
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 18 December 2023
Sec. Genetics of Common and Rare Diseases
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1267368
 Zilong Geng1†
Zilong Geng1† Wenjuan Li2†
Wenjuan Li2† Ping Yang1†
Ping Yang1† Shasha Zhang1
Shasha Zhang1 Shuo Wu1Junhao Xiong1
Shuo Wu1Junhao Xiong1 Kun Sun2Dan Zhu3
Kun Sun2Dan Zhu3 Sun Chen2*
Sun Chen2* Bing Zhang1,2*
Bing Zhang1,2*Left ventricular outflow tract obstruction (LVOTO), a major form of outflow tract malformation, accounts for a substantial portion of congenital heart defects (CHDs). Unlike its prevalence, the genetic architecture of LVOTO remains largely unknown. To unveil the genetic mutations and risk genes potentially associated with LVOTO, we enrolled a cohort of 106 LVOTO patients and 100 healthy controls and performed a whole-exome sequencing (WES). 71,430 rare deleterious mutations were found in LVOTO patients. By using gene-based burden testing, we further found 32 candidate genes enriched in LVOTO patient including known pathological genes such as GATA5 and GATA6. Most variants of 32 risk genes occur simultaneously rather exclusively suggesting polygenic inherence of LVOTO and 14 genes out of 32 risk genes interact with previously discovered CHD genes. Single cell RNA-seq further revealed dynamic expressions of GATA5, GATA6, FOXD3 and MYO6 in endocardium and neural crest lineage indicating the mutations of these genes lead to LVOTO possibly through different lineages. These findings uncover the genetic architecture of LVOTO which advances the current understanding of LVOTO genetics.
Cardiac outflow tract (OFT) abnormalities characterized by an array of structural deviations within the region responsible for facilitating the egress of blood from the heart account for approximately 30% of CHD patients (Srivastava and Olson, 2000). One important category of OFT anomalies is left ventricular outflow tract obstruction (LVOTO), with the stenotic lesions at the region of left ventricular outflow tract (could be valvular, sub-valvular or supravalvular). These stenoses can occur alone or in combination, obstruct the blood flow and induce the overload of left ventricle, if untreated which results in ventricle hypertrophy and systolic failure eventually. The majority of LVOTOs are congenital and account for around 6% of CHD (Gaynor and Elliott, 1993). The congenital LVOTO encompasses a range of conditions, including aortic stenosis (AS), bicuspid aortic valve (BAV), coarctation of the aorta (CoA), and anomalous subaortic stenosis (ASA).
Aortic stenosis represents 3%–6% of CHDs with features of narrowed aortic valve orifice and curbed blood flow from left ventricle to aorta. AS is an important cause of congestive heart failure in neonates and fetal demise (Minners et al., 2020). Bicuspid aortic valve is the most common congenital aortic valve malformation with the presence of only two valve leaflets instead of the normally three. Clinical manifestations of BAV can vary from mild to severe. It is the leading cause of AS in patients under 70 years old though might with subtle symptoms in early years (Pujari and Agasthi, 2023). Coarctation of the aorta accounts for 6%–8% of CHDs, and is often accompanied by BAV (∼60%) (Sinning et al., 2018), aortic arch hypoplasia (∼18%) (Ma et al., 2017), ventricular septal defect (∼13%) and other congenital heart defects.
Genetic mutations had been discovered to play a large role in LVOTO etiology. Previous studies have identified NOTCH1 mutations (p.Arg1108Ter and p.His1505del) in patients with AS and early BAV (Garg et al., 2005). Genetic variants of Cardiac GATA transcription factor GATA4, GATA5, GATA6 and TBX5 were also found to be related to BAV in multiple studies (Bravo-Jaimes and Prakash, 2020; Jiang et al., 2020). A linkage analysis based on family pedigrees reported associations between AS and CoA in chromosomes 2p23, 10q21, and 16p12 (McBride et al., 2009). These studies suggested that there was a shared genetic mechanism across divergent syndromes of LVOTO. Although substantial efforts have been made to uncover LVOTO genetic variants, understanding of the inherent genetic status and mechanism remained largely incomplete especially for the Asian population.
To explore the genetic inherent mechanisms underlying LVOTO of Chinese CHD patients, we performed whole-exome sequencing (WES) analysis on 106 CHD patients with LVOTOs. We discovered rare deleterious variants including missense SNV, microdeletion and insertion in LVOTO patients which are higher than in control subjects. By gene-based burden testing, we identified 32 candidate genes, which were significantly enriched in transcriptional regulation, cardiac development, and neural development. Among 32 candidate risk genes, 14 were found to form an interacting network with known OFT malformation-related genes. Utilizing scRNA-seq and scATAC-seq, we found GATA5 and GATA6 may play essential roles in endocardial-derived OFT lineage, while FOXD3, MYO6 and GATA6 in neural crest-derived lineages. In conclusion, our study uncovers a genetic architecture associated with LVOTO in the Chinese population and offers insights into the pathogenic underpinnings of this complex congenital defect.
Our study enrolled 106 individuals from Han Chinese population diagnosed with LVOTO with clinic syndrome: atrial septal defect (ASD) and ventricular septal defect (VSD). 100 healthy individuals who were ruled out CHD and other birth defects were recruited to the cohort as control. We excluded participants with known chromosomal or syndromic disorders to reduce the statistical errors caused by other confounding factors. This study was approved by the Ethics Committee of Xinhua Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, and conducted in accordance with the principles outlined in the Declaration of Helsinki. All participants or their legal guardians provided written informed consent before being enrolled in the study.
Blood samples were obtained, and genomic DNA was subsequently extracted employing the QIAamp™ DNA Blood Mini kit (Qiagen) in strict accordance with the manufacturer’s recommended protocol. Whole-exome sequencing (WES) was performed utilizing the Agilent SureSelect Target Enrichment kit (V6 58 Mb; Agilent Technologies) for sequence capture, followed by sequencing on the Illumina HiSeq2500 platform. The sequencing aimed to achieve a target depth of 100 fold coverage to ensure accuracy.
To obtain high-quality sequencing data for downstream analysis we used the Genome Analysis Toolkit (GATK) best practice to process the fastq file (McKenna et al., 2010). Firstly, the raw sequencing reads were aligned to the hg19 reference genome using the Burrows-Wheeler Aligner (BWA) (Li and Durbin, 2009). Subsequently, we identified and marked PCR duplicates using the MarkDuplicates tool in GATK. To recalibrate the base quality scores of the aligned reads, we used Base Quality Score Recalibration (BQSR) with the BaseRecalibrator. Next, variant calling was performed using the HaplotypeCaller tool in GATK v4.1.9.0 with default parameters. To ensure that the variant calls were accurate, we performed Variant Quality Score Recalibration (VQSR) using the VariantRecalibrator and ApplyVQSR tools in GATK. Finally, HaplotypeCaller was used to call variants.
To eliminate potential confounding factors due to differences in genetic ancestry among study participants, we performed population stratification analysis on the cohort. We merged the 1000 Genomes Project (1KGP) reference dataset (Clarke et al., 2012) with the data from our cohort. We then performed PCA on the merged dataset using PLINK (Purcell et al., 2007). We plotted the first two principal components (PC1 and PC2) of the PCA results to visualize the genetic structure and population stratification in our cohort and the 1KGP reference panel.
