
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Genet., 09 November 2023
Sec. Genetics of Common and Rare Diseases
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1277948
This article is part of the Research TopicRare Diseases Research and Diagnosis in Low- and Middle-Income CountriesView all 36 articles
 L. Campbell1
L. Campbell1 J. Fredericks2K. Mathivha3P. Moshesh3
J. Fredericks2K. Mathivha3P. Moshesh3 A. Coovadia2
A. Coovadia2 P. Chirwa4
P. Chirwa4 B. Dillon1A. Ghoor2D. Lawrence2L. Nair2N. Mabaso1D. Mokwele1M. Novellie1
B. Dillon1A. Ghoor2D. Lawrence2L. Nair2N. Mabaso1D. Mokwele1M. Novellie1 A. Krause1
A. Krause1 N. Carstens1,5*
N. Carstens1,5*Genetic disorders are significant contributors to infant hospitalization and mortality globally. The early diagnosis of these conditions in infants remains a considerable challenge. Clinical exome sequencing (CES) has shown to be a successful tool for the early diagnosis of genetic conditions, however, its utility in African infant populations has not been investigated. The impact of the under-representation of African genomic data, the cost of testing, and genomic workforce shortages, need to be investigated and evidence-based implementation strategies accounting for locally available genetics expertise and diagnostic infrastructure need to be developed. We evaluated the diagnostic utility of singleton CES in a cohort of 32 ill, South African infants from two State hospitals in Johannesburg, South Africa. We analysed the data using a series of filtering approaches, including a curated virtual gene panel consisting of genes implicated in neonatal-and early childhood-onset conditions and genes with known founder and common variants in African populations. We reported a diagnostic yield of 22% and identified seven pathogenic variants in the NPHS1, COL2A1, OCRL, SHOC2, TPRV4, MTM1 and STAC3 genes. This study demonstrates the utility value of CES in the South African State healthcare setting, providing a diagnosis to patients who would otherwise not receive one and allowing for directed management. We anticipate an increase in the diagnostic yield of our workflow with further refinement of the study inclusion criteria. This study highlights important considerations for the implementation of genomic medicine in under-resourced settings and in under-represented African populations where variant interpretation remains a challenge.
Genetic conditions are significant contributors to infant mortality, morbidity, and hospitalisations globally (Kingsmore et al., 2020). Despite advances in diagnostics with next-generation sequencing (NGS) and microarrays, it remains challenging to make diagnoses in infant populations. Infants often present with atypical or non-specific disease symptoms; many genetic disease phenotypes cannot be distinguished during the neonatal period; and disease progression may often be very rapid, making the identification and diagnosis of genetic conditions difficult (Wilkinson et al., 2016; van Diemen et al., 2017; French et al., 2019).
Clinical and whole exome sequencing (CES and WES), have been widely investigated for their use in diagnosing ill infants, with many studies evidencing their benefit (Petrikin et al., 2015; Smith et al., 2015; Willig et al., 2015; Stark et al., 2016; Meng et al., 2017; van Diemen et al., 2017; Farnaes et al., 2018; French et al., 2019; Kingsmore et al., 2019; Lunke et al., 2020; Wang et al., 2020). While WES targets all protein-coding regions of the genome, CES targets select genes with known disease associations and their flanking splice regions, and has been shown to provide an early, definitive diagnosis, preventing a diagnostic odyssey, and allowing for targeted clinical management (Smith et al., 2015). Virtual panels examining a subset of these clinically relevant genes from WES/CES data can be utilized to minimise the initial time and cost of analysis while offering the potential of comprehensive analysis of additional genes at a later stage with no additional laboratory costs, a key cost consideration for resource-constrained settings. Previous studies suggest that earlier diagnoses promote improved patient care and outcomes by facilitating changes in medical treatment, the early introduction of targeted therapies, increased surveillance, the appropriate initiation of comfort care, the reduction of costly, repeat, and sometimes invasive investigations, and the instigation of cascade testing for family members and counselling for reproductive planning–all measurable utility for the use of diagnostic testing (Rabbani et al., 2012; Splinter et al., 2018; Wright et al., 2018; Lunke et al., 2020).
Despite evidence from numerous research studies illustrating the benefits of CES for the diagnosis and management of ill infants, CES is not accessible in many parts of the world, particularly in low- and middle-income countries (LMICs). In many LMICs, CES is only offered in limited research settings (or not at all) creating larger health disparities in regions where access to primary healthcare and diagnostic services are already poor. This is true across the African continent, with few countries having established genetic services (Kamp et al., 2021). In South Africa, only three of the nine provinces offer genetic services in the State healthcare system, which services more than 80% of the population (Kromberg et al., 2013a; Stats SA, 2022). With limited access to genetic services, many affected by genetic conditions go undiagnosed and untreated. The lack of funding and resources for CES implementation is widely acknowledged by stakeholders, particularly shortages in infrastructure and trained genetics professionals (Kamp et al., 2021; Lumaka et al., 2022). The implementation of CES in the South African State healthcare system needs to be informed by international guidelines and standards, but requires optimisation in the local context to accommodate the scarcity of resources and the limited bioinformatics and genomics capacity (Kamp et al., 2021). The implementation of CES is further complicated by challenges in genetic data interpretation due to the underrepresentation of African populations in genomic databases and literature, African genetic diversity, and the lack of disease registries to document genetic disease prevalence (Kromberg et al., 2013b; Baynam et al., 2020; Lumaka et al., 2022). Despite these challenges, CES may offer significant improvements in the clinical management of vulnerable, underserved South African populations, including ill infants in the neonatal intensive care unit (NICU). Without a genetic diagnosis, infants with genetic conditions are constrained by less accurate risk assessments, prognoses, and specialist referrals; minimal assistance from support organisations and government welfare; and no access to emerging therapies and clinical trials, widening the existing healthcare disparities African populations face in accessing personalised healthcare.
With the shift in focus of genomic medicine to implementation science and translation into clinical practice, studies are needed to determine the benefits and challenges faced in implementing CES in real-world settings (Kingsmore and Cole, 2022). There are many barriers to the implementation of genomic medicine in LMICs which need to be investigated and addressed for genomic medicine and CES to be successfully integrated into global healthcare systems. To meet the World Health Organisation’s sustainable development goal to reduce neonatal and children under-5 mortality, the implementation of adequate genetic services to address the burden of genetic disease, often overshadowed and masked by infectious and communicable disease, is necessary in LMICs. Implementation should address the various barriers faced, including limited infrastructure and technology, shortages in the genetics workforce, poor genomic literacy amongst healthcare providers, poor literacy and education amongst the general population, language barriers and the lack of standardised genomics terminology in local languages, cultural and societal nuances around family structure, cultural beliefs around disease causality, and paucity of information around genetic disease burden in African populations (Kamp et al., 2021). These challenges will vary across LMICs due to population, economic, political, and social diversity, rendering a one-size-fits all implementation approach inappropriate for most settings and making investigations into unique, country-specific challenges necessary (Tiffin, 2014; Kamp et al., 2021; Lumaka et al., 2022).
We performed a scoping study to evaluate the diagnostic utility of singleton CES in a cohort of ill infants suspected of having a genetic condition in the South African State healthcare system through a series of three virtual gene panels. This study provides insight into the implementation of CES for ill infants in an under-resourced LMIC setting, where access to genetic services is limited, the burden of genetic disease is unknown and the interpretation of genomic data remains a considerable challenge.
The study workflow is summarised in Figure 1.
