radiographic presentation of APBD becoming increasingly well-characterized (Mochel et al., 2012; De Winter et al., 2022), the inclusion of GBE1 testing on relevant gene panels, including those for leukodystrophy, ataxia, and peripheral neuropathy, is critical. For those that undergo whole exome sequencing, the limitations of this method have previously been described (Mori et al., 2017; Burdick et al., 2020) and may result in a missed diagnosis—as in cases 1–3. Therefore, until the current pitfalls with whole exome sequencing are resolved, we argue that clinicians should not be completely reliant on negative results from whole exome sequencing. Additionally, the utility of measuring GBE activity testing should not be understated. GBE activity testing can help diagnose APBD and can be conducted using cultured skin fibroblasts, skeletal muscle, leukocytes, and other tissues; diagnosis guidelines previously published include considerations when interpreting GBE activity in different tissues and cells (Koch et al., 2023). For those that are found to have decreased GBE activity and/or be a carrier of a pathogenic GBE1 variant, re-evaluation of genetic data and targeted follow-up testing with the intent of assessing for the presence of commonly missed variants, including the identified deep intronic variant (Koch et al., 2023), is warranted.
Lastly, in light of the increasing number of therapeutic options in development for the treatment of APBD (Kakhlon et al., 2013; Kakhlon et al., 2018; Gumusgoz et al., 2021; Gentry et al., 2022; Gumusgoz et al., 2022) and recently published consensus guidelines (Koch et al., 2023), we piloted the use of PRO surveys to evaluate the domains of pain, fatigue, and quality of life in United States-based patients (cases 1–3). As a conglomerate, the surveys disclosed impairment in all assessed domains. Averaged scores revealed a moderate level of pain severity and pain interference on the BPI; these results were mirrored by responses on the PI-8a, which also indicated a moderate level of interference caused by pain on daily functioning. Regarding fatigue, averaged scores showed moderate levels of fatigue on both the BFI as well as the FACIT-Fatigue compared to the general population. Regarding quality of life, averaged results on the SF-36 showed lower quality of life scores in every domain assessed compared to the general population. The EQ-5D-5L again identified significant impairments in quality of life, with cases 1–3 harboring health index scores ranging from 1.7 to 3.9 and standard deviations lower than population normative values. While the PROs provided useful insights into functional impairment in aggregate, they showed perhaps a greater value at the individual level. Upon review of available medical records, pain was poorly characterized for cases 1 and 2. However, the use of PROs better elucidated this symptom. Similarly, fatigue was a poorly characterized symptom throughout the medical records for all surveyed patients, but the dual use of fatigue-related PROs revealed impairment. To the best of our knowledge, this is the first published implementation of PROs in patients with APBD. Given our limited sample size, our implementation of PROs was intended to be an initial proof of concept to establish use within this patient population, providing preliminary evidence to support the United States Food and Drug Administration guidance regarding the use of PROs to support clinical trials (Food and Drug Administration, 2020). Longitudinal data should be collected and analyzed to further evaluate the true utility of PROs in this population more broadly. In the future, we strongly encourage the use of PROs both in the setting of clinic visits as well the clinical trial space. These metrics provide an opportunity for an enhanced personalized medicine approach for symptom management and can serve as trackable outcomes to monitor therapeutic interventions.
The interdisciplinary, multicentered, retrospective nature of this study inherently has limitations. Over the last decade, MRI of the brain and spinal cord has proven essential in the evaluation and eventual diagnosis of individuals with APBD. Characteristic imaging findings were first described in 2012 (Mochel et al., 2012) and recently expanded on in 2022 (De Winter et al., 2022). While we provided herein the radiological findings reported by the various institutions at which our cases received care, we were unable to independently evaluate primary imaging. Future work should prospectively investigate imaging findings and symptomatology in individuals with APBD from a wide variety of genotypic backgrounds. Moreover, our ability to assess systemic manifestations of disease in our cohort was limited; our cases underwent limited to no evaluation for hepatic or cardiac involvement. Future work should aim to provide a more dedicated analysis of systemic manifestations in APBD. Lastly, the presented data on PROs represent one time point and serve as initial evidence for PRO use in this population while also highlighting their utility in understanding individual disease burden. Longitudinal data across multiple time points will prove essential in the validation of these surveys for APBD.