In this study, we aimed to investigate the role of rare and deleterious variants enriched in the LVOTO patients. To this end, we utilized ANNOVAR (Wang et al., 2010) to annotate the variants. Population frequency databases, Genome Aggregation Database (gnomAD v2.1.1) (Koch, 2020) and Exome Aggregation Consortium (ExAC) were used to assess the frequency of the variants in the general population. In this study, rare variants are defined as having a minor allele frequency (MAF) of less than 1% in both the gnomAD and ExAC databases. Next, to assess the potential functional impact of the identified rare variants, we employed multiple in silico prediction algorithms, including Sorting Intolerant From Tolerant (SIFT), Polymorphism Phenotyping v2 (PolyPhen2), MutationTaster and Combined Annotation Dependent Depletion (CADD). All database downloads were conducted following the protocol provided by the ANNOVAR website (https://annovar.openbioinformatics.org/en/latest/user-guide/). Variants were classified as deleterious if they were predicted to be damaging or deleterious by at least two of the four algorithms. Finally, rare deleterious variants were kept to perform burden test.
Gene based burden test was performed to evaluate the association between the identified rare deleterious variants and the LVOTO phenotype using RVTESTS (Zhan et al., 2016). Then we applied Bonferroni correction on the p-value of burden test to account for multiple testing. Finally, genes with adjusted p-value less than 0.05 were defined as candidate genes enriched with rare deleterious variants.
We carried out functional annotation using the Database for Annotation, Visualization, and Integrated Discovery (DAVID; https://david.ncifcrf.gov) to perform Gene Ontology (GO) term analyses. The GO terms are focused on biological processes to annotate potential function of the gene set.
To investigate the patterns of occurrence of candidate genes in patients, we employed the somaticInteractions function from maftools to conduct co-occurrence statistics between the genes. This method is based on the pair-wise Fisher’s exact test as detailed in the maftools documentation (https://github.com/PoisonAlien/maftools). Then we used the CHDgenes database (http://chdgene.victorchang.edu.au/) to identify known OFT malformation-related genes. Then we investigate the protein-protein interaction (PPI) network using the STRING database (version 11.0; https://string-db.org/). We set the confidence score threshold at 0.7 to ensure a high-quality interaction network and to minimize the inclusion of false-positive interactions. Cytoscape (version 3.9.0; https://cytoscape.org/) was employed for the visualization and analysis of the protein-protein interaction (PPI) network. To identify hub genes within the PPI network, we utilized the cytoHubba plugin based on the PPI topological features.
The processed single-cell RNA-seq and single-cell ATAC-seq datasets were acquired from GSE181346 and https://doi.org/10.5281/zenodo.7063223 respectively (Ameen et al., 2022). The scRNA-seq dataset had been subjected to quality control and preprocessing steps including read alignment, gene assignment for scRNA-seq. We utilized the Seurat package (version 4.0; https://satijalab.org/seurat/) in R and scanpy (Wolf et al., 2018) to conduct downstream analysis, such as normalization and visualization of single-cell RNA sequencing data. Pseudotemporal analysis was conducted using Monocle3 (http://cole-trapnell-lab.github.io/monocle3/) and CellRank (Lange et al., 2022). For the ATAC-seq datasets, cell type-specific marker peaks across various cell populations were identified using the ArchR package (Granja et al., 2021). To perform peak calling in each cell clusters, we used MACS2 (Zhang et al., 2008) for scATAC-seq datasets. Peaks were associated with genes based on the annotations provided in the dataset.
A cohort of 106 LVOTO probands, 69 males (65.09%), 37 females (34.91%) and 100 health controls were enrolled from 2016 to 2023. 82 LVOTO patients (77.36% of total) were at age less than 1 year, 18 patients (16.98%) were at age of 1–3 years and six patients (5.66%) older than 3 years (Table 1). All patients were diagnosed with LVOTO, including BAV (8.49%, n = 9), AS (46.23%, n = 49), and CoA (49.06%, n = 52) (Table 2). Notably, AS and CoA co-occurred in four cases. Several other cardiac abnormalities were observed: 45 atrial septal defects (ASD, 42.45%), 42 ventricular septal defects (VSD, 39.62%), 35 patent ductus arteriosus (PDA, 33.02%) and five mitral stenosis/mitral regurgitation (MS/MR, 4.72%). This was consistent with previous reports that LVOTO is usually complicated with multiple pathological phenotypes outside OFT. The control group consisted of 100 healthy children, confirmed not to have any developmental anomalies.
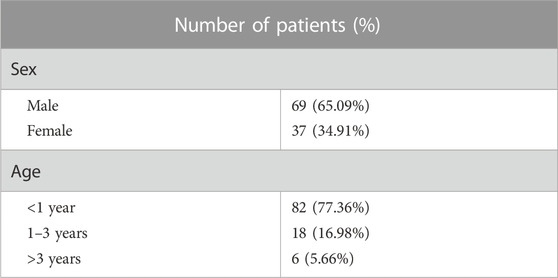
TABLE 1. Demographic characteristics of LVOTO patients.
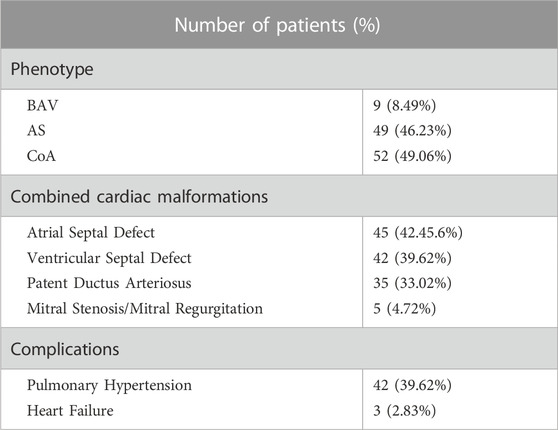
TABLE 2. Diagnosis of the patients with LVOTO.
We applied the Genome Analysis Toolkit (GATK) Best Practices workflow to analyze the whole-exome sequencing data and found a total of 271,462 high-confidence single nucleotide variants (SNVs) and indels in the LVOTO probands. On average, each proband harbored 39,057 Single Nucleotide Variants (SNVs) and 5,597 indels including insertions or deletions, which was significantly higher than the control group with average of 38,764 SNVs and 4,226 indels per subject (Supplementary Figure S1).
To ensure the quality of our analysis, we evaluated the heterozygosity rate, the number of singletons and the ratio of transitions to transversions (Ti/Tv) (Figure 1A). The heterozygous to non-heterozygous ratio in our cohort was 1.3, the transition to transversion ratio was 3.11, which were in agreement to standard expectations for whole-exome sequencing (Wang et al., 2015). The number of singletons averaged at 517 per sample and no apparent outliers were observed. We also performed population stratification analysis by integrating our dataset to 1000 Genomes Project (1KGP) reference panel and found a complete overlap with Chinese Han population, which ruled out the confounding effect from population stratification (Figure 1B).
