
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 19 September 2023
Sec. Genomics of Plants and the Phytoecosystem
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1256627
This article is part of the Research TopicPostharvest Ripening, Senescence, and Technology, Volume IIView all 4 articles
 Girim Park1†
Girim Park1† Durre Shahwar1*†Gaeun Jang1
Durre Shahwar1*†Gaeun Jang1 Jagyeong Shin1Gibeom Kwon2Younjae Kim2Chang Oh Hong3,4Bingkui Jin5Hoytaek Kim6Oakjin Lee7
Jagyeong Shin1Gibeom Kwon2Younjae Kim2Chang Oh Hong3,4Bingkui Jin5Hoytaek Kim6Oakjin Lee7 Younghoon Park1,4*
Younghoon Park1,4*The flesh color of watermelon is an important trait that is determined by carotenoid composition and affects consumers’ fruit desirability. Although a complete dominant control by C locus (Cllcyb) for canary yellow flesh (CY) over red flesh has been reported, red and CY colors frequently appear as a mixed pattern in the same flesh (incomplete canary yellow, ICY) in F1 and inbred lines carrying dominant C alleles. Therefore, we examined the genetic control of the mixed color pattern in ICY using whole-genome resequencing of three ICY (ICY group) and three CY inbred lines (CY group), as well as genetic linkage mapping of an F2 population. The segregation pattern in 135 F2 plants indicated that CY is controlled by a single locus (named C2) dominant over ICY. The whole-genome resequencing of ICY and CY inbred lines revealed an ICY/CY-specific region of approximately 27.60–27.88 Mb on Chr. 2 that was polymorphic between the ICY and CY groups. Our genetic map, using nine cleaved amplified polymorphic sequence markers developed based on the single-nucleotide polymorphisms from the ICY/CY-specific region, confirmed that C2 is located on Chr. 2 and cosegregated with the marker (M7) derived from a non-synonymous single-nucleotide polymorphism of the pentatricopeptide repeat (PPR) gene (ClPPR, Cla97C02G039880). Additionally, 27 watermelon inbred lines of ICY, CY, and red flesh were evaluated using previously reported Cllcyb (C locus)-based markers and our C2 locus-linked ClPPR-based marker (M7). As a result, dominant alleles at the C2 locus were required to produce CY, in addition to dominant alleles at the C locus, while a recessive homozygous genotype at the C locus gave the red flesh irrespective of the genotype at the C2 locus. Using a ClPPR-based cleaved amplified polymorphic sequence developed in this study and Cllcyb-based markers, watermelon cultivars with CY, ICY, and red flesh could be successfully discerned, implying that the combined use of these markers will be efficient for marker-assisted selection of flesh color in watermelon breeding.
Watermelon is a diploid (2n = 2x = 22) vegetative fruit crop belonging to the family Cucurbitaceae and genus Citrullus. The global watermelon cultivation area and production are 4,459,239 ha and 161,837,308 tons, respectively, which account for approximately 10% of the major vegetable production (Food and Agriculture Organization, 2020). Watermelon is native to Africa and has been found in temperate and Mediterranean regions outside Africa (Jeffrey, 1975; Paris, 2015). After more than 4,000 years of domestication, wild-type watermelon species with small fruit size, firm flesh, and bitter taste were improved to modern cultivars such as Citrullus lanatus (Thunb.) Matsum. & Nakai, which have consumable traits, including the large size, thin rind, and sweet red flesh (Paris, 2015). The whole-genome sequencing (WGS) was completed for ‘97103,’ a Chinese cultivar (Ren et al., 2012; Guo et al., 2013), and ‘Charleston Gray,’ a US heirloom (Wu et al., 2019), which revealed an approximate genome size of 410 Mb and 22,500 genes (Cucurbit Genome Database, CuGenDB, http://cucurbitgenomics.org).
The fruit flesh color of watermelon is categorized as red (scarlet or coral red), yellow (canary, salmon, or pale yellow), orange, and white (UPOV). Different flesh colors result from the accumulation of specific carotenoid components in flesh (Grassi et al., 2013). The accumulation of all-trans-lycopene (lycopene) and xanthophylls (zeaxanthin, neoxanthin, and violaxanthin) in the chromoplasts results in red and canary yellow flesh colors, whereas the accumulation of ξ-carotene and prolycopene or ß-carotene results in the salmon yellow and orange flesh colors, respectively (Tadmor et al., 2005; Perkins-Veazie et al., 2006; Bang et al., 2010; Jin et al., 2019). White flesh is resulted from the accumulation of phytofluene, a colorless carotenoid, and light-yellow-colored ξ-carotene (Zhao et al., 2013).
Watermelon flesh color is genetically ascertained by several loci (genes) involved in the carotenoid biosynthesis pathways. According to Henderson (1989), three alleles (Y, yo, and y) at the Y locus are associated with coral red, orange with a high prolycopene content, and salmon yellow flesh color. Coral red, regulated by locus Y, is dominant over the other two colors. Lou (2009) reported that scarlet red and coral red are regulated by the YSCR and Y alleles, respectively, and YSCR was dominant over all other alleles of the Y locus (YSCR > Y > yo > y). Contrarily, a major QTL on Chr. 1 is responsible for orange flesh with a high ß-carotene content, which is dominant to scarlet red flesh (Branham et al., 2017).
The canary yellow flesh (CY) color in watermelon is conferred by the C locus, which is dominant over the red (scarlet or coral red) flesh color (Henderson et al., 1996). The lycopene ß-cyclase (LCYB) gene of the C locus catalyzes the conversion of lycopene to ß-carotene, but its mutant allele suppresses LCYB gene function, leading to the accumulation of lycopene and the appearance of red flesh (Bang et al., 2007; Bang et al., 2014). However, Henderson et al. (1998) described that the homozygous recessive i-C locus results in red flesh irrespective of the presence of the C allele, but the gene responsible for i-C is unknown. In addition, the homozygous recessive py locus prevents the formation of canary yellow flesh but develops pale yellow flesh (Bang et al., 2010). In certain genetic backgrounds, red and yellow colors appear as a mixed pattern in the same flesh. Although Bang et al. (2007) reported a complete dominant control for canary yellow flesh, incomplete yellow (mixed with red) flesh appears frequently in F1 and segregates in the progeny of crosses between red and canary yellow flesh cultivars, which implies that unknown genetic factors other than the C locus may be involved in the determination of canary yellow. Based on our knowledge, there are no research reports on the genetic control of the mixed color pattern in incomplete yellow flesh.
For the marker-assisted selection of fruit flesh color, LCYB gene-based markers have been developed to distinguish red from canary yellow (Bang et al., 2007). Jin et al. (2019) also suggested the development of a CRTISO gene-based marker for orange flesh with high prolycopene content. Recently, Li et al. (2020) and Lee et al. (2021) described the markers distinguishing scarlet and coral red. However, no markers have been reported for selecting incomplete yellow, which is essential for breeding canary yellow- or red-fleshed watermelons.
Whole-genome resequencing (WGRS) based on next-generation sequencing (NGS) provides comprehensive genome sequence variations that are ideal for identifying DNA markers and genes associated with phenotypic variations (Meuwissen and Goddard, 2010; Saidi and Hajibarat, 2020). The WGRS of a large number of watermelon accessions and genome-wide association study (GWAS) revealed sequence polymorphisms and genes for fruit-related traits, such as fruit shape, flesh color, rind patterns (Park et al., 2018; Subburaj et al., 2019), and disease resistances, including gummy stem blight (Lee et al., 2018). The WGRS of near-isogenic lines successfully revealed a locus related to the high lycopene content in a scarlet red-fleshed watermelon cultivar (Lee et al., 2021).
The present study aimed to identify the genetic control responsible for incomplete yellow flesh in watermelon. Using the WGRS and linkage mapping approach, we identified a novel locus named C2 that controls incomplete yellow flesh, which is required for the development of canary yellow flesh in addition to the previously reported locus C. The molecular markers for C2 established in this study will be beneficial for MAS of canary yellow or red-fleshed watermelons.
In the present investigation, three canary yellow-fleshed inbred lines (CY group: PS160, PS188, and PS190) and three incomplete canary yellow-fleshed (red colored mixed in the yellow background) inbred lines (ICY group: PS186, PS187, and PS189) were used for WGRS (Figure 1). The ICY group is further divided into two severely reddish ICY (ICY-H, PS186 and PS189) and one slightly reddish ICY (ICY-L, PS187) fleshed inbred lines depending on the strength of red color mixed in the yellow background. For genetic inheritance and linkage mapping analysis, an ICY-H inbred line PS189 (maternal parent) was crossed with a CY inbred line PS190 (paternal parent) to produce F1 progeny, and then, it was self-pollinated to create a segregating F2 population.

