 Rutaba Gul1,2
Rutaba Gul1,2 Sabika Firasat1*
Sabika Firasat1* Mikkel Schubert2
Mikkel Schubert2 Asmat Ullah2
Asmat Ullah2 Elionora Peña2
Elionora Peña2 Anne C. B. Thuesen2
Anne C. B. Thuesen2 Annete P. Gjesing2Mulazim Hussain3
Annete P. Gjesing2Mulazim Hussain3 Muhammad Tufail1Muhammad Saqib1,4Kiran Afshan1Torben Hansen2*
Muhammad Tufail1Muhammad Saqib1,4Kiran Afshan1Torben Hansen2*- 1Department of Zoology, Faculty of Biological Sciences, Quaid-i-Azam University, Islamabad, Pakistan
- 2Novo Nordisk Foundation Center for Basic Metabolic Research, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark
- 3The Children Hospital, Pakistan Institute of Medical Sciences (PIMS), Islamabad, Pakistan
- 4Department of Zoology, University of Lakki Marwat, Lakki Marwat, Khyber Pakhtunkhwa, Pakistan
Mucopolysaccharidoses (MPSs) are inherited lysosomal storage disorders (LSDs). MPSs are caused by excessive accumulation of mucopolysaccharides due to missing or deficiency of enzymes required for the degradation of specific macromolecules. MPS I-IV, MPS VI, MPS VII, and MPS IX are sub-types of mucopolysaccharidoses. Among these, MPS III (also known as Sanfilippo) and MPS IV (Morquio) syndromes are lethal and prevalent sub-types. This study aimed to identify causal genetic variants in cases of MPS III and MPS IV and characterize genotype-phenotype relations in Pakistan. We performed clinical, biochemical and genetic analysis using Whole Genome Sequencing (WGS) in 14 Pakistani families affected with MPS III or MPS IV. Patients were classified into MPS III by history of aggressive behaviors, dementia, clear cornea and into MPS IV by short trunk, short stature, reversed ratio of upper segment to lower segment with a short upper segment. Data analysis and variant selections were made based on segregation analysis, examination of known MPS III and MPS IV genes, gene function, gene expression, the pathogenicity of variants based on ACMG guidelines and in silico analysis. In total, 58 individuals from 14 families were included in the present study. Six families were clinically diagnosed with MPS III and eight families with MPS IV. WGS revealed variants in MPS-associated genes including NAGLU, SGSH, GALNS, GNPTG as well as the genes VWA3B, BTD, and GNPTG which have not previously associated with MPS. One family had causal variants in both GALNS and BTD. Accurate and early diagnosis of MPS in children represents a helpful step for designing therapeutic strategies to protect different organs from permanent damage. In addition, pre-natal screening and identification of genetic etiology will facilitate genetic counselling of the affected families. Identification of novel causal MPS genes might help identifying new targeted therapies to treat LSDs.
Introduction
Background
Mucopolysaccharidoses (MPS) is a group of disorders belonging to lysosomal storage disorders. MPS are sub categorized into seven types (MPS I, II, III, IV, VI, VII, and IX) mostly inherited as autosomal recessive except for MPS II. Each type is caused by the deficiency of mucopolysaccharide-degrading enzymes. Due to the deficiency of these enzymes, the mucopolysaccharides (glycosaminoglycans) accumulate in different organs of the body including the arteries, skeleton, eyes, joints, ears, skin, teeth, respiratory system, liver, spleen, central nervous system, blood, and bone marrow. This excessive accumulation of mucopolysaccharides result in phenotypes of MPS overlapping with one another, making it difficult to diagnose without use of specific diagnostic approaches like the measurement of enzyme activity in leukocytes or molecular diagnosis (Besley and Wraith, 1997; Chih-Kuang et al., 2002).
Mucopolysaccharidosis type III (MPS III) or Sanfilippo syndrome is a subtype of MPS discovered 50 years ago with an autosomal recessive mode of inheritance. The disease affects 0.3–4.1 cases per 100,000 births (Sanfilippo, 1963; Valstar et al., 2008; Fedele, 2015; Khan et al., 2017). It has been further classified into four categories (MPS type IIIA—D) caused by variants in SGSH, NAGLU, HGSNAT, and GNS respectively. Enzymes encoded by these genes are involved in the degradation of a linear polysaccharide known as heparan sulfate (HS). HS has a crucial role in the development of central nervous system (CNS) which explains why patients suffering from Sanfilippo syndrome develop dementia from early childhood (De Pasquale and Pavone, 2019) and have neurological manifestations such as delayed behavioral and developmental milestones starting from early years of life (Shapiro et al., 2017; Ozkinay et al., 2021).
Mucopolysaccharidosis type IV, also known as Morquio syndrome, was first discovered by a pediatrician Luis Morquio in 1929. It has an autosomal recessive mode of inheritance (Morquio, 1929). It is a rare disorder with prevalence of 1 in 40,000 live births (Nelson et al., 2003). There are two types of Morquio syndrome (type A and type B) caused by pathogenic variants in GALNS and GLB1, respectively. The gene GALNS encodes the GALNS enzyme which has a role in the degradation of glycosaminoglycan including chondroitin-6-sulfate (C6S) and keratin sulfate (KS), while GLB1 encoded by GLB1 is involved in the degradation of keratin sulfate only (Celik et al., 2021). The GLB1 enzyme is also involved in the hydrolyzation of terminal beta galactosyl residues of GM1 gangliosides and glycoproteins (Okada and John, 1968).
Diagnosis of MPS III involves either urinary HS excretion measurement or enzyme measurement from dry blood spot through tandem mass spectrometry or fluorimetry (Seker Yilmaz et al., 2021). The diagnosis of MPS IV involves radiological findings and urinary excretion or liquid chromatography–mass spectrometry (LC-MS) or MS-based method for KS measurements in plasma or urine (Piraud et al., 1993; Martell et al., 2011; Peracha et al., 2018; Chien et al., 2020; Lin et al., 2020). Due to enzymatic pseudo-deficiency, molecular genetic screening of the patients affected with MPS III or MPS IV is the most accurate method to diagnose any subtype of these disorders.
To the best of our knowledge, no prior molecular genetics research has been conducted in Pakistan regarding Sanfilippo cases. However, various international research groups have published studies highlighting different genetic variations found in cases of Pakistani descent across the globe. Notably, the literature includes instances of novel or previously reported variants of Sanfilippo syndrome originating from Pakistani patients. These include a unique deletion that triggers Sanfilippo syndrome type D, specifically the c.1169delA mutation within exon 10, resulting in a premature termination codon (Beesley et al., 2003). Additionally, a new nonsense variant associated with Sanfilippo syndrome type C, identified as p.Ser296Ter in exon 10, as well as a splice site variant designated c.744–2A>G in intron 7, have been documented (Feldhammer et al., 2009a; Feldhammer et al., 2009b). Several studies have been conducted in Pakistan related to cases of Morquio syndrome, revealing a total of seven new variants—namely, p. Phe216Ser, p. Met38Arg, p. Ala291Ser, p. Glu121Argfs*37, p. Tyr294Terfs—alongside two previously reported variants, p. Pro420Arg and p. Arg386Cys (Tomatsu et al., 2004; Morrone et al., 2014; Ullah et al., 2017; Zubaida et al., 2018).
Treatment options for MPS III and IV include enzyme replacement therapy (ERT) (MPS IV A), molecular chaperon therapy (MCT), substrate reduction therapy (SRT), hematopoietic stem cell transplant (HSCT), in vivo adeno-associated viral gene therapy, antioxidant therapy, inhibiting protein aggregation, stop-codon readthrough therapy and anti-inflammatory therapy (Hendriksz et al., 2013; Akyol et al., 2019; Beneto et al., 2020; Seker Yilmaz et al., 2021). In a recent study, recombinant human GALNS enzyme (rhGALNS) was infused with hydrogel like polyethylene glycol to deliver the exogenous enzyme successfully into the fibroblasts for sustainable and longer release (Jain et al., 2020). Molecular genetic testing, prenatal screening, carrier testing, and genetic counselling can help in the prevention of the disease and/or saving organs from permanent degeneration by allowing early diagnosis and appropriate treatment options (where available). This study was designed to evaluate the clinical profiles of Sanfilippo and Morquio syndromes patients from Pakistan, to identify causal gene variants using WGS and to correlate the genotypes with phenotypes.