We describe in detail the clinical course of three compound heterozygotes harboring the identified deep intronic variant in GBE1 as well as four cases from Brazil with unique genotypic profiles. We emphasize the growing presentation of APBD in diverse populations, requiring heightened awareness to ensure proper diagnosis and intervention, and suggest discontinuing the use of discrete classification categories. Lastly, we present the proof-of-concept use of PRO questionnaires to further describe the clinical states of APBD.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
The studies involving humans were approved by the Duke University Health System Institutional Review Board Pro00060753. The study was conducted in accordance with the local legislation and institutional requirements. Cases 1–3 provided their written informed consent to participate in the study and for publication of information and data in the study. Due to language barriers, Cases 4–7 could not provide written informed consent and therefore de-identified data was contributed for them by their care providers in accordance with Federal University of Sao Paulo ethics and privacy policies and the Pro00060753 study protocol. The Federal University of Sao Paulo research ethics committee did not require additional informed written consent from Cases 4–7 to publish de-identified data for this type of scientific report.
MG: conceptualization, data curation, writing–original draft, and writing–review and editing. PS: data curation and writing–review and editing. WP: data curation and writing–review and editing. PK: conceptualization, data curation, supervision, and writing–review and editing. RK: conceptualization, data curation, supervision, and writing–review and editing.
The authors declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors wish to acknowledge the YT and Alice Chen Pediatric Genetics and Genomics Research Center for its encouragement of this work. The authors thank all patients who participated in this study and their families for their ongoing support, time, and commitment to advancing the field of medicine by enrolling in this study. The authors thank Leticia Flores, Erin Huggins, Tracy Boggs, Dr. Lisa Hobson-Webb, and Dr. Mark Waters for their efforts in the implementation of this project. The authors also thank the numerous care teams that have worked tirelessly to improve the lives of patients with APBD.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1282790/full#supplementary-material
Akman, H. O., Kakhlon, O., Coku, J., Peverelli, L., Rosenmann, H., Rozenstein-Tsalkovich, L., et al. (2015). Deep intronic GBE1 mutation in manifesting heterozygous patients with adult polyglucosan body disease. JAMA Neurol. 72 (4), 441–445. doi:10.1001/jamaneurol.2014.4496
Amtmann, D., Cook, K. F., Jensen, M. P., Chen, W.-H., Choi, S., Revicki, D., et al. (2010). Development of A Promis Item Bank to Measure Pain Interference. Pain 150 (1), 173–182. doi:10.1016/j.pain.2010.04.025
Bao, Y., Kishnani, P., Wu, J. Y., and Chen, Y. T. (1996). Hepatic and neuromuscular forms of glycogen storage disease type IV caused by mutations in the same glycogen-branching enzyme gene. J. Clin. Investigation 97 (4), 941–948. doi:10.1172/JCI118517
Beyzaei, Z., Ezgu, F., Geramizadeh, B., Imanieh, M. H., Haghighat, M., Dehghani, S. M., et al. (2021). Clinical and genetic spectrum of glycogen storage disease in Iranian population using targeted gene sequencing. Sci. Rep. 11, 7040. doi:10.1038/s41598-021-86338-4
Billot, S., Hervé, D., Akman, H. O., Froissart, R., Baussan, C., Claeys, K. G., et al. (2013). Acute but transient neurological deterioration revealing adult polyglucosan body disease. J. Neurological Sci. 324 (1-2), 179–182. doi:10.1016/j.jns.2012.10.015
Bruno, C., Cassandrini, D., Assereto, S., Akman, H. O., Minetti, C., and Di Mauro, S. (2007). Neuromuscular forms of glycogen branching enzyme deficiency. Acta Myologica myopathies cardiomyopathies official J. Mediterr. Soc. myology 26 (1), 75–78.
