 Yinfeng Liu1Jie Zheng1
Yinfeng Liu1Jie Zheng1 Yue Xu2Ji Lv1
Yue Xu2Ji Lv1 Zizheng Wu1Kai Feng1Jiani Liu1Weitao Yan1Liguang Wei1
Zizheng Wu1Kai Feng1Jiani Liu1Weitao Yan1Liguang Wei1 Jiangman Zhao2
Jiangman Zhao2 Lisha Jiang2Meng Han1*
Lisha Jiang2Meng Han1*- 1Breast Disease Diagnosis and Treatment Center, The First Hospital of Qinhuangdao, Qinhuangdao, Hebei, China
- 2Shanghai Biotecan Pharmaceuticals Co., Ltd., Shanghai, China
Background: Breast cancer, the most prevalent malignancy in women worldwide, presents diverse onset patterns and genetic backgrounds. This study aims to examine the genetic landscape and clinical implications of rare mutations in Chinese breast cancer patients.
Methods: Clinical data from 253 patients, including sporadic and familial cases, were analyzed. Comprehensive genomic profiling was performed, categorizing identified rare variants according to the American College of Medical Genetics (ACMG) guidelines. In silico protein modeling was used to analyze potentially pathogenic variants’ impact on protein structure and function.
Results: We detected 421 rare variants across patients. The most frequently mutated genes were ALK (22.2%), BARD1 (15.6%), and BRCA2 (15.0%). ACMG classification identified 7% of patients harboring Pathogenic/Likely Pathogenic (P/LP) variants, with one case displaying a pathogenic BRCA1 mutation linked to triple-negative breast cancer (TNBC). Also identified were two pathogenic MUTYH variants, previously associated with colon cancer but increasingly implicated in breast cancer. Variants of uncertain significance (VUS) were identified in 112 patients, with PTEN c.C804A showing the highest frequency. The role of these variants in sporadic breast cancer oncogenesis was suggested. In-depth exploration of previously unreported variants led to the identification of three potential pathogenic variants: ATM c.C8573T, MSH3 c.A2723T, and CDKN1C c.C221T. Their predicted impact on protein structure and stability suggests a functional role in cancer development.
Conclusion: This study reveals a comprehensive overview of the genetic variants landscape in Chinese breast cancer patients, highlighting the prevalence and potential implications of rare variants. We emphasize the value of comprehensive genomic profiling in breast cancer management and the necessity of continuous research into understanding the functional impacts of these variants.
Introduction
Breast cancer (BC) is one of the most prevalent and deadly malignancies in women worldwide. Globally, BC is the most commonly diagnosed cancer among women, with an estimated 2.3 million new cases and nearly 685,000 deaths reported in 2020 (Arnold et al., 2022; Siegel et al., 2023). In China, the incidence rate of BC has been significantly rising, making it an alarming public health concern. It is currently the most common cancer among women in China, with about 367,900 new cases and 97,972 deaths reported in 2020 (Lei et al., 2021). This increasing trend necessitates further research into understanding the risk factors and genetic predispositions underlying BC in the Chinese population.
While environmental and lifestyle factors play a significant role in BC development, a growing body of evidence indicates that genetic susceptibility also contributes to the onset of this disease. Approximately 5%–10% of BC cases are hereditary, often attributed to inherited pathogenic or likely pathogenic (P/LP) variants in highly penetrant predisposition genes (Yoshimura et al., 2022). The BRCA1 and BRCA2 genes are the most well-known and extensively studied of these genes (Yoshimura et al., 2022). Mutations in these genes significantly increase the risk of breast and ovarian cancer (Zhong et al., 2015), and are also related to the prognosis and recurrence of breast cancer (Shah et al., 2016). A study by Zhang et al. found that among 30,223 adult participants of the BioMe Biobank, 218 (0.7%) individuals harbored expected PVs in BRCA1/2 (Abul-Husn et al., 2019). However, mutations in these genes account for only a fraction of hereditary BC cases, suggesting the involvement of other genetic variants. In addition to BRCA1 and BRCA2, several other susceptibility genes have been associated with BC, including TP53, PALB2, CHEK2, ATM and so on (Foulkes, 2021), indicating that the utilization of multi-gene panel testing could potentially confer enhanced benefits to patients (Bono et al., 2021).
Currently the majority of the current understanding of genetic susceptibility to BC is based on studies conducted in Western populations. The genetic landscape of BC in China, a country with a complex population structure and unique lifestyle factors, remains relatively unexplored. Preliminary studies suggest that the prevalence of inherited P/LP variants in highly penetrant predisposition genes among Chinese BC patients may be similar to global estimates. The occurrence of harmful BRCA1/2 mutations in the broad Chinese population has been documented to vary between 0.29% and 1.10% (with BRCA1 variations between 0.02% and 0.34% and BRCA2 variations from 0.11% to 0.27%) (Lei et al., 2022). In the Chinese population, research on susceptibility genes beyond BRCA1 and BRCA2 is still evolving. In a cohort of 7,657 unselected BC patients who tested negative for BRCA1/2 germline mutations, a multigene panel revealed that 29 cases (0.38%) carried harmful RAD51D germline mutations (Chen et al., 2018). A study by Li et al. have suggested the presence of additional susceptibility genes, such as EFEMP1, contributing to BC risk among Chinese women (Li et al., 2016).
Genetic diversity among different population in China may result in different gene-disease associations, emphasizing the need for population-specific genetic studies. In this study, we aim to fill a significant gap in the existing literature by examining the spectrum of hereditary gene variants that may trigger sporadic BC in the northern Chinese population by testing three different gene panels in a total of 253 patients.
Materials and methods
Patients and samples
A total of 253 women with breast cancer were recruited in Department of Breast Surgery of First Hospital of Qinhuangdao, from September 2020 to July 2022. Clinical information of participants was obtained from Electronic Medical Record System and questionnaire. 2–4 mL peripheral blood from each participant was collected by EDTA anticoagulated tube.
DNA extraction and quality control
Genomic DNA (gDNA) of peripheral blood was extracted by QIAamp DNA Blood Mini Kit (QIAGEN GmbH), then quantity and purity were evaluated by Qubit 3.0 and NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Wilmington, United States). 300 ng gDNA per sample was mechanically fragmented using an E220 Focused-ultrasonicator (Covaris, LLC., Massachusetts, United States). The Agilent 2100 Bioanalyzer instrument with Agilent High Sensitivity DNA Kit (Agilent Technologies, Inc., CA, United States) were used for sizing and quantitation of fragmented DNA. The targeted size of fragmented DNA was from 150 to 200 bp.
Library preparation and sequencing
10–100 ng fragmented DNA was used for library construction using the SureSelect XT Low Input Reagent Kits (Agilent Technologies, Inc., CA, United States), including end-repair, dA-tail the 3′end of the DNA fragments, ligating the paired-end adaptor, and pre-amplification. 500–2000ug DNA of the whole genomic libraries were captured using Agilent SureSelect XT custom panel probes and finally amplified. Three panels were used throughout this study (Supplementary Table S1), encompassing 21 genes (97 patients; 38.3%), 37 genes (66 patients; 26.1%) and 64 genes (90 patients, 35.6%). The use of three panels is due to the upgrade in the panel’s detection capability. Patients who were recruited after the upgrade would use the new panel. After quality control and quantification by Agilent 2100 Bioanalyzer and Qubit 3.0, the libraries were sequenced on Illumina Nextseq CN500 platform (Illumina Inc., CA, United States) in PE150 mode. All the sequencing data were upload to SRA database with an accession number PRJNA998571.