Materials and methodology
Ethical approval and patient recruitment
The study was approved by the bioethical committee of Quaid-i-Azam University (BEC-FBS-QAU2019-198), Islamabad, Pakistan.
Patients were recruited and diagnosed based on their presenting clinical profiles (Table 1; Table 3) at outpatient department (OPD), Children Hospital, Pakistan Institute of Medical Sciences (PIMS), Islamabad, Pakistan. Before collecting blood samples from patients and accompanying parents/siblings, informed written consent forms were signed by the participants in accordance with the Declaration of Helsinki (World Medical Association, 2013). The detailed pedigrees were drawn using the information provided by well-informed elders of the family (Figure 1; Figure 2). Localities of families from different regions of Pakistan is mentioned in Figure 3.
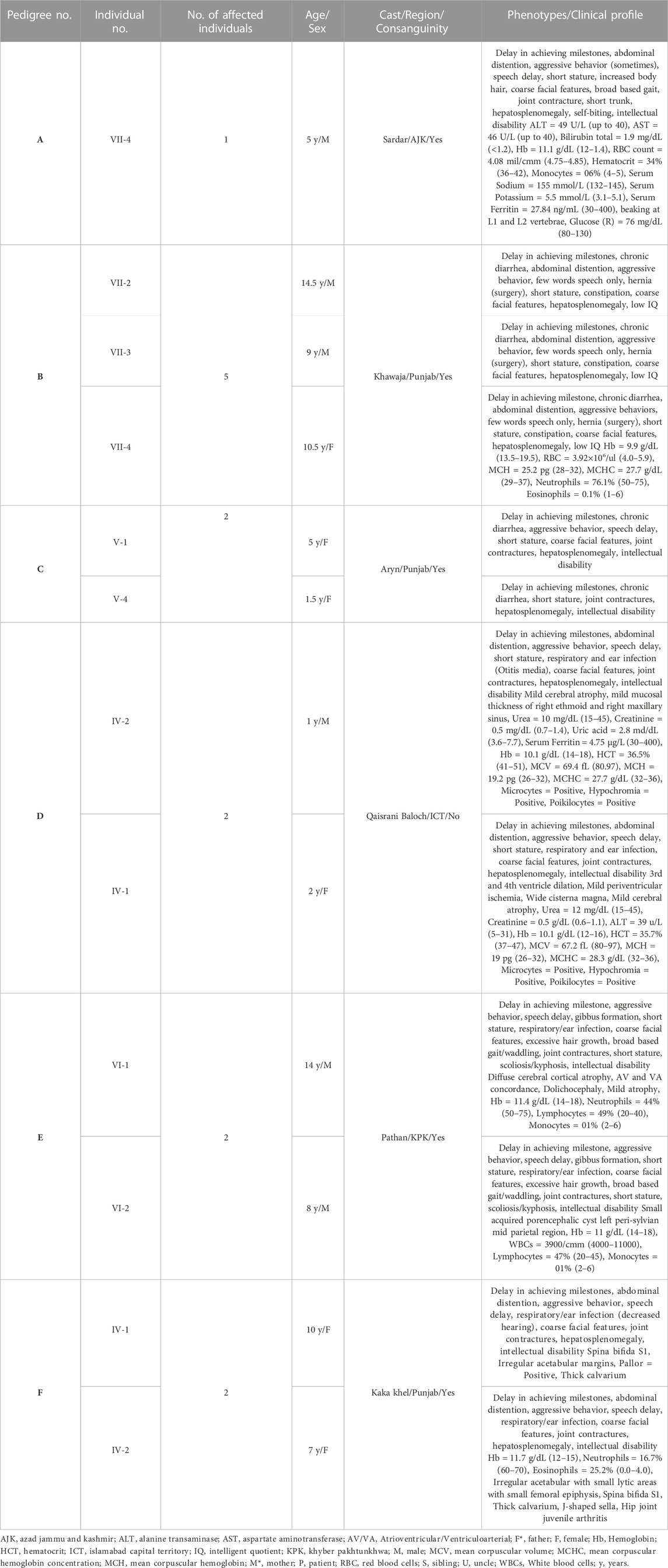
TABLE 1. Clinical details of the patients suspected as Sanfilippo syndrome affected.
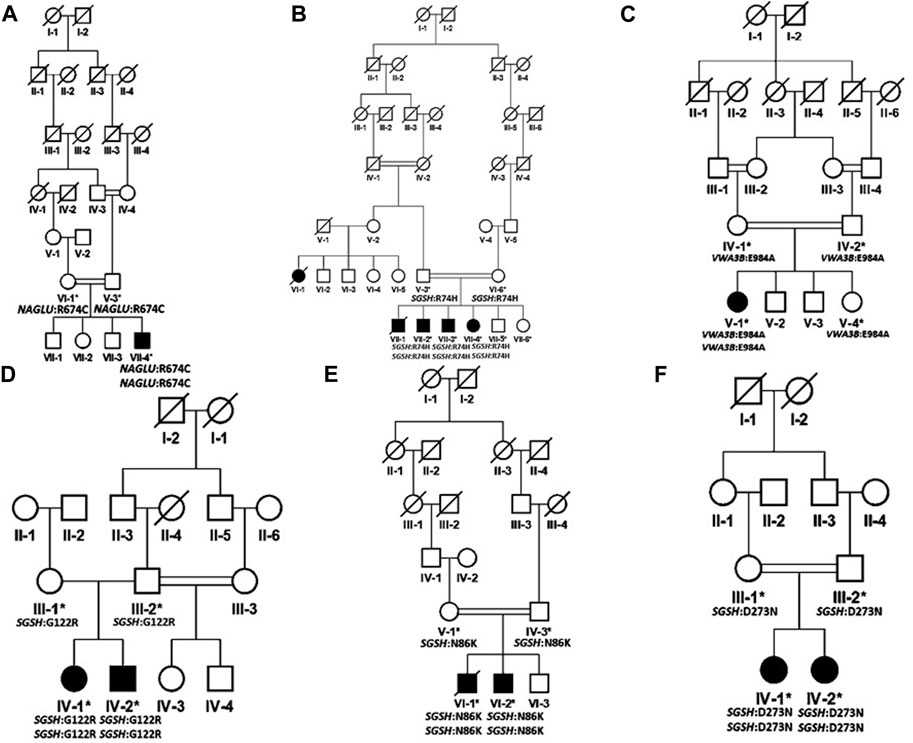
FIGURE 1. Pedigrees of families inheriting MPS III phenotypes (A–F) consistent with autosomal recessive mode of inheritance. Square and circles denote males and females respectively; filled symbols indicate affected individuals and consanguinity is represented by double marriage lines.
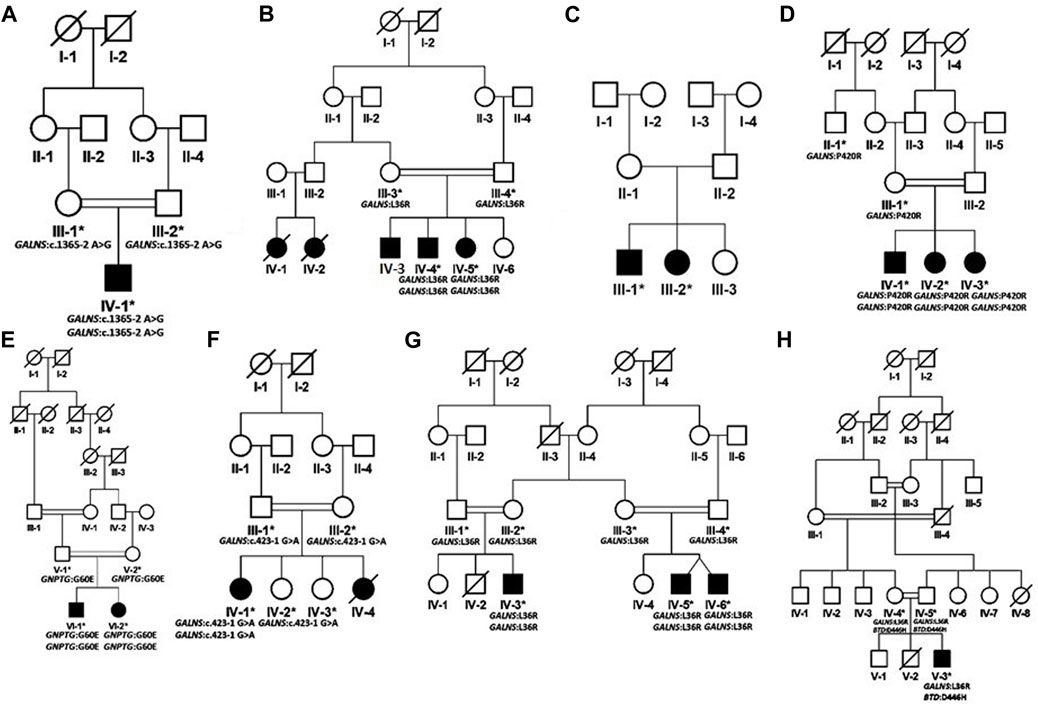
FIGURE 2. Pedigrees of families inheriting MPS IV phenotypes (A–H) consistent with autosomal recessive mode of inheritance. Square and circles denote males and females respectively; filled symbols indicate affected individuals and consanguinity is represented by double marriage lines.