Bruno, C., Servidei, S., Shanske, S., Karpati, G., Carpenter, S., McKee, D., et al. (1993). Glycogen branching enzyme deficiency in adult polyglucosan body disease. Ann. Neurology 33 (1), 88–93. doi:10.1002/ana.410330114
Bruno, C., van Diggelen, O. P., Cassandrini, D., Gimpelev, M., Giuffrè, B., Donati, M. A., et al. (2004). Clinical and genetic heterogeneity of branching enzyme deficiency (glycogenosis type IV). Neurology 63 (6), 1053–1058. doi:10.1212/01.wnl.0000138429.11433.0d
Burdick, K. J., Cogan, J. D., Rives, L. C., Robertson, A. K., Koziura, M. E., Brokamp, E., et al. (2020). Limitations of exome sequencing in detecting rare and undiagnosed diseases. Am. J. Med. Genet. A 182 (6), 1400–1406. doi:10.1002/ajmg.a.61558
Cella, D., Lai, J.-S., Jensen, S. E., Christodoulou, C., Junghaenel, D. U., Reeve, B. B., et al. (2016). PROMIS Fatigue Item Bank had Clinical Validity across Diverse Chronic Conditions. J. Clin. Epidemiol. 73, 128–134. doi:10.1016/j.jclinepi.2015.08.037
Cella, D., Riley, W., Stone, A., Rothrock, N., Reeve, B., Yount, S., et al. (2010). The Patient-Reported Outcomes Measurement Information System (PROMIS) developed and tested its first wave of adult self-reported health outcome item banks: 2005-2008. J. Clin. Epidemiol. 63 (11), 1179–1194. doi:10.1016/j.jclinepi.2010.04.011
Cleeland, C. S., and Ryan, K. M. (1994). Pain assessment: global use of the Brief Pain Inventory. Ann. Acad. Med. Singap. 23 (2), 129–138.
Dainese, L., Monin, M. L., Demeret, S., Brochier, G., Froissart, R., Spraul, A., et al. (2013). Abnormal glycogen in astrocytes is sufficient to cause adult polyglucosan body disease. Gene 515 (2), 376–379. doi:10.1016/j.gene.2012.12.065
De Winter, J., Cypers, G., Jacobs, E., Bossche, S. V., Deconinck, T., De Ridder, W., et al. (2022). Distinct features in adult polyglucosan body disease: a case series. Neuromuscul. Disord. 33 (2), 148–152. doi:10.1016/j.nmd.2022.12.016
Deandrea, S., Montanari, M., Moja, L., and Apolone, G. (2008). Prevalence of undertreatment in cancer pain. A review of published literature. Ann. Oncol. 19 (12), 1985–1991. doi:10.1093/annonc/mdn419
Devlin, N., Brazier, J., Pickard, A. S., and Stolk, E. (2018). 3L, 5L, What the L? A NICE Conundrum. PharmacoEconomics 36 (6), 637–640. doi:10.1007/s40273-018-0622-9
EuroQol Research Foundation, (2019). EQ-5D-5L user Guide. Rotterdam, Netherlands: EuroQol Research Foundation.
Ferguson, I. T., Mahon, M., and Cumming, W. J. K. (1983). An adult case of Andersen's disease - type IV glycogenosis. A clinical, histochemical, ultrastructural and biochemical study. J. Neurological Sci. 60 (3), 337–351. doi:10.1016/0022-510X(83)90144-2
Food and Drug Administration, (2020). “Patient-Reported Outcome Measures: use in medical product development to support labeling claims,” in FDA guidance documents (Oak, Maryland: Food and Drug Administration).
Gentry, M. S., Markussen, K. H., and Donohue, K. J. (2022). Two Diseases—One Preclinical Treatment Targeting Glycogen Synthesis. Neurotherapeutics 19 (3), 977–981. doi:10.1007/s13311-022-01240-9
Grunseich, C., Sarkar, N., Lu, J., Owen, M., Schindler, A., Calabresi, P. A., et al. (2021). Improving the efficacy of exome sequencing at a quaternary care referral centre: novel mutations, clinical presentations and diagnostic challenges in rare neurogenetic diseases. J. Neurology, Neurosurg. Psychiatry 92 (11), 1186–1196. doi:10.1136/jnnp-2020-325437
Gumusgoz, E., Guisso, D. R., Kasiri, S., Wu, J., Dear, M., Verhalen, B., et al. (2021). Targeting Gys1 with AAV-SaCas9 Decreases Pathogenic Polyglucosan Bodies and Neuroinflammation in Adult Polyglucosan Body and Lafora Disease Mouse Models. Neurother. J. Am. Soc. Exp. Neurother. 18 (2), 1414–1425. doi:10.1007/s13311-021-01040-7
Gumusgoz, E., Kasiri, S., Guisso, D. R., Wu, J., Dear, M., Verhalen, B., et al. (2022). AAV-Mediated Artificial miRNA Reduces Pathogenic Polyglucosan Bodies and Neuroinflammation in Adult Polyglucosan Body and Lafora Disease Mouse Models. Neurother. J. Am. Soc. Exp. Neurother. 19 (3), 982–993. doi:10.1007/s13311-022-01218-7
Güngör, D., Schober, A. K., Kruijshaar, M. E., Plug, I., Karabul, N., Deschauer, M., et al. (2013). Pain in adult patients with Pompe disease: a cross-sectional survey. Mol. Genet. Metabolism 109 (4), 371–376. doi:10.1016/j.ymgme.2013.05.021
HealthMeasures, (2023). HealthMeasures PROMIS® Score Cut Points. Available: https://www.healthmeasures.net/score-and-interpret/interpret-scores/promis/promis-score-cut-points (Accessed May 11, 2023).