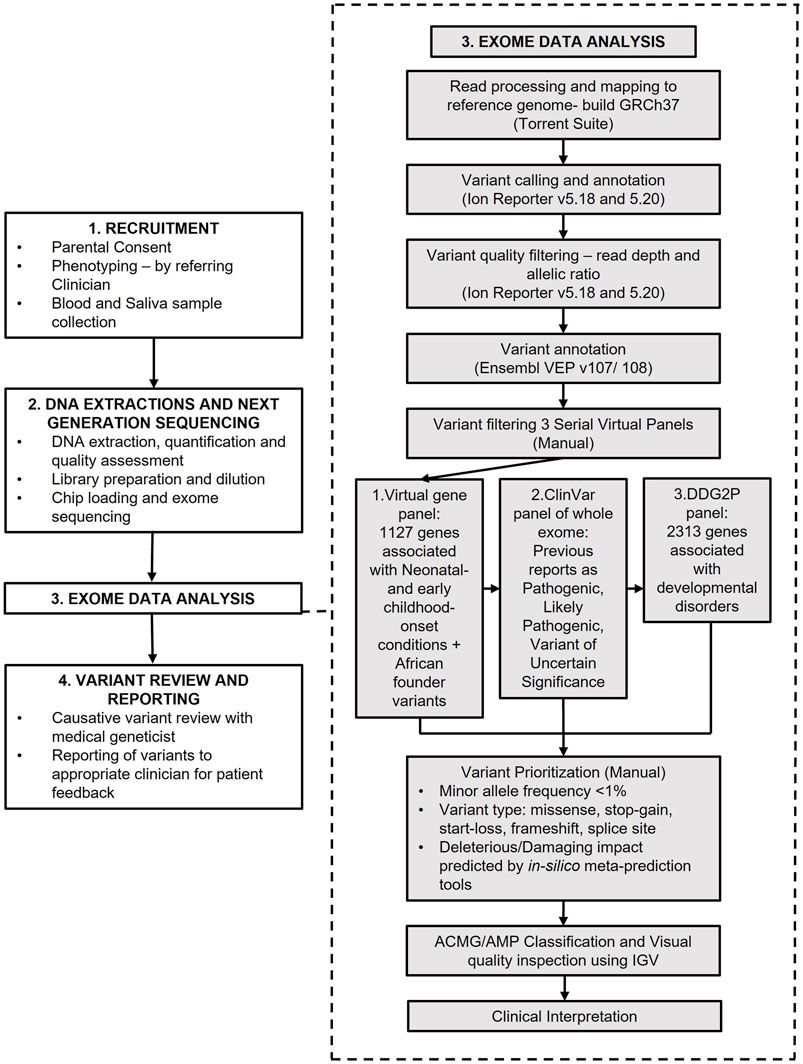
FIGURE 1. Study overview and analysis workflow.
A cohort of thirty-two ill infants, suspected of having a genetic condition, were recruited from two State hospitals in Johannesburg, South Africa: Rahima Moosa Mother and Child Hospital (RMMCH) in Coronationville, and Nelson Mandela Children’s Hospital (NMCH) in Parktown. These hospitals are regional and quaternary hospitals respectively and serve mostly State patients. Participants were referred for genetic testing either directly by neonatologists and paediatricians without consultation with medical geneticists, or by medical geneticists at call out consultations or referral. Clinicians were provided a list of recruitment criteria, highlighting a broad range of phenotypic features that may be suggestive of a genetic cause for an ill infant’s condition. Infants were considered eligible for recruitment if they presented with any of the following: multiple congenital anomalies, a concern for their neurological status, metabolic abnormalities of uncertain cause, dysmorphic features or if they had abnormal growth parameters. Infants were excluded if they had significant teratogen exposure during pregnancy, experienced birthing trauma or asphyxia, or had a diagnosed infection that could be the primary cause of illness. Premature patients were considered eligible for inclusion if their clinical features were not primarily explained by prematurity.
Parents and guardians provided informed consent on behalf of infants for participation in the study after the study was explained to them by a principal investigator or genetic counsellor. A blood sample was collected for DNA extraction and exome testing from each participant. A saliva sample from available parents was banked at consent, however, this study employed exome sequencing of probands only.
Phenotypic information regarding the condition and medical history of the participants was provided by the referring clinician. Clinicians were provided a short format phenotype collection rubric to assist with this as a Redcap form (Supplementary Table S1) (Harris et al., 2009; Harris et al., 2019), and requested to provide any information they believed was relevant to a possible genetic diagnosis.
DNA extraction was performed on the stored participant blood samples using a modified salting-out method (Miller et al., 1988). DNA quality and quantity was assessed using the NanoDrop 2000 (Thermo Fisher Scientific, United States), gel electrophoresis and the Qubit 4.0 system (Thermo Fisher Scientific, United States). Library preparation was performed manually using the Ion Ampliseq Exome RDY library preparation kit, as per the manufacturer’s instruction. DNA libraries were quantified and sized using the TapeStation High Sensitivity D5000 ScreenTape assay (Agilent Technologies, Germany). Diluted libraries were loaded onto Ion 540 sequencing chips for exome sequencing using the automated Ion Chef instrument (Thermo Fisher Scientific, United States). Loaded chips were sequenced in-house on an Ion GeneStudio S5 sequencer (Thermo Fisher Scientific, United States). The laboratory processing time for four samples was approximately 5 days.
Read alignment to the GRCh37 human reference genome was performed on the Ion Torrent Suite software (Thermo Fisher Scientific, United States). Variant calling was subsequently performed using Ion Reporter software (Thermo Fisher Scientific, United States). Variants were annotated using the Ensembl Variant Effect Predictor (VEP), versions 107 and 108 (McLaren et al., 2016).
Variants were filtered and prioritised for manual curation using three sequential filtering approaches, summarised in Figure 1. The first variant filtering approach retained variants in a virtual gene panel consisting of 1,127 genes, curated for their association with neonatal- and early-childhood-onset conditions (Ceyhan-Birsoy et al., 2017; Milko et al., 2019) and genes with known founder variants and common variants in African populations (Krause et al., 2018) (NICU/African) (Supplementary Table S2). If no causative variant was identified, the second variant filtering approach, retaining variants with previous pathogenic, likely pathogenic and variant of uncertain significance (VUS) classifications on ClinVar, including variants with conflicting interpretations, was employed (Landrum et al., 2018). The third filtering strategy, employed if no causative variant was identified from the first two strategies, retained variants in 2,313 genes on the Deciphering Developmental Disorders Genotype to Phenotype (DDG2P) gene panel (Thormann et al., 2019). Filtered variants were then prioritised for manual curation based on the type of variant, population minor allele frequencies, previous pathogenic and likely pathogenic interpretations on ClinVar and predicted deleterious impacts on protein function from in silico prediction tools CADD, REVEL, BayesDel and MetaRNN (Ioannidis et al., 2016; Feng, 2017; Rentzsch et al., 2019; Li et al., 2022). Variants were visually inspected to determine quality using Integrated Genomics Viewer software (IGV) (Robinson et al., 2011).
Prioritised variants were classified according to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) guidelines to determine their pathogenicity (Richards et al., 2015). Filtering, prioritization, manual curation and classification took approximately 6 h to perform per filtering strategy per sample. Pathogenic and likely pathogenic variants were then reviewed by a multidisciplinary team to determine if the diagnosis was appropriate for the participant. Findings were reported in a research report and returned to the referring clinicians and participants. VUSs and incidental findings were not reported in this study. Positive molecular findings were returned to parents by a genetic counsellor or medical geneticist, and appropriate downstream medical intervention and referrals initiated where necessary. Parents of participants with negative findings were invited for further consultation with a medical geneticist to discuss the implications of the negative result and determine an appropriate clinical management route going forward.
Thirty-two infants under the age of two, with a suspicion of a genetic condition were recruited for CES between May 2021 and December 2022 (Table 1). A single participant was enrolled and excluded as the participant had received a blood transfusion and demised before an appropriate amount of time had passed for a blood sample to be drawn. Most participants were referred for genetic testing directly by their treating neonatologist or paediatrician (23) and were not formally assessed by a medical geneticist prior to recruitment. An equal proportion of male and female participants were recruited. Most participants were of African ancestry (26). Only two participants, NE025 and NE035, were born to self-reported consanguineous parents. The most frequent indications for CES were multiple congenital anomalies (9), cardiac defects and abnormalities (6), general dysmorphism (3) and neuromuscular abnormalities (3). No participants had abnormalities detected prenatally on ultrasound or had prenatal genetic investigations, however, fetal anomaly scans are infrequently performed as standard obstetric care in the State healthcare system.