Bioinformatics analysis
Clean data was obtained following filtering adapter, low quality reads and reads with proportion of N>10%. Reads were aligned to the reference human genome (UCSC hg19) (Meyer et al., 2013) using the Burrows-Wheeler Aligner v. 0.7.17 (Li and Durbin, 2009). Next, the Picard and Genome Analysis Toolkit (GATK v.3.7) (McKenna et al., 2010) method was adopted for duplicate removal, local realignment and Base Quality Score Recalibration, and generated the quality statistics, including mapped reads, mean mapping quality and mean coverage. Finally, the GATK HaplotypeCaller was used for SNV and InDel identification.
Variants were annotated using the ANNOVAR software tool (Wang et al., 2010). Annotations for mutation function (including frameshift insertion/deletion, non-frameshift insertion/deletion, synonymous SNV, nonsynonymous SNV, stopgain, stoploss), mutation location [such as exonic, intronic, splicing, upstream, downstream, 3′untranslated region (UTR), 5′UTR and so on], amino acid changes, 1000 Genomes Project data, the Exome Aggregation Consortium (ExAC) data, the NHLBI Exome Sequencing Project (ESP) data and dbSNP reference number were performed. Mutation databases including HGMD (http://www.hgmd.cf.ac.uk/), and ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) were also included in the analysis pipeline. In this study, in order to making the interpretation of the sequence variants much more efficient and conclusive, annotated variants were filtered about mutation location (exonic and splicing region were reserved), mutation function (synonymous SNV and UNKOWN were removed) and allele frequency (≤0.05 AF in 1000 g, ESP and ExAC database).
Germline mutation classification
All mutations were classified according to the American College of Medical Genetics (ACMG) professional practice and guidelines [five-tier mutation: P (Pathogenic); LP (Likely Pathogenic); Variants of uncertain significance (VUS); LB (Likely Benign); and B (Benign)] (Richards et al., 2015). Mutation classification was generated by genetic Counselor and verified by two curators.
Statistical analysis
Descriptive statistics were used to summarize the demographic and clinical characteristics of the participants. Categorical variables were analyzed using Chi-square tests, while continuous variables were examined using Student's t-tests or Mann-Whitney U tests as appropriate. All tests were two-sided, and a p-value <0.05 was considered statistically significant. All statistical analyses were performed using the R statistical software (version 4.0.2).
Results
Patient characteristics
The clinical profiles of 253 patients are detailed in Table 1. Among these, 242 cases were diagnosed with sporadic breast cancer, while the remaining 11 patients presented with a familial history of the disease. The median age at diagnosis across all patients was 58 years, with an interquartile range (IQR) of 48–65 years. The patient cohort was stratified into two groups based on the age at the time of diagnosis: 25 cases were classified as early-onset (diagnosed before 40 years of age), while the remaining 228 were designated as late-onset cases. Notably, three patients were diagnosed with bilateral breast cancer.

TABLE 1. Clinical characteristics of 253 breast cancer.
Frequency of breast cancer predisposition genes with rare variants
All rare variants identified within exonic and splicing regions were filtered based on an allele frequency of ≤0.05, as per the 1000 Genomes Project, ESP, and ExAC databases. A total of 421 rare variants were detected amongst the 253 patients with sporadic BC, consisting of 409 exonic and 12 splicing region variants. The exonic variants were classified into several types: 372 nonsynonymous single-nucleotide variants (SNVs), 7 stopgain, 11 frameshift deletions, 6 frameshift insertions, 5 nonframeshift deletions, 7 nonframeshift insertions, and 1 unidentified deletion. The frequency of rare variants within BC predisposition genes is outlined in Supplementary Table S2 and Figures 1–3. The most frequently mutated genes were ALK (20/90, 22.2%), BARD1 (14/90, 15.6%), and BRCA2 (38/253, 15.0%). We identified 14 unique BRCA2 mutations in 38 cases (Figure 1A), with these mutations exhibiting a mutual exclusivity with MLH1 mutations (Figure 1B, p < 0.1).
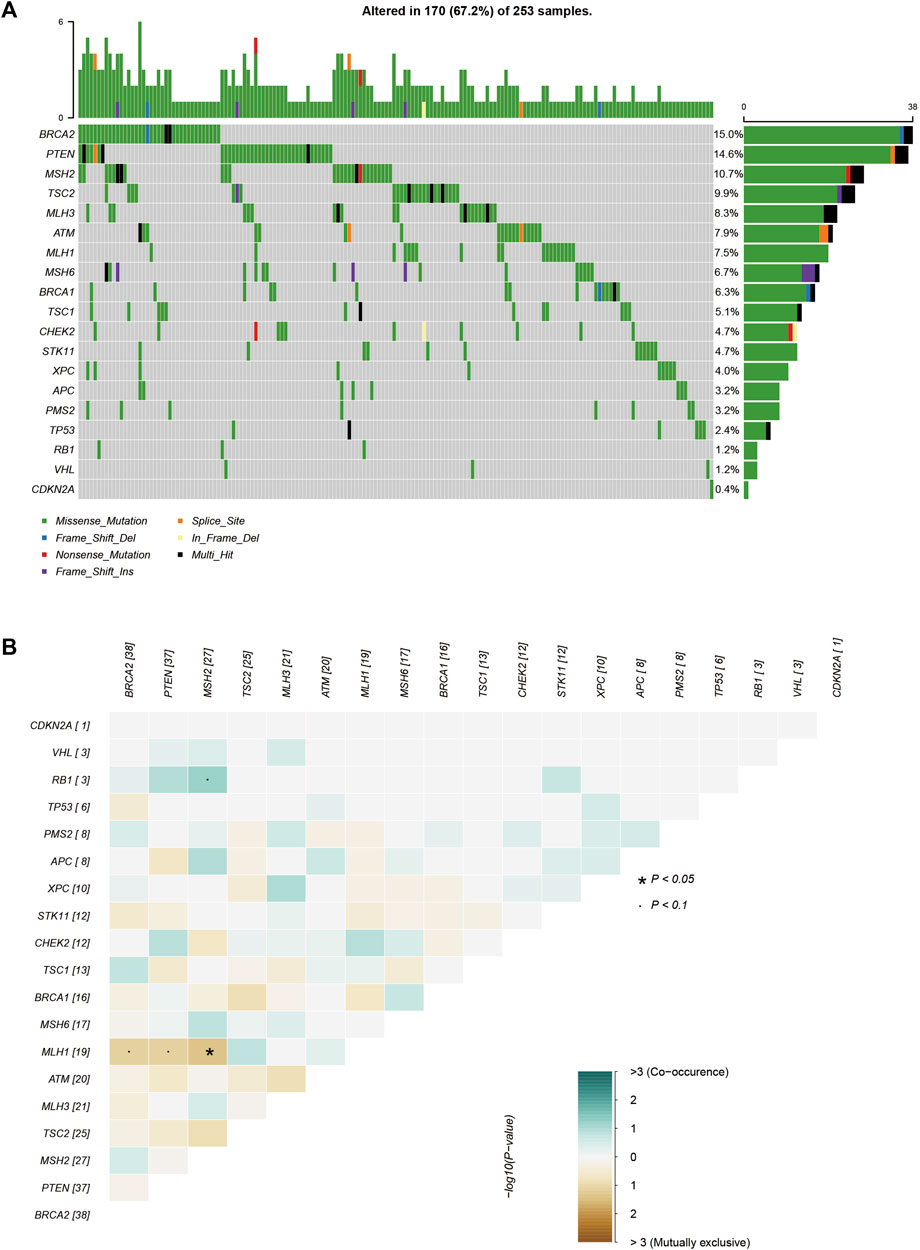
FIGURE 1. The mutations tested by 21-gene-panel in 253 cases. (A) The profile and distribution of these mutations. (B) The correlation analysis of these genes.
