 Charlotte Cunningham-Rundles
Charlotte Cunningham-Rundles Jean-Laurent Casanova
Jean-Laurent Casanova Bertrand Boisson
Bertrand Boisson
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 11 January 2024
Sec. Genetics of Common and Rare Diseases
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1272912
This article is part of the Research TopicRecent Advances in Understanding The Genetics of Immunological DisordersView all 12 articles
Common variable immunodeficiency (CVID) is one of the most common symptomatic groups of inborn errors of immunity. In addition to infections resulting from insufficient levels of immune globulins and antibodies, many patients develop inflammatory or autoimmune conditions, which are associated with increased mortality. This aspect of CVID has been the focus of many studies, and dissecting the clinical phenotypes of CVID, has had the goal of providing biomarkers to identify these subjects, potentially at the time of diagnosis. With the application of whole exome (WES) and whole genome analyses, an increasing number of monogenic causes of CVID have been elucidated. From the standpoint of the practicing physician, an important question is whether the clinical phenotype, particularly the occurrence of autoinflammation of autoimmunity, might suggest the likelihood of identifying a causative mutation, and if possible the gene most likely to underlie CVID. We addressed this question in a patient group of 405 subjects diagnosed with CVID from one medical center.
Common variable immunodefciency (CVID) is one of the more frequently encountered immune defects in clinical practice, with an estimated incidence of about 1 in 20,000. The diagnosis is made in a male or female patient with reduced serum levels of IgG, along with IgA, and/or IgM with documented defects of antibody production to both protein and carbohydrate antigens, and the exclusion of other causes of hypogammaglobulinemia such as physiologic immaturity, medications, malignancy, or protein losses (Bonilla et al., 2016; Registry, 2016; Odnoletkova et al., 2018; Seidel et al., 2019). While considered genetic defects, most newly diagnosed patients are between the ages of 20 and 40 years old. (Resnick et al., 2012a; Gathmann et al., 2014; Odnoletkova et al., 2018). One of the clinical features of the CVID syndrome that has emerged is that about half of these patients have infections as the central manifestation, which can be successfully treated or prevented with antibiotics and immunoglobulins. However, the others also have various apparently non-infectious, autoimmune, autoinflammatory, neoplastic and/or lymphoproliferative manifestations, often associated with systemic immune activation (Wehr et al., 2008; Resnick et al., 2012b; Cols et al., 2016; Smith and Cunningham-Rundles, 2021; Ho and Cunningham-Rundles, 2022). Patients in the second group often have autoimmune or inflammatory features as the initial presentation and primary clinical manifestation, with less obvious susceptibility to significant infectious diseases; these subjects also have increased morbidity and mortality as compared to those with the infection-only phenotype (Chapel et al., 2008; Resnick et al., 2012a). A number of studies have probed reasons for the striking heterogeneity of this CVID patient pool (Wehr et al., 2008; Chapel and Cunningham-Rundles, 2009; Resnick et al., 2012a). These studies have sought biomarkers to identify these subjects, preferably at the time of diagnosis (Ho and Cunningham-Rundles, 2022). Some of these markers include identification of subjects with loss of peripheral isotype switched memory B cells, increased CD21low B cells (<10%), and/or reduced numbers of T cells, especially naïve CD4 T cells (Warnatz et al., 2002; Fevang et al., 2007; Sanchez-Ramon et al., 2008; Wehr et al., 2008; Malphettes et al., 2009; Mouillot et al., 2010). Other markers more recently defined include elevated markers of systemic immune activation: serum lipopolysaccharide binding protein (LBP), sCD14 (Barbosa et al., 2012; Litzman et al., 2012; Fraz et al., 2022) and more recently, serum zonulin and circulating bacterial DNA (Ho et al., 2021). These correlative biomarkers attempt to distinguish many patients with the infection-only clinical phenotype from those with more inflammatory complications, but they do not address the molecular mechanisms.
With the availability of whole exome (WES) and whole genome analyses (WGS), an increasing number of monogenic causes of the CVID phenotype have been elucidated, now accounting for about 25%–30% of subjects (Maffucci et al., 2016; Tuijnenburg et al., 2018; Abolhassani et al., 2020; Ramirez et al., 2021; Rojas-Restrepo et al., 2021). Several recent reports have described the results of genetic analyses of large CVID patient populations, with differences noted due to the location of the populations studied and ethnic background of the patients (Abolhassani et al., 2020; Rojas-Restrepo et al., 2021). The many genes identified in cohorts of subjects diagnosed with CVID, reflect the complex requirements of class switch recombination, B cell antigen signaling, activation, migration, long-term survival, and maturation and retention of antibody-secreting memory B cells into the plasma cell stage (Figure 1). From the standpoint of the practicing physician, an important question is whether the clinical phenotype suggests the possibility of identifying a causative mutation and if so, the gene(s) most likely to underlie the immune defect. Here we address this question in a large patient group from one medical center, encompassing an urban patient population on the East Coast of the United States.
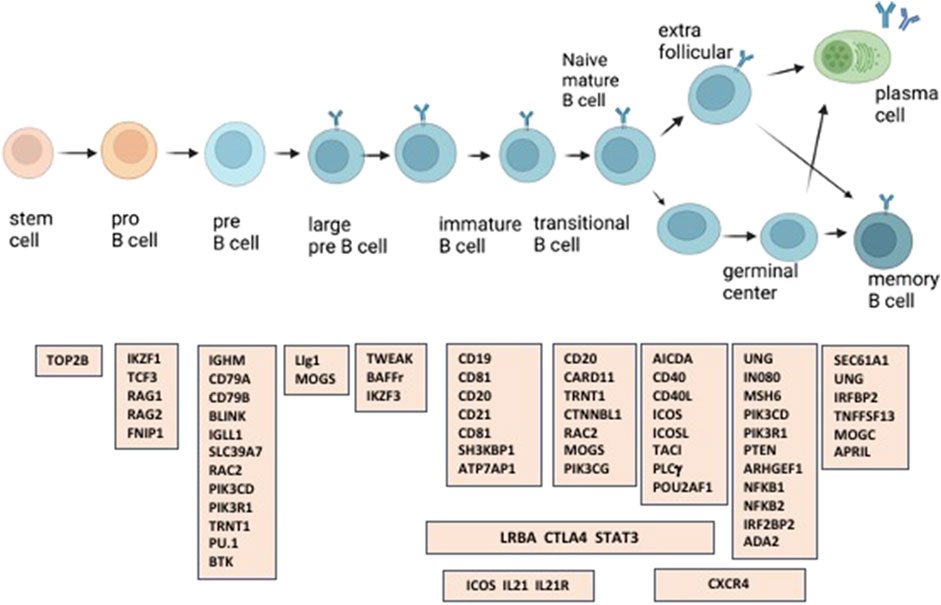
FIGURE 1. A number of the immune defects found in patients with CVID, are in genes involved in the generation and maturation of human B cells.
Subjects were seen in the Immune Deficiency program at the Icahn School of Medicine at Mount Sinai. Patients were diagnosed with CVID using established criteria, including serum IgG and IgA and/or IgM deficiency with proven loss of antibody production (Bonilla et al., 2016; Picard et al., 2018; Seidel et al., 2019). Immunologic and clinical histories were collected from the clinical record and selected manifestations of inflammatory/autoimmune complications were recorded. For purposes of the current study, these sometimes partly overlapping medical conditions are autoimmunity, interstitial lung disease, granulomatous disease identified in one or more tissues, cancer, lymphoma, significant gastrointestinal disease, and previous splenectomy. Ethical permission for these studies was obtained from the Mount Sinai Institutional Review Board, and Informed consent was obtained from all individuals and/or their legal guardians.