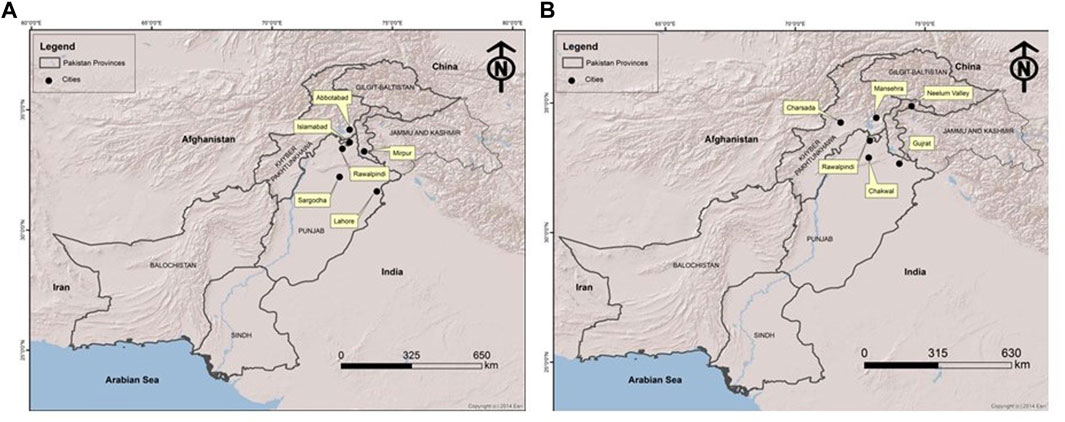
FIGURE 3. Map of Pakistan showing the locations (circles) of cities/towns from where families included in this study were collected. (A) All MPS III collected families. (B) All MPS IV collected families.
In presenting clinical profiles, height of each MPS IV patient was measured and compared to the healthy individual of same age to confirm their stature. Height is plotted on Center for Disease Control and Prevention (CDC) charts, used globally and a height that is two standard deviations below the mean height for age and sex (less than the 3rd percentile) is considered as short stature (Barstow and Rerucha, 2015). Complete skeletal survey through x-ray was performed which showed dysostosis multiplex, fish mouth vertebrae and bullet shaped metacarpals in Morquio syndrome patients.
Sampling and DNA extraction
Peripheral blood samples of 3–5 mL were collected from 58 individuals from 14 families, covering 26 affected and 32 unaffected individuals. The blood samples were collected in 10 mL EDTA vacutainers (BD vacutainer K2 EDTA 18 mg). DNA extraction was performed using the Phenol-Chloroform method and DNA was quantified using a μDrop Plate reader (Multiskan™, Thermo Fisher Scientific, and Waltham, MA, United States).
Whole genome sequencing and variant prioritization
Whole genome sequencing, reads mapping, genotype calling, pre-analysis quality assurance, and annotation were performed as describe previously by Gul et al. (2023).
Briefly, mapping and genotype calling was performed using a modified version of the PALEOMIX pipeline (https://pubmed.ncbi.nlm.nih.gov/24722405/), with initial quality assurance performed using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and MultiQC (https://pubmed.ncbi.nlm.nih.gov/27312411/). Reads were processed using fastp (https://pubmed.ncbi.nlm.nih.gov/30423086/) and mapped against the hg38 human reference using BWA (https://arxiv.org/abs/1303.3997). Alignments were post-processed using samtools (https://pubmed.ncbi.nlm.nih.gov/33590861/) and ‘bwa-postalt.js’ from the BWA-kit. BAMs were calibrated, and genotypes were called and calibrated using GATK (https://pubmed.ncbi.nlm.nih.gov/20644199/). The resulting VCF was annotated using VEP (https://pubmed.ncbi.nlm.nih.gov/27268795/).
Initially, analysis of variants identification was based on MPS-associated genes. Variants in the exomes were extracted from whole genome sequencing data of all individuals. The variants including non-synonymous and splice sites having minor allele frequency (MAF) greater than 0.01 in larger outbred populations were excluded by using gnomAD. In pedigrees with more than one affected individual, variants shared by multiple patients were prioritized. Based on consanguinity and inheritance pattern, homozygous variants were selected and prioritized. During re-analysis, heterozygous variants were also screened for compound heterozygosity. The prioritization was based on segregation, gene function, association with disease and pathogenicity prediction according to ACMG guidelines and in silico analysis (Richards et al., 2015). Genes were annotated according to the following transcripts: NM_000263.4 (NAGLU); NM_144992 (VWA3B); NM_000199.5 (SGSH); NM_000512.5 (GALNS); NM_001281723.2 (BTD); NM_032520.5 (GNPTG).
All variants were found in homozygous state in patients while in heterozygous state in parents. Phenotypically normal siblings in the present families were either homozygous wild type or heterozygous carriers for that particular variant. All identified variants are listed in Table 2 and Table 4. The variants were evaluated using ACMG guidelines and were predicted as pathogenic, likely pathogenic or uncertain significance. To exclude non-disease associated variants, the genomes of more than 100 ethnically matched in-house control samples were screened. The analysis plan for this study was same as mentioned in previously (Gul et al., 2023).
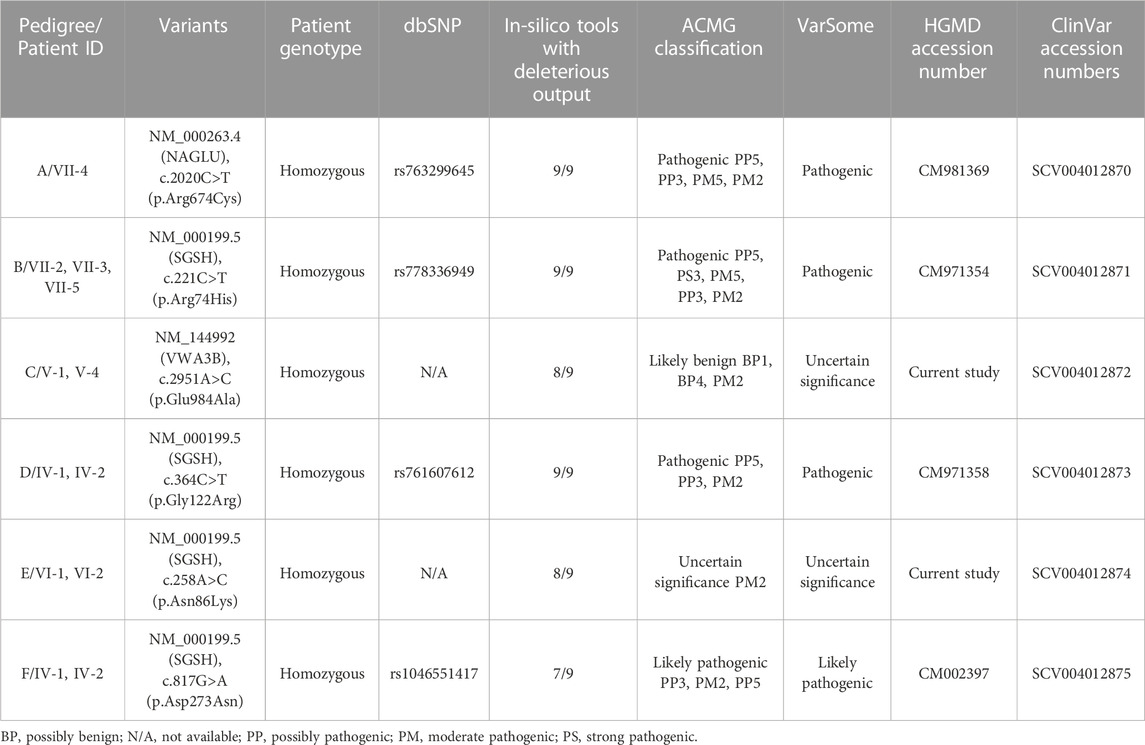
TABLE 2. In-silico analysis and ACMG classification of the identified variants in suspected Sanfilippo syndrome cases.