FIGURE 1. Watermelon inbred lines showing (A) incomplete canary yellow flesh color and (B) complete canary yellow flash color, which were used for whole-genome resequencing in this study. The ICY group is further divided into severely reddish ICY (ICY-H, PS186 and PS189) and slightly reddish ICY (ICY-L, PS187)-fleshed inbred lines. PS189 and PS190 were used to develop an F2 population for genetic mapping of the loci controlling ICY.
Plant cultivation and phenotyping of fruit color were carried out for PS189, PS190, F1 hybrids, and F2 population in a breeding field of the Partner Seed Company (Gimje, Korea). All seeds were sown in spring, 2021, and seedlings were grafted onto the bottle gourd cultivar ‘Bullojangsaeng.’ Grafted seedlings were planted at 30 cm intervals in a plastic film greenhouse and grown following conventional soil cultivation of a single fruit per plant. For phenotyping, fully matured fruits were harvested on the 40th day after self-pollination, and the center of the fruit was cut in the longitudinal direction. Flesh color was scored and photographed as ICY or CY based on the presence or absence of redness in the flesh.
Total genomic DNA was extracted from the first leaf of the seedling for WGRS of the six watermelon inbred lines using the GeneAll Exgene Plant SV mini kit (GeneAll, Seoul, Korea). NGS was performed at Macrogen (Seoul, Korea) by paired-end sequencing (2×, 151 bp) using the HiseqX platform (Illumina, Inc., San Diego, CA, United States).
The raw reads were trimmed to find high-quality clean reads by removing adapter sequences and short reads with a Phred score of <20 and length of <25 bp using cutadapt (v.4.1) (Martin, 2011) and DynamicTrim and LengthSort programs of SolexaQA (v.1.13) (Cox et al., 2010). The cleaned reads were aligned with the watermelon reference genome 97103 v.2 (CuGenDB: http://cucurbitgenomics.org) (Guo et al., 2013) using the BWA (v.0.7.16-r1188) program (Li, 2013). The BAM file created through read mapping was searched for raw sequence variation using the SAMtools (v.0.1.16) program (Li et al., 2009), and the consensus sequence was extracted. An integrated single-nucleotide polymorphism (SNP) matrix between each inbred line was prepared to search for SNPs between the ICY and CY groups (monomorphic between the three inbred lines within each group and polymorphic between the two groups).
A list of unions was constructed using the raw SNP positions obtained by comparing six inbred lines with the reference genome (97103 v.2) as candidates. The SNP loci included in the list were compared with each of the six inbred lines. In the case of missing genotypes at SNP loci within the inbred line, the matrix was completed by imputation from the consensus sequence of the inbred line. The obtained SNPs were classified based on the read rate as homozygous (read rate ≥90%), heterozygous (40%≤ read rate ≤60%), and other (if it could not be distinguished as homozygous or heterozygous). An integrated SNP matrix for each of the six inbred lines was prepared to search for SNPs between the groups of ICY-H, ICY-L, and CY. To identify genomic regions specific to the ICY phenotype, the chromosomal distribution of the SNPs that were monomorphic among the three inbred lines within each group but polymorphic between the two groups was analyzed. Gene annotation information was acquired using watermelon reference genome 97103 v.2.
For the marker conversion of SNPs to a cleaved amplified polymorphic sequence (CAPS), a flanking sequence with a total length of 600 bp was obtained with 300 bp on each side based on the position of each SNP. Restriction enzymes that can differentiate polymorphisms were confirmed using the NEB cutter (v.2.0) program (http://nc2.neb.com/NEBcutter2/index.php). PCR primers for the CAPS were designed based on the franking sequences using the Primer3 (v.0.4.0) program (https://bioinfo.ut.ee/primer3-0.4.0/).
PCR was carried out with 20 µL volume comprising 10 ng of genomic DNA, 1X PCR buffer, 0.1 µM each forward and reverse primer, 0.5 U Taq polymerase, and 0.2 mM dNTPs (Solgent, Daejeon, Korea). The following conditions were used to perform touch-down PCR: 1 cycle at 95°C for 5 min; 10 cycles at 95°C for 15 s, 60°C (decreased by 0.5°C for each cycle) for 30 s, and 72°C for 1 min; and 35 cycles of amplification at 95°C for 15 s, 55°C for 30 s, and 72°C for 1 min. The PCR amplicon was digested using restriction enzymes, in accordance with the manufacturer’s instructions (New England BioLabs Inc., Ipswich, MA, United States). Electrophoresis on a 2% agarose gel at 180 V for around an hour was used to identify the PCR and enzyme-digested products.
The phenotypes of the F2 progeny were classified into CY and ICY according to the presence or absence of redness in the fruit flesh. A chi-squared analysis of the flesh color trait segregation ratio in the F2 population was performed for Mendelian inheritance using the JoinMap (v.5) program (Kyazma, Wageningen, Netherlands). Furthermore, F2 progeny were genotyped by using nine CAPS markers for linkage mapping, and the marker genotypes of each F2 individual, which follows PS189 (ICY) and PS190 (CY), and F1 were scored as ‘a’, ‘b’, and ‘h’, respectively. The phenotype of each F2 individual was scored as ‘a’ for ICY and ‘b’ for CY. The linkage group was created using the JoinMap (v.5) program (Kyazma, Wageningen, Netherlands) based on the Kosambi function and an LOD score of 2.0.
Seeds of a total of 21 watermelon inbreds including 10 CY and 11 red-fleshed lines R were achieved from the Plant Genetics and Breeding Research Center at Pusan National University (Miryang, South Korea) and Partner Seed Company. Genomic DNA was extracted from the first leaf of the seedling using the GeneAll Exgene Plant SV mini kit (GeneAll, Seoul, Korea). Genotyping was performed by using two LCYB gene (C locus)-based markers (Lcyb and Clcyb.600) (Bang et al., 2007) and a C2 locus-linked marker M7 developed in this study.
Whole-genome sequences were analyzed for three watermelon inbred lines with canary yellow flesh and three with incomplete canary yellow flesh mixed with red, and the sequence data (accession number: PRJNA1002084) were deposited to the GenBank of National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/). The number of raw reads produced from each inbred line ranged from 60,736,065 to 77,52,303, with an average of 68,977,937 per line. The total length of the raw reads ranged from 9,171,145,815 bp to 11,705,565,753 bp, with an average of 10,415,668,487 bp. The average Q30, or the ratio of reads with a Phred score of 30 or higher indicating read quality, was 89.44%. The genome coverage, which represents the ratio of the total read length and reference genome size of a watermelon, was in the fold range of 45.58×–55.09×, with an average of 49.01×.
After trimming the raw reads, the average length of the reads ranged from 103.44 to 109.54 bp, with an average of 106.65 bp, and the total length of trimmed reads ranged from 5,689,784,468 bp to 7,133,026,915 bp with an average of 6,389,653,991 bp. The ratio between trimmed reads and raw reads (trimmed/raw (%)) ranged from 58.97% to 63.76%, with an average of 61.43%. After trimming, the range of genome coverage of the six inbred lines was 26.83 × –33.07×, with an average of 30.06 × (Table 1).
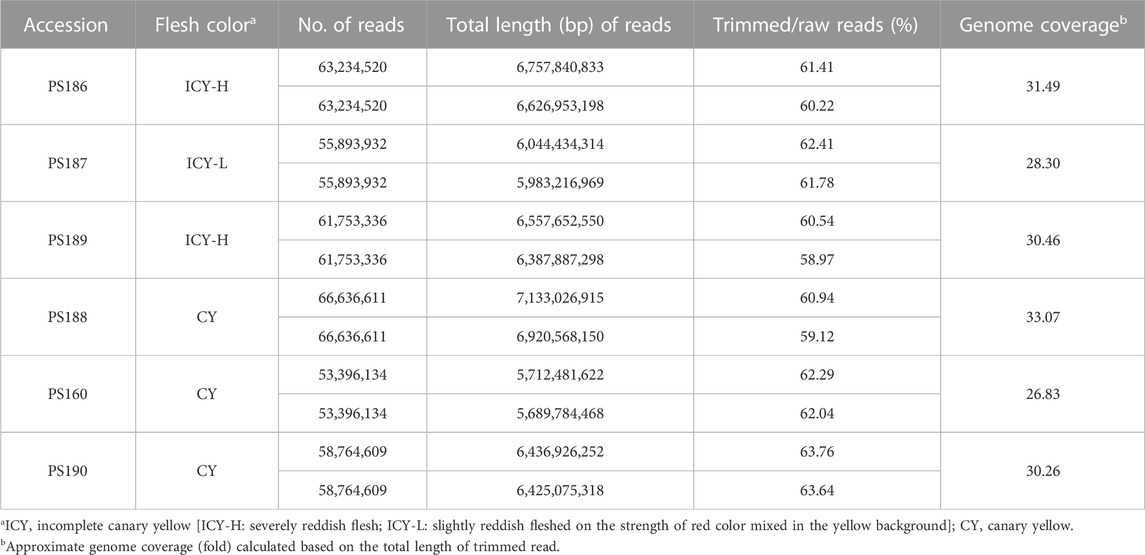
TABLE 1. Summary of the whole-genome resequencing data for three incomplete canary yellow and three canary yellowfleshed watermelon inbred lines.