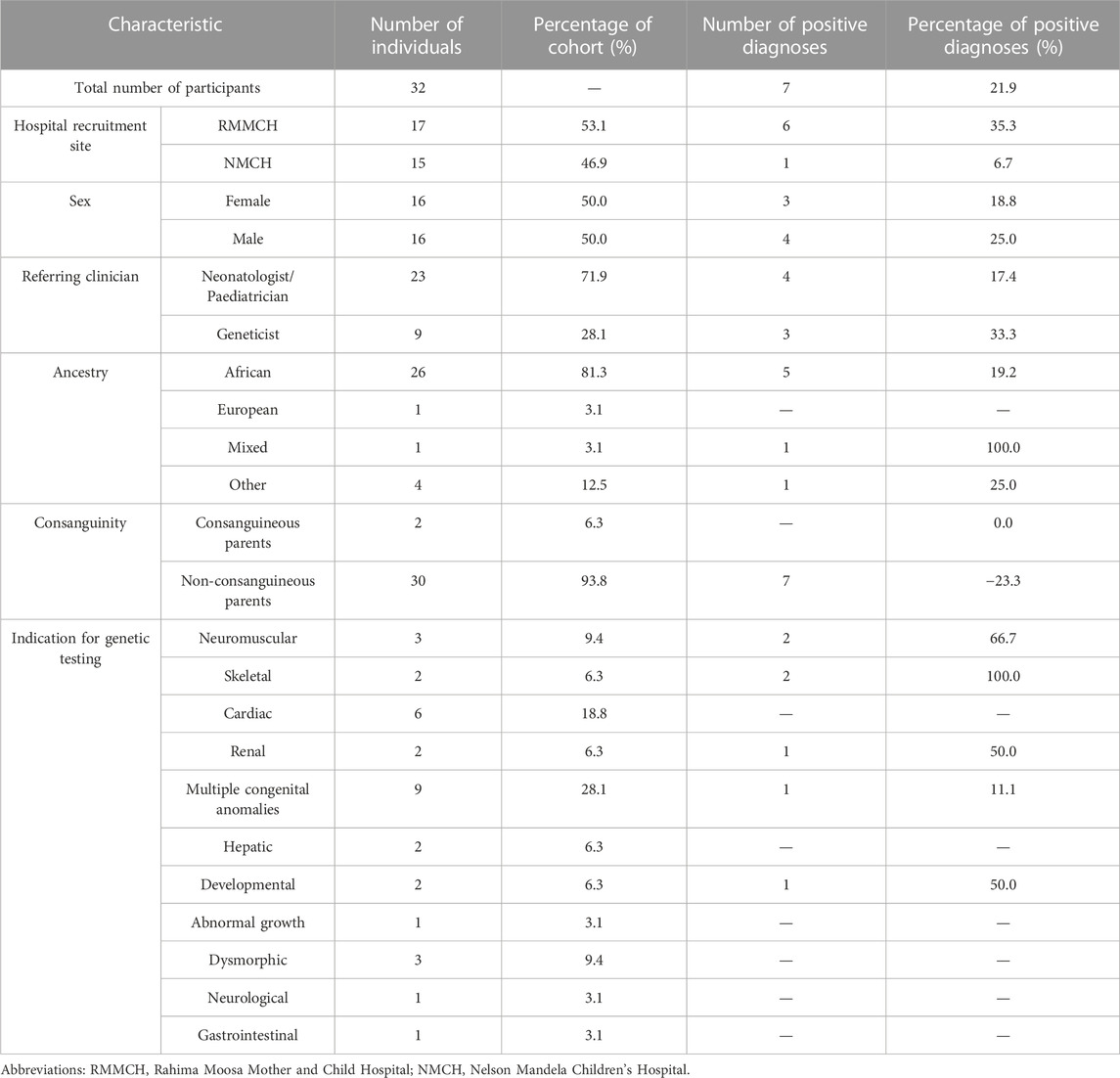
TABLE 1. Demographics and clinical characteristics of the patient cohort referred for clinical exome sequencing.
Singleton CES was performed on all participants with an average coverage of 134%X and 90% uniformity. A molecular diagnosis was confirmed for 7 of the 32 recruited participants, resulting in a diagnostic yield of 22% (Table 2). Five of the identified causative variants were in genes in our NICU/African virtual panel.
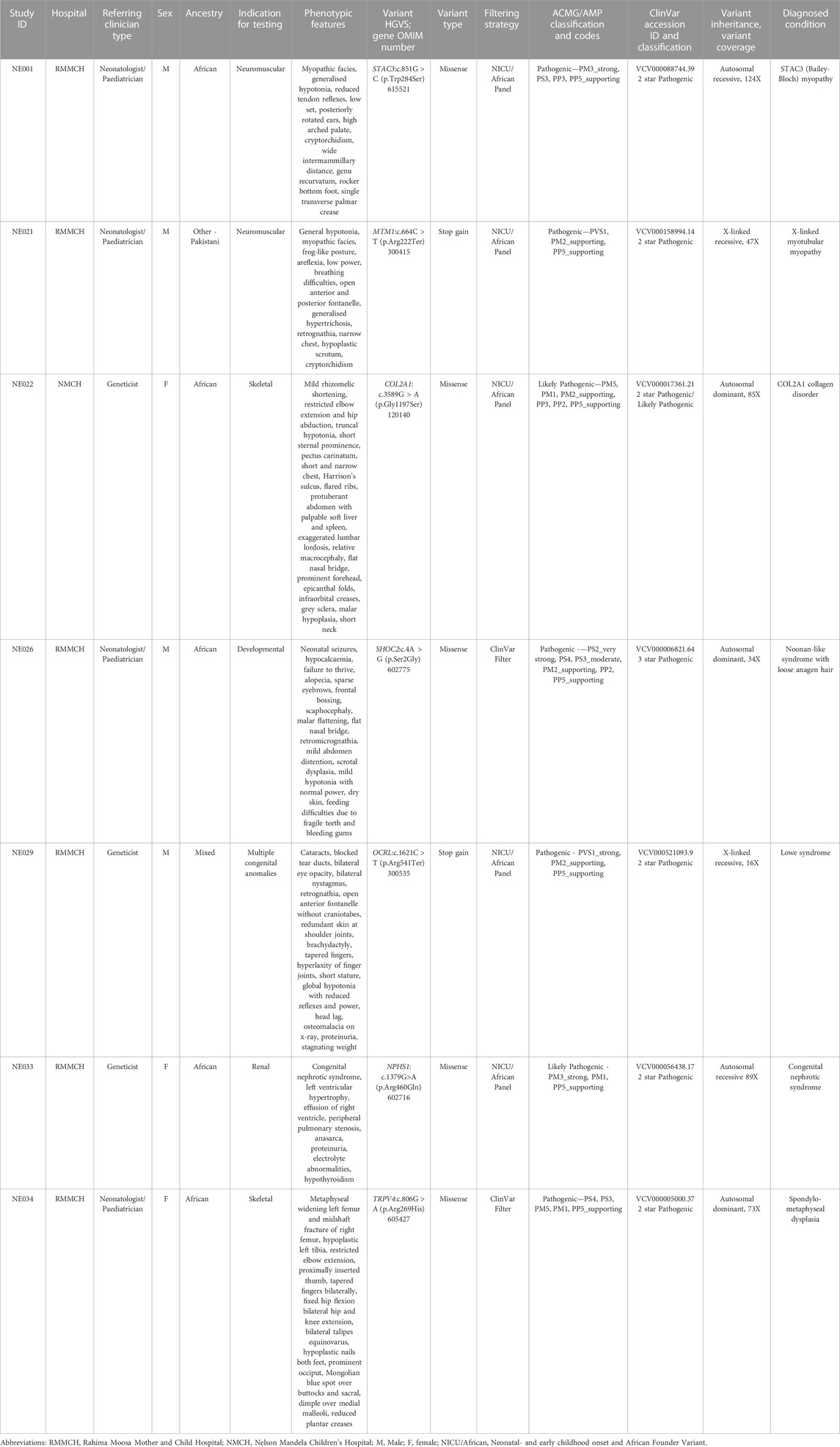
TABLE 2. Positive infant diagnoses through clinical exome sequencing using three filtering strategies.