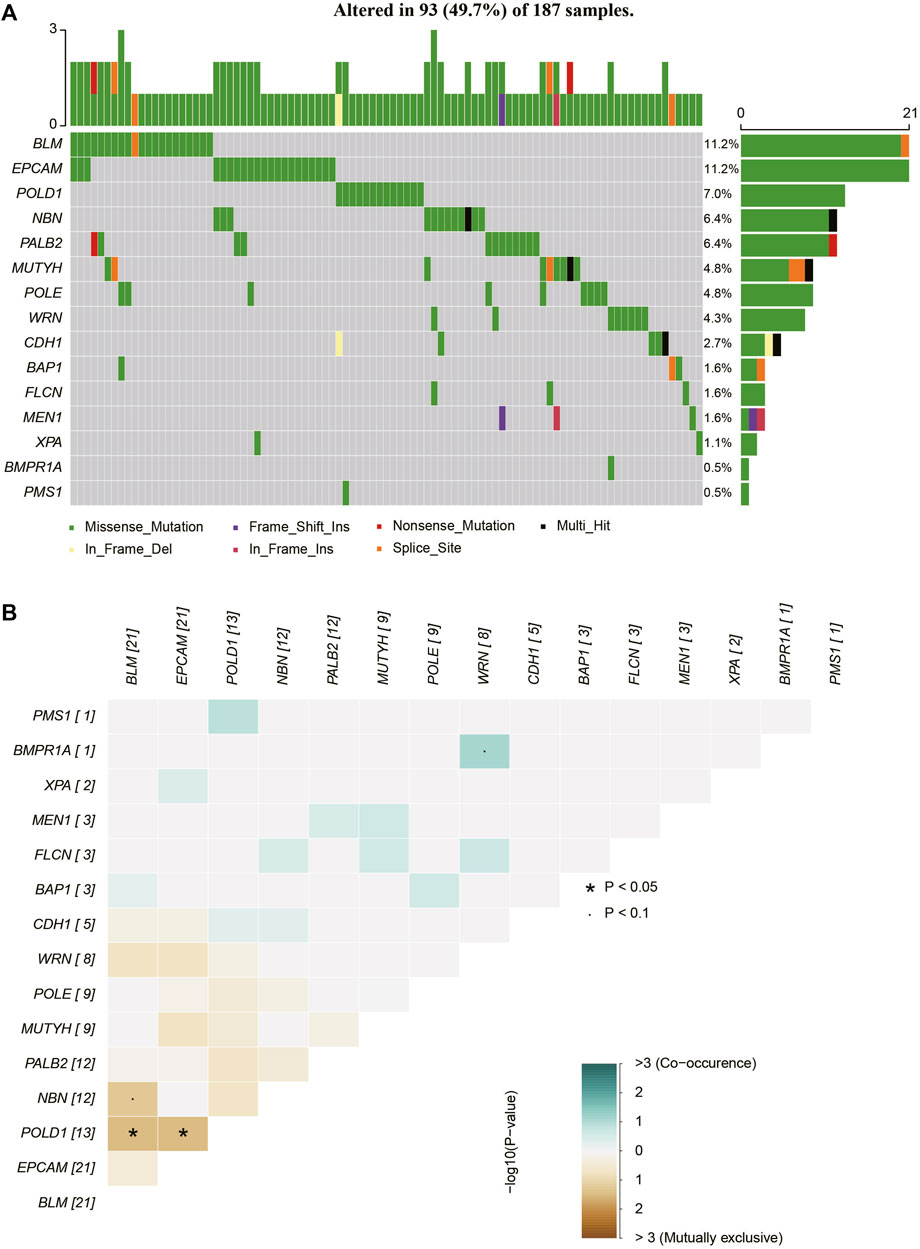
FIGURE 2. The mutations tested by 37-gene-panel in 187 cases. (A) The profile and distribution of these mutations. (B) The correlation analysis of these genes.
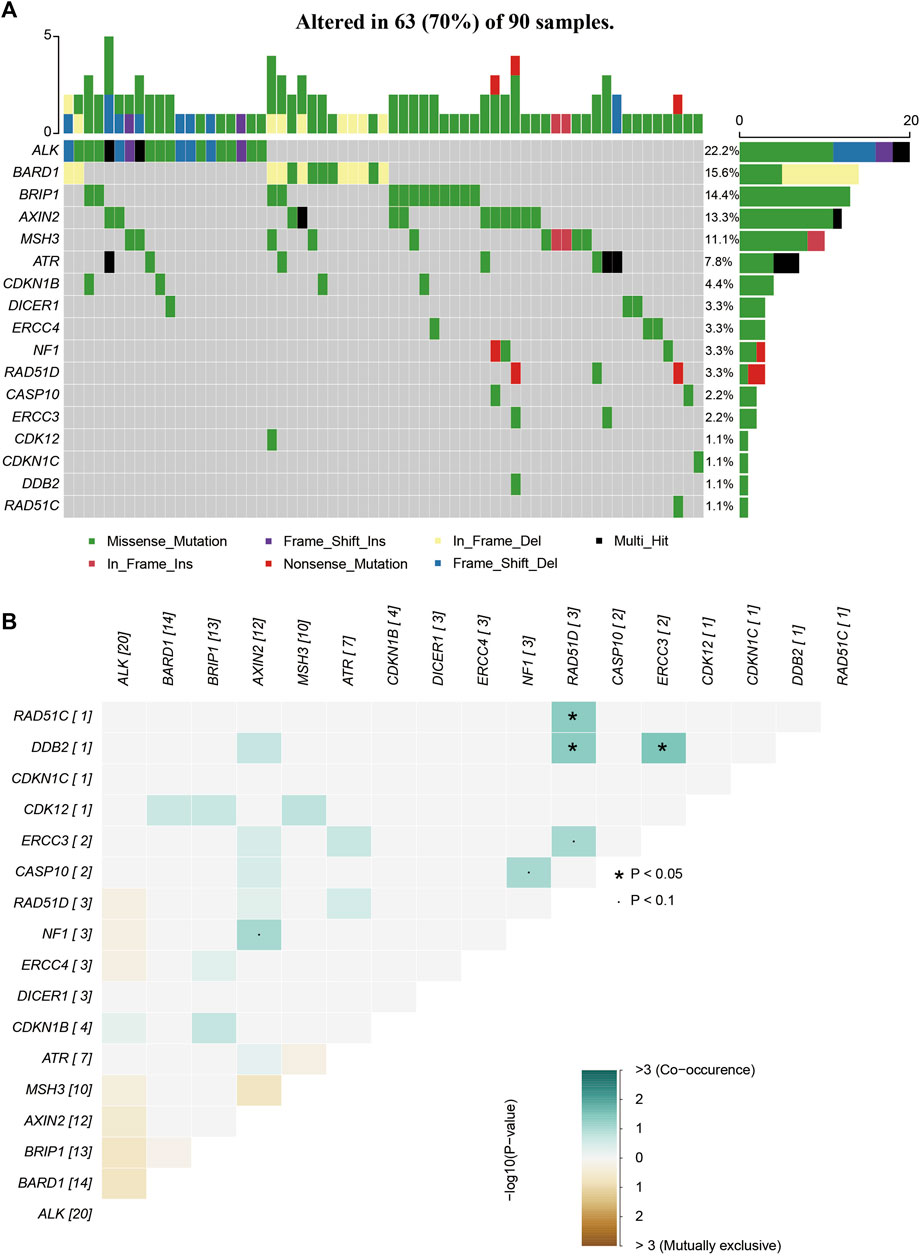
FIGURE 3. The mutations tested by 64-gene-panel in 90 cases. (A)The profile and distribution of these mutations. (B) The correlation analysis of these genes.