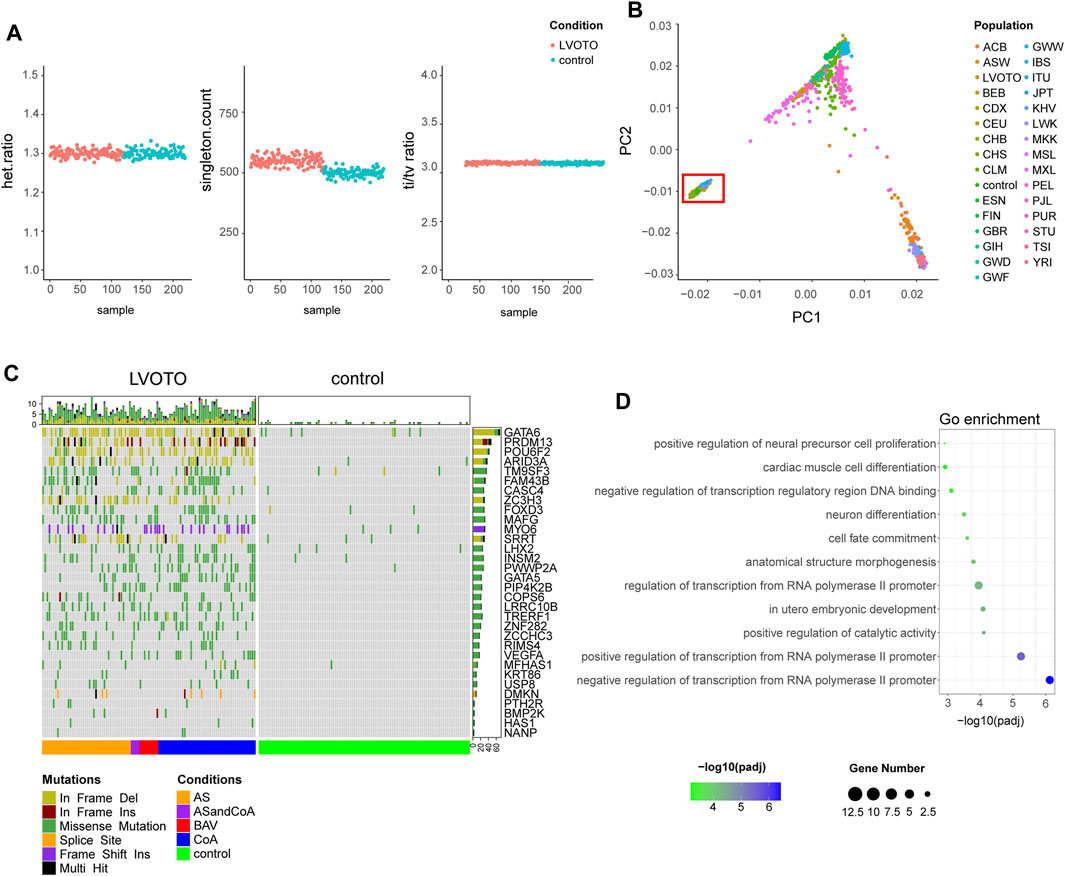
FIGURE 1. Identification of candidate genes with rare deleterious variants. (A) Quality control of variants in LVOTO patients and controls, including heterozygous mutation rate, singleton statistics, and transition-to-transversion ratio. (B) Population stratification comparison with the 1000 Genomes Project. The red box indicates the LVOTO patients, control subjects, and Chinese samples from the 1000 Genomes Project. (C) Landscape of rare deleterious variants in LVOTO-associated candidate genes based on burden test. (D) Functional enrichment analysis of the candidate gene set.
Compared with common diseases, LVOTO is rare and prone to be caused by rare variants. Therefore, we focused on the rare variants. To refine our identification and on potentially pathogenic variants, we adhered to the guideline provided by the American College of Medical Genetics and Genomics (ACMG). By leveraging GATK and ANNOVAR platform, we identified 106,476 rare deleterious variants with minor allele frequency (MAF) less than 1% in both the Genome Aggregation Database (gnomAD) and the Exome Aggregation Consortium (ExAC) population databases. 71,430 rare deleterious variants were discovered in LVOTO patient cohort, with each patient carrying an average of 4,600 rare deleterious SNVs and 301 indels. In contrast, the control group exhibited 52,989 such variants, with each individual carrying an average 4,477 rare deleterious SNVs and 265 indels (Supplementary Figure S1). This disparity suggested the potential contribution of these rare variants to the pathophysiology of LVOTO.
In order to identify genes enriched with rare deleterious SNVs and indels in LVOTO patients, we performed a gene-based burden test using RVTESTS program. We defined LVOTO risk genes as those with p-value less than 0.05 adjusted by multiple testing correction. As a result, 32 candidate genes with 572 rare deleterious variants in LVOTO patients versus 32 in controls were potentially associated with LOVTO pathogenesis (Figure 1C, Supplementary Tables S1, S2). The variants of GATA6, PRDM13, POU6F2, ARID3A, ZC3H3 and SRRT were predominantly in a form of frame deletion, in MYO6 were frameshift insertions, and in the rest were the missense mutations. Functional enrichment analysis indicated these 32 genes are involved in transcription regulation, embryonic development and cardiac muscle cell differentiation (Figure 1D). Interestingly, neural cell differentiation term was also enriched, indicating some of LVOTO risky genes were from neural crest lineage that plays an important role in OFT septation and aortic arch patterning.
Among 32 candidate genes, GATA5 (p = 0.0096 in burden test) and GATA6 (p = 6.22E-10 in burden test) had been known to be associated with LVOTO. GATA5 and GATA6 are the zinc finger transcription factors specifically binding to GATA DNA motif and play critical roles in regulating early cardiac gene expression and cardiac cell differentiation. Previous human genetics study showed that GATA5 and GATA6 were associated with BAV and AS (Shi et al., 2014; Bravo-Jaimes and Prakash, 2020). Animal model study illustrated that loss of GATA5 in mice resulted in BAV (Laforest et al., 2011). Herein, we identified a c.40G>C missense mutation of GATA5 in 22 out of 106 patients, which resulted in alanine to proline change at position 14 (p.Ala14Pro) in a N terminal disordered region. Among these 22 patients, 17 had CoA combined with VSD and AVSD and five patients had AS. This variant had not been reported before to be associated with outflow tract malformation in western population. Same mutation observed in a substantial portion of our small cohort indicated c.40G>C might be a founder mutation specifically affected Chinese Han people. We also found a heterozygous c.993_998 microdeletion of GATA6 which causes the deletion of Histidine at position 332 and 333 (p.His332_His333del) in a disordered region (Supplementary Figure S2). Similar to GATA5, 58 patients harbored this deletion, indicating it may be a founder variant in Chinese Han population as well.
VEGFA (Vascular Endothelial Growth Factor-A) is an essential member of the vascular endothelial growth factor (VEGF) family. Nonsense mutations in VEGFA have been discerned in patients diagnosed with LVOTO (Zhao et al., 2010). Lack of OFT septation is seen in Vegfa120/120 mouse model expressing a short form of VEGFA 120 (Plein et al., 2015). Several pathways such as BMP and SEMA3C regulate OFT development by interacting with VEGFA. Here, we found VEGFA is enriched in LVOTO patient cohorts as 17 variants were discovered in LVOTO versus none in controls (p = 0.041). Among them, a c.329G>T missense mutation causing arginine to leucine substitution at position 139 (p.Arg110Leu) were found in 15 patients. Intriguingly, within this cohort, 11 patients were diagnosed with CoA, 3 with BAV, and one presented with a combination of CoA and AS. Interestingly, none with AS only. These well-recognized candidate genes association with LVOTO, certified the effectiveness of our gene-based burden testing approach.
For complex diseases, the genetic risk genes usually do not exert their roles alone but in concert with other genes in a regulatory network. Therefore, we conducted a protein-protein interaction network analysis using STRING to assess the interaction between our candidate genes and known disease-causal genes of outflow tract malformations and obstructive lesions quoted from the congenital heart disease risk gene database (http://chdgene.victorchang.edu.au/, Supplementary Table S3). 49 OFT risky genes including NOTCH1, GATA4 and NKX2-5 were found to be connected with 14 LVOTO risk genes (Figure 2A). Among them, we identified five key hub genes including GATA5, GATA6 and VEGFA. These genes demonstrated the high degree of connections, interacted with multiple quoted CHD genes, including NKX2-5, HAND2, and GATA4.