Whole exome sequencing: Genetic evaluation was done by whole exome sequencing (WES) as previously described (Maffucci et al., 2016; Picard et al., 2018; Maffucci et al., 2019). For this genomic DNA was extracted from peripheral blood mononuclear cells and sheared with a Covaris S2 Ultrasonicator. An adaptor-ligated library was prepared with the Paired-End Sample Prep kit V1 (Illumina). Exome capture was performed with the SureSelect Human All Exon kit (Agilent Technologies). Massively parallel sequencing was performed on a HiSeq 2,500 (Illumina), which generates 100-base reads. Sequences were aligned for variant calling and annotation with the human genome reference sequence (hg19 build) using BWA aligner (Li and Durbin, 2009). Downstream processing was performed with the Genome analysis toolkit (GATK) (McKenna et al., 2010), SAMtools (Li et al., 2009), and Picard Tools (http://picard.sourceforge.net/). A GATK UnifiedGenotyper and a GATK IndelGenotyperV2 were used to identify substitution and indel variant calls, respectively. Calls with a read coverage of ≤2x and a Phred-scaled single-nucleotide polymorphism (SNP) quality of ≤20 were filtered out. All variants were annotated with the GATK Genomic Annotator (Broad Institute). Heterozygous and homozygous variants were excluded if the allele frequencies in the general population were greater than 0.01% or 1.0%, respectively, in the Exome Aggregation Consortium database (ExAC, Broad Institute) and Genome Aggregation Database (gnomAD, Broad Institute) in respect with genetic model tested. This filtering strategy excluded polymorphic variants from consideration. Familial segregation was studied when samples were available. Other candidate mutations were confirmed by examining read alignment in the Integrated Genomics Viewer (IGV; Broad Institute). All confirmed mutations were subsequently analyzed using computational predictors of mutation severity including Sift (Ng and Henikoff, 2003), Polyphen 2 (Adzhubei et al., 2010). and Combined Annotation Dependent Depletion (CADD) (Kircher et al., 2014) and were compared with the gene-specific mutation significance cutoff (MSC) (Itan et al., 2016). Variants with CADD scores below the gene-specific MSC were excluded with the exception of CXCR4, LIG1, LRBA and NFKB2, 4 genes with a very high MSC (>32), but known to be causal of CVID. Confirmed variations were also screened through the Human Gene Mutation Database (Stenson et al., 2003) to identify published disease-associated variations. In a number of cases, the variants selected were tested for pathogenicity, and if not, categorized (as likely benign, VUS - Variant of Uncertain Significance, or likely pathogenic.). The pathogenicity of all disease attributable gene variants was evaluated using the updated guideline for interpretation of molecular sequencing by the American College of Medical Genetics and Genomics (ACMG) considering the allele frequency, computational data, immunological/functional data, familial segregation and parental data and clinical phenotyping (Richards et al., 2015).
Using a targeting panel of genes: More recently, patient exomes were also examined for mutations in 429 genes associated with a primary immune deficiency disease (Invitae Diagnostics), when faster results were needed and insurance or other payment for this service was available. For inheritance questions, the targeted sequence method was used in particular as it has the Clinical Laboratory Improvement Amendments (CLIA) certification needed for clinical decision analysis. As the targeted panel contains only a defined panel of genes, the above WES method was also used on these same samples so that the data could be verified, and so that additional queries for new genes could be undertaken at a later date.
For evaluating the significance of genetics as related to clinical observations, Graphpad Prism was used; a p-value of less than 0.05 was considered significant.
The Mount Sinai cohort of 405 genetically-tested CVID patients included 26 cases in which a family history was noted (Table 1). The age range of patients was 5–77 years, with median age of 44 years; 187 were female and 218 were male. Of these subjects, most were of European descent, 22 were Hispanic, 12 were Black, and 4 were Asian.
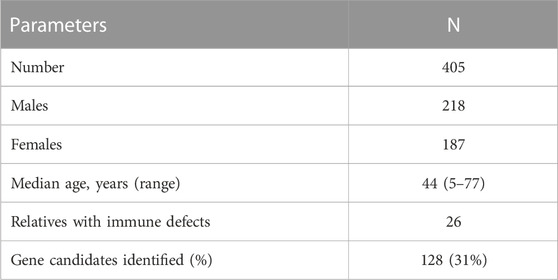
TABLE 1. CVID subjects.
In this cohort, 125 of 405 subjects (31%) had mutations considered deleterious while the remainder (280) had no clear genetic abnormality identified. As reported previously, around 10% of our patients (42 subjects) have variants in the TNFRSF13B/TACI gene (Transmembrane Activator and CAML Interactor) most of these known to be functionally deleterious (Salzer and Grimbacher, 2021). These included compound heterozygous mutations in 5 subjects, and homozygous mutations in one subject. These variants would be considered associated but not causative of CVID. (Table 2). Sixteen other subjects had autosomal dominant (AD) NFKB1 deficiency (Nuclear Factor Kappa B Subunit 1), and 7 others had NFKB2 (Nuclear Factor Kappa B Subunit 2) defects, both viewed as causative of this immune defect (Chen et al., 2013; Tuijnenburg et al., 2018; Li et al., 2021). More than one subject had mutations in heterozygous genes previously found in subjects with a CVID phenotype: IRF2BP2, CTLA4 and IKZF1 (in 6 subjects each), TCF3 (in 5 subjects), BACH2 (in 4 subjects), and in STAT3, and PIK3CD (3 subjects for each). Three other subjects had autosomal recessive (AR) deficiency of LRBA. Four adult subjects with infections, autoimmunity and mild retardation had mutations in KMT2D (Lysine Methyltransferase 2), a gene associated with Kabuki syndrome. Two sisters and the son of one of them, with no warts and moderate neutropenia, but severe autoimmune thrombocytopenia and autoimmune hemolytic anemia, had frameshift mutations in CXCR4 (C-X-C Motif Chemokine Receptor 4), a gene associated with WHIM syndrome (warts, hypogammaglobulinemia, infections, and myelokathexis) (Maffucci et al., 2016; Abolhassani et al., 2020). Note that as in other reports, genes identified with previously un-identified X-immune linked defects were also noted in this cohort, BTK, CD40L, IKBKG and WAS (Table 2). Complicating the genetics is that in 15 subjects studied, more than one heterozygous variant, predicted to be deleterious, was identified. This included 8 subjects with at least one TACI variants, but additional variants in other autosomal dominant or recessive genes were also noted, for example, in TBX1, TCF3, IL10-RA, NFKB2, NBAS, RAG1, RAG2 or a DiGeorge chromosome 22q deletion (Table 3). Further information related to minor allele frequency, and predicted deleterious effects on the selected variants are included in Methods and Supplementary Table S1.