The read mapping of the six inbred lines resulted in an average of 98.48% of all reads being mapped. The number of homozygous SNPs to the reference genome for each inbred line ranged from 154,049 to 189,966, with an average of 174,409 (Table 2). To identify the genomic region for flesh color associated with ICY, we first detected 244,982 and 148,999 SNPs that were monomorphic within the inbred lines of each ICY-H and CY group, respectively. Then, there were a total of 98,602 SNPs that could be compared between the ICY-H and CY groups. Among them, 1,821 homozygous SNPs that were polymorphic between the two groups were selected. Furthermore, among those SNPs, 645 and 1,169 SNPs that were monomorphic between ICY-H and ICY-L groups and between CY and ICY-L groups were selected, respectively. Finally, a total of 283 SNPs that were polymorphic between the two SNP groups (645 vs 1,169 SNPs) were selected as the SNPs specific to ICY and CY (polymorphic regions between ICY and CY groups) (Table 2; Figure 2; Supplementary Table S1).
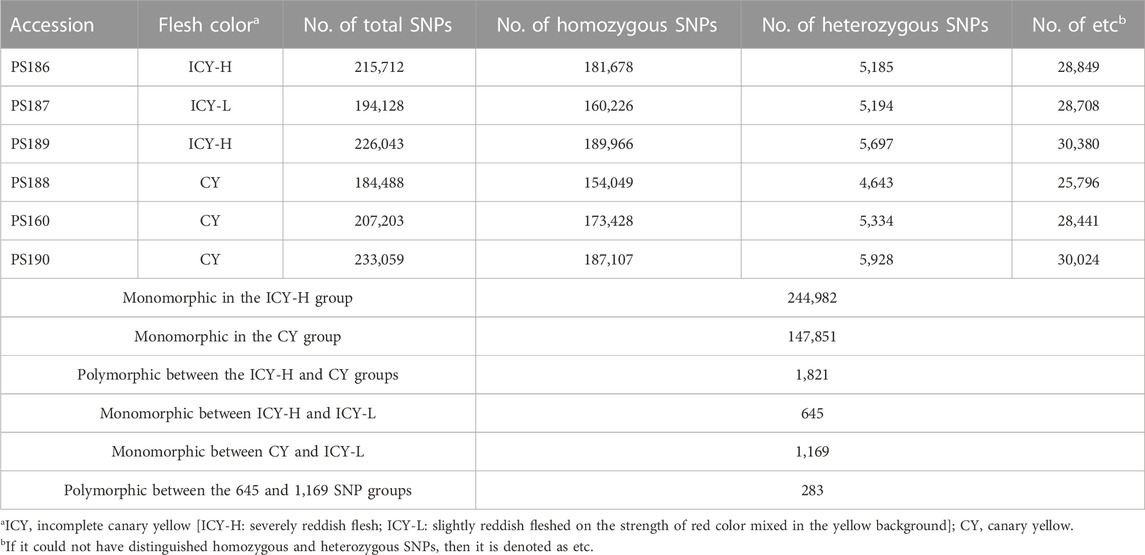
TABLE 2. Statistics of SNP detection of three incomplete canary yellow and three canary yellowfleshed watermelon inbred lines confirmed through mapping to the reference genome.
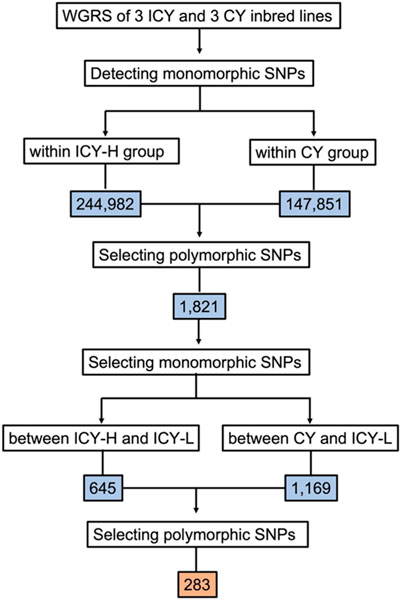
FIGURE 2. Flow chart showing sequential steps for selecting the single-nucleotide polymorphisms (SNPs) that were polymorphic between incomplete canary yellow-fleshed (ICY group) and canary yellow-fleshed watermelon inbred lines (CY group). WGRS, whole-genome resequencing; ICY-H, severely reddish ICY; and ICY-L, slightly reddish ICY. The number of SNPs is presented in the colored box.
Analysis of the chromosomal distribution of 283 SNPs revealed the clusters of SNPs largely on three genomic regions: Chr. 2 (27.60—27.88 Mb, 272 SNPs), Chr. 5 (27.24—27.25 Mb, 3 SNPs), and Chr. 9 (18.29—18.30 Mb, 8 SNPs) (Figure 3). The majority of these SNPs, specifically 272 out of 283, are densely clustered within the 27.60–27.88 Mb genomic region on Chr. 2. Additionally, two other notable but smaller clusters can be observed on Chr. 5 between the 27.24–27.25 Mb genomic region with three SNPs and on Chr. 9 from 18.29–18.30 Mb with eight SNPs (Figure 3). In the region of Chr. 2, 272 SNPs were recorded in both genic and intergenic regions. A total of 14 genes were annotated in the genomic region of Chr. 2 with 37 SNPs. Among these SNPs, 29 were located in the intron, while eight were in the exon (Supplementary Table S1). Of the eight SNPs identified in the exon region, five non-synonymous SNPs were found in five genes (Table 3; Supplementary Table S2). A total of 17 indels were found in 11 genes, all of which were located in the intron region (Supplementary Table S2). However, there were no genes with variations in the corresponding region of Chr. 5 and 9, but three and eight SNPs, respectively, were found in the intergenic region.
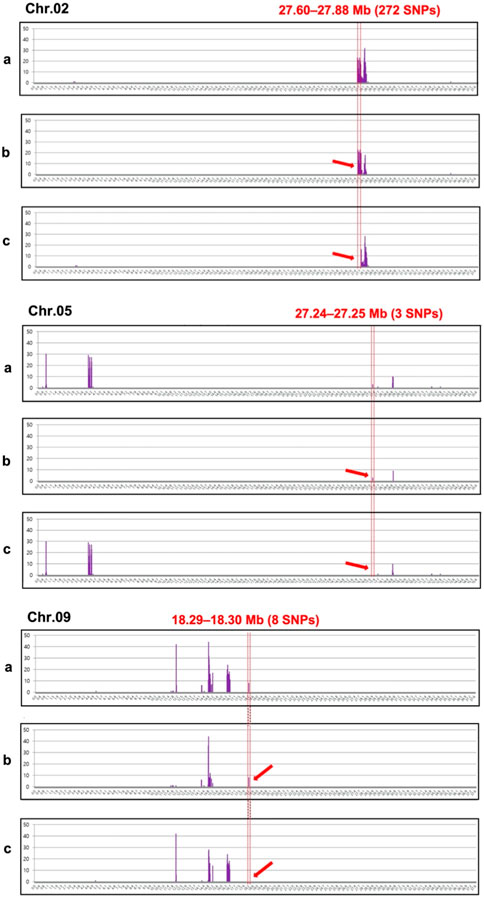
FIGURE 3. Chromosomal distribution of 283 single-nucleotide polymorphisms (SNPs) showing polymorphisms between the incomplete canary yellow-fleshed watermelon inbred line group (ICY group) and canary yellow-fleshed inbred line group (CY group). The number of SNPs (Y-axis) was counted at window sliding of every 4 Mb (X-axis) for each chromosome and represented as a vertical bar graph in violet. Three clusters of SNPs (ICY/CY-specific regions) indicating an association with the ICY and CY flesh were detected on Chr. 2, 5, and 9 and depicted using red dotted lines. The red arrows indicate the ICY/CY-specific regions that are delimitated by the red dotted line. (a) SNPs that were polymorphic between the severely reddish ICY (ICY-H) and CY group; (b) SNPs that were monomorphic between the slightly reddish ICY (ICY-L) and ICY-H group; and (c) SNPs that were monomorphic between the ICY-L and CY groups.