ACMG level distribution of rare variants
Out of the 253 patients examined, 17 cases (17/253, 7%) carried pathogenic/likely pathogenic (P/LP) variants. A total of 97 patients (97/253, 38%) exhibited at least one variant of uncertain significance (VUS), without any P/LP variants, while 109 cases (109/253, 43%) harbored at least one benign/likely benign (B/LB) variant without any P/LP variants or VUS (Figure 4A). Notably, in 30 BC patients (30/253, 12%), no rare variants were detected.
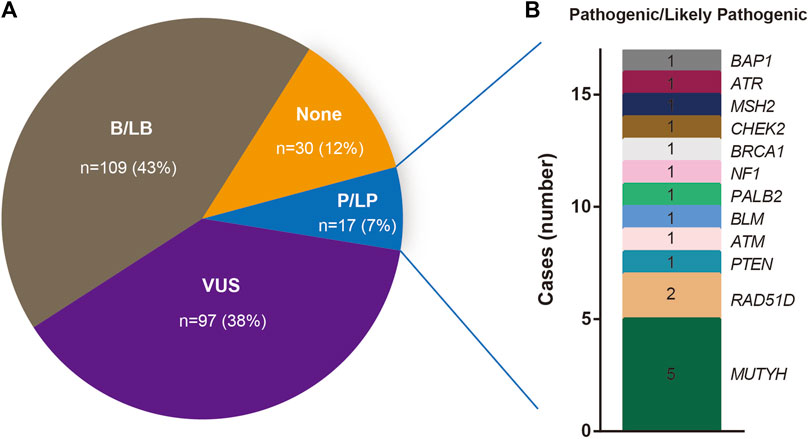
FIGURE 4. (A) Percentage of Beneficial/likely Beneficial (B/LB), Variants of uncertain significance (VUS), Pathogenic/Likely pathogenic (P/LP) and not recorded (none) mutations. (B) The number of the Genes that carry P/LP mutations.
Distribution of pathogenic and likely pathogenic variants
The spectrum and details of the 17 identified pathogenic/likely pathogenic (P/LP) variants are depicted in Figure 4B and Table 2. To be specific, these P/LP variants include NF1 c.C1381T, RAD51D c.C562T, PALB2 c.C2257T, MUTYH c.850-2A>G, BLM c.960-1G>A, PTEN c.211-2A>T, ATM c.2921+1G>T, BRCA1 c.5015delT, CHEK2 c.C409T, MSH2 c.C1566A, RAD51D c.C184T, ATR c.2320delA, BAP1 c.123-2A>C, MUTYH c.C689T, ATM c.1236-2A>T. Of these, only one P/LP variant was found in a BRCA gene (specifically BRCA1), which was a frameshift deletion (BRCA1 c.5015delT) classified as a PV according to the American College of Medical Genetics and Genomics (ACMG). This variant was absent in the 1000 Genomes Project, ESP, and ExAC databases. The patient harboring this BRCA1 PV had no family history of the disease and was initially diagnosed with triple-negative breast cancer (TNBC) at age 48, stage IIA. These findings further substantiate the potential pathogenicity of the BRCA1 c.5015delT, variant in sporadic BC cases. Additionally, of the 14 rare BRCA2 variants, only one missense variant was classified as a variant of uncertain significance (VUS), with the remaining 13 variants categorized as benign or likely benign (B/LB).
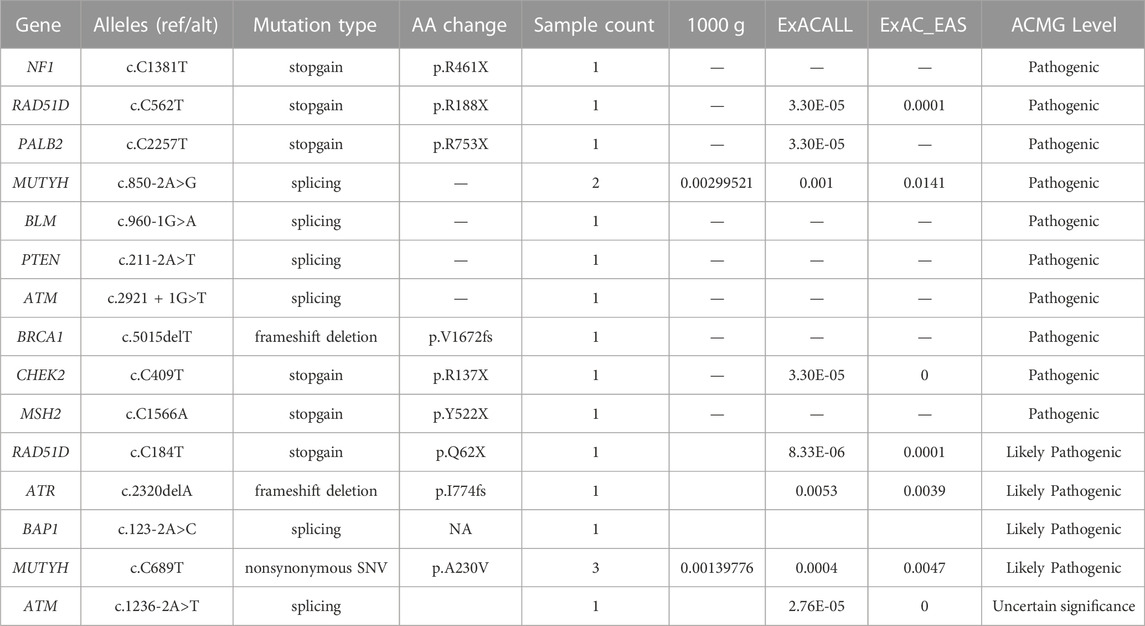
TABLE 2. Pathogenic and likely pathogenic variants identified in multi-gene panel testing.