Two participants received a severe myopathy diagnosis. A homozygous STAC3 variant (c.851G>C; p.Trp284Ser) was identified in participant NE001. This variant has been associated with a diagnosis of STAC3 myopathy (Horstick et al., 2013; Telegrafi et al., 2017; Waldrop et al., 2017; Zaharieva et al., 2018; Schoonen et al., 2019; Ravenscroft et al., 2021; Saleh et al., 2021). Multiple in silico prediction tools predict this variant to be deleterious to STAC3 function, confirmed in zebrafish models, which show a deficiency in skeletal muscle excitation-contraction coupling as a result of this missense mutation (Horstick et al., 2013; Linsley et al., 2017). A hemizygous, nonsense MTM1 variant (c.664C>T; p.Arg222Ter), affecting the functional myotubularin phosphatase domain of the MTM1 protein, was identified in participant NE021. This variant has been associated with severe X-linked myotubular myopathy (Laporte et al., 1997; Tanner et al., 1999; Biancalana et al., 2003; Longo et al., 2016). Both diagnosed myopathies had severe disease presentations, resulting in the demise of both infants within a week of life. Parents of these infants were offered prenatal testing for future pregnancies.
A pathogenic, heterozygous missense variant in COL2A1 (c.3589G > A; p.Gly1197Ser) was identified in participant NE022. This variant occurs in a predicted missense constrained gene and has predicted deleterious effects on type II collagen stability and structure (Beck et al., 2000). Other amino acid substitutions at the same position, p.Gly1197Ala and p.Gly1197Arg, have previously been reported as pathogenic. Mutations in the COL2A1 gene have been associated with a spectrum of overlapping skeletal dysplasias, therefore, this participant received a broad diagnosis of a COL2A1 collagen disorder (Cole et al., 1993; Nishimura et al., 2005; Terhal et al., 2015). Further assessments are needed to refine this diagnosis. This participant will need to be monitored for eye abnormalities, cervical instability and have their respiratory function assessed by the appropriate medical specialties.
Participant NE026 was diagnosed with Noonan-like syndrome with loose anagen hair caused by a heterozygous SHOC2 variant (c.4A > G, p.Ser2Gly). This variant, carrying a three-star ClinVar classification, has been shown to alter MAPK activation in in vitro studies and animal models (Cordeddu et al., 2009; Hoban et al., 2012; Gripp et al., 2013; Gargano et al., 2014; Choi et al., 2015). This participant will be referred for neurodevelopmental and cardiac assessments, as cardiac defects and psychomotor delay are frequently observed in this condition.
A hemizygous, nonsense OCRL variant (c.1621C > T, p.Arg541Ter) was identified in participant NE029, associated with X-linked recessive Lowe (Oculo-cerebro-renal) syndrome. This specific premature termination variant has been observed in other Lowe syndrome patients with similar clinical presentations (Hichri et al., 2011; Yang et al., 2014; Recker et al., 2015). This participant will require ophthalmology treatment for cataracts and glaucoma, as well as electrolyte monitoring for renal tubular acidosis. The mother of Participant NE029 received carrier screening by Sanger sequencing, confirming that she was not a carrier for the identified pathogenic variant.
Participant NE033 received a diagnosis of severe congenital nephrotic syndrome (Finnish type), harbouring a homozygous variant in the NPHS1 gene (c.1379G > A, p.Arg460Gln). This variant has been reported in a homozygous state in multiple congenital nephrotic syndrome cases (Beltcheva et al., 2001; Schoeb et al., 2010; Warejko et al., 2018; Mann et al., 2019). Participant NE033 presented with end stage renal disease and was unable to access renal replacement therapy in South Africa. Due to a poor prognosis and limited available medical intervention, this family received counselling on comfort care.
A heterozygous missense variant in TRPV4 (c.806G > A, p.Arg269His) was identified in participant NE034. This variant has been extensively reported in the literature for its association with autosomal dominant neuromuscular disease and skeletal dysplasia (Auer-Grumbach et al., 2010; Deng et al., 2010; Landoure et al., 2010; Zimon et al., 2010; Echaniz-Laguna et al., 2014; Louis et al., 2014; Biasini et al., 2016; Fleming and Quan, 2016; Jedrzejowska et al., 2019). Multiple functional studies support this variant’s gain of function role in increasing calcium channel activity, resulting in cell death and axonal degeneration (Deng et al., 2010; Landoure et al., 2010; Fecto et al., 2011; Klein et al., 2011; Takahashi et al., 2014). Participant NE034 presented clinically with only the skeletal features of TRPV4-associated disease and received a diagnosis of spondylometaphyseal dysplasia. As the neuropathic symptoms of this disease have been reported to occur after the neonatal period, this participant will need to be monitored for the development of these symptoms (Fleming and Quan, 2016). This participant will require physical therapy to maintain lower limb function. Pulmonary function and cervical spine stability will also be monitored in this participant.
This study generated valuable insights into the practicalities and utility of implementing CES in a NICU setting in South Africa. As the developed world moves toward rapid testing for ill infants, challenges in the implementation of genomic medicine in African and LMICs exacerbates existing healthcare disparities due to the slow uptake of these newer technologies, particularly in Sub-Saharan Africa, where most NGS testing, even in the research setting, is performed outside of the African continent (Baine-Savanhu et al., 2023). These challenges require careful consideration for the implementation of cutting-edge genomic services and for health equity to be achieved (Kingsmore and Cole, 2022).
We identified a genetic cause in seven ill infants with a range of clinical disease presentations, achieving a minimum diagnostic yield of 22%. Many studies globally have assessed the utility of CES in infant populations, showing impressive diagnostic yields of up to 70% (Clark et al., 2018), however, there is an absence of African individuals in these studies and low representation from LMICs. Patients of African ancestry are less likely to receive a diagnosis using current analytical strategies, as shown in the Deciphering Developmental Disorders study, creating a significant healthcare disparity for these populations (Wright et al., 2023). A tailored implementation strategy is needed for successful uptake of CES in Africa and in LMICs.
Comprehensive analysis strategies offer a higher probability of identifying a causative variant, however, these strategies are more time and labour intensive and have higher VUS rates (Stark and Ellard, 2022). A curated virtual panel of carefully selected genes, as demonstrated in this study, is an effective first approach for exome data analysis. Five of the seven causative variants identified in this study were in genes in our custom NICU/African panel, illustrating its utility for infant cohorts. The simplification of analysis pipelines and strategies in LMIC settings is necessary to deliver a result in a timely manner and with a low dependence on computational infrastructure and bioinformatics capacity. More comprehensive analysis pipelines, such as whole exome analysis, and the investigation of CNVs and structural variants could be deployed next for the remaining undiagnosed participants (Splinter et al., 2018). Undiagnosed participants may also benefit from reanalysis to incorporate updates to the virtual gene panel, new variant information, novel gene-disease associations, updated phenotypic data and bioinformatic advancements (Schobers et al., 2022).
The implementation of CES across the African continent and in LMICs is significantly under-investigated and many of the challenges associated with CES implementation into genetic service are exacerbated in an African setting. The significant barriers to the implementation of genomic medicine include a lack of infrastructure and support for clinical translation, the scarcity of information regarding African genetic epidemiology, poor genomic literacy among healthcare workers, few formally trained genetics professionals, a lack of investment by governments and international funders, the low cost effectiveness in establishing genetic services, and the fear of widening the existing disparities by only benefiting those who can afford access to these technologies (Jongeneel et al., 2022). These challenges, many seen in this study, need to be addressed when planning implementation strategies for CES in our context.