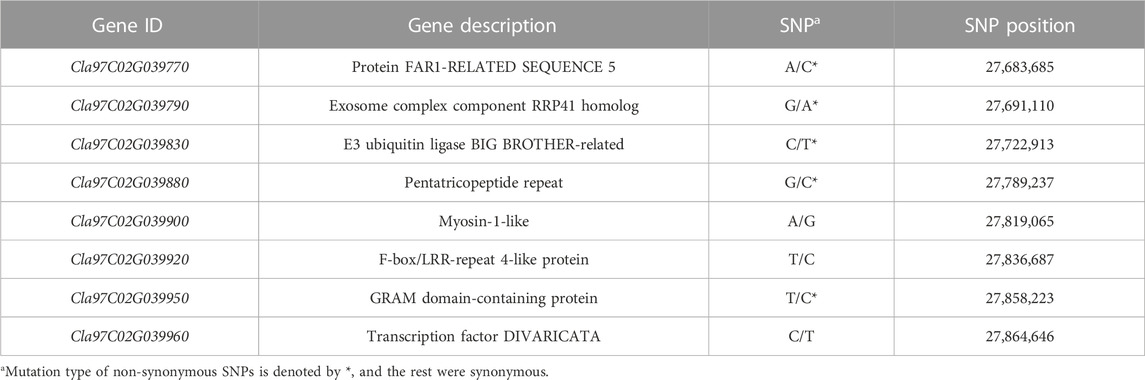
TABLE 3. List of annotated genes with significant SNPs in the polymorphic region between the incomplete canary yellow-fleshed watermelon inbred lines group (ICY group) and canary yellow-fleshed inbred line group (CY group) of Chr. 2
The F1 progeny from the cross between PS189 (ICY-H) and PS190 (CY) showed a CY flesh color that was close to that of PS190 (Figure 4). In the F2 population of 135 individuals, 94 showed CY flesh color and 41 exhibited ICY flesh color, which was consistent with the Mendelian ratio of 3:1 (X2 = 2.076, p-value = 0.149). The fruit flesh color of the F1 plants and their segregation ratio in the F2 population indicated that CY was dominant over ICY and was regulated by a single locus. The locus was named C2 in this study.

FIGURE 4. Fruit flesh color of (A) inbred lines PS189 and PS190 and their F1 and (B) F2 plants. Segregating for flesh color: ICY, incomplete canary yellow; CY, canary yellow.
For genetic linkage map construction, the cleaved amplified polymorphic sequence (CAPS) markers (M1 to M9) were designed based on nine SNPs located within the region of Chr. 2 polymorphic between ICY and CY groups (ICY/CY-specific region) (Table 4). Of the nine SNPs used for marker development, four were located in the genic region and two were a missense mutation (non-synonymous), causing amino acid sequence substitution. The remaining five SNPs were in intergenic regions. These CAPS markers were genotyped on 135 F2 plants (Supplementary Table S3), and a single linkage group (13.5 cM) composed of nine CAPS markers and the C2 locus was constructed (Figure 5). In this linkage group, all CAPS markers were mapped in accordance with their physical genomic orders and the C2 locus was cosegregating with three markers M6, M7, and M8. Among these three cosegregating markers, M7 (Figure 6) was a non-synonymous SNP-based marker for the gene (Cla97C02G039880) encoding pentatricopeptide repeat protein (PPR). These results confirmed that the C2 locus exists in the ICY/CY-specific region of Chr. 2, and Cla97C02G039880 (ClPPR) is a putative candidate gene for C2.
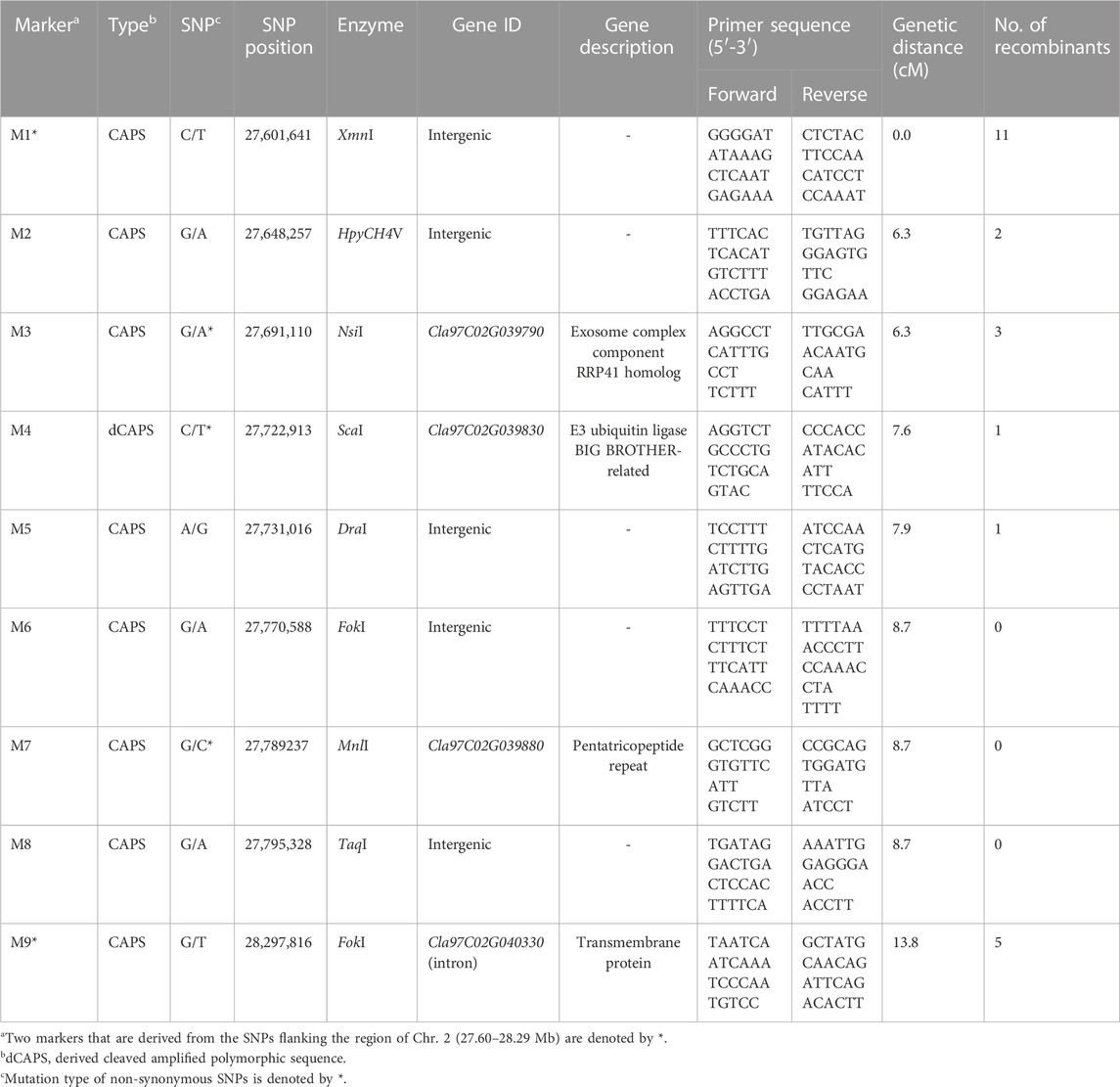
TABLE 4. Information of cleaved amplified polymorphic sequence markers designed from the single-nucleotide polymorphisms in the region of Chr. 2 (27.60–28.29 Mb) polymorphic between incomplete canary yellow and canary yellow flesh in watermelon.