Hellmann, M. A., Kakhlon, O., Landau, E. H., Sadeh, M., Giladi, N., Schlesinger, I., et al. (2015). Frequent misdiagnosis of adult polyglucosan body disease. J. Neurology 262 (10), 2346–2351. doi:10.1007/s00415-015-7859-4
Hussain, A., Armistead, J., Gushulak, L., Kruck, C., Pind, S., Triggs-Raine, B., et al. (2012). The adult polyglucosan body disease mutation GBE1 c.1076A>C occurs at high frequency in persons of Ashkenazi Jewish background. Biochem. Biophys. Res. Commun. 426 (2), 286–288. doi:10.1016/j.bbrc.2012.08.089
Janecke, A. R., Dertinger, S., Ketelsen, U.-P., Bereuter, L., Simma, B., Müller, T., et al. (2004). Neonatal type IV glycogen storage disease associated with "null" mutations in glycogen branching enzyme 1. J. Pediatr. 145 (5), 705–709. doi:10.1016/j.jpeds.2004.07.024
Janssen, M. F., Bonsel, G. J., and Luo, N. (2018). Is EQ-5D-5L Better Than EQ-5D-3L? A Head-to-Head Comparison of Descriptive Systems and Value Sets from Seven Countries. PharmacoEconomics 36 (6), 675–697. doi:10.1007/s40273-018-0623-8
Janssen, M. F., Pickard, A. S., Golicki, D., Gudex, C., Niewada, M., Scalone, L., et al. (2013). Measurement properties of the EQ-5D-5L compared to the EQ-5D-3L across eight patient groups: a multi-country study. Qual. Life Res. 22 (7), 1717–1727. doi:10.1007/s11136-012-0322-4
Jiang, R., Janssen, M. F. B., and Pickard, A. S. (2021). US population norms for the EQ-5D-5L and comparison of norms from face-to-face and online samples. Qual. Life Res. 30 (3), 803–816. doi:10.1007/s11136-020-02650-y
Kakhlon, O., Ferreira, I., Solmesky, L. J., Khazanov, N., Lossos, A., Alvarez, R., et al. (2018). Guaiacol as a drug candidate for treating adult polyglucosan body disease. JCI insight 3 (17), e99694–e99694. doi:10.1172/jci.insight.99694
Kakhlon, O., Glickstein, H., Feinstein, N., Liu, Y., Baba, O., Terashima, T., et al. (2013). Polyglucosan neurotoxicity caused by glycogen branching enzyme deficiency can be reversed by inhibition of glycogen synthase. J. Neurochem. 127 (1), 101–113. doi:10.1111/jnc.12277
Kiely, B. T., Koch, R. L., Flores, L., Burner, D., Kaplan, S., and Kishnani, P. S. (2022). A novel approach to characterize phenotypic variation in GSD IV: reconceptualizing the clinical continuum. Front. Genet. 13, 992406. doi:10.3389/fgene.2022.992406
Klein, C. J., Boes, C. J., Chapin, J. E., Lynch, C. D., Campeau, N. G., Dyck, P. J. B., et al. (2004). Adult polyglucosan body disease: case description of an expanding genetic and clinical syndrome. Muscle Nerve 29 (2), 323–328. doi:10.1002/mus.10520
Koch, R. L., Soler-Alfonso, C., Kiely, B. T., Asai, A., Smith, A. L., Bali, D. S., et al. (2023). Diagnosis and management of glycogen storage disease type IV, including adult polyglucosan body disease: A clinical practice resource. Mol. Genet. Metabolism 138 (3), 107525. doi:10.1016/j.ymgme.2023.107525
Lai, J.-S., Cella, D., Choi, S., Junghaenel, D. U., Christodoulou, C., Gershon, R., et al. (2011). How Item Banks and Their Application Can Influence Measurement Practice in Rehabilitation Medicine: A PROMIS Fatigue Item Bank Example. Archives Phys. Med. Rehabilitation 92 (10), S20–S27. doi:10.1016/j.apmr.2010.08.033
Li, K. K., Harris, K., Hadi, S., and Chow, E. (2007). What Should be the Optimal Cut Points for Mild, Moderate, and Severe Pain? J. Palliat. Med. 10 (6), 1338–1346. doi:10.1089/jpm.2007.0087