More than 70% of our infant cohort was referred for genetic testing directly by a neonatologist or paediatrician. Appropriate phenotyping and a predictive clinical diagnosis from a medical geneticist can direct CES data interpretation and contribute to a higher diagnostic yield (Trujillano et al., 2017; Gubbels et al., 2020). The limited formally trained genetics workforce in South Africa makes input from trained geneticists prior to genetic testing a virtually impossible task (Kromberg et al., 2013a). The engagement and upskilling of primary care medical specialities is necessary for CES implementation in under-resourced settings, to allow the few medical geneticists and genetic counsellors to be most efficiently utilised (Dragojlovic et al., 2020; Chou et al., 2021).
The lack of representation of African and Sub-Saharan African populations in publicly available databases and limited understanding of African genetic disease epidemiology makes CES data analysis and interpretation complex. A representative reference genome which considers African population diversity and disease epidemiology is essential in identifying disease-causing variants in a timely manner. A better understanding of genetic disease in African populations is key to improved management and service of our populations. This study allowed us to investigate genetic disease epidemiology in an underrepresented African population. The variant identified in participant NE001, diagnosed with STAC3 myopathy, has the highest carrier rate in the Genome Aggregation Database (GnomAD, accessed June 2023) in individuals of African/African American ancestry (Zaharieva et al., 2018; Gromand et al., 2022). Unpublished research from the South African National Health Laboratory Service revealed a frequency of 20% for this variant in a homozygous or compound heterozygous state in SMN1 negative African patients with myopathic phenotypes (Mhlongu et al., 2022). Despite its prevalence in African-ancestry patients, STAC3 testing has only been recognized as a common myopathy and introduced into diagnostic service in the South African State system within the past year.
Singleton CES was utilised in this study to elucidate the potential cause of disease in our infant cohort. Despite its recognised diagnostic superiority, trio-sequencing is costly to consider in LMICs, where limited financial resources must effectively serve large populations {Baine-Savanhu et al., 2023 #416} (Clark et al., 2018; Kingsmore et al., 2019). Trio-sequencing is thus not affordable at present in LMICs and the benefit versus cost requires further investigation. Complex family dynamics and social issues are other key considerations for African populations. In this cohort, only 31% of participants had both parents available at enrolment due to a variety of social factors, including: admission at hospitals far away from their residence with no access to transportation; the inability to get time off work for hospital visits; some participants were African immigrants who were not in South Africa as a complete family unit; and many participants were born to single-parent households and families. A diagnosis was still achievable in some participants in this study with singleton sequencing, illustrating the effectiveness and utility of this tool as a first-tier strategy, despite difficulties in documenting de novo mutations and reoccurrence risk. For effective implementation of genomic services in Africa, these social challenges need to be embraced to prevent the exclusion of patients who would greatly benefit from access to these services.
Approximately 80% of participants did not receive a molecular diagnosis in this study. The workflow offers a starting strategy in the diagnostic trajectory of these participants, who would benefit from further investigations to fill in gaps not addressed in our virtual gene panels. Refinement of the inclusion and exclusion criteria may provide more clarity to referring clinicians as to which infants would most benefit from CES, allowing for better utilisation of the scarce resources for testing. More comprehensive phenotyping may also provide more guidance in participant screening and in variant interpretation for a more cost- and time-effective implementation strategy.
In conclusion, this study yielded a definitive diagnosis for seven families affected by a genetic condition with minimal access to genetic testing in the South African State healthcare system, providing the first evidence of the diagnostic utility of CES for ill infants in an under-resourced NICU setting in Africa. Our experience can be used as a point of departure to develop feasible, local CES implementation strategies, utilising appropriately curated virtual panels to provide clinically actionable findings within an appropriate timeframe. Optimisation of an appropriate gatekeeping strategy that leverages locally available genetics capacity may increase our diagnostic yield and minimise inappropriate testing.
The data presented in the study are deposited into ClinVar, Organisation ID 508172 and can be accessed at https://www.ncbi.nlm.nih.gov/clinvar/submitters/508172/.
The studies involving humans were approved by the University of the Human Research Ethical Committee (Medical) at the University of the Witwatersrand, Johannesburg, South Africa. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
LC: Data curation, Formal Analysis, Investigation, Methodology, Project administration, Writing–original draft, Writing–review and editing. JF: Conceptualization, Investigation, Project administration, Writing–review and editing. KM: Investigation, Project administration, Writing–review and editing. PM: Investigation, Project administration, Writing–review and editing. AC: Conceptualization, Project administration, Writing–review and editing. PC: Conceptualization, Project administration, Writing–review and editing. BD: Investigation, Writing–review and editing. AG: Investigation, Writing–review and editing. DL: Investigation, Writing–review and editing. LN: Investigation, Writing–review and editing. NM: Investigation, Writing–review and editing. DM: Investigation, Writing–review and editing. MN: Investigation, Writing–review and editing. AK: Conceptualization, Data curation, Formal Analysis, Investigation, Project administration, Resources, Supervision, Writing–original draft, Writing–review and editing. NC: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Writing–original draft, Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded by a South African National Research Foundation Thuthuka Grant (Grant Number: 1903204240019). LC was supported by a National Research Foundation Block Grant.
We acknowledge and thank the participants and their families and caregivers; the staff of the neonatal and paediatric intensive care units and wards at the recruiting hospitals, Rahima Moosa Mother and Child Hospital and Nelson Mandela Children’s Hospital; and the medical geneticists and genetic counselling interns of the Division of Human Genetics, National Health Laboratory Service and School of Pathology, Faculty of Health Sciences, University of the Witwatersrand.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1277948/full#supplementary-material