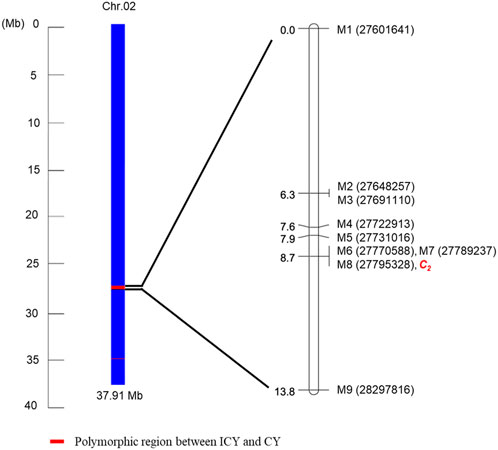
FIGURE 5. Schematic representation of Chr. 2 showing the ICY/CY-specific region [polymorphic region (red) between the incomplete canary yellow-fleshed watermelon inbred line group (ICY group) and canary yellow-fleshed inbred line group (CY group)]. A genetic linkage map was constructed using seven cleaved amplified polymorphic sequence markers developed from the SNPs (Table 5) in the ICY/CY-specific region, two CAPS markers developed from the SNPs flanking the ICY/CY-specific region, and C2 locus controlling ICY and CY in an F2 population.
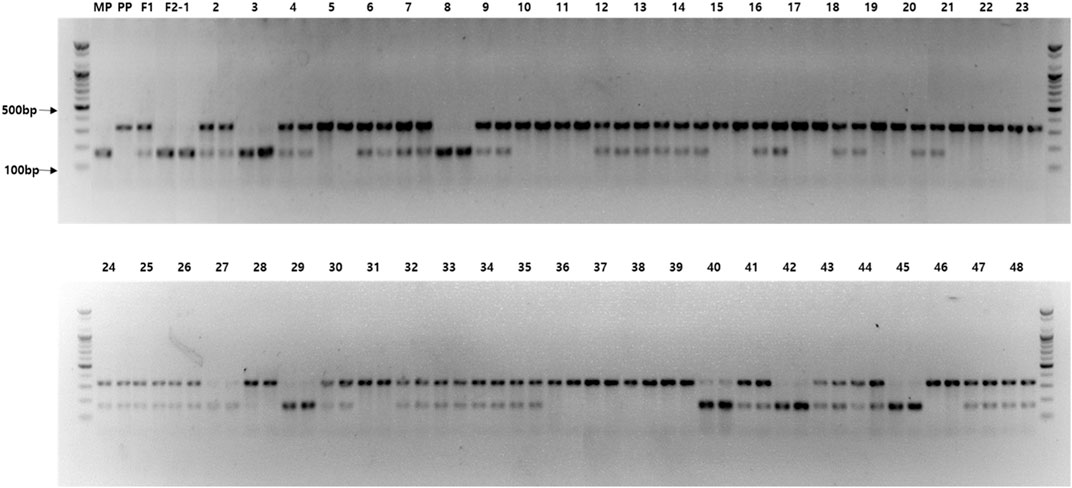
FIGURE 6. Agarose gel image exhibiting the genotyping results for the Cla97C02G039880 (ClPPR) gene-based cleaved amplified polymorphic sequence (M7) that were cosegregating with the incomplete canary yellow (ICY) and canary yellow (CY) flesh color in F2 watermelon plants. M, 100 bp size marker; MP, maternal parent (PS189); PP, paternal parent (PS190); F1, progeny from cross between MP and PP; and F2—1–48, F2 plants (two PCR replicates).
A total of 28 watermelon accessions, including 13 inbred lines with CY, 11 with red fruit flesh, and PS186, PS187, and PS189 with ICY, were genotyped using the M7 marker and two previous LCYB gene (C locus)-based markers, Lcyb and Clcyb.600, discerning CY and red flesh color (Bang et al., 2007; Bang et al., 2014) (Table 5). As a result, all CY lines showed the dominant homozygous genotype for all LCYB gene-based markers (CC) and the C2-linked M7 (C2C2). In contrast, the accessions with red flesh showed either dominant homozygosity or recessive homozygosity marker genotype (c2c2) for C2-linked markers, but all showed recessive homozygosity (cc) for the two LCYB gene-based markers. For PS186, PS187, and PS189 with ICY, LCYB gene-based markers were genotyped as dominant homozygous, whereas the C2-linked markers were genotyped as recessive homozygous. Based on the results, it is likely that dominant alleles at both the C locus and the C2 locus are required to produce canary yellow flesh, while a recessive homozygous genotype at the C locus will give the red flesh irrespective of the genotype at the C2 locus. This suggests that the recessive allele at the C2 locus (c2) is in an epistatic relationship over the C locus gene, meaning that the effect of the LCYB gene for canary yellow flesh is partially masked by c2.
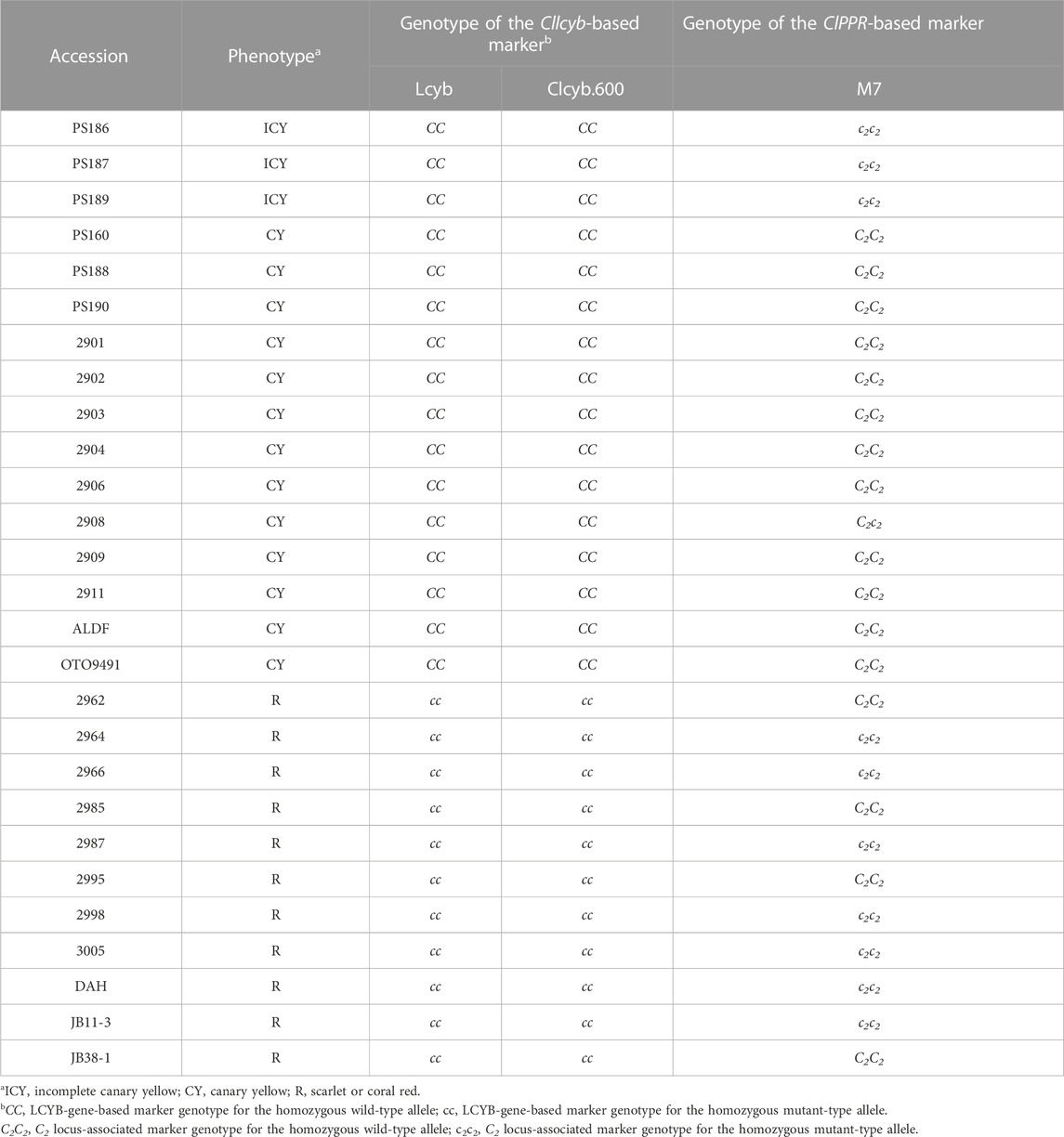
TABLE 5. Evaluation of 27 watermelon inbred lines using Cllcyb gene (C locus)-based markers and C2 locus-linked ClPPR gene-based marker.