Protein modeling and protein-protein interaction
To generate the complete three-dimensional (3D) structures of BTD, SGSH, VWA3B, GNTGP, GALNS, and NAGLU proteins I-TASSER (Iterative Threading Assembly Refinement) (https://zhanggroup.org/I-TASSER/) was utilized. This is a bioinformatics tool that generates 3D protein structures using hierarchical techniques, first searching the PDB library for templates and then building the models using iterative template fragment assembly simulation. The degree of appropriateness of all structures were verified through VERIFY3D and ERRAT. UCSF Chimera software 1.14 was used to inspect all the structures graphically (Supplementary Figure S1).
Evolutionary conservation analysis
The degree of evolutionary conservation of the amino acids was calculated using the ConSurf (https://consurf.tau.ac.il/consurf_index.php) server to maintain the structural integrity and function of protein, yielding a conservation rating from 1-9, classified as variable, intermediate, or highly conserved protein site. Furthermore, properties of residues were categorized as either exposed (e), buried (b), highly conserved and exposed (functional, f), or highly conserved and buried (s) (Supplementary Figure S2). As a result, any change in the highly conserved area has a significant impact on the protein’s integral structure and function.
Results
Clinical characterization
Based on clinical and phenotypic features, 11 patients (6 males and 5 females) with average age of ±7.2 years from six families were diagnosed with Sanfilippo syndrome (MPS III). The other 15 patients (9 males and 6 females) from 8 families were labeled as suspected cases of MPS IV with average age of ±7.3 years. MPS III patients showed a wide range of overlapping features like delay in achieving milestones, aggressive behavior, abdominal distention, increased body hair growth and intellectual disability (Table 1). MPS IV patients showing overlapping features like short stature, coarse facial features, scoliosis/kyphosis, gibbus formation and many others (Table 3). All families of MPS III show parental consanguinity except family D having intra-cast marriage without any direct blood relation among parents (Figure 1D). For MPS IV all families show consanguinity except family C (Figure 2C).
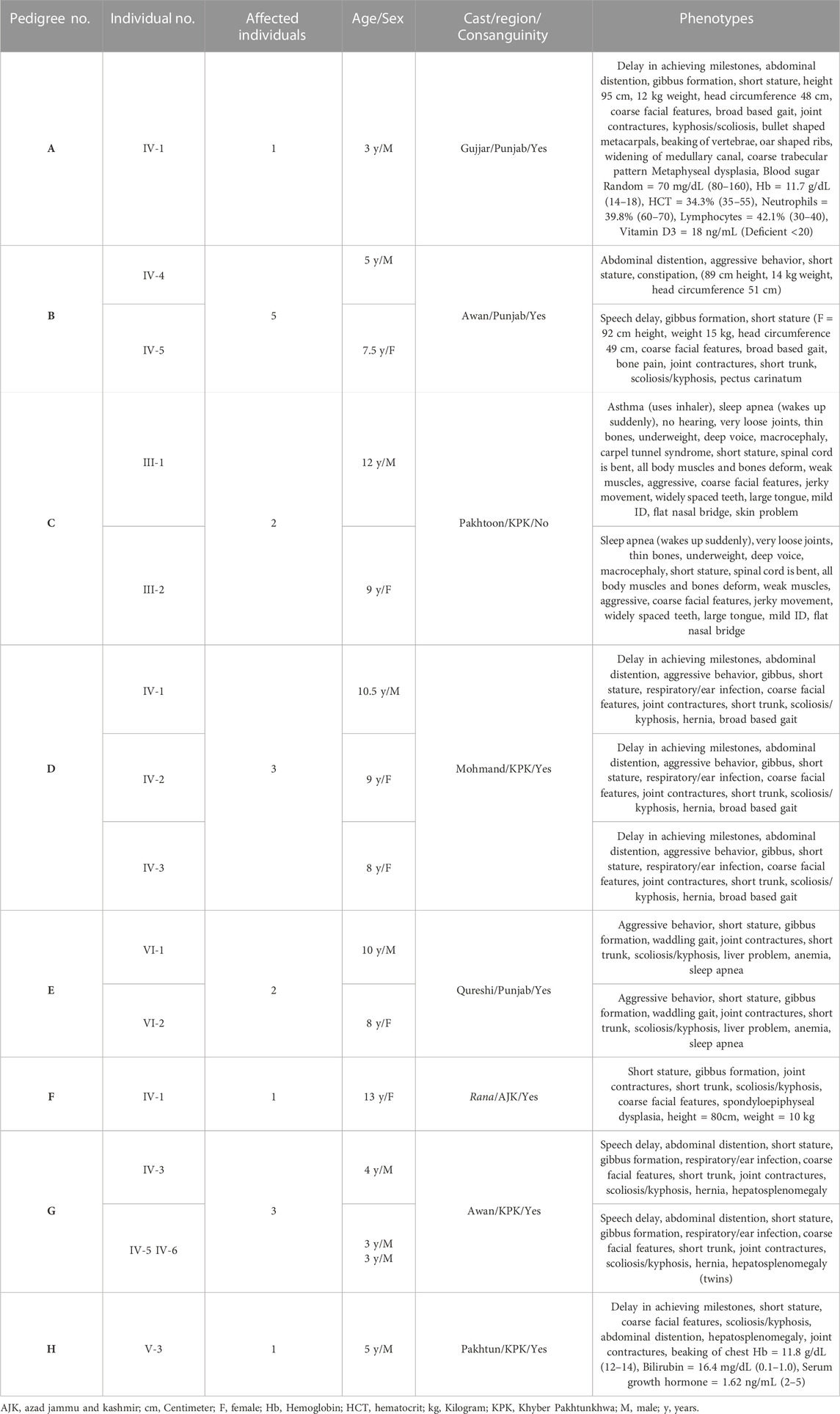
TABLE 3. Clinical details of the patients suspected as Morquio syndrome affected.
Genetic characterization
Among 14 families diagnosed with MPS, 6 novel and 6 reported variants were identified, where 11 families carried variants in MPS-associated genes (NAGLU, SGSH, GALNS), two families carried variants in two genes (VWA3B, GNPTG) that were not previously associated with MPS phenotypes, while one family remained genetically undiagnosed. In family H we found dual molecular diagnosis (GALNS and BTD genes involvement). In family C (MPS IV), initial analysis of coding regions in patients did not reveal any homozygous or compound heterozygous pathogenic variant(s).
In silico analysis
Pathogenicity prediction
The identified variants were predicted to be deleterious/pathogenic by various mutation prediction tools like MutationTaster, I-TASSER, SIFT, Polyphen-2, CADD, VarSome, VARSEAK and PROVEAN (Table 2; Table 4). Generated structures were selected based on their high C-score and Tm-score. Wild-type structure of proteins (BTD, SGSH, VWA3B, GNPTG, NAGLU, and GALNS) were retrieved from Alpha-fold. Comparison between the wild-type and mutant type was performed and the mutated structures showed significant structural change. The altered structure may result in a loss of function, altered interactions with ligands or neighboring proteins, or altered stability of the proteins. None of the identified variants were found in homozygous state in any human genome variation databases including ExAC, gnomAD, 1000 Genome Project, dbSNP and healthy control samples of same ethnicity.