While MUTYH mutations are typically associated with colon cancer, increasing evidence suggests their pathogenic potential in BC (Chen et al., 2020; Felicio et al., 2021). In this study, two pathogenic/likely pathogenic (P/LP) MUTYH variants were identified in five BC patients (Table 2). The MUTYH c.850-2A>G variant, located in the splicing region, was found in two patients in our cohort. This variant exhibited allele frequencies of 0.003, 0.001, and 0.014 in the 1000 Genomes Project, ExAC_ALL, and ExAC_EAS databases, respectively. Classified as a PV by ACMG guidelines, it is commonly associated with MYH-associated polyposis. Among the two patients carrying this variant, one, a 49-year-old patient diagnosed with Luminal A breast cancer, had no family history of the disease. The other, a 63-year-old patient diagnosed with basal-like breast cancer, had a sister who was also a BC patient. The MUTYH c.C689T variant, found within the exonic region, was identified in three patients. This variant displayed allele frequencies of 0.0014, 0.0004, and 0.0047 in the 1000 Genomes Project, ExAC_ALL, and ExAC_EAS databases, respectively. While the ACMG guidelines list MUTYH c.C689T as a likely pathogenic variant (LPV), the ClinVar database classifies it as likely benign. Notably, none of the three patients harboring this variant had a family history of BC.
Distribution of variants of uncertain significance in our cohort
A total of 97 distinct variants, annotated as variants of uncertain significance (VUS) and located across 39 different genes, were identified in 112 patients, as detailed in Supplementary Table S3. Of these patients, one individual harbored four VUS, nine patients carried three VUS, 29 patients exhibited two VUS, and the remaining 73 patients presented with a single VUS. Notably, the PTEN c.C804A variant displayed the highest frequency, appearing in 36 patients within our cohort. The allele frequencies of this variant were found to be 0.0009 and 0.0006 in the ExAC_ALL and ExAC_EAS databases, respectively. This significant contrast suggests a potentially critical role for this variant in the oncogenesis of sporadic BC. Furthermore, the ALK c.2760_2766del variant was found in six patients in our cohort. This variant’s allele frequencies in the ExAC_ALL and ExAC_EAS databases were 0.00008 and 0.0001, respectively. Intriguingly, all patients carrying the ALK c.2760_2766del mutation also harbored at least one other genetic mutation.
Reclassification of ATM c.1236-2A>T variant as likely pathogenic based on ACMG criteria and HGMD data
The ATM c.1236-2A>T variant, currently classified as a variant of uncertain significance (VUS) by the American College of Medical Genetics and Genomics (ACMG), has been associated with cancer according to the high-confidence Human Gene Mutation Database (HGMD). Moreover, this variant is predicted to undergo nonsense-mediated decay, thus satisfying the PVS1 criterion for pathogenicity as per ACMG guidelines. Its low frequency in the ExAC_EAS database and its location within a highly conserved region further fulfill the PM2 and PP3 criteria, respectively. Accordingly, we propose a reclassification of ATM c.1236-2A>T as a LPV.
Implications of rare mutations for their downstream protein structures
To identify significant rare mutations within the Chinese population, we conducted an in-depth investigation of all yet-to-be-reported variants without a reference number. Any variant that was classified as Benign or Tolerate by the predictive software SIFT, Polyphen2_HDIV, and Polyphen2_HVAR, failed to reach the threshold (ClinPred >0.95, and + CADD-based scores >25), located outside a functional domain, or were present in patients who already had P/LP variants were excluded from further analysis. The filtering process identified three rare variants for further investigation, specifically ATM c.C8573T, CDKN1C c.C221T, and MSH3 c.A2723T. To elucidate their biological implications, we performed in silico protein modeling analysis.
The ATM c.C8573T variant resides in the Phosphatidylinositol 3- and 4-kinase domain, as illustrated in Figure 5A. This mutation engenders a nonsynonymous Threonine to Isoleucine substitution at the conserved position 2858, suggesting potential functional impact on the resultant protein (Figure 5B). 3D protein model prediction and energy change analysis indicate that the Threonine/Isoleucine substitution resulted in a reduced inter-atomic distance in the mutated domain (Figures 5C, D). This corresponds to a mutation Cutoff Scanning Matrix (mCSM) protein stability score of ΔΔG = −0.367 kcal/mol, indicating a potential decrease in stability.
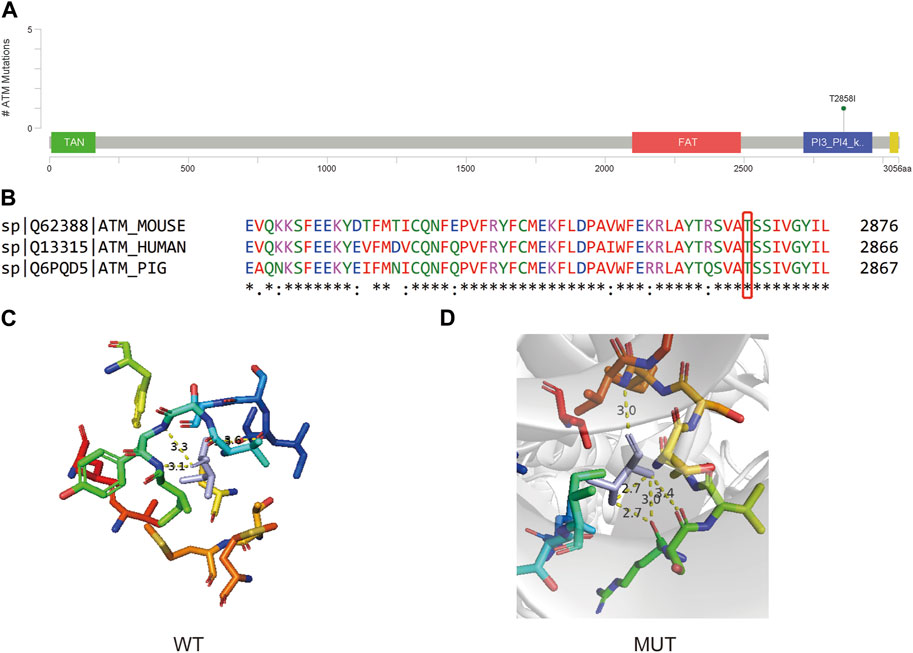
FIGURE 5. (A) Lollipop plot showing the location of ATM c.C8573T in ATM gene. (B) Protein conservation assessment of the amino acid affected by ATM c.C8573T, which is emphasize by a red box. (C,D) The in silico protein model of wildtype (C) and mutated type (D).
The CDKN1C c.C221T variant is situated in the Cyclin_dependent kinase inhibitor domain (Figure 6A), leading to a Proline to Leucine nonsynonymous mutation at a conserved position 74 (Figure 6B). Analysis of the predicted 3D protein model reveals a covalent linkage between the proline’s carbon atom and the protein structure, maintaining an intact aromatic ring in the wild-type protein (Figures 6C, D). However, this mutation disrupts the aromatic ring structure, resulting in a decrease in protein stability (ΔΔG = −0.618 kcal/mol).