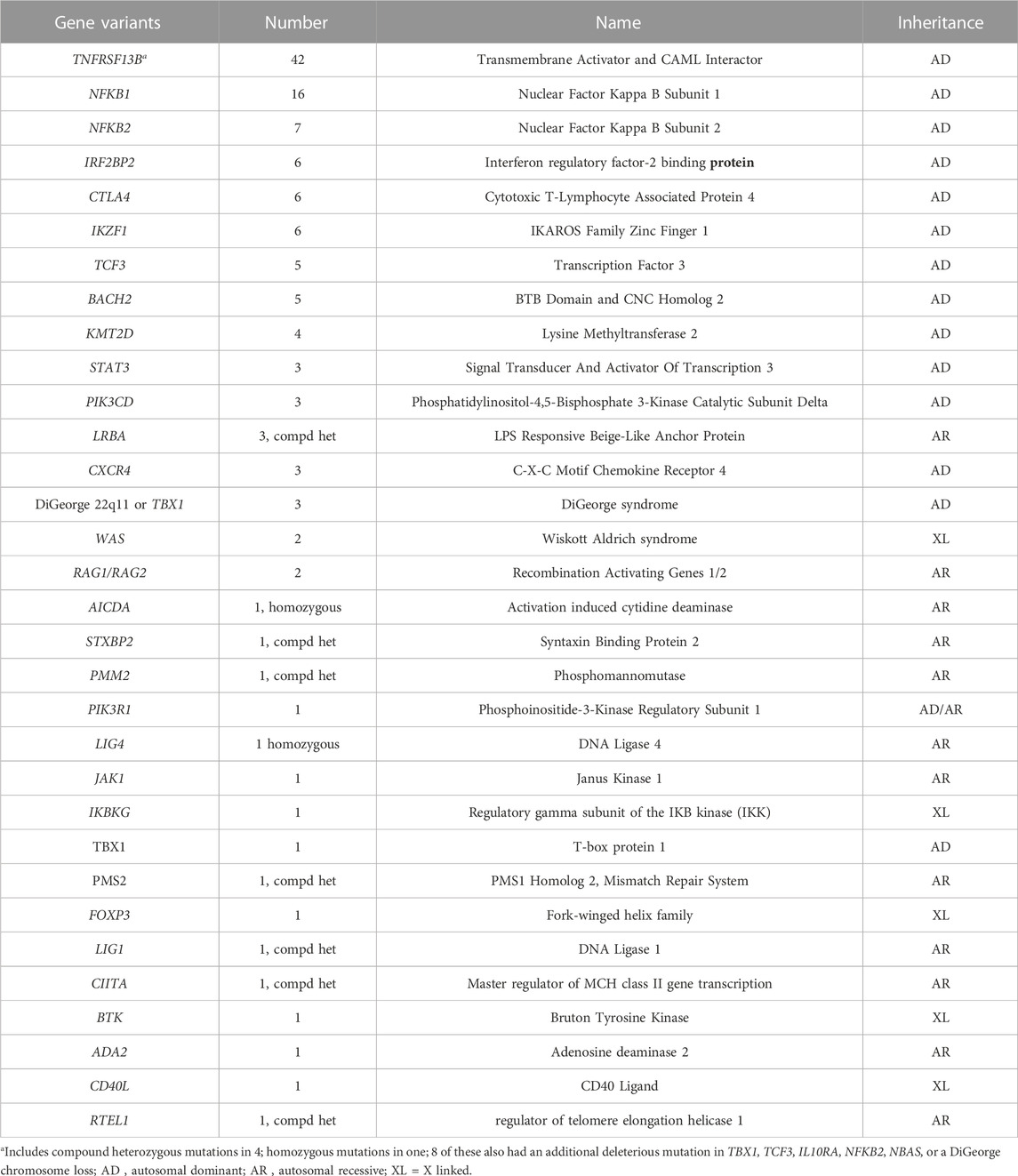
TABLE 2. Gene variants identified - 125 subjects (31% of the group).
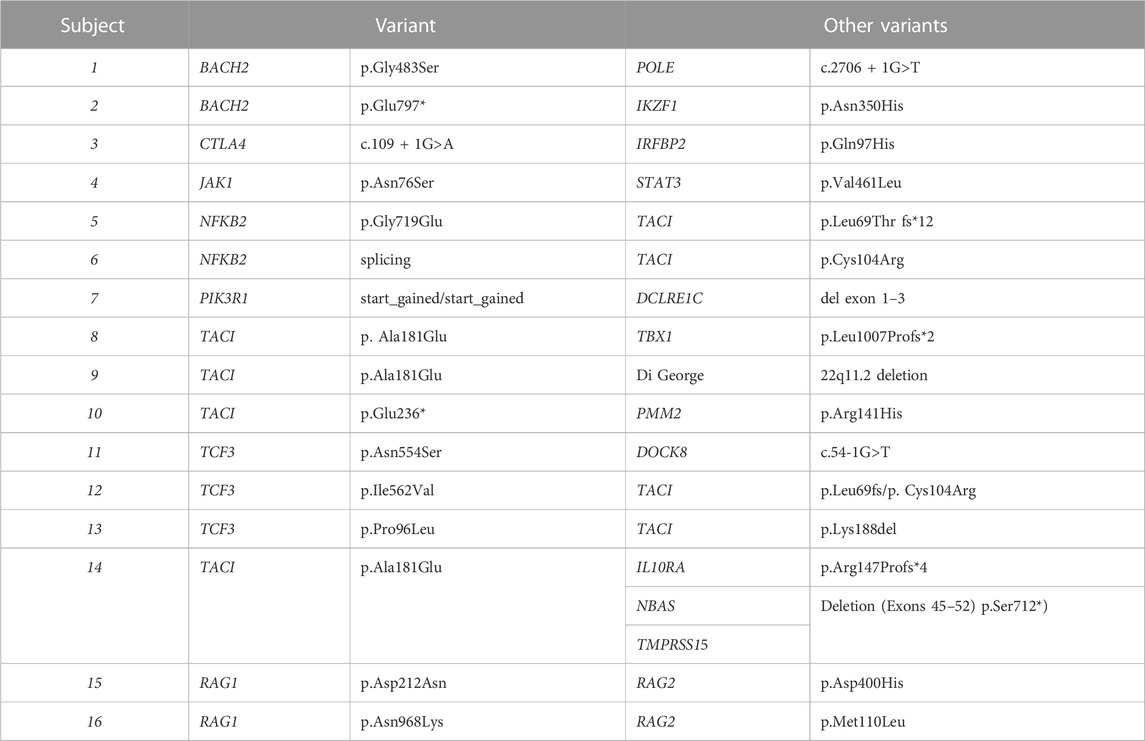
TABLE 3. Subjects with more than one gene variant.
We then examined if subjects with autoinflammatory, autoimmune, lymphoproliferative, neoplastic, granulomatous infiltrates, and/or gastrointestinal complications were more likely to have mutations in one or more of the genes identified in CVID, in contrast to others for whom a gene was not identified. Table 4 outlines the results for this cohort, dividing subjects according to whether or not a gene defect was identified in subjects with autoimmunity, significant pulmonary, gastrointestinal disease or liver disease, biopsy-demonstrated granulomatous disease, previous splenectomy, cancer, or lymphoma. Various forms of autoimmunity were noted in 151 subjects (37%) of the 405 group genetically tested, with no sex predominance. Of the 125 subjects with genes identified, 59 (47%) had autoimmunity, while of the larger group of 281 subjects with no gene noted, 32% had autoimmunity, suggesting some enrichment of this clinical feature in those with any known gene association; however these differences were not statistically significant. We also examined the types of autoimmunity in each group, in those with or without identified gene variants. Tables 5, 6 show these data. However, for both sets of patients, the most prominent autoimmune conditions were cytopenias, particularly thrombocytopenia, hemolytic anemia (or both, i.e., Evan’s syndrome) or, in fewer numbers, neutropenia (Figure 2). The mutations found in those with autoimmunity are included in Supplementary Table S2. Note that of these, 14 had mutations in the TACI gene (2 with compound heterozygous variants, one with homozygosity) and 4 others were in association with additional deleterious variants). Five of the subjects with autoimmunity had mutations in NFKB1, 4 had variants in CTLA4, IKZF1, or in STAT3, and with other genes noted in other subjects.