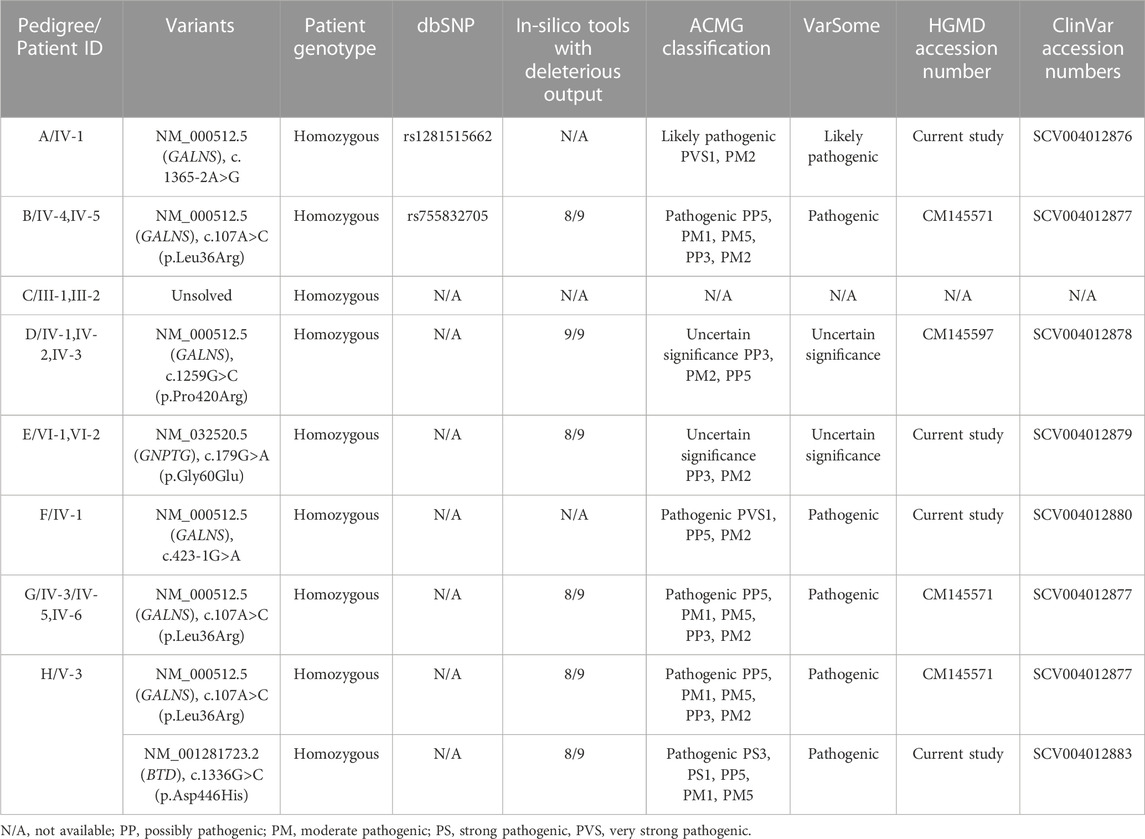
TABLE 4. In-silico analysis and ACMG classification of the identified variants in suspected Morquio syndrome cases.
Evolutionary conservation analysis
The evolutionary conservation analysis of candidate genes showed that, in BTD protein, p.Asp446His is an average (partially conserved) and exposed residue according to the NACSES. In the protein SGSH, the Asn86Lys is highly conserved and buried residue. In protein VWA3B, the Glu984Ala is highly conserved, exposed and functional residue. In protein GNPTG, the Gly60Glu is highly conserved, buried and structural residue according to NACSES. In the protein NAGLU, Arg674Cys is highly conserved, exposed and functional residue according to NACSES.
Discussion
In 14 families with phenotypically diagnosed MPS III (A-F) or MPS IV (A-H) we report that 11 families carried pathogenic variants in known MPS-associated genes. Two families had novel mutations in genes previously not associated with MPS and one family remains genetically undiagnosed. Additionally, one family (family H) had pathogenic variants in both GALNS and BTD segregating with the phenotype.
Mucopolysaccharidoses III
The initial recruitment and diagnosis of six families with Sanfilippo syndrome (MPS III) were based on their clinical profiles and phenotypes (Table 1).
We report two likely causal variations: One identified in NAGLU (family A) co-segregating with MPS III type B and the other in SGSH (families B, D, E and F) co-segregating with MPS III type A and VWA3B (Family C).
In family A, a homozygous missense variant c. 2020C>T (p. Arg674Cys) was found in exon 6 of NAGLU gene. This variant was first reported in compound heterozygous state by Zhao et al. (1998) in two different families affected with end-stage neurological debilitation and moderate mental retardation. Another missense variant at the same codon (c. 2021G>A (p. Arg674His)) in compound heterozygous state was reported by Zhao et al. (1996) in an Arab family. This locus is the mutational hot spot for substitution of Arginine. The locus has CpG dinucleotide involvement (Zhao et al., 1996). Currently studied patient showed more severe phenotypes including delay in achieving milestone, abdominal distention, aggressive behavior, speech delay, short stature, increased body hair, coarse facial features, broad-based gait, joint contracture, short trunk, hepatosplenomegaly, self-biting and intellectual disability. The interfamilial variability in the severity of phenotypes might be due to homozygous substitution of Arginine at amino acid position 674 in the present patient and/or different ethnicities of the families reported by Zhao et al. (1996), Zhao et al. (1998). Furthermore, such variability in clinical presentations amongst patients harboring the same genetic variant have also been reported for other LSD subtypes, indicating the contribution of epigenetic modifications, environmental factors and/or genetic modifiers that is yet to be elucidated (Davidson et al., 2018; Sheth et al., 2022).
In family B, five individuals were affected including three males and two females. One female (VI-1) and one male (VII-1) patient had died at the age of 12 and 14 years respectively. All available patients were homozygous for the SGSH c. 221C>T (p. Arg74His) variant. The parents were heterozygous, while two healthy live siblings including a female (VII-5; 21 years) and a male (VII-4; 15 years) were wild type carriers. This pathogenic variant was first identified in compound heterozygous form in the Polish population (Bunge et al., 1997). The same codon is altered for another substitution where Arginine is replaced by Cysteine (c. 222G>A, p.Arg74Cys) (Bunge et al., 1997) resulting in a similar phenotype. This locus is likely to be the mutational hot spot for the disease. This residue is conserved among all mammalian sulfatases and stabilizes the active site of the enzyme (Bunge et al., 1997). The substitution might cause loss of ionic interactions formed by positively charged arginine; also, difference in sizes causes loss of interactions with other residues and domains.
In family C, a novel VWA3B variant (c.2951A>C; p.Glu984Ala, exon 22) was identified. The variant causes loss of interactions and loss of hydrogen bonds thus affecting the correct folding of protein (HOPE analysis https://www3.cmbi.umcn.nl/hope/).
Previously, four variants including three nonsense and one missense have been reported in VWA3B underlying neurological phenotypes including intellectual disability, cerebellar ataxia and autism spectrum disorder as per HGMD (Accessed on 20-May-2022). Affected individuals in the present study showed MPS III overlapping phenotypes including delay in achieving milestones, chronic diarrhea, aggressive behavior, speech delay, short stature, coarse facial features, joint contractures, hepatosplenomegaly, and intellectual disability. Variability in the phenotypes of previous reported cases and the patients in the present study might be due to different position of the variant, familial background, and/or the impact of SNPs in modifier genes.
In family D, the homozygous missense variant SGSH c. 364C>T (p. Gly122Arg) in exon 4 was associated with MPS III phenotype in both patients. This variant was first reported in 1997 in Dutch and Arab patients (Bunge et al., 1997). Insertion of a charged residue Arginine at amino acid position 122 of SGSH will cause distortion in the structure and might cause disruption of correct folding of protein. Clinical details and pathogenicity of the variant is described in Table 1 and Table 2, respectively. There were two affected individuals in the family, one sister and one brother born to a non-consanguineous couple, without any previous family history for the disease but all the successive marriages in previous generations are intra-cast marriages (Figure 1D).
In family E, a homozygous missense SGSH c. 258A>C (p. Asn86Lys) variant was identified. This variant is novel and has not previously been reported to cause Sanfilippo syndrome. The family had two affected males, born to a first-degree cousin couple (Figure 1E). One patient VI-1 died a month after sample collection at the age of 15 years. Asparagine at amino acid 86 is highly conserved in SGSH in different orthologues. The wild-type residue forms a hydrogen bond with methionine at position 88, the difference in size of wild type and mutated residues causes loss of interaction. The variation is in the catalytic domain that is important for the activity of the enzyme and interact with another domain which might affect the function of protein. As per ACMG guidelines, the variant was classified as a variant of uncertain significance (Table 2).