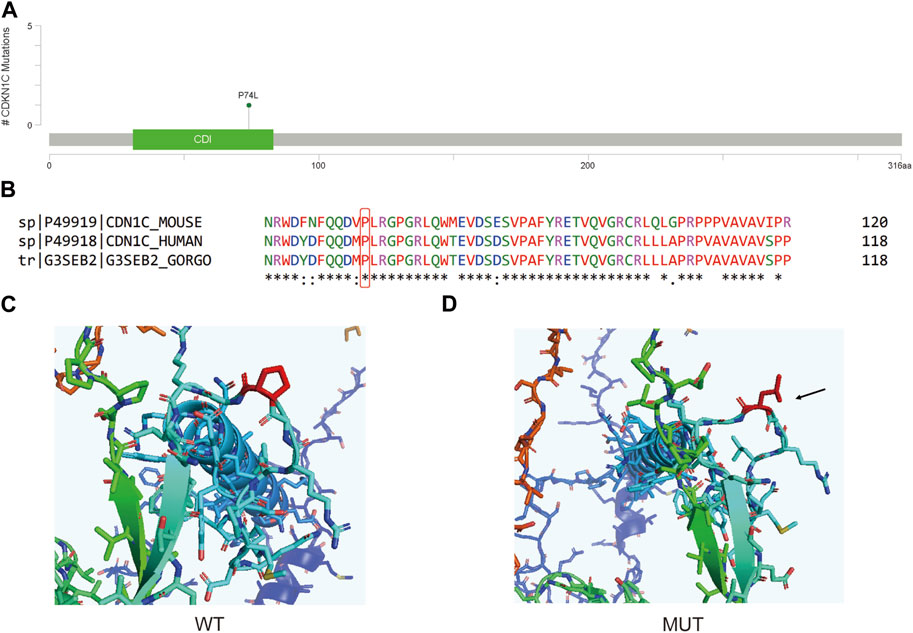
FIGURE 6. (A) Lollipop plot showing the location of CDKN1C c.C221T in CDKN1C gene. (B) Protein conservation assessment of the amino acid affected by CDKN1C c.C221T, which is emphasize by a red box. (C,D) The in silico protein model of wildtype (C) and mutated type (D).
The MSH3 c.A2723T variant is located within the MutS domain V (Figure 7A) and introduces a Glutamine to Leucine nonsynonymous mutation at protein position 908 (Figure 7B). This mutation, occurring at a conserved site, results in an unstable protein structure (ΔΔG = −0.324 kcal/mol) and alters the inter-atomic distances between several amino acids (Figures 7C, D).
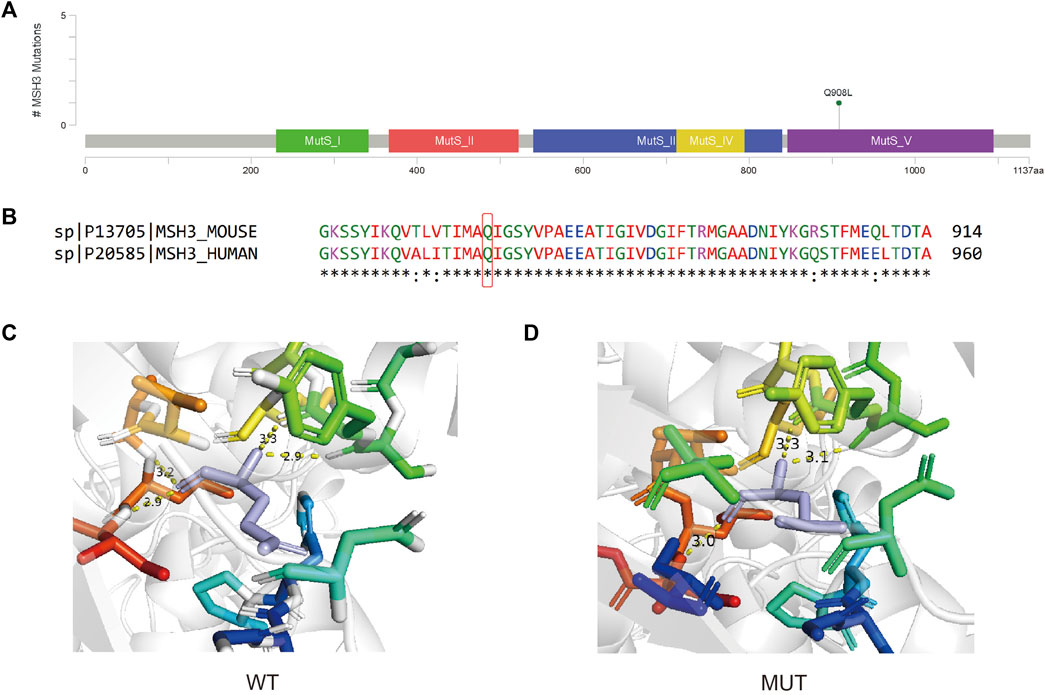
FIGURE 7. (A) Lollipop plot showing the location of MSH3 c.A2723T in MSH3 gene. (B) Protein conservation assessment of the amino acid affected by MSH3 c.A2723T, which is emphasize by a red box. (C,D) The in silico protein model of wildtype (C) and mutated type (D).
Discussion
In this study, we investigated the genetic landscape of BC in a cohort of 253 patients, identifying significant genetic variants that may contribute to disease susceptibility and progression. Our results, which highlight the heterogeneity and complexity of BC genetics, underscore the potential for personalized medicine in its management.
Most cases in our cohort (northern Chinese population) are sporadic BC, and the top 3 frequent mutated genes in were MUTYH (5/17, 22.2%), BARD1 (14/90, 15.6%) and BRCA2 (38/253, 15.0%), and the frequency of top 2 P/LP variants are MUTYH (5/187, 2.26%) and RAD51D (2/90, 2.22%). In Yi’s research, out of the 27 individuals studied, 9 (constituting 13.6% of the sample population) were identified as carriers of the TP53 gene mutation, 5 (representing 7.6%) carried the MSH6 mutation, and another 5 (equivalent to 7.6%) were found to carry the BRCA1 mutation (Yi et al., 2019), while no P/LP variants were found. Another research on Chinese population focused on the prevalence and characteristics of BRCA1/2 germline mutations, and BRCA1/2 mutation shows low frequency (2%) in sporadic BC (Zhang et al., 2016). Through the analysis of our multi-omics TNBC cohort consisting of 325 individuals, Ma et al. mapped the range of germline variants in TNBC to assess their biological and clinical impacts, with the most common mutations identified in the genes BRCA1 (7.4%), RAD51D (2.8%), and BRCA2 (2.2%) (Ma et al., 2021). A research by Su et al. also reported the frequency of RAD51D is 1.3% in high-risk BC patients (Su et al., 2021). In Hongkong population, RAD51D is one of the most common mutation gene (0.8%) (Kwong et al., 2020). The frequency of MUTYH in Chen’s research is 0.7% (Chen et al., 2020), while in Jian’s research, it is 1.7% (Jian et al., 2017). By testing 30 cancer susceptible genes in 384 Chinese subjects with 2 high-risk factors, Lang et al. reported that both MUTHY and RAD51D have pathogenic/likely-pathogenic, with frequency of 2.9% and 0.5% (Lang et al., 2020). All these results suggest that genes that carry P/LP variants and their frequency are slightly different among Chinese population in different area, and may be affected by the high-risk factors of BC, indicating that the multi-gene test screening for BC patients are important any may found new target for the early screening or therapies.