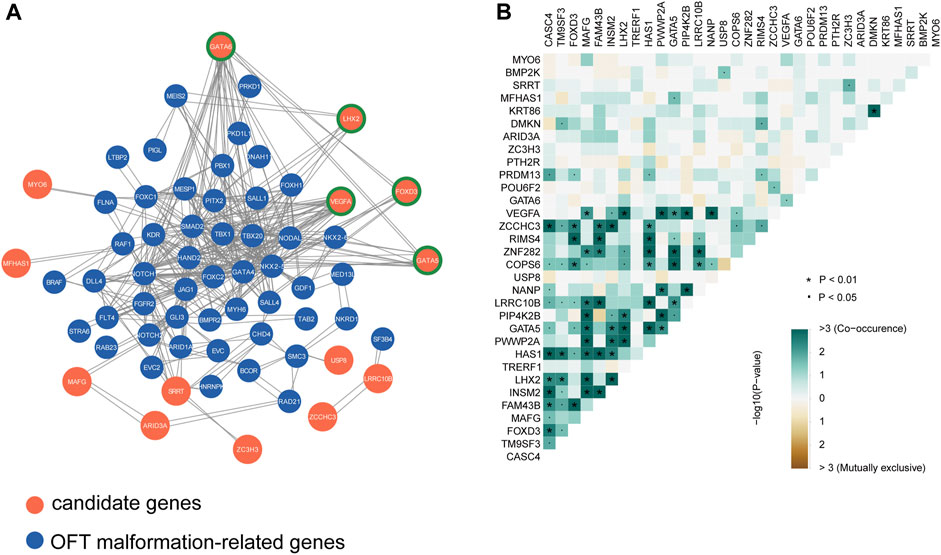
FIGURE 2. Interactions analysis among candidate genes and known CHD genes. (A)Protein-protein interaction network of candidate genes and known OFT abnormality risk genes, highlighting the potential functional relationships and molecular pathways shared by these genes in the context of outflow tract development and dysfunction. Node genes are marked with a green border. (B) Co-occurrence statistics of LVOTO-associated candidate genes, demonstrating the co-occurrence of these genes in patients with LVOTO.
FOXD3, a member of the forkhead box (FOX) family transcription factors, also had pleiotropic connections with GATA6, NKX2-5 and SALL4. FOXD3 is expressed in cardiac neural crest cells (Stankunas et al., 2010). These cells migrate and contribute to the formation and remodeling of the pharyngeal arch arteries and the septation of the cardiac outflow tract (Yamagishi, 2021). Deletion of Foxd3 in zebrafish results in significant losses of distinct neural crest derivatives. A subset of Foxd3 mutant mouse exhibited severe cardiac neural crest defects and absence of the outflow tract septation (Teng et al., 2008). In our cohort, FOXD3 rare deleterious variants were highly enriched in LVOTO patients (p < 0.05 in burden test). Among these variants, we discovered a previously unreported missense mutation c.803C>G in 23 LVOTO patients, which resulted in an alanine to glycine substitution at position 268 (p.Ala268Gly). Importantly, this mutation was not observed in our control group. This evidence suggested that FOXD3 might be a LVOTO-causal gene in Chinese population.
LHX2 is another gene in our 32 candidates gene list interacting with known CHD genes. LHX2 is a LIM domain-containing transcription factor specifically involving in pharyngeal mesoderm development. Knockout of this gene in mouse results in pharyngeal muscle defects, as well as DiGeorge syndrome-like phenotypes such as variety of outflow tract malformations and tetralogy of Fallot (Harel et al., 2012). In our cohort, we discovered a missense mutation c.92C>T in LHX2 in 22 individuals. This mutation results in an amino acid substitution from serine to phenylalanine at position 31 (p.Ser31Phe).
The candidate genes may have two distinct modes of action in pathogenesis: the first, with minor pathogenicity, and simultaneous occurrence in different probands; the second, with strong pathogenicity, mutually exclusive occurrence in probands (Fahed et al., 2013). Therefore, we conducted a concurrency analysis of rare deleterious variants in 32 risky genes to reveal their genetic behaviors in LVOTO (Figure 2B). The results demonstrated that compared to mutual exclusion, much more concurrency (>3) was observed between 32 genes (p < 0.01) indicating a polygenic inheritance in LVOTO pathogenesis. Notably, we found strong co-occurrence of GATA5 with LHX2 and VEGFA with LHX2 in LVOTO, suggesting a synergetic role of LHX2 with VEGFA and GATA5 in LVOTO pathogenesis.
To gain insight into the spatiotemporal expression profile of the candidate genes, we analyzed the previously published single-cell transcriptomic dataset of human embryonic cardiac development at 6PCW (post-conception weeks), 8PCW, 12PCW and 18PCW (Ameen et al., 2022). The cells were grouped into 14 clusters by using UMAP dimensionality reduction method. Major cell types including myocytes, fibroblast, endothelium, smooth muscle and endocardium were all obtained (Figure 3A). As reported in mouse studies, GATA6 was highly expressed in endocardium, myocardium, smooth muscle progenitor and fibroblast, while GATA5 expression is much limited to endocardium and epicardium (Laforest and Nemer, 2011). FOXD3 was specifically expressed in neural crest cells while DMKN more specifically in epicardium. There were several genes such as CASC4, TM9SF3, USP8 and COPS6 ubiquitously expressed in all tissues, but their functions in OFT remain ambiguous (Figure 3B).
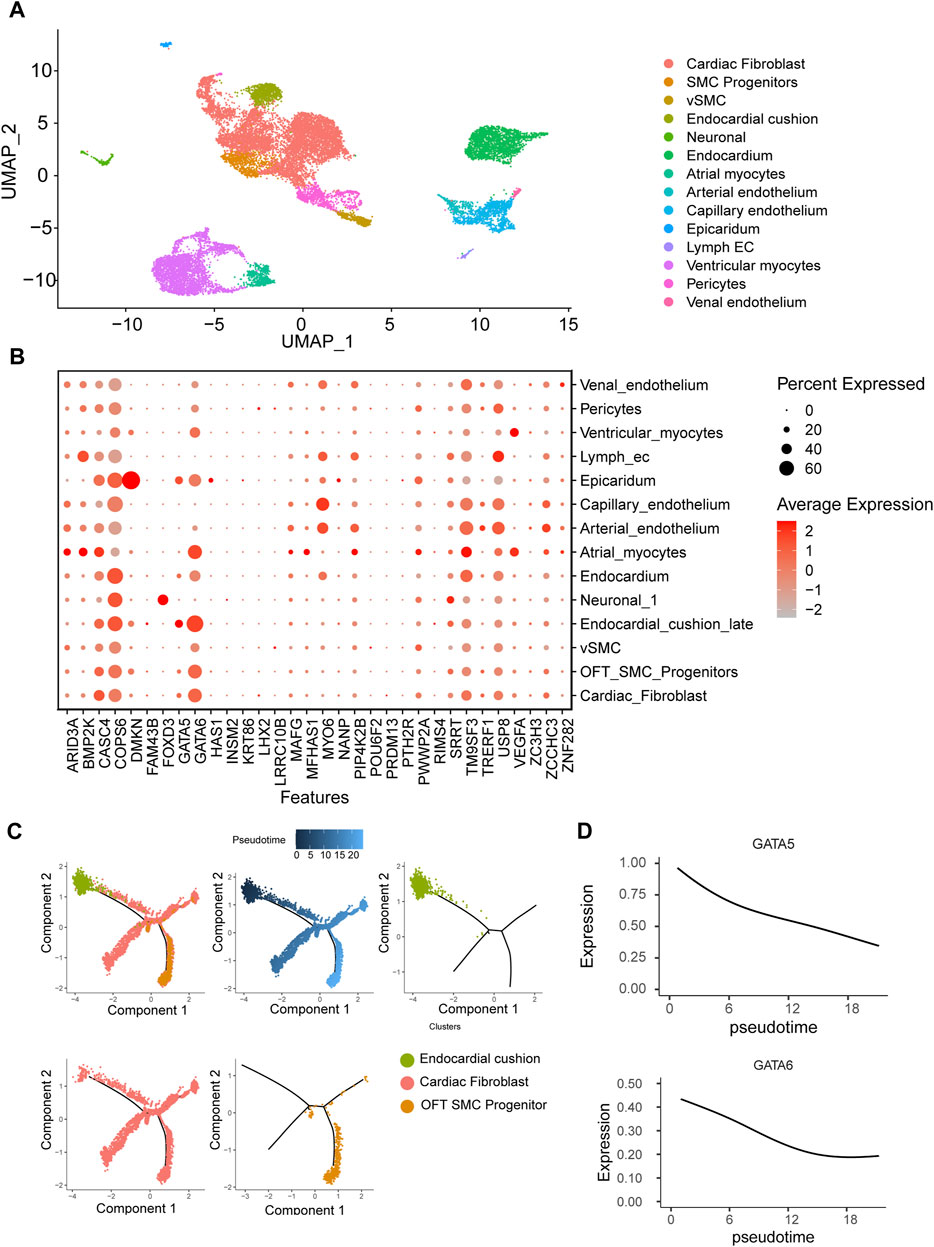
FIGURE 3. Human embryonic heart single-cell transcriptome analysis. (A) Clustering of human embryonic heart single cells. (B) Expression patterns of candidate genes across distinct cell populations. (C) Pseudotemporal analysis of endothelial-to-mesenchymal transition process of endocardial cushion cells. (D) Dynamic expression patterns of GATA5 and GATA6 along the pseudotime trajectory.