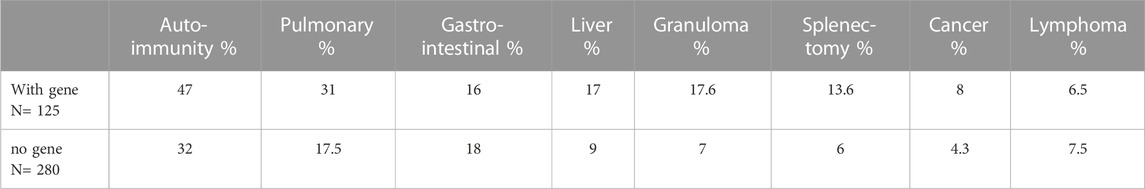
TABLE 4. Percentage of complications in each group.
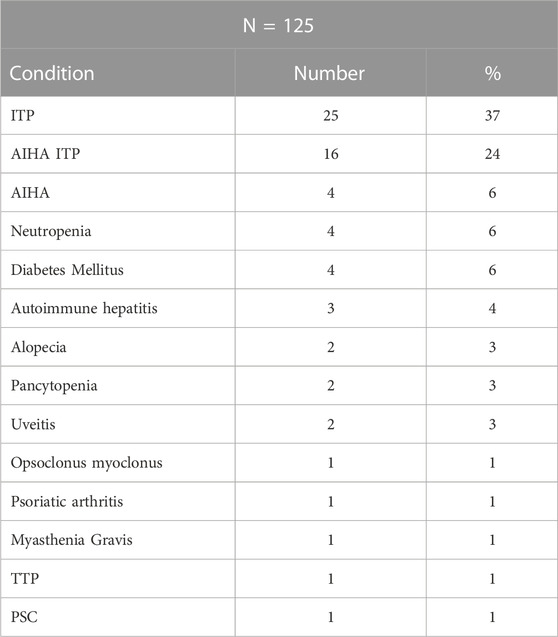
TABLE 5. Autoimmunity: Gene identified.
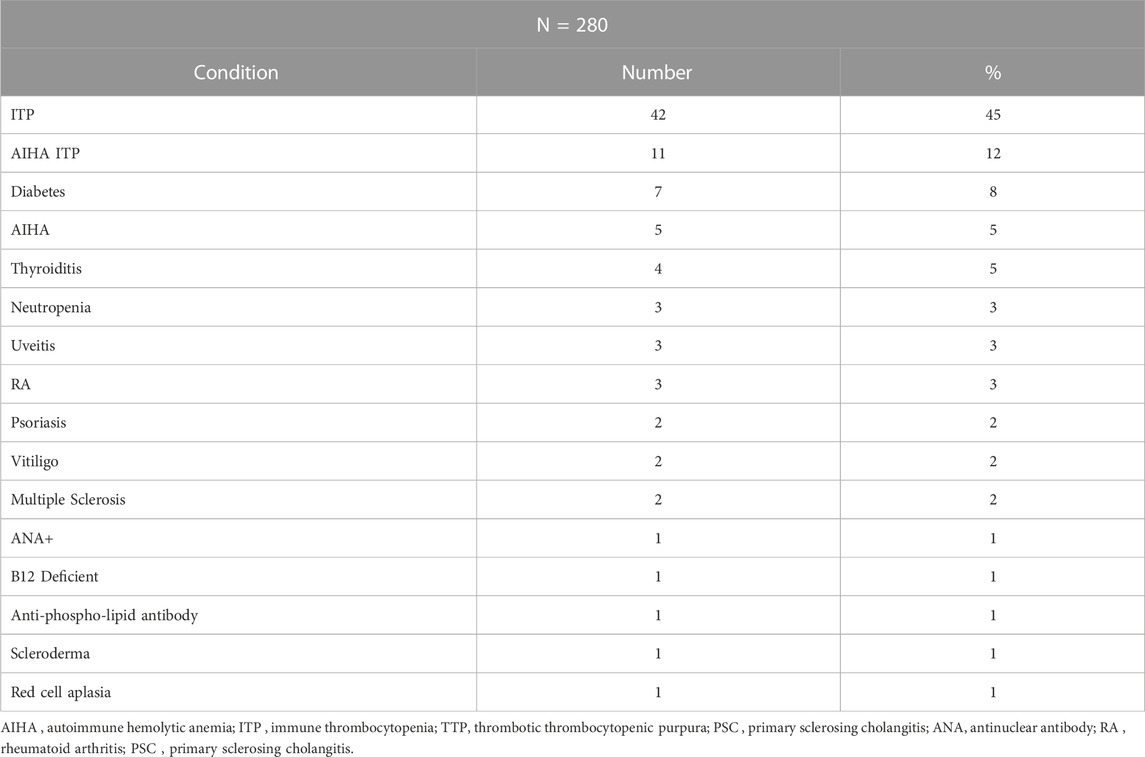
TABLE 6. Autoimmunity: No gene identified.
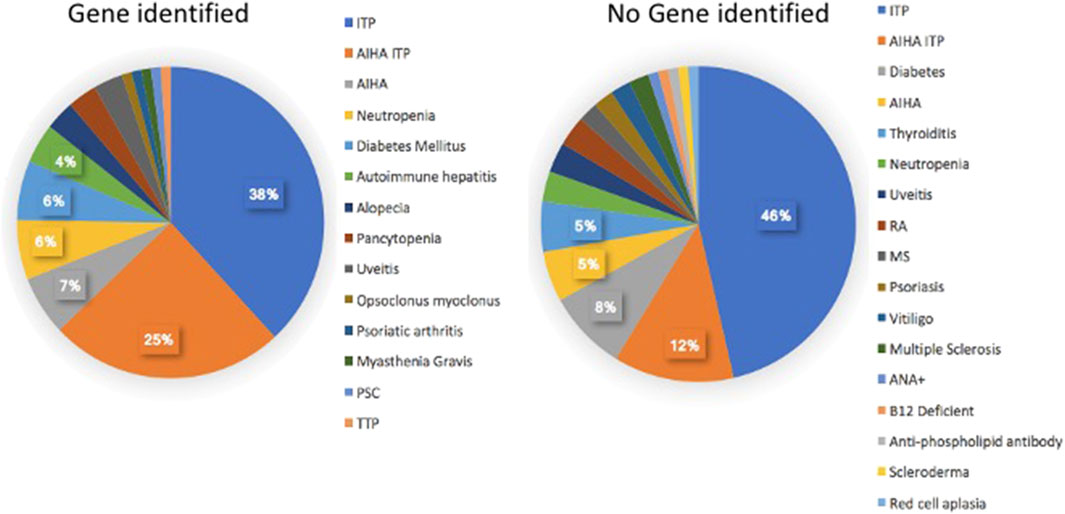
FIGURE 2. The kinds of autoimmune conditions found in patients with, and without identified genetic defects are similar.