ATM is a gene that involved in DNA double-strand break repair pathways, and usually the PVs in ATM are considered to be associated with a moderate risk of BC (Graffeo et al., 2022). The Phosphatidylinositol 3- and 4-kinase domain (also called ATM kinase domain) is a kinase domain that shares significantly homologous structure to that of PI3K, and activated ATM protein use this kinase activity to phosphorylate a series of downstream targets that are essential for DNA-damage repair (Phan and Rezaeian, 2021). The instability of the kinase domain caused by ATM c.C8573T (p.T2858I) may affect this domain and eventually the dysfunction of down-stream DNA-damage response. Although currently there is no record for this site in Clinvar, the nearby annotated mutation sites show the potential influence on the function of this protein. When only considering pathogenic, likely pathogenic, benign, and likely pathogenic, ATM c.8564G>T (p.S2855I) and ATM c.8565T>A/G (p.S2855R) are the closest mutation site near ATM c.C8573T, and are Likely pathogenic and Pathogenic/Likely pathogenic, respectively. A total of 14 Pathogenic/Likely pathogenic or Likely pathogenic and 1 Likely benign were found in this domain. A mutation on the same amino acid site ATM: c.8572A>G (p.T2858A) is annotated as uncertain significance. Hence, based on the current information, though the significance of ATM c.C8573T is not clearly identified, but based on the nearby mutation and the fact that it is the only mutation in that patient of our study, it may serve as a rare PV in at least Chinese population.
CDKN1C, is a gene that codes for the Cyclin-dependent kinase inhibitor p57Kip2. This protein can obstruct the interaction domain on cyclins, preventing ATP binding and catalytic function, which in turn leads to the inhibition of the cyclin/CDK complex and slows down cell growth (Lai et al., 2021). As a gene associated with suppressing tumor growth, CDKN1C is linked to a variety of human cancers and Beckwith-Wiedemann Syndrome. Prior research has attempted to explore the relationship between CDKN1C methylation and BC (Zohny et al., 2017). However, how CDKN1C variants may affect BC oncogenesis is not well-studied. By searching the nearby variants of CDKN1C c.C221T, only one pathogenic, one likely pathogenic and one conflicting interpretations of pathogenicity annotation were found in this domain, and the conditions were all annotated to Beckwith-Wiedemann syndrome. The patient carrying this variant is a 57-year-old female with Luminal B breast cancer on stage IA. Though this variant is the only one she carries based on our testing panel, whether this variant somehow led to breast remain unclear.
MSH3 is a crucial contributor to the mismatch repair (MMR) pathways, which is an essential biological process that exerts significant control over cell cycle regulation and apoptosis, thereby mitigating various forms of DNA damage (Brown et al., 2003). In the absence of appropriate repair mechanisms, these mismatches may increase spontaneous mutation rates, ultimately fostering microsatellite repeat instability in cells and promoting carcinogenesis. Some potentially functional variants of MSH3 may influence the DNA repair capacity and thereby predispose individuals to a variety of cancers. For example, rs26279 (MSH3 c.3133G>A) has been frequently studied and implicated in carcinogenesis in recent years (Miao et al., 2015). However, the potential influence of MSH3 c.A2723T variant have not been recorded. Another mutation on the same site MSH3 A2723C (p.Gln908Pro) is related to Hereditary cancer-predisposing syndrome but is considered uncertain significance. The same situation can be found on all mutations that around this site. Hence, MSH3 c.A2723T may cause a potential effect on carcinogenesis of BC, but further studies are still needed.
While our study provides valuable insights into the mutation frequency among women with BC in northern Chinese population, it is not without limitations. First, our study focuses on a specific geographical location, which could limit the generalizability of our findings to other population groups. This is particularly important given the genetic heterogeneity of BC. Second, our sample size relatively small when considering the broad spectrum of BC patients globally, which might limit the statistical power of our results. Third, our study relies on genomic DNA extracted from peripheral blood and not from the tumor itself, which could potentially miss tumor-specific mutations and underestimate intra-tumor heterogeneity. Lastly, while we utilized stringent filtering criteria to identify relevant mutations, the potential for false-positive or false-negative findings remains, due to the inherent complexities of genomic analysis. Despite these limitations, our study lays crucial groundwork for further research on genetic predisposition to BC in this specific population.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA998571.
Ethics statement
The studies involving humans were approved by the Medical Ethics Committee of the First Hospital of Qinhuangdao. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
YL: Conceptualization, Writing–original draft. JZhe: Formal analysis, Writing–original draft. YX: Data curation, Writing–original draft. JLv: Resources, Writing–original draft. ZW: Resources, Writing–original draft. KF: Investigation, Writing–original draft. JLi: Data curation, Writing–original draft. WY: Data curation, Writing–original draft. LW: Formal analysis, Writing–original draft. JZha: Project administration, Writing–review and editing. LJ: Data analysis, Writing–original draft. MH: Writing–review and editing.
Funding
The authors declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We extend our sincere thanks to all the patients who participated in this study. Special appreciation is given to our colleague, Huihui Jiang, for her academic help and advice.
Conflict of interest
Authors YX, JZha, and LJ were employed by Shanghai Biotecan Pharmaceuticals Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1271710/full#supplementary-material
References
Abul-Husn, N. S., Soper, E. R., Odgis, J. A., Cullina, S., Bobo, D., Moscati, A., et al. (2019). Exome sequencing reveals a high prevalence of BRCA1 and BRCA2 founder variants in a diverse population-based biobank. Genome Med. 12 (1), 2. doi:10.1186/s13073-019-0691-1
Arnold, M., Morgan, E., Rumgay, H., Mafra, A., Singh, D., Laversanne, M., et al. (2022). Current and future burden of breast cancer: global statistics for 2020 and 2040. Breast 66, 15–23. doi:10.1016/j.breast.2022.08.010
Bono, M., Fanale, D., Incorvaia, L., Cancelliere, D., Fiorino, A., Calò, V., et al. (2021). Impact of deleterious variants in other genes beyond BRCA1/2 detected in breast/ovarian and pancreatic cancer patients by NGS-based multi-gene panel testing: looking over the hedge. ESMO Open 6 (4), 100235. doi:10.1016/j.esmoop.2021.100235