One of major cell source for OFT is endocardium-derived cells through endothelial-to-mesenchymal transition (EndMT) that contributes to mesenchymal valves and OFT cushion (Ma et al., 2005). Interestingly, scRNA-seq uncovered two isolated populations of endocardium. One is in spatial proximity to myocardial fibroblast cells and outflow tract smooth muscle progenitor clusters, suggesting a close transcriptomic relationship among them. We conducted a pseudotime analysis and observed a decreasing expression of GATA5 and GATA6 from endocardium to fibroblast cells (Figures 3C, D), indicating the roles of GATA5 and GATA6 in the EndMT process. To further elucidate the genomic binding profiles and downstream effectors of GATA5 and GATA6 in human endocardial lineage, we analyzed scATAC-seq dataset (GSE181346) for human cardiac development (Ameen et al., 2022). Using MACS2, we called peaks for each cell population, obtaining a total of 2,352 peaks in endocardial lineage. We identified a high prevalence of GATA5 and GATA6 (Figure 4A) by motif enrichment analysis. GO term analysis of GATA5 and GATA6 motif-associated genes included outflow tract morphogenesis, endocardial cushion to mesenchymal transition, indicating GATA5 and GATA6 regulated the expression of these genes that contributed to endocardial lineage and OFT development (Figure 4B).
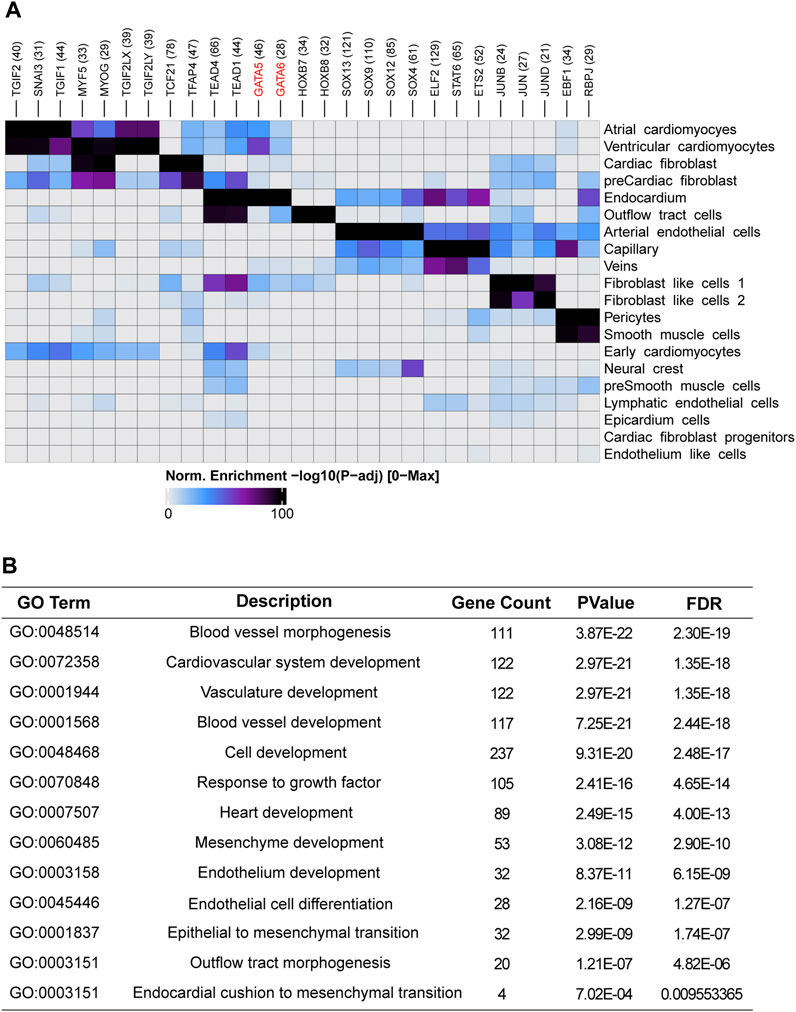
FIGURE 4. Single-cell accessibility analysis of the human embryonic heart. (A) Transcription factor motif enrichment analysis in cell-type-specific accessible genomic regions for different cell populations within the human embryonic heart. (B) Functional enrichment analysis of GATA5 and GATA6 potential target genes in endocardium.
Apart from the endocardial lineage, neural crest cells are another major progenitor cell lineage that populated outflow tract. We analyzed the scRNA-seq specific for neural crest lineage from E8.5 to E10.5 in mouse (De Bono et al., 2023). We found that neural crest cells were consisted of four clusters: early migrated NCC, mesenchymal NCC, Cardiac NCC of OFT and neural progenitors on specific markers (Figure 5A). Early migrated neural crest cells were abundant in E8.5 but declined gradually from E9.5. The mesenchymal NCCs were first increased and then decreased, while the cardiac NCC were gradually increased (Figure 5B). We found FOXD3 highly expressed in the early migrated NCC but nearly diminished in other lineages, indicating FOXD3 plays a role in the early stage of NCC progenitor differentiation (Figure 5C). GATA6, rather than GATA5, was also expressed in NCC and increased its expression following NCC development (it was low in early migrated NCC but high in later cardiac NCC) (Figure 5D).