Auer-Grumbach, M., Olschewski, A., Papic, L., Kremer, H., Mcentagart, M. E., Uhrig, S., et al. (2010). Alterations in the ankyrin domain of TRPV4 cause congenital distal SMA, scapuloperoneal SMA and HMSN2C. Nat. Genet. 42, 160–164. doi:10.1038/ng.508
Baine-Savanhu, F., Macaulay, S., Louw, N., Bollweg, A., Flynn, K., Molatoli, M., et al. (2023). Identifying the genetic causes of developmental disorders and intellectual disability in Africa: a systematic literature review. Front. Genet. 14, 1137922. doi:10.3389/fgene.2023.1137922
Baynam, G. S., Groft, S., van der Westhuizen, F. H., Gassman, S. D., du Plessis, K., Coles, E. P., et al. (2020). A call for global action for rare diseases in Africa. Nat. Genet. 52, 21–26. doi:10.1038/s41588-019-0552-2
Beck, K., Chan, V. C., Shenoy, N., Kirkpatrick, A., Ramshaw, J. A., and Brodsky, B. (2000). Destabilization of osteogenesis imperfecta collagen-like model peptides correlates with the identity of the residue replacing glycine. Proc. Natl. Acad. Sci. U. S. A. 97, 4273–4278. doi:10.1073/pnas.070050097
Beltcheva, O., Martin, P., Lenkkeri, U., and Tryggvason, K. (2001). Mutation spectrum in the nephrin gene (NPHS1) in congenital nephrotic syndrome. Hum. Mutat. 17, 368–373. doi:10.1002/humu.1111
Biancalana, V., Caron, O., Gallati, S., Baas, F., Kress, W., Novelli, G., et al. (2003). Characterisation of mutations in 77 patients with X-linked myotubular myopathy, including a family with a very mild phenotype. Hum. Genet. 112, 135–142. doi:10.1007/s00439-002-0869-1
Biasini, F., Portaro, S., Mazzeo, A., Vita, G., Fabrizi, G. M., Taioli, F., et al. (2016). TRPV4 related scapuloperoneal spinal muscular atrophy: report of an Italian family and review of the literature. Neuromuscul. Disord. 26, 312–315. doi:10.1016/j.nmd.2016.02.010
Ceyhan-Birsoy, O., Machini, K., Lebo, M. S., Yu, T. W., Agrawal, P. B., Parad, R. B., et al. (2017). A curated gene list for reporting results of newborn genomic sequencing. Genet. Med. 19, 809–818. doi:10.1038/gim.2016.193
Choi, J. H., Oh, M. Y., Yum, M. S., Lee, B. H., Kim, G. H., and Yoo, H. W. (2015). Moyamoya syndrome in a patient with Noonan-like syndrome with loose anagen hair. Pediatr. Neurol. 52, 352–355. doi:10.1016/j.pediatrneurol.2014.11.017
Chou, A. F., Duncan, A. R., Hallford, G., Kelley, D. M., and Dean, L. W. (2021). Barriers and strategies to integrate medical genetics and primary care in underserved populations: a scoping review. J. Community Genet. 12, 291–309. doi:10.1007/s12687-021-00508-5
Clark, M. M., Stark, Z., Farnaes, L., Tan, T. Y., White, S. M., Dimmock, D., et al. (2018). Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom Med. 3, 16. doi:10.1038/s41525-018-0053-8
Cole, W. G., Hall, R. K., and Rogers, J. G. (1993). The clinical features of spondyloepiphyseal dysplasia congenita resulting from the substitution of glycine 997 by serine in the alpha 1(II) chain of type II collagen. J. Med. Genet. 30, 27–35. doi:10.1136/jmg.30.1.27
Cordeddu, V., Di Schiavi, E., Pennacchio, L. A., Ma'ayan, A., Sarkozy, A., Fodale, V., et al. (2009). Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat. Genet. 41, 1022–1026. doi:10.1038/ng.425
Deng, H. X., Klein, C. J., Yan, J., Shi, Y., Wu, Y., Fecto, F., et al. (2010). Scapuloperoneal spinal muscular atrophy and CMT2C are allelic disorders caused by alterations in TRPV4. Nat. Genet. 42, 165–169. doi:10.1038/ng.509
Dragojlovic, N., Borle, K., Kopac, N., Ellis, U., Birch, P., Adam, S., et al. (2020). The composition and capacity of the clinical genetics workforce in high-income countries: a scoping review. Genet. Med. 22, 1437–1449. doi:10.1038/s41436-020-0825-2
Echaniz-Laguna, A., Dubourg, O., Carlier, P., Carlier, R. Y., Sabouraud, P., Péréon, Y., et al. (2014). Phenotypic spectrum and incidence of TRPV4 mutations in patients with inherited axonal neuropathy. Neurology 82, 1919–1926. doi:10.1212/WNL.0000000000000450
Farnaes, L., Hildreth, A., Sweeney, N. M., Clark, M. M., Chowdhury, S., Nahas, S., et al. (2018). Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization. NPJ Genom Med. 3, 10. doi:10.1038/s41525-018-0049-4
Fecto, F., Shi, Y., Huda, R., Martina, M., Siddique, T., and Deng, H. X. (2011). Mutant TRPV4-mediated toxicity is linked to increased constitutive function in axonal neuropathies. J. Biol. Chem. 286, 17281–17291. doi:10.1074/jbc.M111.237685
Feng, B. J. (2017). PERCH: a unified framework for disease gene prioritization. Hum. Mutat. 38, 243–251. doi:10.1002/humu.23158
Fleming, J., and Quan, D. (2016). A case of congenital spinal muscular atrophy with pain due to a mutation in TRPV4. Neuromuscul. Disord. 26, 841–843. doi:10.1016/j.nmd.2016.09.013
French, C. E., Delon, I., Dolling, H., Sanchis-Juan, A., Shamardina, O., Mégy, K., et al. (2019). Whole genome sequencing reveals that genetic conditions are frequent in intensively ill children. Intensive Care Med. 45, 627–636. doi:10.1007/s00134-019-05552-x
Gargano, G., Guidotti, I., Balestri, E., Vagnarelli, F., Rosato, S., Comitini, G., et al. (2014). Hydrops fetalis in a preterm newborn heterozygous for the c.4A>G SHOC2 mutation. Am. J. Med. Genet. A 164A, 1015–1020. doi:10.1002/ajmg.a.36376
Gripp, K. W., Zand, D. J., Demmer, L., Anderson, C. E., Dobyns, W. B., Zackai, E. H., et al. (2013). Expanding the SHOC2 mutation associated phenotype of Noonan syndrome with loose anagen hair: structural brain anomalies and myelofibrosis. Am. J. Med. Genet. A 161A, 2420–2430. doi:10.1002/ajmg.a.36098
Gromand, M., Gueguen, P., Pervillé, A., Ferroul, F., Morel, G., Harouna, A., et al. (2022). STAC3 related congenital myopathy: a case series of seven Comorian patients. Eur. J. Med. Genet. 65, 104598. doi:10.1016/j.ejmg.2022.104598
Gubbels, C. S., VanNoy, G. E., Madden, J. A., Copenheaver, D., Yang, S., Wojcik, M. H., et al. (2020). Prospective, phenotype-driven selection of critically ill neonates for rapid exome sequencing is associated with high diagnostic yield. Genet. Med. 22, 736–744. doi:10.1038/s41436-019-0708-6
Harris, P. A., Taylor, R., Minor, B. L., Elliott, V., Fernandez, M., O'Neal, L., et al. (2019). The REDCap consortium: building an international community of software platform partners. J. Biomed. Inf. 95, 103208. doi:10.1016/j.jbi.2019.103208
Harris, P. A., Taylor, R., Thielke, R., Payne, J., Gonzalez, N., and Conde, J. G. (2009). Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J. Biomed. Inf. 42, 377–381. doi:10.1016/j.jbi.2008.08.010
Hichri, H., Rendu, J., Monnier, N., Coutton, C., Dorseuil, O., Poussou, R. V., et al. (2011). From Lowe syndrome to Dent disease: correlations between mutations of the OCRL1 gene and clinical and biochemical phenotypes. Hum. Mutat. 32, 379–388. doi:10.1002/humu.21391
Hoban, R., Roberts, A. E., Demmer, L., Jethva, R., and Shephard, B. (2012). Noonan syndrome due to a SHOC2 mutation presenting with fetal distress and fatal hypertrophic cardiomyopathy in a premature infant. Am. J. Med. Genet. A 158A, 1411–1413. doi:10.1002/ajmg.a.35318
Horstick, E. J., Linsley, J. W., Dowling, J. J., Hauser, M. A., McDonald, K. K., Ashley-Koch, A., et al. (2013). Stac3 is a component of the excitation-contraction coupling machinery and mutated in Native American myopathy. Nat. Commun. 4, 1952. doi:10.1038/ncomms2952