In family F, the identified pathogenic variant was SGSH c. 817G>A (p. Asp273Asn) in exon 7. This variant was reported in UK in 2000 (Beesley et al., 2000). The variant was found in homozygous state in both patients (IV-1, IV-2) and heterozygous in their parents. Both affected individuals were females and born to a first-degree cousin couple (Figure 1F). This variant is located in a CpG dinucleotide site which is the mutational hot spot of the gene (Beesley et al., 2000). As per HOPE analysis, the wild-type residue is negatively charged, and therefore, interaction with calcium ions is lost with this substitution, reducing the stability of the enzyme. The wild type residue Asp273 also forms hydrogen bonds with Asp31 and Asp32 and salt bridges with Arg74, Leu123, and Arg282, which might be lost due to substitution of Asp273 with a neutral Asparagine residue, disturbing the ionic interactions. As per ACMG guidelines the variant is classified as likely pathogenic (Table 2).
Functional lysosomes are important for autophagy to regulate the quality of cytoplasm by eliminating cellular macromolecular aggregates. If autophagy is disturbed due to compromised or absent lysosomal enzyme it may lead to inappropriate storage of material in different cells. This storage interferes with normal cell function and affects many organ systems including brain, viscera, bone and cartilage causing various diseases including lysosomal storage disorders (LSDs), neurodegenerative diseases and cancers (Saha et al., 2018; Scerra et al., 2022). Interestingly, in present study we identified missense disease-causing variants in all MPS-III diagnosed families, such variants could have little or no impact on the enzymatic activity of the mutant protein but may cause folding or tertiary structure alterations. Misfolded protein/s are retained in the endoplasmic reticulum for subsequent degradation thus causing enzyme deficiency and compromise in autophagy (Mohamed et al., 2017; Valencia et al., 2021). Therefore, neurological symptoms are one of the most common phenotypes observed in MPS III because neurons being post-mitotic cells could not dilute damaged cell organelles and protein aggregates (Valencia et al., 2021; Scerra et al., 2022). No therapeutic approach has been successful in reverting the symptoms of MPS III and neurological manifestations caused by it. Some of the common therapies tested include enzyme replacement therapy (ERT), substrate reduction therapy (SRT), pharmacological chaperon therapy (PCT) and hematopoietic stem cell transplant (HSCT) (Valstar et al., 2010; Beneto et al., 2020; Penon-Portmann et al., 2023). Some of the therapies applying on cell lines and animal models for treating MPS III include over expression of TFEB (Lotfi et al., 2018), master regulator of lysosomal biogenesis (Bajaj et al., 2019) and coenzyme Q10 (Matalonga et al., 2014).
Mucopolysaccharidoses IV
Eight families (A-H) were recruited on the basis of their MPS IV (Morquio syndrome type A) phenotypes. In six families (A, B, D, F, G, H) identification of GALNS mutations confirmed the diagnosis. In family E, a variant in the mucolipidosis-associated gene GNPTG segregated with the phenotype. No disease-causing variant could be identified in family C.
In family A, the patient had a novel homozygous GALNS splice site variant g.38845A>G (chr16:88884534T>C); c.1365–2A>G (intron 12). A splice site prediction tool varSEAK (https://varseak.bio/) predicted the identified splice site variant with class 5 (splicing effect). The identified variant is predicted to activate a cryptic site 34 nucleotide upstream of 3′splice site leading to the production of an abnormal protein.
Three families B, G and H shared a highly prevalent GALNS c.107T>G; p.Leu36Arg missense variant. Previously, two different variations (c. 107T>G; p. Leu36Arg; c. 107T>C, p.Leu36Pro) were identified on the same codon to cause Morquio syndrome type A in Asian-multiethnic and Mexican populations respectively (Tomatsu et al., 2005; Morrone et al., 2014). The variation (p.Leu36Arg) is in the catalytic domain, which can disturb the function of the enzyme. It may also hinder the correct protein folding due to the charged arginine residue leading to the loss of hydrophobic interactions. All the three families in the present study showed consanguinity and homozygosity of the variant in patients, while parents were heterozygous carriers (Figures 2B,G,H). Clinical examination demonstrated intra and interfamilial variability in the phenotypes of patients carrying the same variant (Table 3). The variability of phenotypes in this study might be due to different ethnicities of affected families. Additionally, family H showed dual molecular diagnosis as another homozygous missense novel variant in BTD gene, c. 1336G>C; p. Asp446His causing biotinidase deficiency (EC 3.5.1.12) segregated with the phenotype. This enzyme is involved in the recycling of biotin bound to protein by releasing biotin and lysine (Pispa, 1965; Wolf et al., 1985). The wild-type residue Asp at 446 forms hydrogen bonds with Gly423, Tyr456 and Gln458, and a salt bridge with Arg544, but because of the substitution, hydrogen bonding and ionic interaction will be lost.
In family D, there were three affected individuals, two females and a male born to first-degree cousin parents (Figure 2D). A homozygous GALNS c. 1259G>C; p. Pro420Arg missense variant segregated with disease. Previously, Morrone et al. (2014) identified the same variant to cause MPS IV in Asian-multiethnic population. Ullah et al. (2017) reported a consanguineous Pakistani family affected with MPS IV segregating same disease-causing variant, i.e., p. Pro420Arg where substitution of proline residue with arginine distorts the protein conformation (Ullah et al., 2017).
In family E, a novel homozygous GNPTG c.179G>A; p.Gly60Glu missense variant was identified. Variants in GNPTG cause mucolipidoses type III (ML III). This gene encodes the gamma subunit of enzyme N-acetylglucosamine-1-phosphotransferase (EC 2.7.8.17) responsible for catalyzing the first step in synthesis of a mannose 6-phosphate lysosomal recognition marker (Braulke et al., 2008; Qian et al., 2010). The clinical profile for ML III includes phenotypes like short stature, scoliosis and joint contractures (Tiede et al., 2005; Bargal et al., 2006), which were also present in the patients in the present study (Table 3). As the initial diagnosis in the present study was based on clinical phenotypes, and the two disorders (MPS and ML) have overlapping phenotypes, we could not rule out the possibility of dual molecular diagnosis. Next-generation sequencing techniques should be used for the correct diagnosis of these complex overlapping phenotypes. Glycine at amino acid 60 in the GNPTG protein is highly conserved, so its substitution is probably damaging to the protein. The mutant residue is negatively charged so its incorporation will lead ligands and other residues of same charge to be repelled.
In family F, there were two affected individuals, one living while the other died at the age 14. A novel homozygous splice acceptor site GALNS c.423-1G>A variant was found in the patient. The parents were first degree cousins (Figure 2F). The identified variant is predicted to activate a cryptic site 1 nucleotide downstream of 3′splice site leading to a frameshift mutation (varSEAK Online - Splice Site Prediction Version 2.1).
Therapies for MPS IV include enzyme replacement therapy which is currently working only for type A not for type B. Some therapies in experimental phases include substrate reduction therapy, hematopoietic stem cell transplant, gene therapy, anti-oxidant therapy, inhibiting protein aggregation, stope codon read through therapy and anti-inflammatory therapy (Akyol et al., 2019; Seker Yilmaz et al., 2021; Penon-Portmann et al., 2023). In a recent therapy, the delivery of recombinant human GALNS enzyme (rhGALNS) into fibroblasts was achieved by infusing it with a hydrogel, such as polyethylene glycol. This innovative approach ensures sustainable and prolonged release of the exogenous enzyme. However, it is important to consider that this treatment might be costly and require frequent administration (Jain et al., 2020).
From Pakistan, very little knowledge about the genetic basis of MPS IV has revealed a common variant p.Leu36Arg in 3/8 families sequenced during current study, and another variant p.Pro420Arg reported formerly by Ullah et al., 2017, also found in family D with three affected siblings.