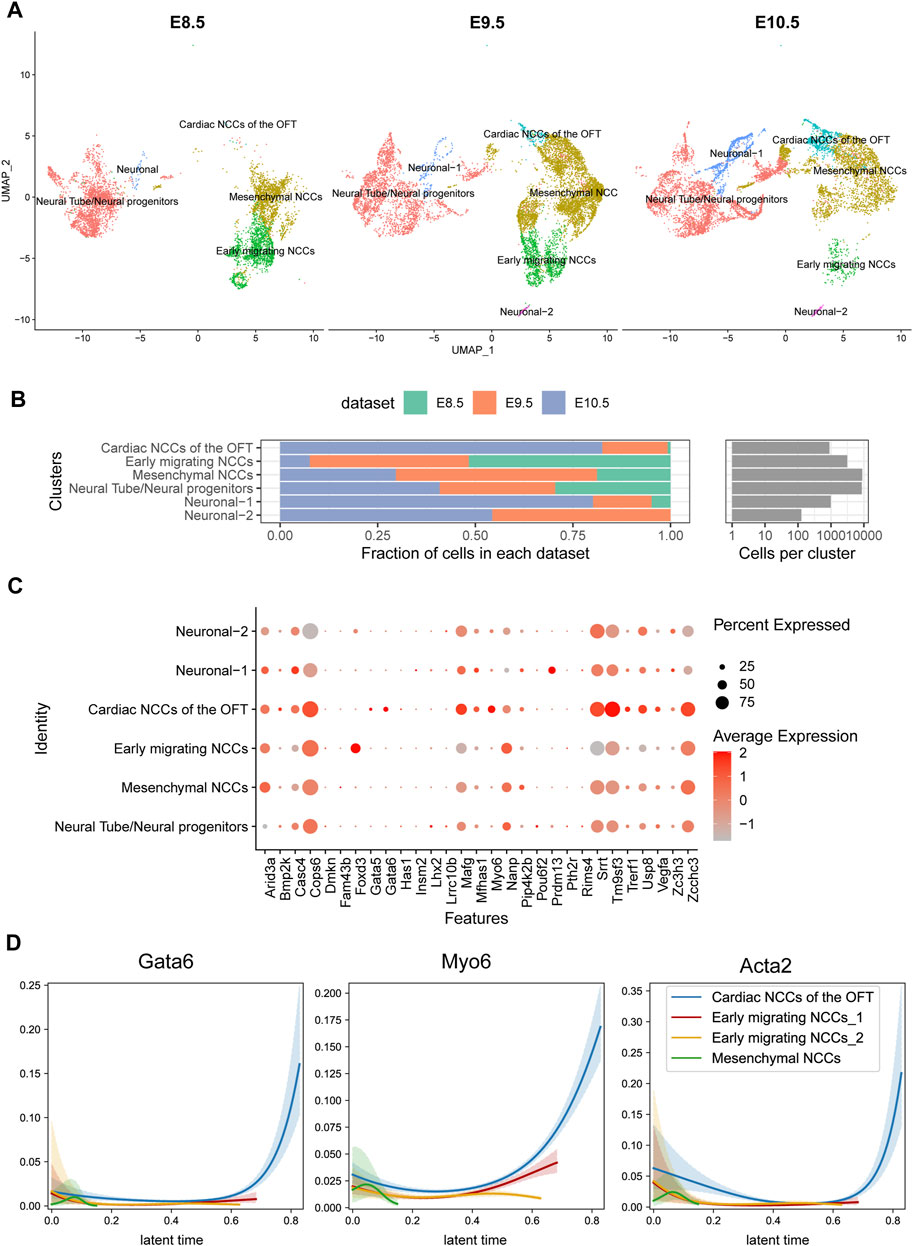
FIGURE 5. Temporal analysis of single-cell transcriptomics of cardiac neural crest cells in mouse embryos. (A) Single-cell clustering within the pharyngeal region at E8.5, E9.5, and E10.5 time points. (B) Proportion statistics of each cell type at each time point. (C) Expression patterns of candidate genes across cell populations. (D) Gata6 and Myo6 expression pattern in CNCCs migrating and differentiation process.
One of 32 candidate genes, MYO6 is a member of myosin family functioning in a variety of intracellular processes such as vesicular membrane trafficking and cell migration whose expression was also increased during neural crest development. Loss of MYO6 leads to the deafness in human and animal (Melchionda et al., 2001; Ahmed et al., 2003). A study suggested that MYO6 mutations in humans were also associated with cardiomyopathies (Hegan et al., 2015). In our cohort, we identified a duplication mutation c.2751dup, resulting in a frameshift change of p.Gln918ThrfsTer24 in 27 LVOTO patients. This mutation, documented as pathogenic in ClinVar, has previously been associated with deafness. However, its association with CHD had not been previously reported. The dynamic expression of MYO6 in neural crest suggested that a part of OFT malformation may be attributed to functional abnormality of MYO6 in neural crest.
In this study, we constructed a LVOTO cohort and performed whole exome sequencing to identify 71,430 rare deleterious SNVs and indels in 106 LVOTO patients. Utilizing a gene-based burden test, 32 candidate genes including GATA5, GATA6, FOXD3, LHX2, MYO6 were identified as possibly associated with LVOTO pathogenesis. Among the candidate genes, GATA5 and GATA6 have been reported to collaboratively regulate the development of the outflow tract (Laforest and Nemer, 2011; Gharibeh et al., 2018). Interestingly, we identified recurrent variants c.40G>C missense mutation in GATA5 (20.75%, n = 22) and c.993_998 microdeletion in GATA6 (54.71%, n = 58), both of which were enriched in LVOTO patients and not reported in western populations’ LVOTO cohorts. Besides, VEGFA, a key gene for vascular development, was identified as another LVOTO candidate gene. In the cohort, we observed 15 LVOTO patients harboring c.329G>T missense mutations. The recurrence of these mutations in our cohort suggests that they may be the founder mutations in the Han Chinese LVOTO population.
Apart from novel SNVs, our study also identified the novel candidate genes FOXD3 and LHX2. Although these genes had not been previously reported to associate with LVOTO, they have been implicated in neural crest and pharyngeal mesoderm development (Stewart et al., 2006; Harel et al., 2012). The identification of mutant genes in neural crest also has a key role in OFT development, which cannot be revealed by lineage tracing approach as in mouse. Additionally, we identified MYO6 as LVOTO genes that was previously reported to be associated with artery disease (Slavin et al., 2011). Of note, we identified a high impact frameshift mutation c.2751dup in MYO6 in 31 LVOTO patients, a variant that is associated with deafness (Kwon et al., 2014; Garcia-Garcia et al., 2020). However, there were no deafness observed in these 31 patients. This implies the incomplete penetrance of MYO6 in deafness onset, or possibly cooperated with distinct genetic modifiers to cause deafness or CHD respectively. Further investigations are required to clarify the underlying mechanism. It is noteworthy that despite the occurrence of incomplete penetrance in disease association analysis, the gene-based burden testing approach utilized in our study has demonstrated robustness, even in the face of diseases with moderate incomplete penetrance (Guo et al., 2016). This underscores the potential of this approach in uncovering genetic contributors to complex disorders, paving the way for more comprehensive genetic studies that could further elucidate the intricate mechanisms underlying LVOTO and similar conditions.
By scRNA-seq, we found LVOTO genes dynamically expressed mainly in two lineages. In the endocardium-derived lineage, GATA5 and GATA6 expressed in the early stage and then quickly declined during the process of EndMT. In the cardiac neural crest cell lineage, GATA6, MYO6, and FOXD3 were progressively upregulated along with the migration and differentiation of neural crest cells. Moreover, an elevation in LHX2 expression was observed only in an early progenitors stage. These results illuminate the dynamic transcriptional landscape of candidate genes during OFT development and underscore their distinct functions. This also highlights the complexity of OFT malformation and LVOTO pathogenesis.