Significant pulmonary disease (interstitial lung disease, numerous nodules, and/or known granulomatous disease or respiratory failure) was observed in 39 subjects. Of these (31%) had an identified gene defect, as compared to 17.5% of those with no gene noted, suggesting a slight but not significant enrichment of significant respiratory disease in those with an identified gene, p = 0.053. Of those with significant lung disease, 12 subjects had a TACI variant, 4 had NFKB2, 4 had NFKB1 and 3 others had PI3KCD variants. Mutations in the genes, BACH2, KMT2D, LRBA, LIG4, STAT3 FOXP3 and CASP8 were identified in other patients as outlined (Supplementary Table S3). Thirteen of these subjects had been diagnosed as having granulomatous lymphocytic infiltrates in the lungs (GLILD) (Bates et al., 2004). Overall, the tissue diagnosis of granulomatous disease (in lymph nodes, lung skin, or liver) was noted in 22 (17.7%) of the subjects with defects in known genes (BACH2, CTLA4, KMT2D, LRBA, PIK3R1, STAT3, TACI, TCF3 or WAS.) This was significantly different from the 20 other subjects (7.5%) with granulomatous infiltrations) with no known gene defect, (p = 0.046).
The diagnosis of cancer or lymphoma was made in 18 patients (14.5%) with an identified gene defect, including 9 lymphoid malignancies and 6 other cancers, with the genetic changes noted for each (Table 7). Cancer or lymphoma was also diagnosed in 32 other subjects (11%) with no known genetic variants (not significantly different.) Gastrointestinal and/or liver disease were noted in similar proportions in each group, with the genes noted in Supplementary Table S4. Splenectomy, usually done for treatment of cytopenias, had been performed in 34 patients overall, with 17 (13.7%) of these in subjects with known genetic variants, as opposed to 17 others (6%) with no known genetic association (not significantly different.) Of the splenectomized subjects, 7 had TACI gene variants; others included NFKB1 in 4, LRBA, TCF3, STAT3, NFKB2, CTLA4 and RAG1/RAG2.
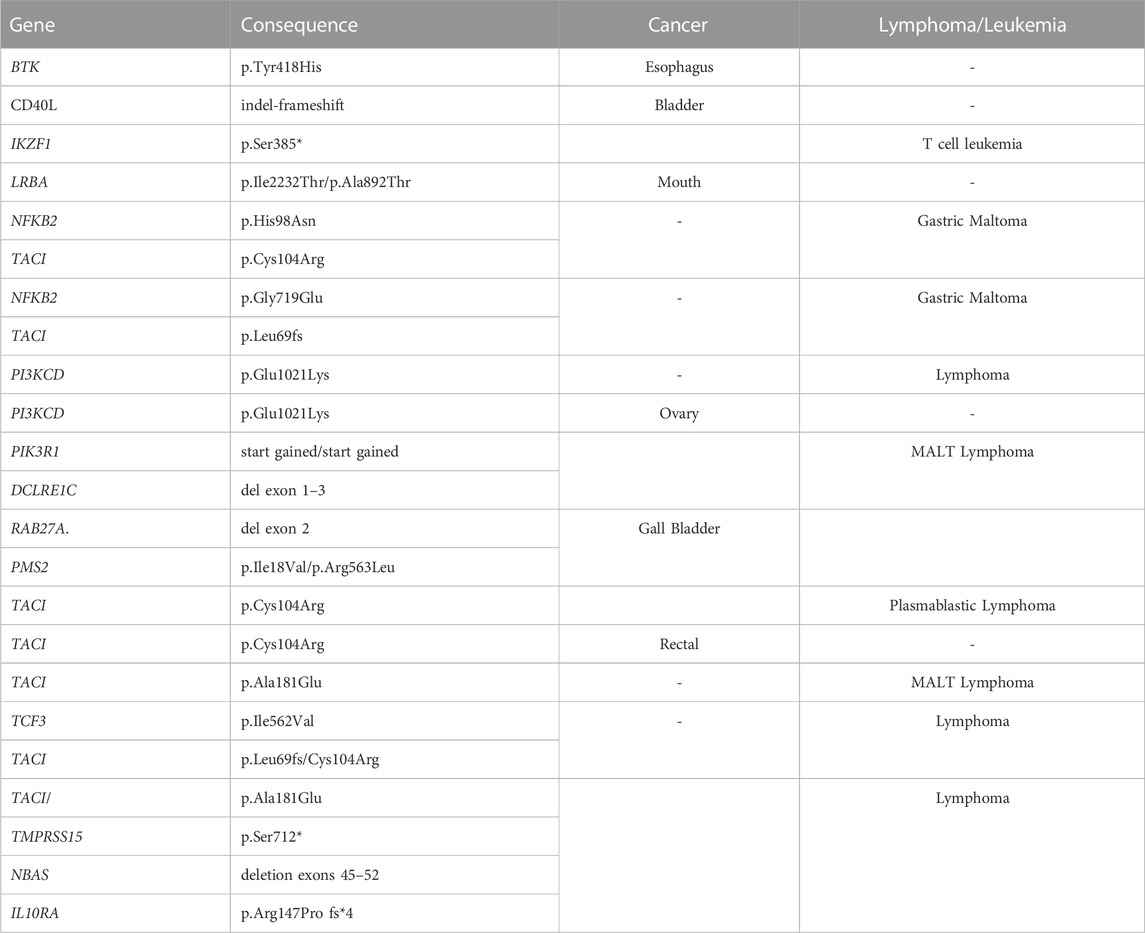
TABLE 7. Gene defects in cancers and lymphoma.
We also considered if those with genetic variants might have specific types of unusual infections. However, a history of infections with Herpes Zoster, Candida sp, Giardia, Clostridia difficile, Helicobacter pylori, Norovirus, Campylobacter, Herpes simplex, or more unusual infections with atypical mycobacteria, mycoplasma, histoplasmosis, or cryptococcus, were seen in subjects with and without genetic variants.
A number of previous studies have outlined the clinical phenotypes of large patient groups with antibody deficiency who have mutations in selected genes, including the TACI gene (Salzer et al., 2005; Zhang et al., 2007; Salzer et al., 2009), CTLA4 (Schwab et al., 2018), NFKB1 (Lorenzini et al., 2020), NFKB2 (Klemann et al., 2019), STAT3 (Fabre et al., 2019), PI3KCD (Jamee et al., 2020), or LRBA (Habibi et al., 2019). These studies describe the infectious, autoimmune and inflammatory characteristics of patients with these specific inborn errors of immunity. Here, the genetic analyses of CVID patients from one large cohort were examined to determine if the clinical complications might lead the physician to suspect a genetic defect in one or more of the previously established causal genes. In the current cohort of 405 subjects, 125 (31%) had an identifiable causative or associated genetic variant, similar to other investigated cohorts (Abolhassani et al., 2020; Rojas-Restrepo et al., 2021), however leaving the majority of patients without a known genetic cause. Autoimmunity was one of the commonest conditions for this group of patients with 37% of the patients having one or more of these conditions. Of these, 47% of them carried a predicted deleterious variant, while for those with no gene noted, 32% had autoimmunity. The autoimmune conditions noted were similar for each group, with cytopenias being the most prevalent manifestation, resulting in splenectomy in a number of subjects. While patients with mutations in CTLA4, IKZF1, STAT3 and LRBA were in the autoimmune group, the largest number had variants in the TACI gene. While variants in the TACI gene are not thought to be disease causing, they are commonly associated with autoimmunity in CVID (Salzer et al., 2005; Zhang et al., 2007), possibly explaining this enrichment. If subjects with a TACI variant are excluded, 35% of subjects with a known gene were noted to have autoimmunity, more similar to those for whom no gene was identified (at 32%) as illustrated (Figure 3) The autoimmune cytopenia (ITP or AIHA) resulted in splenectomy in 34 patients, and 7 of the 17 with a known gene, had a TACI variant. Significant respiratory disease was also more common in those subjects with a known gene defect (at 31%). Excluding the 8 subjects with a TACI variant, 25% of these subjects had this complication, more similar to those with no known genetic background (17.5%). Granulomatous disease was overall, significantly enriched in those with genes identified. In this group, 17.6% had this complication if they had TACI variants; if these are excluded, the percentage was 14%, as opposed to 7% of those with no noted genes. Other complications, such as gastrointestinal, substantial liver disease or cancer appeared in subjects with and without identified genetic defects in similar numbers (Figure 3).