In Pakistan, the burden of recessively inherited lethal genetic disorders like Sanfilippo and Morquio syndromes is higher due to custom of intra-familial marriages, but the precise diagnosis is lacking due to unavailability of state-of-the-art diagnosis facilities (Hussain and Bittles, 1998; Shahid et al., 2020; Shahzadi et al., 2020; Gul et al., 2023). However, to ensure the health of children suffering from such life-threatening disorders, timely diagnosis and treatment is essential. Patients without proper medical care are affected by multiple medical conditions and progressive tissue degeneration. Therefore, early, and correct diagnosis of such disorders should be performed using targeted gene sequencing and/or WGS. Furthermore, molecular genetic diagnosis will help to provide premarital carrier diagnosis, genetic counselling and to devise prenatal screening tests to address this deficit in the foreseeable future.
Strength of Study: This study details extensive work from a highly consanguineous population of Pakistan, showing the need of genetic exploration for more detailed analysis related to lethal genetic disorders like Sanfilippo and Morquio syndromes.
Limitation of Study: The initial diagnosis of the patients included in this study, was performed based on clinical and radiographic examinations; thus, misdiagnosis of the patients could not be ruled out due to overlapping phenotypes of MPS, VWA3B-related phenotypes and ML. Furthermore, the effect of the identified variants on protein structure and function is based on in silico tools, and future functional studies are needed to validate the prediction/s.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statement
The studies involving humans were approved by the Bioethical Review board, Quaid-i-Azam University, Islamabad, Pakistan (BEC-FBS-QAU2019-198) and Shaheed Zulfiqar Ali Bhutto Medical University, Islamabad, Pakistan. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
RG: Conceptualization, Formal Analysis, Methodology, Writing–original draft. SF: Conceptualization, Supervision, Writing–review and editing. MS: Data curation, Writing–review and editing. AU: Conceptualization, Formal Analysis, Methodology, Writing–original draft, Writing–review and editing. EP: Formal Analysis, Writing–review and editing. AT: Formal Analysis, Project administration, Writing–review and editing. AG: Conceptualization, Writing–original draft, Writing–review and editing. MH: Conceptualization, Investigation, Supervision, Writing–review and editing. MT: Investigation, Software, Visualization, Writing–review and editing. MS: Investigation, Methodology, Writing–review and editing. KA: Supervision, Writing–review and editing. TH: Conceptualization, Funding acquisition, Supervision, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded by Center for Basic Metabolic Research (CBMR) Novo Nordisk Foundation (NNF) through the Challenge Program (NNF18OC0033950) and Higher Education Commission, Islamabad, Pakistan under International Research Support Initiative Program (IRSIP) (1-8/HEC/HRD/2020/10579).
Acknowledgments
We would like to thank all participating families, staff and colleagues who helped us during this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1254909/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Three-dimensional (3D) of wild-type and mutants of BTD, SGSH, VWA3B, GNTGP. The zoom-in part represents the mutated residue of each protein. Mutated proteins show a significant change in their structure.
SUPPLEMENTARY FIGURE S2 | Evolutionary conservation of mutated residue of BTD, SGSH, VWA3B, and GNTGP. The red boxes represent wild-type amino acids that might be altered by pathogenic mutations of high risk. The conservation scale has a score range of 1–9, with conserved sites receiving ratings in the range of 7–9. The tool illustrated the properties of residue that is either exposed (e), buried (b), highly conserved and exposed (functional, ,(F) or highly conserved and buried (b).
References
Akyol, M. U., Alden, T. D., Amartino, H., Ashworth, J., Belani, K., Berger, K. I., et al. (2019). Recommendations for the management of MPS IVA: systematic evidence-and consensus-based guidance. Orphanet J. rare Dis. 14 (1), 137–225. doi:10.1186/s13023-019-1074-9
Bajaj, L., Lotfi, P., Pal, R., Ronza, A. D., Sharma, J., and Sardiello, M. (2019). Lysosome biogenesis in health and disease. J. Neurochem. 148 (5), 573–589. doi:10.1111/jnc.14564
Bargal, R., Zeigler, M., Abu-Libdeh, B., Zuri, V., Mandel, H., Neriah, Z. B., et al. (2006). When mucolipidosis III meets mucolipidosis II: GNPTA gene mutations in 24 patients. Mol. Genet. metabolism 88 (4), 359–363. doi:10.1016/j.ymgme.2006.03.003
Barstow, C., and Rerucha, C. (2015). Evaluation of short and tall stature in children. Am. Fam. Physician 92 (1), 43–50.
Beesley, C. E., Burke, D., Jackson, M., Vellodi, A., Winchester, B. G., and Young, E. P. (2003). Sanfilippo syndrome type D: identification of the first mutation in the nacetylglucosamine-6-sulphatase gene. J. Med. Genet. 40 (3), 192–194. doi:10.1136/jmg.40.3.192
Beesley, C. E., Young, E. P., Vellodi, A., and Winchester, B. G. (2000). Mutational analysis of sanfilippo syndrome type A (MPS IIIA): identification of 13 novel mutations. J. Med. Genet. 37 (9), 704–707. doi:10.1136/jmg.37.9.704
Benetó, N., Vilageliu, L., Grinberg, D., and Canals, I. (2020). Sanfilippo syndrome: molecular basis, disease models and therapeutic approaches. Int. J. Mol. Sci. 21 (21), 7819. doi:10.3390/ijms21217819
Besley, G. T. N., and Wraith, J. E. (1997). Lysosomal disorders. Curr. Paediatr. 7 (2), 128–134. doi:10.1016/s0957-5839(97)80195-9
Braulke, T., Pohl, S., and Storch, S. (2008). Molecular analysis of the GlcNac-1-phosphotransferase. J. Inherit. metabolic Dis. 31 (2), 253–257. doi:10.1007/s10545-008-0862-5
Bunge, S., Ince, H., Steglich, C., Kleijer, W. J., Beck, M., Zaremba, J., et al. (1997). Identification of 16 sulfamidase gene mutations including the common R74C in patients with mucopolysaccharidosis type IIIA (Sanfilippo A). Hum. Mutat. 10 (6), 479–485. doi:10.1002/(SICI)1098-1004(1997)10:6<479:AID-HUMU10>3.0.CO;2-X
Celik, B., Tomatsu, S. C., Tomatsu, S., and Khan, S. A. (2021). Epidemiology of mucopolysaccharidoses update. Diagnostics 11 (2), 273. doi:10.3390/diagnostics11020273
Chien, Y. H., Lee, N. C., Chen, P. W., Yeh, H. Y., Gelb, M. H., Chiu, P. C., et al. (2020). Newborn screening for morquio disease and other lysosomal storage diseases: results from the 8-plex assay for 70,000 newborns. Orphanet J. rare Dis. 15 (1), 38–47. doi:10.1186/s13023-020-1322-z
Chih-Kuang, C., Shuan-Pei, L., Shyue-Jye, L., and &Tuen-Jen, W. (2002). MPS screening methods, the berry spot and acid turbidity tests, cause a high incidence of false-negative results in sanfilippo and morquio syndromes. J. Clin. laboratory analysis 16 (5), 253–258. doi:10.1002/jcla.10051
Davidson, B. A., Hassan, S., Garcia, E. J., Tayebi, N., and Sidransky, E. (2018). Exploring genetic modifiers of gaucher disease: the next horizon. Hum. Mutat. 39 (12), 1739–1751. doi:10.1002/humu.23611
De Pasquale, V., and Pavone, L. M. (2019). Heparan sulfate proteoglycans: the sweet side of development turns sour in mucopolysaccharidoses. Biochimica Biophysica Acta (BBA)-Molecular Basis Dis. 1865 (11), 165539. doi:10.1016/j.bbadis.2019.165539
Fedele, A. O. (2015). Sanfilippo syndrome: causes, consequences, and treatments. Appl. Clin. Genet. 8, 269–281. doi:10.2147/TACG.S57672
Feldhammer, M., Durand, S., Mrázová, L., Boucher, R. M., Laframboise, R., Steinfeld, R., et al. (2009). Sanfilippo syndrome type C: mutation spectrum in the heparan sulfate acetyl-CoA: alpha-glucosaminide N-acetyltransferase (HGSNAT) gene. Hum. Mutat. 30 (6), 918–925. doi:10.1002/humu.20986
Feldhammer, M., Durand, S., Mrazova, L., van Diggelen, O. P., Hrebicek, M., Kmoch, S., et al. (2009). a. 54. Sanfilippo syndrome type C: novel mutations in the HGSNAT gene. Mol. Genet. Metabolism 96 (2), S24S24.