Li, S.-C., Chen, C.-M., Goldstein, J. L., Wu, J.-Y., Lemyre, E., Burrow, T. A., et al. (2010). Glycogen storage disease type IV: novel mutations and molecular characterization of a heterogeneous disorder. J. Inherit. Metabolic Dis. 33 (3), S83–S90. doi:10.1007/s10545-009-9026-5
Liu, D., Weng, J.-S., Ke, X., Wu, X.-Y., and Huang, S.-T. (2022). The relationship between cancer-related fatigue, quality of life and pain among cancer patients. Int. J. Nurs. Sci. 10 (1), 111–116. doi:10.1016/j.ijnss.2022.12.006
Mendoza, T. R., Wang, X. S., Cleeland, C. S., Morrissey, M., Johnson, B. A., Wendt, J. K., et al. (1999). The rapid assessment of fatigue severity in cancer patients: use of the Brief Fatigue Inventory. Cancer 85 (5), 1186–1196. doi:10.1002/(SICI)1097-0142(19990301)85:5<1186:AID-CNCR24>3.0.CO;2-N
Mochel, F., Schiffmann, R., Steenweg, M. E., Akman, H. O., Wallace, M., Sedel, F., et al. (2012). Adult polyglucosan body disease: natural history and key magnetic resonance imaging findings. Ann. Neurology 72 (3), 433–441. doi:10.1002/ana.23598
Mori, M., Haskell, G., Kazi, Z., Zhu, X., DeArmey, S. M., Goldstein, J. L., et al. (2017). Sensitivity of whole exome sequencing in detecting infantile- and late-onset Pompe disease. Mol. Genet. Metab. 122 (4), 189–197. doi:10.1016/j.ymgme.2017.10.008
National Center for Biotechnology Information, (2023a). VCV000002777.46 - ClinVar - NCBI. Available: https://www.ncbi.nlm.nih.gov/clinvar/variation/2777/ (Accessed August 17, 2023).
National Center for Biotechnology Information, (2023b). VCV000225145.6 - ClinVar - NCBI. Available: https://www.ncbi.nlm.nih.gov/clinvar/variation/225145/ (Accessed August 17, 2023).
National Center for Biotechnology Information, (2023c). VCV000346785.11 - ClinVar - NCBI. Available: https://www.ncbi.nlm.nih.gov/clinvar/variation/346785/ (Accessed August 17, 2023).
National Center for Biotechnology Information, (2023d). VCV000374517.33 - ClinVar - NCBI. Available: https://www.ncbi.nlm.nih.gov/clinvar/variation/374517/ (Accessed August 17, 2023).
National Center for Biotechnology Information, (2023e). VCV000520678.12 - ClinVar - NCBI. Available: https://www.ncbi.nlm.nih.gov/clinvar/variation/520678/ (Accessed August 17, 2023).
National Center for Biotechnology Information, (2023f). VCV002152670.1 - ClinVar - NCBI. Available: https://www.ncbi.nlm.nih.gov/clinvar/variation/2152670/ (Accessed August 17, 2023).
Peress, N. S., DiMauro, S., and Roxburgh, V. A. (1979). Adult polysaccharidosis. Clinicopathological, ultrastructural, and biochemical features. Arch. Neurol. 36 (13), 840–845. doi:10.1001/archneur.1979.00500490054009
Pickard, A. S., Law, E. H., Jiang, R., Pullenayegum, E., Shaw, J. W., Xie, F., et al. (2019). “United States Valuation of EQ-5D-5L Health States Using an International Protocol,” in Value in health (Boston, MA, USA: ISPOR–The Professional Society for Health Economics and Outcomes Research).
Postler, E., Sindern, E., Vorgerd, M., Schmitz, I., Malin, J. P., and Müller, K. M. (2002). Fatal cardiomyopathy in adult in polyglucosan body disease. Pathologe 23 (3), 229–234. doi:10.1007/s00292-002-0533-5
Rand Corporation, (2023). RAND Corporation 36-Item Short Form Survey (SF-36) Scoring Instructions. Available: https://www.rand.org/health-care/surveys_tools/mos/36-item-short-form/scoring.html (Accessed April 29, 2023).