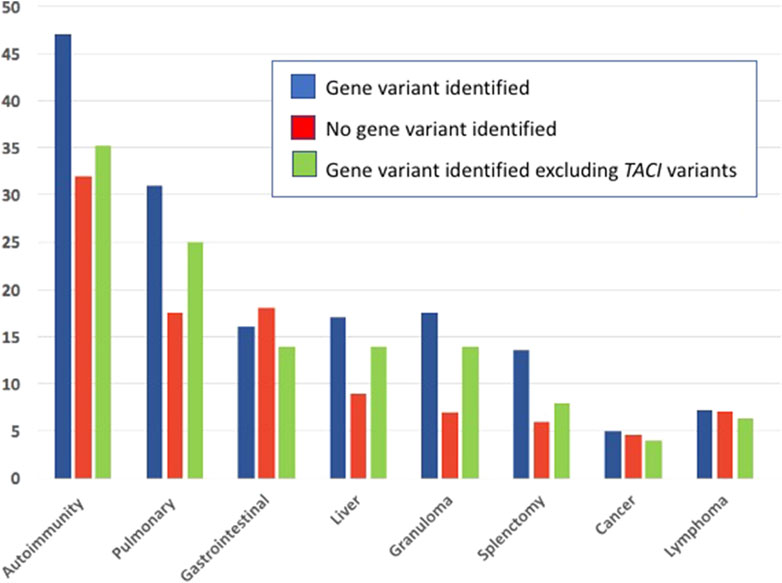
FIGURE 3. The medical complications noted in subjects with identified genetic defects, those with no known gene defects and subjects with a gene variant, but minus those with defects in the TNFRSF13B TACI receptor.
As the data presented here was gathered over a decade, a question that might arise if the DNA samples tested over time, using WES or the targeted panel, would lead to different results. We did not find this difficulty, but using the two methods led to confirmation. In addition, the accumulation of genetic data obtained by WES on all samples, also allowed for continued surveying for newly reported mutational differences. This allowed for updating as new genes contributing to the CVID phenotype were identified, and allowed all samples to be examined by the same parameters. The targeting panel was particularly useful for rapid analysis or inheritance questions, but even in these cases, WES was still performed. Limitations of this study include the fairly narrow demographic background of the subjects referred, a known reason for genetic differences in any CVID patient population (Abolhassani et al., 2020; Rojas-Restrepo et al., 2021), and the possibility of incomplete clinical information as these data were collected over varying periods of time. A further limitation is that confirmation of the pathogenicity of the identified variants depends on previous publications, in vitro assays where available, and the genetic methods used to assign the likelihood of a variant of dexterous variant exerting a deleterious change.
Overall, we conclude that CVID subjects with currently identifiable gene variants, either associated with or causative of this immune defect, appear to have an increased numbers of autoimmune manifestations, more significant respiratory disease and granulomatous changes in pathology; some of these differences can be attributed to co-existence of TACI variants as a genetic modifier. However, numerous other patients with no genetic basis yet discovered, have similar medical histories. It is possible that aside from genetics, these different clinical manifestations result from metabolic, environmental factors or epigenetic causes (Del Pino-Molina et al., 2019; Jorgensen et al., 2019; Macpherson et al., 2019; Ho et al., 2021; Rodriguez-Ubreva et al., 2022; Macpherson et al., 2023). While the majority of patients who carry the “CVID” diagnosis do not yet have a clarified molecular cause, the genetic discoveries in antibody defects continue to reveal the complex immunologic pathways needed to initiate and sustain normal B cell development and the long-term maintenance of B cell memory. With further exploration, more digenic or even polygenic causes of CVID are likely to be dissected, considering the intersecting immunologic pathways.
The datasets presented in this study are available upon request.
The studies involving humans were approved by the Mount Sinai School of Medicine. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participantsʼ legal guardians/next of kin.
CC-R: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. J-LC: Data curation, Funding acquisition, Investigation, Methodology, Resources, Software, Supervision, Writing–review and editing. BB: Formal Analysis, Funding acquisition, Investigation, Methodology, Resources, Software, Supervision, Validation, Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was partially supported by the NIH NIAID, AI-061093, AI-08603. The Laboratory of Human Genetics of Infectious Diseases was supported by the Howard Hughes Medical Institute, the Rockefeller University, the St. Giles Foundation, the National Institutes of Health (NIH) (P01AI061093), the French National Research Agency (ANR) under the “Investments for the Future” program (ANR-10-IAHU-01), the French Foundation for Medical Research (FRM; EQU201903007798), the Square Foundation, Grandir—Fonds de solidarité pour l’enfance, the Fondation du Souffle, the SCOR Corporate Foundation for Science, Institut National de la Santé et de la Recherche Médicale (INSERM), and the University of Paris Cité.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1272912/full#supplementary-material
Abolhassani, H., Hammarstrom, L., and Cunningham-Rundles, C. (2020). Current genetic landscape in common variable immune deficiency. Blood 135 (9), 656–667. doi:10.1182/blood.2019000929
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7 (4), 248–249. doi:10.1038/nmeth0410-248
Barbosa, R. R., Silva, S. P., Silva, S. L., Tendeiro, R., Melo, A. C., Pedro, E., et al. (2012). Monocyte activation is a feature of common variable immunodeficiency irrespective of plasma lipopolysaccharide levels. Clin. Exp. Immunol. 169 (3), 263–272. doi:10.1111/j.1365-2249.2012.04620.x
Bates, C. A., Ellison, M. C., Lynch, D. A., Cool, C. D., Brown, K. K., and Routes, J. M. (2004). Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J. Allergy Clin. Immunol. 114 (2), 415–421. doi:10.1016/j.jaci.2004.05.057
Bonilla, F. A., Barlan, I., Chapel, H., Costa-Carvalho, B. T., Cunningham-Rundles, C., de la Morena, M. T., et al. (2016). International consensus document (ICON): common variable immunodeficiency disorders. J. Allergy Clin. Immunol. Pract. 4 (1), 38–59. doi:10.1016/j.jaip.2015.07.025
Chapel, H., and Cunningham-Rundles, C. (2009). Update in understanding common variable immunodeficiency disorders (CVIDs) and the management of patients with these conditions. Br. J. Haematol. 145 (6), 709–727. doi:10.1111/j.1365-2141.2009.07669.x
Chapel, H., Lucas, M., Lee, M., Bjorkander, J., Webster, D., Grimbacher, B., et al. (2008). Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood 112 (2), 277–286. doi:10.1182/blood-2007-11-124545
Chen, K., Coonrod, E. M., Kumanovics, A., Franks, Z. F., Durtschi, J. D., Margraf, R. L., et al. (2013). Germline mutations in NFKB2 implicate the noncanonical NF-κB pathway in the pathogenesis of common variable immunodeficiency. Am. J. Hum. Genet. 93 (5), 812–824. doi:10.1016/j.ajhg.2013.09.009
Cols, M., Rahman, A., Maglione, P. J., Garcia-Carmona, Y., Simchoni, N., Ko, H. M., et al. (2016). Expansion of inflammatory innate lymphoid cells in patients with common variable immune deficiency. J. Allergy Clin. Immunol. 137 (4), 1206–1215. doi:10.1016/j.jaci.2015.09.013
Del Pino-Molina, L. R.-U. J., Torres Canizales, J., Coronel-Díaz, M., Kulis, M., Martín-Subero, J. I., van der Burg, M., et al. (2019). Impaired CpG demethylation in common variable immunodeficiency associates with B cell phenotype and proliferation rate. Front. Immunol. 10, 878. doi:10.3389/fimmu.2019.00878
Fabre, A., Marchal, S., Barlogis, V., Mari, B., Barbry, P., Rohrlich, P. S., et al. (2019). Clinical aspects of STAT3 gain-of-function germline mutations: a systematic Review. J. Allergy Clin. Immunol. Pract. 7 (6), 1958–1969. doi:10.1016/j.jaip.2019.02.018