Brown, K. D., Rathi, A., Kamath, R., Beardsley, D. I., Zhan, Q., Mannino, J. L., et al. (2003). The mismatch repair system is required for S-phase checkpoint activation. Nat. Genet. 33 (1), 80–84. doi:10.1038/ng1052
Chen, B., Zhang, G., Li, X., Ren, C., Wang, Y., Li, K., et al. (2020). Comparison of BRCA versus non-BRCA germline mutations and associated somatic mutation profiles in patients with unselected breast cancer. Aging (Albany NY) 12 (4), 3140–3155. doi:10.18632/aging.102783
Chen, X., Li, Y., Ouyang, T., Li, J., Wang, T., Fan, Z., et al. (2018). Associations between RAD51D germline mutations and breast cancer risk and survival in BRCA1/2-negative breast cancers. Ann. Oncol. 29 (10), 2046–2051. doi:10.1093/annonc/mdy338
Felicio, P. S., Grasel, R. S., Campacci, N., de Paula, A. E., Galvão, H. C. R., Torrezan, G. T., et al. (2021). Whole-exome sequencing of non-BRCA1/BRCA2 mutation carrier cases at high-risk for hereditary breast/ovarian cancer. Hum. Mutat. 42 (3), 290–299. doi:10.1002/humu.24158
Foulkes, W. D. (2021). The ten genes for breast (and ovarian) cancer susceptibility. Nat. Rev. Clin. Oncol. 18 (5), 259–260. doi:10.1038/s41571-021-00491-3
Graffeo, R., Rana, H. Q., Conforti, F., Bonanni, B., Cardoso, M. J., Paluch-Shimon, S., et al. (2022). Moderate penetrance genes complicate genetic testing for breast cancer diagnosis: ATM, CHEK2, BARD1 and RAD51D. Breast 65, 32–40. doi:10.1016/j.breast.2022.06.003
Jian, W., Shao, K., Qin, Q., Wang, X., Song, S., and Wang, X. (2017). Clinical and genetic characterization of hereditary breast cancer in a Chinese population. Hered. Cancer Clin. Pract. 15, 19. doi:10.1186/s13053-017-0079-4
Kwong, A., Shin, V. Y., Chen, J., Cheuk, I. W. Y., Ho, C. Y. S., Au, C. H., et al. (2020). Germline mutation in 1338 BRCA-negative Chinese hereditary breast and/or ovarian cancer patients: clinical testing with a multigene test panel. J. Mol. Diagn 22 (4), 544–554. doi:10.1016/j.jmoldx.2020.01.013
Lai, J., Lin, X., Cao, F., Mok, H., Chen, B., and Liao, N. (2021). CDKN1C as a prognostic biomarker correlated with immune infiltrates and therapeutic responses in breast cancer patients. J. Cell Mol. Med. 25 (19), 9390–9401. doi:10.1111/jcmm.16880
Lang, G. T., Shi, J. X., Huang, L., Cao, A. Y., Zhang, C. H., Song, C. G., et al. (2020). Multiple cancer susceptible genes sequencing in BRCA-negative breast cancer with high hereditary risk. Ann. Transl. Med. 8 (21), 1417. doi:10.21037/atm-20-2999
Lei, H., Zhang, M., Zhang, L., Hemminki, K., Wang, X. J., and Chen, T. (2022). Overview on population screening for carriers with germline BRCA mutation in China. Front. Oncol. 12, 1002360. doi:10.3389/fonc.2022.1002360
Lei, S., Zheng, R., Zhang, S., Wang, S., Chen, R., Sun, K., et al. (2021). Global patterns of breast cancer incidence and mortality: a population-based cancer registry data analysis from 2000 to 2020. Cancer Commun. (Lond) 41 (11), 1183–1194. doi:10.1002/cac2.12207
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, X., Quan, Y., Tang, C., and Chen, Y. (2016). Association between genetic variants of EGF-containing fibulin-like extracellular matrix protein1 gene and sporadic breast cancer in a Chinese Han population. Eur. J. Gynaecol. Oncol. 37 (1), 80–85. Available at: https://article.imrpress.com/journal/EJGO/37/1/pii/2016001/80-85.pdf
Ma, D., Chen, S. Y., Ren, J. X., Pei, Y. C., Jiang, C. W., Zhao, S., et al. (2021). Molecular features and functional implications of germline variants in triple-negative breast cancer. J. Natl. Cancer Inst. 113 (7), 884–892. doi:10.1093/jnci/djaa175
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20 (9), 1297–1303. doi:10.1101/gr.107524.110
Meyer, L. R., Zweig, A. S., Hinrichs, A. S., Karolchik, D., Kuhn, R. M., Wong, M., et al. (2013). The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res. 41, D64–D69. doi:10.1093/nar/gks1048
Miao, H. K., Chen, L. P., Cai, D. P., Kong, W. J., and Xiao, L. (2015). MSH3 rs26279 polymorphism increases cancer risk: a meta-analysis. Int. J. Clin. Exp. Pathol. 8 (9), 11060–11067.
Phan, L. M., and Rezaeian, A. H. (2021). ATM: main features, signaling pathways, and its diverse roles in DNA damage response, tumor suppression, and cancer development. Genes (Basel) 12 (6), 845. doi:10.3390/genes12060845
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Shah, P. D., Patil, S., Dickler, M. N., Offit, K., Hudis, C. A., and Robson, M. E. (2016). Twenty-one-gene recurrence score assay in BRCA-associated versus sporadic breast cancers: differences based on germline mutation status. Cancer 122 (8), 1178–1184. doi:10.1002/cncr.29903
Siegel, R. L., Miller, K. D., Wagle, N. S., and Jemal, A. (2023). Cancer statistics, 2023. CA Cancer J. Clin. 73 (1), 17–48. doi:10.3322/caac.21763
Su, Y., Yao, Q., Xu, Y., Yu, C., Zhang, J., Wang, Q., et al. (2021). Characteristics of germline non-BRCA mutation status of high-risk breast cancer patients in China and correlation with high-risk factors and multigene testing suggestions. Front. Genet. 12, 674094. doi:10.3389/fgene.2021.674094
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38 (16), e164. doi:10.1093/nar/gkq603
Yi, D., Xu, L., Luo, J., You, X., Huang, T., Zi, Y., et al. (2019). Germline TP53 and MSH6 mutations implicated in sporadic triple-negative breast cancer (TNBC): a preliminary study. Hum. Genomics 13 (1), 4. doi:10.1186/s40246-018-0186-y
Yoshimura, A., Imoto, I., and Iwata, H. (2022). Functions of breast cancer predisposition genes: implications for clinical management. Int. J. Mol. Sci. 23 (13), 7481. doi:10.3390/ijms23137481
Zhang, J., Sun, J., Chen, J., Yao, L., Ouyang, T., Li, J., et al. (2016). Comprehensive analysis of BRCA1 and BRCA2 germline mutations in a large cohort of 5931 Chinese women with breast cancer. Breast Cancer Res. Treat. 158 (3), 455–462. doi:10.1007/s10549-016-3902-0
Zhong, Q., Peng, H. L., Zhao, X., Zhang, L., and Hwang, W. T. (2015). Effects of BRCA1-and BRCA2-related mutations on ovarian and breast cancer survival: a meta-analysis. Clin. Cancer Res. 21 (1), 211–220. doi:10.1158/1078-0432.CCR-14-1816
Keywords: breast cancer, multi-gene panels, rare genetic variants, in silico protein modeling, pathogenic/likely pathogenic variants
Citation: Liu Y, Zheng J, Xu Y, Lv J, Wu Z, Feng K, Liu J, Yan W, Wei L, Zhao J, Jiang L and Han M (2023) Multigene testing panels reveal pathogenic variants in sporadic breast cancer patients in northern China. Front. Genet. 14:1271710. doi: 10.3389/fgene.2023.1271710
Received: 16 August 2023; Accepted: 16 October 2023;
Published: 09 November 2023.
Edited by:
Daniele Fanale, Azienda Ospedaliera Universitaria Policlinico Paolo Giaccone, ItalyReviewed by:
Chiara Brando, University of Palermo, ItalyEdvīns Miklaševičs, Riga Stradiņš University, Latvia
Copyright © 2023 Liu, Zheng, Xu, Lv, Wu, Feng, Liu, Yan, Wei, Zhao, Jiang and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meng Han, bWVuZzY4NTI3QDEyNi5jb20=