Ritchie, E. K., Cella, D., Fabbiano, F., Pigneux, A., Kanda, Y., Ivanescu, C., et al. (2023). Patient-reported outcomes from the phase 3 ADMIRAL trial in patients with FLT3-mutated relapsed/refractory AML. Leukemia Lymphoma 64, 938–950. doi:10.1080/10428194.2023.2186731
Robitaille, Y., Carpenter, S., Karpati, G., and DiMauro, S. D. (1980). A distinct form of adult polyglucosan body disease with massive involvement of central and peripheral neuronal processes and astrocytes: a report of four cases and a review of the occurrence of polyglucosan bodies in other conditions such as Lafora's disease and normal ageing. Brain 103 (2), 315–336. doi:10.1093/brain/103.2.315
Rothrock, N. E., Hays, R. D., Spritzer, K., Yount, S. E., Riley, W., and Cella, D. (2010). Relative to the General US Population, Chronic Diseases are Associated with Poorer Health-Related Quality of Life as Measured by the Patient-Reported Outcomes Measurement Information System (PROMIS). J. Clin. Epidemiol. 63 (11), 1195–1204. doi:10.1016/j.jclinepi.2010.04.012
Schwartz, L. R., Lu, Q., Liu, R., Kornreich, R., Edelman, L. L., Birch, A. H., et al. (2020). Estimating the Prevalence of the Adult Polyglucosan Body Disease at the Gene Level for Ashkenazi Jews in the United States. Am. J. Rare Disord. Diagnosis Ther. 3 (1), 004–008. doi:10.37871/ajrddt.id17
Serlin, R. C., Mendoza, T. R., Nakamura, Y., Edwards, K. R., and Cleeland, C. S. (1995). When is cancer pain mild, moderate or severe? Grading pain severity by its interference with function. Pain 61 (2), 277–284. doi:10.1016/0304-3959(94)00178-H
Shahid, A., Wilkinson, K., Marcu, S., and Shapiro, C. (2012). STOP, THAT, and one hundred other Sleep Scales. Berlin, Germany: Springer.
Sindern, E., Ziemssen, F., Ziemssen, T., Podskarbi, T., Shin, Y., Brasch, F., et al. (2003). Adult polyglucosan body disease: A postmortem correlation study. Neurology 61 (2), 263–265. doi:10.1212/01.WNL.0000073144.96680.CB
Souza, P. V. S., Badia, B. M. L., Farias, I. B., Pinto, W. B. V. D. R., Oliveira, A. S. B., Akman, H. O., et al. (2021). GBE1-related disorders: adult polyglucosan body disease and its neuromuscular phenotypes. J. Inherit. Metabolic Dis. 44 (3), 534–543. doi:10.1002/jimd.12325
Sullivan, M. A., Nitschke, S., Skwara, E. P., Wang, P., Zhao, X., Pan, X. S., et al. (2019). Skeletal Muscle Glycogen Chain Length Correlates with Insolubility in Mouse Models of Polyglucosan-Associated Neurodegenerative Diseases. Cell Rep. 27, 1334–1344. doi:10.1016/J.CELREP.2019.04.017
Tan, G., Jensen, M. P., Thornby, J. I., and Shanti, B. F. (2004). Validation of the brief pain inventory for chronic nonmalignant pain. J. Pain 5 (2), 133–137. doi:10.1016/j.jpain.2003.12.005
Ware, J. E., and Sherbourne, C. D. (1992). The MOS 36-ltem Short-Form Health Survey (SF-36): I. Conceptual Framework and Item Selection. Med. Care 30 (6), 473–483. doi:10.1097/00005650-199206000-00002
Keywords: adult polyglucosan body disease, glycogen storage disease type IV, whole exome sequencing, patient-reported outcome, polyglucosan body neuropathy
Citation: Gayed MM, Sgobbi P, Pinto WBVDR, Kishnani PS and Koch RL (2023) Case report: Expanding the understanding of the adult polyglucosan body disease continuum: novel presentations, diagnostic pitfalls, and clinical pearls. Front. Genet. 14:1282790. doi: 10.3389/fgene.2023.1282790
Received: 24 August 2023; Accepted: 26 September 2023;
Published: 18 December 2023.
Edited by:
Ivan Martinez Duncker, Universidad Autónoma del Estado de Morelos, MexicoReviewed by:
Patryk Lipiński, Children’s Memorial Health Institute (IPCZD), PolandCopyright © 2023 Gayed, Sgobbi, Pinto, Kishnani and Koch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Priya S. Kishnani, cHJpeWEua2lzaG5hbmlAZHVrZS5lZHU=
†These authors have contributed equally to this work and share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.