Differences in the flesh color of watermelons are mostly due to the composition and amount of carotenoids. The selection of specific flesh color is an important breeding target in terms of the consumer’s preferences and nutritional aspects. Previous studies have suggested that those genes involved in the metabolism of carotenoids are associated with various colors of watermelon flesh. The canary yellow flesh (CY) is regulated by the dominant C locus, and a homozygous recessive mutation of C results in red (scarlet and coral red) flesh (Bang et al., 2010). Molecular studies have characterized the C locus as the gene Cllcyb (Cla97C04G070940) encoding LCYB (Wang P. et al., 2019; Wang C. et al., 2019; Zhang et al., 2020), and gene-based markers have been used for the MAS of canary yellow and red flesh. However, some watermelon inbred lines with the Cllcyb marker genotype of dominant homozygosity (CC) have been found to show a mixture of CY and red flesh (incomplete canary yellow, ICY). Although it has been suggested that environmental variables might contribute to ICY (Bang et al., 2007), no genetic factors that are involved in the expression of ICY are currently known.
In this study, to identify possible genetic factors determining ICY and CY, we compared genome sequences of six ICY and CY inbred lines to identify genomic regions specific to this trait (ICY-specific genomic region) and mapped this region using an F2 population. A whole-genome sequence comparison of the ICY and CY inbred groups identified a genomic region containing 66 genes on Chr. 2 that was polymorphic between these groups. Genetic inheritance analysis using an F2 population indicated that ICY is controlled by the homozygous recessive alleles at a single locus C2, named in this study. Genetic linkage mapping using a set of CAPS markers derived from the SNPs in the ICY-specific genomic region showed that these markers are tightly linked to C2. In this map, a CAPS (M7) derived from a non-synonymous SNP in the gene encoding PPR cosegregated with the flesh color of 135 F2 plants, possibly implying that ClPPR (Cla97C02G039880) may be a putative candidate gene for C2. Furthermore, genotyping of 28 watermelon accessions using the two Cllcyb gene-based markers and ClPPR-based CAPS markers (M7) indicated that all CY inbreds require dominant alleles for both C and C2 and that c2 is dominantly epistatic to C.
Bang et al. (2010) identified that the pale yellow flesh color in watermelon is due to a locus distinct from i-C, indicating the involvement of at least two loci in the variation of canary yellow, pale yellow, and red flesh. This resonates with our findings on the influence of gene interactions in the expression of ICY. Bang et al. (2010) also found that the expression of pale yellow flesh required a dominant allele at the C locus, analogous to the necessity for that allele in the expression of ICY, with the presence of a necessary allele at the C2 locus. We observed that all CY plants demonstrated dominant homozygosity or heterozygosity for both the LCYB and the C2 locus (PPR gene). However, the presence of a recessive homozygosity at the C2 locus (c2c2) indicates an epistatic relationship over the C locus gene, partially masking the effect of the LCYB gene for canary yellow flesh. In essence, our study is consistent with the multi-gene influences and epistatic relationships found by Bang et al. (2010), highlighting the complex gene interactions influencing watermelon flesh color.
PPRs are a diverse group of proteins predominantly present in plants (Lurin et al., 2004), play crucial roles in organelle formation, plant growth and development (Sun et al., 2018), and expression of cytoplasmic male sterility (CMS) (Jo et al., 2016), and affect fruit development and flesh color formation (Eriksson et al., 2004). Moreover, PPRs are linked to the early chloroplast formation and plastid gene expression (Pyo et al., 2013). Protein deficiency of PPR generally caused embryo development delay in maize (Sun et al., 2019; Yuan et al., 2019), abnormal chloroplast development, and aberrant pigmentation in seeds in Arabidopsis (Bryant et al., 2011). The significance of PPR proteins lies in their role as essential proteins that bind to RNA and can regulate gene expression in multiple post-transcriptional processes, including shearing, RNA editing, splicing, degradation, and translation in the chloroplasts (Wang et al., 2021a), plastids (Wang et al., 2021b), and mitochondria (Binder et al., 2013). The PPR protein family has been categorized into two subfamilies, namely, P type and PPR-like long and short (PLS) type (Lurin et al., 2004). Several studies have demonstrated that PPR proteins belonging to these subfamilies, such as the P-type PPR protein ACM1 (Wang et al., 2021b) and the PLS-type PPR protein MORF9 (Yan et al., 2017), have an impact on plastid development. Some PPR members in the P-subfamily of watermelon (ClPPR) had high expression levels in the fruit flesh, fruit rind, or both rind and flesh tissues, indicating that they might be crucial in coordinating the development of flesh and rind in watermelons (Subburaj et al., 2020). In contrast, the majority of subgroups in the PLS-subfamily showed preferential expression in the rind; high expression levels were observed at all fruit development stages for rind, but only at 10-day post anthesis (DPA) for flesh. Subburaj et al. (2020) suggested that these PLS-subfamily genes may play significant roles in the fruit rind and early fruit flesh development of watermelon. Taken together, these outcomes suggest that the ClPPR genes are likely involved in fruit development of watermelon including chloroplast-to-chromoplast transition in fruit flesh (Subburaj et al., 2020).
Subburaj et al. (2020) identified and characterized 422 ClPPR genes in the watermelon genome, and out of them, four ClPPR genes are associated with orange, two genes with yellow, and one gene with red flesh color among marker genotypes and phenotypes of orange, yellow, and red flesh color in all surveyed lines. Various studies suggested that ClaPPR11 co-localized with ß-carortene-related QTL on Chr.1 (Branham et al., 2017), whereas ClaPPR140 co-localized with lycopene-related QTL on Chr.4 (Liu et al., 2015). However, ClaPPR25 and ClaPPR95 were not co-localized with any QTL for flesh color. Similarly, another PPR member ClPPR (Cla97C06G122120) on Chr. 6 has been also reported as a candidate gene for the locus Cyf that regulates the carotenoid accumulation level in watermelon (Liu et al., 2023). The mutant allele of Cyf was responsible for pale yellow flesh (Liu et al., 2023). However, the molecular mechanism for genetic control of the PPR on fruit flesh color formation is not yet fully elucidated. PPR proteins have so far been discovered to be cis-element-recognizing site-specific RNA factors in the plastid (Okuda et al., 2006). PPR (CRR4) specifically those nucleotide sequences nearby cis-acting element of the ndhD-1 site, allowing the scaffold for RNA editing (conversion of C to U) and likely making it easier for the putative RNA editing enzyme to access these sequences (Okuda et al., 2006). The deficiency of a single PPR protein can have an impact on RNA editing at multiple locations in the gene, consequently influencing gene expression within the plastid (Okuda et al., 2006). In a recent study by Galpaz et al. (2018), it was found that CmPPR1 participates in RNA editing processes within plastids, which are responsible for the accumulation of carotenoids and chlorophyll, consequently giving rise to white, orange, and green flesh formation in melon. It has also been suggested that the CmPPR1 gene might be involved in retrograde signaling from plastids to the nucleus, influencing the expression of plastid-targeted genes, exhibiting that PPR proteins are involved in the variation of fruit flesh color (Galpaz et al., 2018).
Plastids can be categorized into three groups based on their pigments: chloroplasts, chromoplasts, and leucoplasts. The development of chloroplasts through the involvement of PPR proteins has been reported by numerous studies (Bryant et al., 2011; Pyo et al., 2013). The ClPPR gene might be involved in chromoplast development, carotenoid accumulation, and flesh color regulation as well (Liu et al., 2023). However, there is limited research available on the role of PPR proteins in chromoplast development and the formation of flesh color in watermelon. Notably, there are noticeable differences between globular and crystalloid chromoplasts in terms of their structure and color. The presence of red flesh in watermelon is primarily associated with the presence of crystalloid plastid structures, whereas white, yellow, or orange flesh indicates the presence of globular chromoplasts (Fang et al., 2020). These structural differences are likely attributed to variations in pigment components. In a study by Fang et al. (2020), a direct relationship was observed between the formation of crystalloid chromoplasts and lycopene accumulation. Conversely, excessive accumulation of violaxanthin, lutein, γ-carotene, and ß-carotene led to the development of globular chromoplasts, and they have a potential role in the regulation of carotenoid accumulation and flesh color. The chromoplast’s plastoglobulus serves as a specialized structure responsible for the mechanism of producing and storing carotenoids (Brehelin and Kessler, 2008). The PPR gene may specifically influence the number of plastoglobuli, thereby contributing to differences in flesh color. Fang et al. (2020) observed a progressive increase in the accumulation of carotenoids during the fruit flesh development of pale yellow-fleshed ‘COS’ and canary yellow-fleshed ‘PI 635597’watermelon varieties. This increase in carotenoid accumulation was accompanied by an increase in both the quantity and size of chromoplasts and plastoglobuli. Importantly, they reported that the size and number of plastoglobuli were significantly greater in PI 635597 than those in COS, suggesting that the enlarged size and number of plastoglobuli may contribute to the formation of canary yellow flesh color.