Fevang, B., Yndestad, A., Sandberg, W. J., Holm, A. M., Muller, F., Aukrust, P., et al. (2007). Low numbers of regulatory T cells in common variable immunodeficiency: association with chronic inflammation in vivo. Clin. Exp. Immunol. 147 (3), 521–525. doi:10.1111/j.1365-2249.2006.03314.x
Fraz, M. S. A., Michelsen, A. E., Moe, N., Aalokken, T. M., Macpherson, M. E., Nordoy, I., et al. (2022). Raised serum markers of T cell activation and exhaustion in granulomatous-lymphocytic interstitial lung disease in common variable immunodeficiency. J. Clin. Immunol. 42 (7), 1553–1563. doi:10.1007/s10875-022-01318-1
Gathmann, B., Mahlaoui, N., Ceredih, , Gerard, L., Oksenhendler, E., Warnatz, K., et al. (2014). Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J. Allergy Clin. Immunol. 134 (1), 116–126. doi:10.1016/j.jaci.2013.12.1077
Habibi, S., Zaki-Dizaji, M., Rafiemanesh, H., Lo, B., Jamee, M., Gamez-Diaz, L., et al. (2019). Clinical, immunologic, and molecular spectrum of patients with LPS-responsive beige-like anchor protein deficiency: a systematic Review. J. Allergy Clin. Immunol. Pract. 7 (7), 2379–2386. doi:10.1016/j.jaip.2019.04.011
Ho, H. E., and Cunningham-Rundles, C. (2022). Seeking relevant biomarkers in common variable immunodeficiency. Front. Immunol. 13, 857050. doi:10.3389/fimmu.2022.857050
Ho, H. E., Radigan, L., Bongers, G., El-Shamy, A., and Cunningham-Rundles, C. (2021). Circulating bioactive bacterial DNA is associated with immune activation and complications in common variable immunodeficiency. JCI Insight 6 (19), e144777. doi:10.1172/jci.insight.144777
Itan, Y., Shang, L., Boisson, B., Ciancanelli, M. J., Markle, J. G., Martinez-Barricarte, R., et al. (2016). The mutation significance cutoff: gene-level thresholds for variant predictions. Nat. Methods 13 (2), 109–110. doi:10.1038/nmeth.3739
Jamee, M., Moniri, S., Zaki-Dizaji, M., Olbrich, P., Yazdani, R., Jadidi-Niaragh, F., et al. (2020). Clinical, immunological, and genetic features in patients with activated PI3Kδ syndrome (APDS): a systematic Review. Clin. Rev. Allergy Immunol. 59 (3), 323–333. doi:10.1007/s12016-019-08738-9
Jorgensen, S. F., Fevang, B., and Aukrust, P. (2019). Autoimmunity and inflammation in CVID: a possible crosstalk between immune activation, gut microbiota, and epigenetic modifications. J. Clin. Immunol. 39 (1), 30–36. doi:10.1007/s10875-018-0574-z
Kircher, M., Witten, D. M., Jain, P., O'Roak, B. J., Cooper, G. M., and Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46 (3), 310–315. doi:10.1038/ng.2892
Klemann, C., Camacho-Ordonez, N., Yang, L., Eskandarian, Z., Rojas-Restrepo, J. L., Frede, N., et al. (2019). Clinical and immunological phenotype of patients with primary immunodeficiency due to damaging mutations in NFKB2. Front. Immunol. 10, 297. doi:10.3389/fimmu.2019.00297
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25 (16), 2078–2079. doi:10.1093/bioinformatics/btp352
Li, J., Lei, W. T., Zhang, P., Rapaport, F., Seeleuthner, Y., Lyu, B., et al. (2021). Biochemically deleterious human NFKB1 variants underlie an autosomal dominant form of common variable immunodeficiency. J. Exp. Med. 218 (11), e20210566. doi:10.1084/jem.20210566
Litzman, J., Nechvatalova, J., Xu, J., Ticha, O., Vlkova, M., and Hel, Z. (2012). Chronic immune activation in common variable immunodeficiency (CVID) is associated with elevated serum levels of soluble CD14 and CD25 but not endotoxaemia. Clin. Exp. Immunol. 170 (3), 321–332. doi:10.1111/j.1365-2249.2012.04655.x
Lorenzini, T., Fliegauf, M., Klammer, N., Frede, N., Proietti, M., Bulashevska, A., et al. (2020). Characterization of the clinical and immunologic phenotype and management of 157 individuals with 56 distinct heterozygous NFKB1 mutations. J. Allergy Clin. Immunol. 146 (4), 901–911. doi:10.1016/j.jaci.2019.11.051
Macpherson, M. E., Halvorsen, B., Yndestad, A., Ueland, T., Mollnes, T. E., Berge, R. K., et al. (2019). Impaired HDL function amplifies systemic inflammation in common variable immunodeficiency. Sci. Rep. 9 (1), 9427. doi:10.1038/s41598-019-45861-1
Macpherson, M. E., Skarpengland, T., Hov, J. R., Ranheim, T., Vestad, B., Dahl, T. B., et al. (2023). Increased plasma levels of triglyceride-enriched lipoproteins associate with systemic inflammation, lipopolysaccharides, and gut dysbiosis in common variable immunodeficiency. J. Clin. Immunol. 43, 1229–1240. doi:10.1007/s10875-023-01475-x
Maffucci, P., Bigio, B., Rapaport, F., Cobat, A., Borghesi, A., Lopez, M., et al. (2019). Blacklisting variants common in private cohorts but not in public databases optimizes human exome analysis. Proc. Natl. Acad. Sci. U. S. A. 116 (3), 950–959. doi:10.1073/pnas.1808403116
Maffucci, P., Filion, C. A., Boisson, B., Itan, Y., Shang, L., Casanova, J. L., et al. (2016). Genetic diagnosis using whole exome sequencing in common variable immunodeficiency. Front. Immunol. 7, 220. doi:10.3389/fimmu.2016.00220
Malphettes, M., Gerard, L., Carmagnat, M., Mouillot, G., Vince, N., Boutboul, D., et al. (2009). Late-onset combined immune deficiency: a subset of common variable immunodeficiency with severe T cell defect. Clin. Infect. Dis. 49 (9), 1329–1338. doi:10.1086/606059
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20 (9), 1297–1303. doi:10.1101/gr.107524.110
Mouillot, G., Carmagnat, M., Gerard, L., Garnier, J. L., Fieschi, C., Vince, N., et al. (2010). B-cell and T-cell phenotypes in CVID patients correlate with the clinical phenotype of the disease. J. Clin. Immunol. 30 (5), 746–755. doi:10.1007/s10875-010-9424-3
Ng, P. C., and Henikoff, S. (2003). SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 31 (13), 3812–3814. doi:10.1093/nar/gkg509
Odnoletkova, I., Kindle, G., Quinti, I., Grimbacher, B., Knerr, V., Gathmann, B., et al. (2018). The burden of common variable immunodeficiency disorders: a retrospective analysis of the European Society for Immunodeficiency (ESID) registry data. Orphanet J. Rare Dis. 13 (1), 201. doi:10.1186/s13023-018-0941-0
Picard, C., Bobby Gaspar, H., Al-Herz, W., Bousfiha, A., Casanova, J. L., Chatila, T., et al. (2018). International union of immunological societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J. Clin. Immunol. 38 (1), 96–128. doi:10.1007/s10875-017-0464-9
Ramirez, N. J., Posadas-Cantera, S., Caballero-Oteyza, A., Camacho-Ordonez, N., and Grimbacher, B. (2021). There is no gene for CVID - novel monogenetic causes for primary antibody deficiency. Curr. Opin. Immunol. 72, 176–185. doi:10.1016/j.coi.2021.05.010
Registry, E. (2016). Working definitions for clinical diagnosis of PID, 1–25. Available at: https://esidorg/Working-Parties/Registry/Diagnosis-criteria.