Ioannidis, N. M., Rothstein, J. H., Pejaver, V., Middha, S., McDonnell, S. K., Baheti, S., et al. (2016). REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99, 877–885. doi:10.1016/j.ajhg.2016.08.016
Jedrzejowska, M., Dębek, E., Kowalczyk, B., Halat, P., Kostera-Pruszczyk, A., Ciara, E., et al. (2019). The remarkable phenotypic variability of the p.Arg269HiS variant in the TRPV4 gene. Muscle Nerve 59, 129–133. doi:10.1002/mus.26346
Jongeneel, C. V., Kotze, M. J., Bhaw-Luximon, A., Fadlelmola, F. M., Fakim, Y. J., Hamdi, Y., et al. (2022). A view on genomic medicine activities in Africa: implications for policy. Front. Genet. 13, 769919. doi:10.3389/fgene.2022.769919
Kamp, M., Krause, A., and Ramsay, M. (2021). Has translational genomics come of age in Africa? Hum. Mol. Genet. 30, R164–R173. doi:10.1093/hmg/ddab180
Kingsmore, S. F., Cakici, J. A., Clark, M. M., Gaughran, M., Feddock, M., Batalov, S., et al. (2019). A randomized, controlled trial of the analytic and diagnostic performance of singleton and trio, rapid genome and exome sequencing in ill infants. Am. J. Hum. Genet. 105, 719–733. doi:10.1016/j.ajhg.2019.08.009
Kingsmore, S. F., and Cole, F. S. (2022). The role of genome sequencing in neonatal intensive care units. Annu. Rev. Genomics Hum. Genet. 23, 427–448. doi:10.1146/annurev-genom-120921-103442
Kingsmore, S. F., Henderson, A., Owen, M. J., Clark, M. M., Hansen, C., Dimmock, D., et al. (2020). Measurement of genetic diseases as a cause of mortality in infants receiving whole genome sequencing. NPJ Genom Med. 5, 49. doi:10.1038/s41525-020-00155-8
Klein, C. J., Shi, Y., Fecto, F., Donaghy, M., Nicholson, G., McEntagart, M. E., et al. (2011). TRPV4 mutations and cytotoxic hypercalcemia in axonal Charcot-Marie-Tooth neuropathies. Neurology 76, 887–894. doi:10.1212/WNL.0b013e31820f2de3
Krause, A., Seymour, H., and Ramsay, M. (2018). Common and founder mutations for monogenic traits in sub-saharan african populations. Annu. Rev. Genomics Hum. Genet. 19, 149–175. doi:10.1146/annurev-genom-083117-021256
Kromberg, J. G., Sizer, E. B., and Christianson, A. L. (2013a). Genetic services and testing in South Africa. J. Community Genet. 4, 413–423. doi:10.1007/s12687-012-0101-5
Kromberg, J. G., Wessels, T. M., and Krause, A. (2013b). Roles of genetic counselors in South Africa. J. Genet. Couns. 22, 753–761. doi:10.1007/s10897-013-9606-2
Landoure, G., Zdebik, A. A., Martinez, T. L., Burnett, B. G., Stanescu, H. C., Inada, H., et al. (2010). Mutations in TRPV4 cause Charcot-Marie-Tooth disease type 2C. Nat. Genet. 42, 170–174. doi:10.1038/ng.512
Landrum, M. J., Lee, J. M., Benson, M., Brown, G. R., Chao, C., Chitipiralla, S., et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067. doi:10.1093/nar/gkx1153
Laporte, J., Guiraud-Chaumeil, C., Vincent, M. C., Mandel, J. L., Tanner, S. M., Liechti-Gallati, S., et al. (1997). Mutations in the MTM1 gene implicated in X-linked myotubular myopathy. ENMC international consortium on myotubular myopathy. European neuro-muscular center. Hum. Mol. Genet. 6, 1505–1511. doi:10.1093/hmg/6.9.1505
Li, C., Zhi, D., Wang, K., and Liu, X. (2022). MetaRNN: differentiating rare pathogenic and rare benign missense SNVs and InDels using deep learning. Genome Med. 14, 115. doi:10.1186/s13073-022-01120-z
Linsley, J. W., Hsu, I. U., Groom, L., Yarotskyy, V., Lavorato, M., Horstick, E. J., et al. (2017). Congenital myopathy results from misregulation of a muscle Ca2+ channel by mutant Stac3. Proc. Natl. Acad. Sci. U. S. A. 114, E228–E236. doi:10.1073/pnas.1619238114
Longo, G., Russo, S., Novelli, G., Sangiuolo, F., and D'Apice, M. R. (2016). Mutation spectrum of the MTM1 gene in XLMTM patients: 10 years of experience in prenatal and postnatal diagnosis. Clin. Genet. 89, 93–98. doi:10.1111/cge.12674
Louis, E. D., Hernandez, N., Ottman, R., Ionita-Laza, I., and Clark, L. N. (2014). Essential tremor in a charcot-marie-tooth type 2C kindred does not segregate with the TRPV4 R269H mutation. Case Rep. Neurol. 6, 1–6. doi:10.1159/000357665
Lumaka, A., Carstens, N., Devriendt, K., Krause, A., Kulohoma, B., Kumuthini, J., et al. (2022). Increasing African genomic data generation and sharing to resolve rare and undiagnosed diseases in Africa: a call-to-action by the H3Africa rare diseases working group. Orphanet J. Rare Dis. 17, 230. doi:10.1186/s13023-022-02391-w
Lunke, S., Eggers, S., Wilson, M., Patel, C., Barnett, C. P., Pinner, J., et al. (2020). Feasibility of ultra-rapid exome sequencing in critically ill infants and children with suspected monogenic conditions in the australian public health care system. JAMA 323, 2503–2511. doi:10.1001/jama.2020.7671
Mann, N., Braun, D. A., Amann, K., Tan, W., Shril, S., Connaughton, D. M., et al. (2019). Whole-exome sequencing enables a precision medicine approach for kidney transplant recipients. J. Am. Soc. Nephrol. 30, 201–215. doi:10.1681/ASN.2018060575
Mclaren, W., Gil, L., Hunt, S. E., Riat, H. S., Ritchie, G. R. S., Thormann, A., et al. (2016). The Ensembl variant effect predictor. Genome Biol. 17, 122. doi:10.1186/s13059-016-0974-4
Meng, L., Pammi, M., Saronwala, A., Magoulas, P., Ghazi, A. R., Vetrini, F., et al. (2017). Use of exome sequencing for infants in intensive care units: ascertainment of severe single-gene disorders and effect on medical management. JAMA Pediatr. 171, e173438. doi:10.1001/jamapediatrics.2017.3438
Mhlongu, F., Bhengu, L., Dillon, B., Essop, F., and Krause, A. (2022). STAC3 disorder: frequent genetic cause of congenital hypotonia in African patients. Johannesburg, South Africa: University of the Witwatersrand Faculty of Health Sciences Research Day.
Milko, L. V., O'Daniel, J. M., DeCristo, D. M., Crowley, S. B., Foreman, A. K. M., Wallace, K. E., et al. (2019). An age-based framework for evaluating genome-scale sequencing results in newborn screening. J. Pediatr. 209, 68–76. doi:10.1016/j.jpeds.2018.12.027
Miller, S. A., Dykes, D. D., and Polesky, H. F. (1988). A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16, 1215. doi:10.1093/nar/16.3.1215
Nishimura, G., Haga, N., Kitoh, H., Tanaka, Y., Sonoda, T., Kitamura, M., et al. (2005). The phenotypic spectrum of COL2A1 mutations. Hum. Mutat. 26, 36–43. doi:10.1002/humu.20179
Petrikin, J. E., Willig, L. K., Smith, L. D., and Kingsmore, S. F. (2015). Rapid whole genome sequencing and precision neonatology. Semin. Perinatol. 39, 623–631. doi:10.1053/j.semperi.2015.09.009
Rabbani, B., Mahdieh, N., Hosomichi, K., Nakaoka, H., and Inoue, I. (2012). Next-generation sequencing: impact of exome sequencing in characterizing Mendelian disorders. J. Hum. Genet. 57, 621–632. doi:10.1038/jhg.2012.91
Ravenscroft, G., Clayton, J. S., Faiz, F., Sivadorai, P., Milnes, D., Cincotta, R., et al. (2021). Neurogenetic fetal akinesia and arthrogryposis: genetics, expanding genotype-phenotypes and functional genomics. J. Med. Genet. 58, 609–618. doi:10.1136/jmedgenet-2020-106901
Recker, F., Zaniew, M., Böckenhauer, D., Miglietti, N., Bökenkamp, A., Moczulska, A., et al. (2015). Characterization of 28 novel patients expands the mutational and phenotypic spectrum of Lowe syndrome. Pediatr. Nephrol. 30, 931–943. doi:10.1007/s00467-014-3013-2