In conclusion, we identified a novel locus, C2, which is required for CY flesh color in watermelon in addition to the previously known C locus cloned as the Cllcyb. Our genetic mapping results indicated that C2 is located on Chr. 2, and ClPPR (Cla97C02G039880) encoding a member of PPR protein is a putative candidate gene for the locus. Using a ClPPR-based CAPS developed in this study and Cllcyb-based markers, watermelon cultivars with CY, ICY, and red flesh could be successfully discerned, implying that the combined use of these markers will be efficient for MAS of flesh color in watermelon breeding. Nevertheless, the molecular mechanism for the involvement of ClPPR in carotenoid synthesis and its interaction with the Cllcyb remains to be elucidated.
The WGRS datasets presented in this study can be found in online repositories. This data can be found here: National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/), accession number PRJNA1002084.
GP: data curation, formal analysis, methodology, software, and writing–original draft. DS: data curation, formal analysis, software, visualization, writing–original draft, and writing–review and editing. GJ: data curation, formal analysis, and writing–review and editing. JS: data curation, formal analysis, and writing–review and editing. GK: data curation, formal analysis, and writing–review and editing. YK: conceptualization, methodology, supervision, and writing–review and editing. CH: visualization and writing–review and editing. BJ: data curation, formal analysis, and writing–review and editing. HK: funding acquisition and writing–review and editing. OL: funding acquisition and writing–review and editing. YP: conceptualization, formal analysis, funding acquisition, investigation, methodology, supervision, visualization, writing–original draft, and writing–review and editing.
The authors declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by a grant (project no. PJ014846012020) from the National Institute of Horticultural Herbal Sciences, Rural Development Administration.
Authors GK and YK were employed by the company Partner Seeds Co., Ltd., and Author BJ was employed by the company UNELL Biotechnology Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1256627/full#supplementary-material
Bang, H., Davis, A. R., Kim, S., Leskovar, D. I., and King, S. R. (2010). Flesh color inheritance and gene interactions among canary yellow, pale yellow, and red watermelon. J. Am. Soc. Horti. Sci. 135 (4), 362–368. doi:10.21273/JASHS.135.4.362
Bang, H., Kim, S., Leskovar, D., and King, S. (2007). Development of a codominant CAPS marker for allelic selection between canary yellow and red watermelon based on SNP in lycopene β-cyclase (LCYB) gene. Mol. Breed. 20, 63–72. doi:10.1007/s11032-006-9076-4
Bang, H., Yi, G., Kim, S., Leskovar, D., and Patil, B. S. (2014). Watermelon lycopene β-cyclase: promoter characterization leads to the development of a PCR marker for allelic selection. Euphy 200, 363–378. doi:10.1007/s10681-014-1158-5
Binder, S., Stoll, K., and Stoll, B. (2013). P-class pentatricopeptide repeat proteins are required for efficient 5′ end formation of plant mitochondrial transcripts. RNA Biol. 10 (9), 1511–1519. doi:10.4161/rna.26129
Branham, S., Vexler, L., Meir, A., Tzuri, G., Frieman, Z., Levi, A., et al. (2017). Genetic mapping of a major codominant QTL associated with β-carotene accumulation in watermelon. Mol. Breed. 37 (146), 1–13. doi:10.1007/s11032-017-0747-0
Bréhélin, C., and Kessler, F. (2008). The plastoglobule: A bag full of lipid biochemistry tricks. Photochem. Photobiol. 84 (6), 1388–1394. doi:10.1111/j.1751-1097.2008.00459.x
Bryant, N., Lloyd, J., Sweeney, C., Myouga, F., and Meinke, D. (2011). Identification of nuclear genes encoding chloroplast-localized proteins required for embryo development in Arabidopsis. Plant Phys. 155 (4), 1678–1689. doi:10.1104/pp.110.168120
Cox, M. P., Peterson, D. A., and Biggs, P. J. (2010). SolexaQA: at-a-glance quality assessment of illumina second-generation sequencing data. BMC bioinfo 11 (1), 485–486. doi:10.1186/1471-2105-11-485
Eriksson, E. M., Bovy, A., Manning, K., Harrison, L., Andrews, J., De Silva, J., et al. (2004). Effect of the Colorless non-ripening mutation on cell wall biochemistry and gene expression during tomato fruit development and ripening. Plant Physio 136 (4), 4184–4197. doi:10.1104/pp.104.045765
Fang, X., Liu, S., Gao, P., Liu, H., Wang, X., Luan, F., et al. (2020). Expression of ClPAP and ClPSY1 in watermelon correlates with chromoplast differentiation, carotenoid accumulation, and flesh color formation. Sci. Horticul. 270, 109437. doi:10.1016/j.scienta.2020.109437
Food and Agriculture Organization (2020). Crops. http://www.fao.org/faostat/en/#data/QC/(Accessed August 12, 2022).
Galpaz, N., Gonda, I., Shem-Tov, D., Barad, O., Tzuri, G., Lev, S., et al. (2018). Deciphering genetic factors that determine melon fruit-quality traits using RNA-Seq-based high-resolution QTL and eQTL mapping. Plant J. 94 (1), 169–191. doi:10.1111/tpj.13838
Grassi, S., Piro, G., Lee, J. M., Zheng, Y., Fei, Z., Dalessandro, G., et al. (2013). Comparative genomics reveals candidate carotenoid pathway regulators of ripening watermelon fruit. BMC geno 14 (781), 1–20. doi:10.1186/1471-2164-14-781
Guo, S., Zhang, J., Sun, H., Salse, J., Lucas, W. J., Zhang, H., et al. (2013). The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat. Genet. 45 (1), 51–58. doi:10.1038/ng.2470
Henderson, W. R. (1989). Inheritance of orange flesh color in watermelon. Cucurbit Genet. Coop. 12, 59–63.
Henderson, W. R., Scott, G. H., and Wehner, T. C. (1998). Interaction of flesh color genes in watermelon. J. Hered. 89 (1), 50–53. doi:10.1093/jhered/89.1.50
Henderson, W. R., Scott, G. H., and Wehner, T. C. (1996). Interaction of genes for flesh color in watermelon. HortScience 31 (4), 602d–602d. doi:10.21273/HORTSCI.31.4.602d
Jeffrey, C. (1975). Further notes on Cucurbitaceae: III: some southern african taxa. Kew Bull. 30, 475–493. doi:10.2307/4103075
Jin, B., Lee, J., Kweon, S., Cho, Y., Choi, Y., Lee, S. J., et al. (2019). Analysis of flesh color-related carotenoids and development of a CRTISO gene-based DNA marker for prolycopene accumulation in watermelon. Horti., Env. Biotech 60, 399–410. doi:10.1007/s13580-019-00139-3
Jo, Y. D., Ha, Y., Lee, J. H., Park, M., Bergsma, A. C., Choi, H. I., et al. (2016). Fine mapping of Restorer-of-fertility in pepper (Capsicum annuum L.) identified a candidate gene encoding a pentatricopeptide repeat (PPR)-containing protein. Theo. Appl. genet. 129, 2003–2017. doi:10.1007/s00122-016-2755-6
Lee, E. S., Kim, J., Hong, J. P., Kim, D. S., Kim, M., Huh, Y. C., et al. (2018). Development of HRM markers based on SNPs identified from next generation resequencing of susceptible and resistant parents to gummy stem blight in watermelon. Korean Soc. Breed. Sci. 50 (4), 424–433. doi:10.9787/KJBS.2018.50.4.424
Lee, S., Park, G., Choi, Y., Park, S., Kim, H., Lee, O., et al. (2021). Whole-genome resequencing of near-isogenic lines reveals a genomic region associated with high trans-lycopene contents in watermelon. Plants 11 (1), 8. doi:10.3390/plants11010008
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997.