Resnick, E. S., Moshier, E. L., Godbold, J. H., and Cunningham-Rundles, C. (2012a). Morbidity and mortality in common variable immune deficiency over 4 decades. Blood 119 (7), 1650–1657. doi:10.1182/blood-2011-09-377945
Resnick, E. S., Moshier, E. L., Godbold, J. H., and Cunningham-Rundles, C. (2012b). Morbidity and mortality in common variable immune deficiency over 4 decades. Blood 119 (7), 1650–1657. doi:10.1182/blood-2011-09-377945
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Rodriguez-Ubreva, J., Arutyunyan, A., Bonder, M. J., Del Pino-Molina, L., Clark, S. J., de la Calle-Fabregat, C., et al. (2022). Single-cell Atlas of common variable immunodeficiency shows germinal center-associated epigenetic dysregulation in B-cell responses. Nat. Commun. 13 (1), 1779. doi:10.1038/s41467-022-29450-x
Rojas-Restrepo, J., Caballero-Oteyza, A., Huebscher, K., Haberstroh, H., Fliegauf, M., Keller, B., et al. (2021). Establishing the molecular diagnoses in a cohort of 291 patients with predominantly antibody deficiency by targeted next-generation sequencing: experience from a monocentric study. Front. Immunol. 12, 786516. doi:10.3389/fimmu.2021.786516
Salzer, U., Bacchelli, C., Buckridge, S., Pan-Hammarstrom, Q., Jennings, S., Lougaris, V., et al. (2009). Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood 113 (9), 1967–1976. doi:10.1182/blood-2008-02-141937
Salzer, U., Chapel, H. M., Webster, A. D., Pan-Hammarstrom, Q., Schmitt-Graeff, A., Schlesier, M., et al. (2005). Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat. Genet. 37 (8), 820–828. doi:10.1038/ng1600
Salzer, U., and Grimbacher, B. (2021). TACI deficiency - a complex system out of balance. Curr. Opin. Immunol. 71, 81–88. doi:10.1016/j.coi.2021.06.004
Sanchez-Ramon, S., Radigan, L., Yu, J. E., Bard, S., and Cunningham-Rundles, C. (2008). Memory B cells in common variable immunodeficiency: clinical associations and sex differences. Orlando, Fla: Clinical immunology.
Schwab, C., Gabrysch, A., Olbrich, P., Patino, V., Warnatz, K., Wolff, D., et al. (2018). Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J. Allergy Clin. Immunol. 142 (6), 1932–1946. doi:10.1016/j.jaci.2018.02.055
Seidel, M. G., Kindle, G., Gathmann, B., Quinti, I., Buckland, M., van Montfrans, J., et al. (2019). The European society for immunodeficiencies (ESID) registry working definitions for the clinical diagnosis of inborn errors of immunity. J. Allergy Clin. Immunol. Pract. 7, 1763–1770. doi:10.1016/j.jaip.2019.02.004
Smith, T., and Cunningham-Rundles, C. (2021). Lymphoid malignancy in common variable immunodeficiency in a single-center cohort. Eur. J. Haematol. 107 (5), 503–516. doi:10.1111/ejh.13687
Stenson, P. D., Ball, E. V., Mort, M., Phillips, A. D., Shiel, J. A., Thomas, N. S., et al. (2003). Human gene mutation database (HGMD): 2003 update. Hum. Mutat. 21 (6), 577–581. doi:10.1002/humu.10212
Tuijnenburg, P., Lango Allen, H., Burns, S. O., Greene, D., Jansen, M. H., Staples, E., et al. (2018). Loss-of-function nuclear factor κB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans. J. Allergy Clin. Immunol. 142, 1285–1296. doi:10.1016/j.jaci.2018.01.039
Warnatz, K., Denz, A., Drager, R., Braun, M., Groth, C., Wolff-Vorbeck, G., et al. (2002). Severe deficiency of switched memory B cells (CD27(+)IgM(-)IgD(-)) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood 99 (5), 1544–1551. doi:10.1182/blood.v99.5.1544
Wehr, C., Kivioja, T., Schmitt, C., Ferry, B., Witte, T., Eren, E., et al. (2008). The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood 111 (1), 77–85. doi:10.1182/blood-2007-06-091744
Zhang, L., Radigan, L., Salzer, U., Behrens, T. W., Grimbacher, B., Diaz, G., et al. (2007). Transmembrane activator and calcium-modulating cyclophilin ligand interactor mutations in common variable immunodeficiency: clinical and immunologic outcomes in heterozygotes. J. Allergy Clin. Immunol. 120 (5), 1178–1185. doi:10.1016/j.jaci.2007.10.001
Keywords: common variable immunodeficiency, genetics, autoimmunity, lung disease, granulomatous disease, cancer, lymphoma
Citation: Cunningham-Rundles C, Casanova J-L and Boisson B (2024) Genetics and clinical phenotypes in common variable immunodeficiency. Front. Genet. 14:1272912. doi: 10.3389/fgene.2023.1272912
Received: 04 August 2023; Accepted: 09 November 2023;
Published: 11 January 2024.
Edited by:
Che Kang Lim, Singapore General Hospital, SingaporeReviewed by:
Intan Juliana Abd Hamid, University of Science Malaysia (USM), MalaysiaCopyright © 2024 Cunningham-Rundles, Casanova and Boisson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charlotte Cunningham-Rundles, Y2hhcmxvdHRlLmN1bm5pbmdoYW0tcnVuZGxlc0Btc3NtLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.