NOTCH1 is a well-established risk gene for LVOTO. However, our burden test did not identify the significant SNV enrichment of NOTCH1 and other seven previously reported risk genes (Supplementary Figure S3). A reasonable explanation might be that, as a complex disease, the pathogenesis of LVOTO is not solely attributed to a single gene. NOTCH1, as the most extensively studied gene associated with LVOTO, accounts for only 6% of patients with LVOT malformations (Roifman et al., 2021). The relatively small size of our cohort or geographical bias of patient enrollment could also influence these findings. A smaller cohort size often leads to reduced statistical power, which can limit our ability to detect and accurately interpret genetic associations and trends. Especially for gene-based burden analyses, analyzing dominant disorders with high locus heterogeneity typically requires larger cohort sizes (Guo et al., 2016). Therefore, future studies should aim to include larger cohorts, which would provide more robust statistical analyses. Besides, the genetic diversity between our Han Chinese cohort and the Western population could be another reason for the failed identification of NOTCH1 as significantly enriched. Additionally, it is important to mention that there are another 26 genes identified by our association test that may contribute to LVOTO, but were not discussed in detail within this paper. For example, a recent study highlighted an association of BMP2K to OFT development (Dai et al., 2013). One study of our group demonstrated MAFG as a key regulatory factor that plays a crucial role in the modulation of VEGFA expression and angiogenesis (Wang et al., 2019). These genes could potentially possess crucial information to understand the genetic landscape of LVOTO. Moreover, our results suggested LVOTO has a complex genetic architecture and cooperative gene function manner (Figure 2B), therefore, it will become particularly important to clarify the collective gene interacting network for sake of ultimately understanding the genetic pathogenesis of LVOTO.
In sum, our study provides a comprehensive investigation of the genetic architecture of LVOTO in Han Chinese population and new insights into the potential molecular mechanisms underlying the pathogenesis of outflow tract malformations. Our study identified new rare deleterious variants and genes possibly contributing to LVOTO pathogenesis. Further studies are warranted to validate these SNVs and genes and elucidate the underlying molecular mechanisms.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
The studies involving humans were approved by Ethics Committee of Xinhua Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
ZG: Data curation, Formal Analysis, Methodology, Visualization, Writing–original draft, Writing–review and editing. WL: Data curation, Resources, Writing–original draft, Writing–review and editing. PY: Data curation, Validation, Writing–review and editing. SZ: Conceptualization, Supervision, Validation, Writing–original draft. SW: Data curation, Writing–review and editing. JX: Data curation, Writing–review and editing. KS: Resources, Writing–review and editing. DZ: Resources, Writing–review and editing. SC: Conceptualization, Resources, Supervision, Writing–review and editing. BZ: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing.
The authors declare financial support was received for the research, authorship, and/or publication of this article. This work is supported by the BZ’s grants from National Foundation of Distinguished Young Scholar (82225006), National Key Research and Development Program of China (2020YFA0803800,2020YFA0803802), the Innovation Program of the Shanghai Municipal Education Commission (2021-01-07-00-02-E00088), WLA Program of Shanghai Science and Technology Commission (21dz2210202, 21dz2210200), and SJTU STAR Award (YG2021ZD11, YG2022ZD023).
We express profound gratitude to the patients for their active participation in this study. We sincerely thank the medical staff in Shanghai Children’s Medical Center for their invaluable contribution in supplying the clinical samples.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1267368/full#supplementary-material
Ahmed, Z. M., Morell, R. J., Riazuddin, S., Gropman, A., Shaukat, S., Ahmad, M. M., et al. (2003). Mutations of MYO6 are associated with recessive deafness, DFNB37. Am. J. Hum. Genet. 72 (5), 1315–1322. doi:10.1086/375122
Ameen, M., Sundaram, L., Shen, M., Banerjee, A., Kundu, S., Nair, S., et al. (2022). Integrative single-cell analysis of cardiogenesis identifies developmental trajectories and non-coding mutations in congenital heart disease. Cell 185 (26), 4937–4953.e23. doi:10.1016/j.cell.2022.11.028
Bravo-Jaimes, K., and Prakash, S. K. (2020). Genetics in bicuspid aortic valve disease: where are we? Prog. Cardiovasc Dis. 63 (4), 398–406. doi:10.1016/j.pcad.2020.06.005
Clarke, L., Zheng-Bradley, X., Smith, R., Kulesha, E., Xiao, C., Toneva, I., et al. (2012). The 1000 Genomes Project: data management and community access. Nat. Methods 9 (5), 459–462. doi:10.1038/nmeth.1974
Dai, X., Jiang, W., Zhang, Q., Xu, L., Geng, P., Zhuang, S., et al. (2013). Requirement for integrin-linked kinase in neural crest migration and differentiation and outflow tract morphogenesis. BMC Biol. 11, 107. doi:10.1186/1741-7007-11-107
De Bono, C., Liu, Y., Ferrena, A., Valentine, A., Zheng, D., and Morrow, B. E. (2023). Single-cell transcriptomics uncovers a non-autonomous Tbx1-dependent genetic program controlling cardiac neural crest cell development. Nat. Commun. 14 (1), 1551. doi:10.1038/s41467-023-37015-9
Fahed, A. C., Gelb, B. D., Seidman, J. G., and Seidman, C. E. (2013). Genetics of congenital heart disease: the glass half empty. Circ. Res. 112 (4), 707–720. doi:10.1161/CIRCRESAHA.112.300853
Garcia-Garcia, G., Berzal-Serrano, A., Garcia-Diaz, P., Villanova-Aparisi, R., Juarez-Rodriguez, S., de Paula-Vernetta, C., et al. (2020). Improving the management of patients with hearing loss by the implementation of an NGS panel in clinical practice. Genes (Basel) 11 (12), 1467. doi:10.3390/genes11121467
Garg, V., Muth, A. N., Ransom, J. F., Schluterman, M. K., Barnes, R., King, I. N., et al. (2005). Mutations in NOTCH1 cause aortic valve disease. Nature 437 (7056), 270–274. doi:10.1038/nature03940
Gaynor, J. W., and Elliott, M. J. (1993). Congenital left ventricular outflow tract obstruction. J. Heart Valve Dis. 2 (1), 80–93.