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J., and Kircher, M. (2019). CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894. doi:10.1093/nar/gky1016
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. doi:10.1038/nbt.1754
Saleh, S., Beyyumi, E., Al Kaabi, A., Hertecant, J., Barakat, D., Al Dhaheri, N. S., et al. (2021). Spectrum of neuro-genetic disorders in the United Arab Emirates national population. Clin. Genet. 100, 573–600. doi:10.1111/cge.14044
Schobers, G., Schieving, J. H., Yntema, H. G., Pennings, M., Pfundt, R., Derks, R., et al. (2022). Reanalysis of exome negative patients with rare disease: a pragmatic workflow for diagnostic applications. Genome Med. 14, 66. doi:10.1186/s13073-022-01069-z
Schoeb, D. S., Chernin, G., Heeringa, S. F., Matejas, V., Held, S., Vega-Warner, V., et al. (2010). Nineteen novel NPHS1 mutations in a worldwide cohort of patients with congenital nephrotic syndrome (CNS). Nephrol. Dial. Transpl. 25, 2970–2976. doi:10.1093/ndt/gfq088
Schoonen, M., Smuts, I., Louw, R., Elson, J. L., van Dyk, E., Jonck, L. M., et al. (2019). Panel-based nuclear and mitochondrial next-generation sequencing outcomes of an ethnically diverse pediatric patient cohort with mitochondrial disease. J. Mol. Diagn 21, 503–513. doi:10.1016/j.jmoldx.2019.02.002
Smith, L. D., Willig, L. K., and Kingsmore, S. F. (2015). Whole-exome sequencing and whole-genome sequencing in critically ill neonates suspected to have single-gene disorders. Cold Spring Harb. Perspect. Med. 6, a023168. doi:10.1101/cshperspect.a023168
Splinter, K., Adams, D. R., Bacino, C. A., Bellen, H. J., Bernstein, J. A., Cheatle-Jarvela, A. M., et al. (2018). Effect of genetic diagnosis on patients with previously undiagnosed disease. N. Engl. J. Med. 379, 2131–2139. doi:10.1056/NEJMoa1714458
Stark, Z., and Ellard, S. (2022). Rapid genomic testing for critically ill children: time to become standard of care? Eur. J. Hum. Genet. 30, 142–149. doi:10.1038/s41431-021-00990-y
Stark, Z., Tan, T. Y., Chong, B., Brett, G. R., Yap, P., Walsh, M., et al. (2016). A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet. Med. 18, 1090–1096. doi:10.1038/gim.2016.1
Takahashi, N., Hamada-Nakahara, S., Itoh, Y., Takemura, K., Shimada, A., Ueda, Y., et al. (2014). TRPV4 channel activity is modulated by direct interaction of the ankyrin domain to PI(4,5)P₂. Nat. Commun. 5, 4994. doi:10.1038/ncomms5994
Tanner, S. M., Schneider, V., Thomas, N. S., Clarke, A., Lazarou, L., and Liechti-Gallati, S. (1999). Characterization of 34 novel and six known MTM1 gene mutations in 47 unrelated X-linked myotubular myopathy patients. Neuromuscul. Disord. 9, 41–49. doi:10.1016/s0960-8966(98)00090-x
Telegrafi, A., Webb, B. D., Robbins, S. M., Speck-Martins, C. E., FitzPatrick, D., Fleming, L., et al. (2017). Identification of STAC3 variants in non-Native American families with overlapping features of Carey-Fineman-Ziter syndrome and Moebius syndrome. Am. J. Med. Genet. A 173, 2763–2771. doi:10.1002/ajmg.a.38375
Terhal, P. A., Nievelstein, R. J. A. J., Verver, E. J. J., Topsakal, V., van Dommelen, P., Hoornaert, K., et al. (2015). A study of the clinical and radiological features in a cohort of 93 patients with a COL2A1 mutation causing spondyloepiphyseal dysplasia congenita or a related phenotype. Am. J. Med. Genet. A 167A, 461–475. doi:10.1002/ajmg.a.36922
Thormann, A., Halachev, M., McLaren, W., Moore, D. J., Svinti, V., Campbell, A., et al. (2019). Flexible and scalable diagnostic filtering of genomic variants using G2P with Ensembl VEP. Nat. Commun. 10, 2373. doi:10.1038/s41467-019-10016-3
Tiffin, N. (2014). Unique considerations for advancing genomic medicine in African populations. Per Med. 11, 187–196. doi:10.2217/pme.13.105
Trujillano, D., Bertoli-Avella, A. M., Kumar Kandaswamy, K., Weiss, M. E., Köster, J., Marais, A., et al. (2017). Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur. J. Hum. Genet. 25, 176–182. doi:10.1038/ejhg.2016.146
Van Diemen, C. C., Kerstjens-Frederikse, W. S., Bergman, K. A., de Koning, T. J., Sikkema-Raddatz, B., van der Velde, J. K., et al. (2017). Rapid targeted genomics in critically ill newborns. Pediatrics 140, e20162854. doi:10.1542/peds.2016-2854
Waldrop, M. A., Boue, D. R., Sites, E., Flanigan, K. M., and Shell, R. (2017). Clinicopathologic conference: a newborn with hypotonia, cleft palate, micrognathia, and bilateral club feet. Pediatr. Neurol. 74, 11–14. doi:10.1016/j.pediatrneurol.2017.01.026
Wang, H., Qian, Y., Lu, Y., Qin, Q., Lu, G., Cheng, G., et al. (2020). Clinical utility of 24-h rapid trio-exome sequencing for critically ill infants. NPJ Genom Med. 5, 20. doi:10.1038/s41525-020-0129-0
Warejko, J. K., Tan, W., Daga, A., Schapiro, D., Lawson, J. A., Shril, S., et al. (2018). Whole exome sequencing of patients with steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol. 13, 53–62. doi:10.2215/CJN.04120417
Wilkinson, D. J., Barnett, C., Savulescu, J., and Newson, A. J. (2016). Genomic intensive care: should we perform genome testing in critically ill newborns? Arch. Dis. Child. Fetal Neonatal Ed. 101, F94–F98. doi:10.1136/archdischild-2015-308568
Willig, L. K., Petrikin, J. E., Smith, L. D., Saunders, C. J., Thiffault, I., Miller, N. A., et al. (2015). Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir. Med. 3, 377–387. doi:10.1016/S2213-2600(15)00139-3
Wright, C. F., Campbell, P., Eberhardt, R. Y., Aitken, S., Perrett, D., Brent, S., et al. (2023). Genomic diagnosis of rare pediatric disease in the United Kingdom and Ireland. N. Engl. J. Med. 388, 1559–1571. doi:10.1056/NEJMoa2209046
Wright, C. F., Fitzpatrick, D. R., and Firth, H. V. (2018). Paediatric genomics: diagnosing rare disease in children. Nat. Rev. Genet. 19, 325. doi:10.1038/nrg.2018.12
Yang, Y., Muzny, D. M., Xia, F., Niu, Z., Person, R., Ding, Y., et al. (2014). Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 312, 1870–1879. doi:10.1001/jama.2014.14601
Zaharieva, I. T., Sarkozy, A., Munot, P., Manzur, A., O'Grady, G., Rendu, J., et al. (2018). STAC3 variants cause a congenital myopathy with distinctive dysmorphic features and malignant hyperthermia susceptibility. Hum. Mutat. 39, 1980–1994. doi:10.1002/humu.23635
Keywords: clinical-exome, infant, diagnostic, South Africa, LMIC, implementation
Citation: Campbell L, Fredericks J, Mathivha K, Moshesh P, Coovadia A, Chirwa P, Dillon B, Ghoor A, Lawrence D, Nair L, Mabaso N, Mokwele D, Novellie M, Krause A and Carstens N (2023) The implementation and utility of clinical exome sequencing in a South African infant cohort. Front. Genet. 14:1277948. doi: 10.3389/fgene.2023.1277948
Received: 15 August 2023; Accepted: 23 October 2023;
Published: 09 November 2023.
Edited by:
Atanu Kumar Dutta, All India Institute of Medical Sciences, Kalyani (AIIMS Kalyani), IndiaReviewed by:
Volkan Okur, New York Genome Center, United StatesCopyright © 2023 Campbell, Fredericks, Mathivha, Moshesh, Coovadia, Chirwa, Dillon, Ghoor, Lawrence, Nair, Mabaso, Mokwele, Novellie, Krause and Carstens. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: N. Carstens, bmFkaWEuY2Fyc3RlbnNAbXJjLmFjLnph
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.