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinfor 25 (16), 2078–2079. doi:10.1093/bioinformatics/btp352
Li, N., Shang, J., Wang, J., Zhou, D., Li, N., and Ma, S. (2020). Discovery of the genomic region and candidate genes of the scarlet red flesh color (Yscr) locus in watermelon (Citrullus lanatus L.). Fronti. Plant Sci. 11, 116. doi:10.3389/fpls.2020.00116
Liu, S., Gao, P., Wang, X., Davis, A. R., Baloch, A. M., and Luan, F. (2015). Mapping of quantitative trait loci for lycopene content and fruit traits in Citrullus lanatus. Euphy 202, 411–426. doi:10.1007/s10681-014-1308-9
Liu, S., Liu, M., Cao, Y., Xu, Y., Liu, H., Zhu, Q., et al. (2023). Identification of chromosome region and candidate genes for canary-yellow flesh (Cyf) locus in watermelon (Citrullus lanatus). Plant Sci. 329, 111594. doi:10.1016/j.plantsci.2023.111594
Lou, L. (2009). Inheritance of fruit characteristics in watermelon [Citrullus lanatus (thunb.) Matsum. and Nakai]. Releigh, North Carolina: M.S Thesis, North Carolina State University.
Lurin, C., Andreés, C., Aubourg, S., Bellaoui, M., Bitton, F., Bruyere, C., et al. (2004). Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell 16 (8), 2089–2103. doi:10.1105/tpc.104.022236
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17 (1), 10–12. doi:10.14806/ej.17.1.200
Meuwissen, T., and Goddard, M. (2010). Accurate prediction of genetic values for complex traits by whole-genome resequencing. Genet 185 (2), 623–631. doi:10.1534/genetics.110.116590
Okuda, K., Nakamura, T., Sugita, M., Shimizu, T., and Shikanai, T. (2006). A pentatricopeptide repeat protein is a site recognition factor in chloroplast RNA editing. J. Bio. Chem. 281 (49), 37661–37667. doi:10.1074/jbc.M608184200
Paris, H. S. (2015). Origin and emergence of the sweet dessert watermelon, Citrullus lanatus. Ann. bot. 116 (2), 133–148. doi:10.1093/aob/mcv077
Park, G., Kim, J., Jin, B., Yang, H. B., Park, S. W., Kang, S. C., et al. (2018). Genome-wide sequence variation in watermelon inbred lines and its implication for marker-assisted breeding. Horti. Sci.and Techno. 36 (2), 280–291. doi:10.12972/kjhst.20180028
Perkins-Veazie, P., Collins, J. K., Davis, A. R., and Roberts, W. (2006). Carotenoid content of 50 watermelon cultivars. J. agri. food chem. 54 (7), 2593–2597. doi:10.1021/jf052066p
Pyo, Y. J., Kwon, K. C., Kim, A., and Cho, M. H. (2013). Seedling Lethal1, a pentatricopeptide repeat protein lacking an E/E+ or DYW domain in Arabidopsis, is involved in plastid gene expression and early chloroplast development. Plant Physio 163 (4), 1844–1858. doi:10.1104/pp.113.227199
Ren, Y., Zhao, H., Kou, Q., Jiang, J., Guo, S., Zhang, H., et al. (2012). A high resolution genetic map anchoring scaffolds of the sequenced watermelon genome. PLoS One 7 (1), e29453. doi:10.1371/journal.pone.0029453
Saidi, A., and Hajibarat, Z. (2020). Application of Next Generation Sequencing, GWAS, RNA seq, WGRS, for genetic improvement of potato (Solanum tuberosum L) under drought stress. Biocat. Agricul. Biotech. 29, 101801. doi:10.1016/j.bcab.2020.101801
Subburaj, S., Lee, K., Jeon, Y., Tu, L., Son, G., Choi, S., et al. (2019). Whole genome resequencing of watermelons to identify single nucleotide polymorphisms related to flesh color and lycopene content. PLoS One 14 (10), e0223441. doi:10.1371/journal.pone.0223441
Subburaj, S., Tu, L., Lee, K., Park, G. S., Lee, H., Chun, J. P., et al. (2020). A genome-wide analysis of the pentatricopeptide repeat (PPR) gene family and PPR-derived markers for flesh color in watermelon (Citrullus lanatus). Genes 11 (10), 1125. doi:10.3390/genes11101125
Sun, F., Xiu, Z., Jiang, R., Liu, Y., Zhang, X., Yang, Y. Z., et al. (2019). The mitochondrial pentatricopeptide repeat protein EMP12 is involved in the splicing of three nad2 introns and seed development in maize. J. exp. Bot. 70 (3), 963–972. doi:10.1093/jxb/ery432
Sun, Y., Huang, J., Zhong, S., Gu, H., He, S., and Qu, L. J. (2018). Novel DYW-type pentatricopeptide repeat (PPR) protein BLX controls mitochondrial RNA editing and splicing essential for early seed development of Arabidopsis. J. Genet. Geno. 45 (3), 155–168. doi:10.1016/j.jgg.2018.01.006
Tadmor, Y. A. A. K. O. V., King, S. T. E. P. H. E. N., Levi, A., Davis, A., Meir, A. Y. A. L. A., Wasserman, B. O. R. I. S., et al. (2005). Comparative fruit colouration in watermelon and tomato. Food Res. interna. 38 (8-9), 837–841. doi:10.1016/j.foodres.2004.07.011
Wang, C., Qiao, A., Fang, X., Sun, L., Gao, P., Davis, A. R., et al. (2019b). Fine mapping of lycopene content and flesh color related gene and development of molecular marker–assisted selection for flesh color in watermelon (Citrullus lanatus). Front. Plant Sci. 10, 1240. doi:10.3389/fpls.2019.01240
Wang, P., Wang, S., Chen, Y., Xu, X., Guang, X., and Zhang, Y. (2019a). Genome-wide analysis of the MADS-box gene family in Watermelon. Comput. Bio. chem. 80, 341–350. doi:10.1016/j.compbiolchem.2019.04.013
Wang, X., An, Y., Li, Y., and Xiao, J. (2021b). A PPR protein ACM1 is involved in chloroplast gene expression and early plastid development in Arabidopsis. Intern. J. Mol. Sci. 22 (5), 2512. doi:10.3390/ijms22052512
Wang, X., An, Y., Qi, Z., and Xiao, J. (2021a). PPR protein early chloroplast development 2 is essential for chloroplast development at the early stage of Arabidopsis development. Plant Sci. 308, 110908. doi:10.1016/j.plantsci.2021.110908
Wu, S., Wang, X., Reddy, U., Sun, H., Bao, K., Gao, L., et al. (2019). Genome of ‘Charleston Gray’, the principal American watermelon cultivar, and genetic characterization of 1,365 accessions in the US National Plant Germplasm System watermelon collection. Plant Biotech. J. 17 (12), 2246–2258. doi:10.1111/pbi.13136
Yan, J., Zhang, Q., Guan, Z., Wang, Q., Li, L., Ruan, F., et al. (2017). MORF9 increases the RNA-binding activity of PLS-type pentatricopeptide repeat protein in plastid RNA editing. Nat. Plants 3 (5), 17037–17038. doi:10.1038/nplants.2017.37
Yuan, N., Wang, J., Zhou, Y., An, D., Xiao, Q., Wang, W., et al. (2019). EMB-7L is required for embryogenesis and plant development in maize involved in RNA splicing of multiple chloroplast genes. Plant Sci. 287, 110203. doi:10.1016/j.plantsci.2019.110203
Zhang, J., Sun, H., Guo, S., Ren, Y., Li, M., Wang, J., et al. (2020). Decreased protein abundance of lycopene β-cyclase contributes to red flesh in domesticated watermelon. Plant Physio 183 (3), 1171–1183. doi:10.1104/pp.19.01409
Keywords: Citrullus lanatus, flesh color, pentatricopeptide repeat, marker-assisted selection, watermelon
Citation: Park G, Shahwar D, Jang G, Shin J, Kwon G, Kim Y, Hong CO, Jin B, Kim H, Lee O and Park Y (2023) Identification of a novel locus C2 controlling canary yellow flesh color in watermelons. Front. Genet. 14:1256627. doi: 10.3389/fgene.2023.1256627
Received: 11 July 2023; Accepted: 29 August 2023;
Published: 19 September 2023.
Edited by:
Purushothaman Natarajan, West Virginia State University, United StatesReviewed by:
Jian Zhang, Beijing Academy of Agriculture and Forestry Sciences, ChinaCopyright © 2023 Park, Shahwar, Jang, Shin, Kwon, Kim, Hong, Jin, Kim, Lee and Park. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Younghoon Park, eXBhcmtAcHVzYW4uYWMua3I=; Durre Shahwar, ZHNoOTJAcHVzYW4uYWMua3I=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.