Gharibeh, L., Komati, H., Bosse, Y., Boodhwani, M., Heydarpour, M., Fortier, M., et al. (2018). GATA6 regulates aortic valve remodeling, and its haploinsufficiency leads to right-left type bicuspid aortic valve. Circulation 138 (10), 1025–1038. doi:10.1161/CIRCULATIONAHA.117.029506
Granja, J. M., Corces, M. R., Pierce, S. E., Bagdatli, S. T., Choudhry, H., Chang, H. Y., et al. (2021). ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 53 (3), 403–411. doi:10.1038/s41588-021-00790-6
Guo, M. H., Dauber, A., Lippincott, M. F., Chan, Y. M., Salem, R. M., and Hirschhorn, J. N. (2016). Determinants of power in gene-based burden testing for monogenic disorders. Am. J. Hum. Genet. 99 (3), 527–539. doi:10.1016/j.ajhg.2016.06.031
Harel, I., Maezawa, Y., Avraham, R., Rinon, A., Ma, H. Y., Cross, J. W., et al. (2012). Pharyngeal mesoderm regulatory network controls cardiac and head muscle morphogenesis. Proc. Natl. Acad. Sci. U. S. A. 109 (46), 18839–18844. doi:10.1073/pnas.1208690109
Hegan, P. S., Lanahan, A. A., Simons, M., and Mooseker, M. S. (2015). Myosin VI and cardiomyopathy: left ventricular hypertrophy, fibrosis, and both cardiac and pulmonary vascular endothelial cell defects in the Snell's waltzer mouse. Cytoskelet. Hob. 72 (8), 373–387. doi:10.1002/cm.21236
Jiang, W. F., Xu, Y. J., Zhao, C. M., Wang, X. H., Qiu, X. B., Liu, X., et al. (2020). A novel TBX5 mutation predisposes to familial cardiac septal defects and atrial fibrillation as well as bicuspid aortic valve. Genet. Mol. Biol. 43 (4), e20200142. doi:10.1590/1678-4685-GMB-2020-0142
Koch, L. (2020). Exploring human genomic diversity with gnomAD. Nat. Rev. Genet. 21 (8), 448. doi:10.1038/s41576-020-0255-7
Kwon, T. J., Oh, S. K., Park, H. J., Sato, O., Venselaar, H., Choi, S. Y., et al. (2014). The effect of novel mutations on the structure and enzymatic activity of unconventional myosins associated with autosomal dominant non-syndromic hearing loss. Open Biol. 4 (7), 140107. doi:10.1098/rsob.140107
Laforest, B., Andelfinger, G., and Nemer, M. (2011). Loss of Gata5 in mice leads to bicuspid aortic valve. J. Clin. Invest. 121 (7), 2876–2887. doi:10.1172/JCI44555
Laforest, B., and Nemer, M. (2011). GATA5 interacts with GATA4 and GATA6 in outflow tract development. Dev. Biol. 358 (2), 368–378. doi:10.1016/j.ydbio.2011.07.037
Lange, M., Bergen, V., Klein, M., Setty, M., Reuter, B., Bakhti, M., et al. (2022). CellRank for directed single-cell fate mapping. Nat. Methods 19 (2), 159–170. doi:10.1038/s41592-021-01346-6
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Ma, L., Lu, M. F., Schwartz, R. J., and Martin, J. F. (2005). Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development 132 (24), 5601–5611. doi:10.1242/dev.02156
Ma, Z. L., Yan, J., Li, S. J., Hua, Z. D., Yan, F. X., Wang, X., et al. (2017). Coarctation of the aorta with aortic arch hypoplasia: midterm outcomes of aortic arch reconstruction with autologous pulmonary artery patch. Chin. Med. J. Engl. 130 (23), 2802–2807. doi:10.4103/0366-6999.215279
McBride, K. L., Zender, G. A., Fitzgerald-Butt, S. M., Koehler, D., Menesses-Diaz, A., Fernbach, S., et al. (2009). Linkage analysis of left ventricular outflow tract malformations (aortic valve stenosis, coarctation of the aorta, and hypoplastic left heart syndrome). Eur. J. Hum. Genet. 17 (6), 811–819. doi:10.1038/ejhg.2008.255
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20 (9), 1297–1303. doi:10.1101/gr.107524.110
Melchionda, S., Ahituv, N., Bisceglia, L., Sobe, T., Glaser, F., Rabionet, R., et al. (2001). MYO6, the human homologue of the gene responsible for deafness in Snell's waltzer mice, is mutated in autosomal dominant nonsyndromic hearing loss. Am. J. Hum. Genet. 69 (3), 635–640. doi:10.1086/323156
Minners, J., Rossebo, A., Chambers, J. B., Gohlke-Baerwolf, C., Neumann, F. J., Wachtell, K., et al. (2020). Sudden cardiac death in asymptomatic patients with aortic stenosis. Heart 106 (21), 1646–1650. doi:10.1136/heartjnl-2019-316493
Plein, A., Calmont, A., Fantin, A., Denti, L., Anderson, N. A., Scambler, P. J., et al. (2015). Neural crest-derived SEMA3C activates endothelial NRP1 for cardiac outflow tract septation. J. Clin. Invest. 125 (7), 2661–2676. doi:10.1172/JCI79668
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81 (3), 559–575. doi:10.1086/519795
Roifman, M., Chung, B. H. Y., Reid, D. M., Teitelbaum, R., Martin, N., Nield, L. E., et al. (2021). Heterozygous NOTCH1 deletion associated with variable congenital heart defects. Clin. Genet. 99 (6), 836–841. doi:10.1111/cge.13948
Shi, L. M., Tao, J. W., Qiu, X. B., Wang, J., Yuan, F., Xu, L., et al. (2014). GATA5 loss-of-function mutations associated with congenital bicuspid aortic valve. Int. J. Mol. Med. 33 (5), 1219–1226. doi:10.3892/ijmm.2014.1700
Sinning, C., Zengin, E., Kozlik-Feldmann, R., Blankenberg, S., Rickers, C., von Kodolitsch, Y., et al. (2018). Bicuspid aortic valve and aortic coarctation in congenital heart disease-important aspects for treatment with focus on aortic vasculopathy. Cardiovasc Diagn Ther. 8 (6), 780–788. doi:10.21037/cdt.2018.09.20
Slavin, T. P., Feng, T., Schnell, A., Zhu, X., and Elston, R. C. (2011). Two-marker association tests yield new disease associations for coronary artery disease and hypertension. Hum. Genet. 130 (6), 725–733. doi:10.1007/s00439-011-1009-6
Srivastava, D., and Olson, E. N. (2000). A genetic blueprint for cardiac development. Nature 407 (6801), 221–226. doi:10.1038/35025190
Stankunas, K., Ma, G. K., Kuhnert, F. J., Kuo, C. J., and Chang, C. P. (2010). VEGF signaling has distinct spatiotemporal roles during heart valve development. Dev. Biol. 347 (2), 325–336. doi:10.1016/j.ydbio.2010.08.030
Stewart, R. A., Arduini, B. L., Berghmans, S., George, R. E., Kanki, J. P., Henion, P. D., et al. (2006). Zebrafish foxd3 is selectively required for neural crest specification, migration and survival. Dev. Biol. 292 (1), 174–188. doi:10.1016/j.ydbio.2005.12.035
Teng, L., Mundell, N. A., Frist, A. Y., Wang, Q., and Labosky, P. A. (2008). Requirement for Foxd3 in the maintenance of neural crest progenitors. Development 135 (9), 1615–1624. doi:10.1242/dev.012179
Wang, J., Raskin, L., Samuels, D. C., Shyr, Y., and Guo, Y. (2015). Genome measures used for quality control are dependent on gene function and ancestry. Bioinformatics 31 (3), 318–323. doi:10.1093/bioinformatics/btu668
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38 (16), e164. doi:10.1093/nar/gkq603
Wang, S., Chen, J., Garcia, S. P., Liang, X., Zhang, F., Yan, P., et al. (2019). A dynamic and integrated epigenetic program at distal regions orchestrates transcriptional responses to VEGFA. Genome Res. 29 (2), 193–207. doi:10.1101/gr.239053.118
Wolf, F. A., Angerer, P., and Theis, F. J. (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19 (1), 15. doi:10.1186/s13059-017-1382-0
Yamagishi, H. (2021). Cardiac neural crest. Cold Spring Harb. Perspect. Biol. 13 (1), a036715. doi:10.1101/cshperspect.a036715
Zhan, X., Hu, Y., Li, B., Abecasis, G. R., and Liu, D. J. (2016). RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinformatics 32 (9), 1423–1426. doi:10.1093/bioinformatics/btw079
Zhang, Y., Liu, T., Meyer, C. A., Eeckhoute, J., Johnson, D. S., Bernstein, B. E., et al. (2008). Model-based analysis of ChIP-seq (MACS). Genome Biol. 9 (9), R137. doi:10.1186/gb-2008-9-9-r137
Keywords: left ventricular outflow tract obstruction, outflow tract malformation, congenital heart defect, whole-exome sequencing, gene-based burden test
Citation: Geng Z, Li W, Yang P, Zhang S, Wu S, Xiong J, Sun K, Zhu D, Chen S and Zhang B (2023) Whole exome sequencing reveals genetic landscape associated with left ventricular outflow tract obstruction in Chinese Han population. Front. Genet. 14:1267368. doi: 10.3389/fgene.2023.1267368
Received: 26 July 2023; Accepted: 29 November 2023;
Published: 18 December 2023.
Edited by:
Francesco Vetrini, Indiana University Bloomington, United StatesReviewed by:
Cagri Gulec, Istanbul University, TürkiyeCopyright © 2023 Geng, Li, Yang, Zhang, Wu, Xiong, Sun, Zhu, Chen and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sun Chen, Y2hlbnN1bkB4aW5odWFtZWQuY29tLmNu; Bing Zhang, YmluZ3poYW5nQHNqdHUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.