Gul, R., Firasat, S., Schubert, M., Ullah, A., Peña, E., Thuesen, A. C., et al. (2023). Identifying the genetic causes of phenotypically diagnosed Pakistani mucopolysaccharidoses patients by whole genome sequencing. Front. Genet. 14, 1128850. doi:10.3389/fgene.2023.1128850
Hendriksz, C. J., Harmatz, P., Beck, M., Jones, S., Wood, T., Lachman, R., et al. (2013). Review of clinical presentation and diagnosis of mucopolysaccharidosis IVA. Mol. Genet. metabolism 110 (1-2), 54–64. doi:10.1016/j.ymgme.2013.04.002
Hussain, R., and Bittles, A. H. (1998). The prevalence and demographic characteristics of consanguineous marriages in Pakistan. J. Biosoc. Sci. 30 (2), 261–275. doi:10.1017/s0021932098002612
Jain, E., Flanagan, M., Sheth, S., Patel, S., Gan, Q., Patel, B., et al. (2020). Biodegradable polyethylene glycol hydrogels for sustained release and enhancedstability of rhGALNS enzyme. Drug Deliv. Transl. Res. 10 (5), 1341–1352. doi:10.1007/s13346-020-00714-7
Khan, S. A., Peracha, H., Ballhausen, D., Wiesbauer, A., Rohrbach, M., Gautschi, M., et al. (2017). Epidemiology of mucopolysaccharidoses. Mol. Genet. metabolism 121 (3), 227–240. doi:10.1016/j.ymgme.2017.05.016
Lin, H. Y., Lee, C. L., Chang, C. Y., Chiu, P. C., Chien, Y. H., Niu, D. M., et al. (2020). Survival and diagnostic age of 175 Taiwanese patients with mucopolysaccharidoses (1985–2019). Orphanet J. Rare Dis. 15 (1), 314–411. doi:10.1186/s13023-020-01598-z
Lotfi, P., Tse, D. Y., Di Ronza, A., Seymour, M. L., Martano, G., Cooper, J. D., et al. (2018). Trehalose reduces retinal degeneration, neuroinflammation and storage burden caused by a lysosomal hydrolase deficiency. Autophagy 14 (8), 1419–1434. doi:10.1080/15548627.2018.1474313
Matalonga, L., Arias, A., Coll, M. J., Garcia-Villoria, J., Gort, L., and Ribes, A. (2014). Treatment effect of coenzyme Q 10 and an antioxidant cocktail in fibroblasts of patients with Sanfilippo disease. J. Inherit. metabolic Dis. 37 (3), 439–446. doi:10.1007/s10545-013-9668-1
Martell, L., Lau, K., Mei, M., Burnett, V., Decker, C., and Foehr, E. D. (2011). Biomarker analysis of morquio syndrome: identification of disease state and drug responsive markers. Orphanet J. Rare Dis. 6 (1), 84–10. doi:10.1186/1750-1172-6-84
Mohamed, F. E., Al-Gazali, L., Al-Jasmi, F., and Ali, B. R. (2017). Pharmaceutical chaperones and proteostasis regulators in the therapy of lysosomal storage disorders: current perspective and future promises. Front. Pharmacol. 8, 448. doi:10.3389/fphar.2017.00448
Morquio, L. (1929). Sur une forme de dystrophie osseuse familiale. Bull. Soc. Pediatr. Paris 27, 145–152.
Morrone, A., Tylee, K. L., Al-Sayed, M., Brusius-Facchin, A. C., Caciotti, A., Church, H. J., et al. (2014). Molecular testing of 163 patients with Morquio A (Mucopolysaccharidosis IVA) identifies 39 novel GALNS mutations. Mol. Genet. metabolism 112 (2), 160–170. doi:10.1016/j.ymgme.2014.03.004
Nelson, J., Crowhurst, J., Carey, B., and Greed, L. (2003). Incidence of the mucopolysaccharidoses in western Australia. Am. J. Med. Genet. Part A 123 (3), 310–313. doi:10.1002/ajmg.a.20314
Okada, S., and O'Brien, J. S. (1968). Generalized gangliosidosis: beta-galactosidase deficiency. Science 160 (3831), 1002–1004. doi:10.1126/science.160.3831.1002
Ozkinay, F., Emecen, D. A., Kose, M., Isik, E., Bozaci, A. E., Canda, E., et al. (2021). Clinical and genetic features of 13 patients with mucopolysaccarhidosis type IIIB: description of two novel NAGLU gene mutations. Mol. Genet. Metabolism Rep. 27, 100732. doi:10.1016/j.ymgmr.2021.100732
Penon-Portmann, M., Blair, D. R., and Harmatz, P. (2023). Current and new therapies for mucopolysaccharidoses. Pediatr. Neonatol. 64, S10–S17. doi:10.1016/j.pedneo.2022.10.001
Peracha, H., Sawamoto, K., Averill, L., Kecskemethy, H., Theroux, M., Thacker, M., et al. (2018). Molecular genetics and metabolism, special edition: diagnosis, diagnosis and prognosis of mucopolysaccharidosis IVA. Mol. Genet. metabolism 125 (1-2), 18–37. doi:10.1016/j.ymgme.2018.05.004
Piraud, M., Boyer, S., Mathieu, M., and Maire, I. (1993). Diagnosis of mucopolysaccharidoses in a clinically selected population by urinary glycosaminoglycan analysis: a study of 2,000 urine samples. Clin. Chim. acta 221 (1-2), 171–181. doi:10.1016/0009-8981(93)90031-x
Qian, Y., Lee, I., Lee, W. S., Qian, M., Kudo, M., Canfield, W. M., et al. (2010). Functions of the alpha, beta, and gamma subunits of UDP-GlcNAc:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase. J. Biol. Chem. 285 (5), 3360–3370. doi:10.1074/jbc.M109.068650
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Saha, S., Panigrahi, D. P., Patil, S., and Bhutia, S. K. (2018). Autophagy in health and disease: A comprehensive review. Biomed. Pharmacother. 104, 485–495. doi:10.1016/j.biopha.2018.05.007
Sanfilippo, S. J., Podosin, R., Langer, L., and Good, R. A. (1963). Mental retardation associated with acid mucopolysacchariduria (heparitin sulfate type). J. Pediatr. 63, 837–838. doi:10.1016/s0022-3476(63)80279-6
Scerra, G., De Pasquale, V., Scarcella, M., Caporaso, M. G., Pavone, L. M., and D'Agostino, M. (2022). Lysosomal positioning diseases: beyond substrate storage. Open Biol. 12 (10), 220155. doi:10.1098/rsob.220155
Seker Yilmaz, B., Davison, J., Jones, S. A., and Baruteau, J. (2021). Novel therapies for mucopolysaccharidosis type III. J. Inherit. Metabolic Dis. 44 (1), 129–147. doi:10.1002/jimd.12316
Shahid, M., Firasat, S., Satti, H. S., Satti, T. M., Ghafoor, T., Sharif, I., et al. (2020). Screening of the FANCA gene mutational hotspots in the Pakistani fanconi anemia patients revealed 19 sequence variations. Congenit. Anomalies 60 (1), 32–39. doi:10.1111/cga.12331
Shahzadi, M., Firasat, S., Kaul, H., Afshan, K., Afzal, R., and Naz, S. (2020). Genetic mapping of autosomal recessive microspherophakia to chromosome 14q24. 3 in a consanguineous Pakistani family and screening of exon 36 of LTBP2 gene. JPMA. J. Pak. Med. Assoc. 70 (3), 515–518. doi:10.5455/JPMA.302440
Shapiro, E. G., Jones, S. A., and Escolar, M. L. (2017). Developmental and behavioral aspects of mucopolysaccharidoses with brain manifestations—Neurological signs and symptoms. Mol. Genet. metabolism 122, 1–7. doi:10.1016/j.ymgme.2017.08.009
Sheth, J., Nair, A., and Jee, B. (2022). Lysosomal storage disorders: from biology to the clinic with reference to India. Lancet Regional Health-Southeast Asia 2022.
Tiede, S., Muschol, N., Reutter, G., Cantz, M., Ullrich, K., and &Braulke, T. (2005). Missense mutations in N-acetylglucosamine-1-phosphotransferase alpha/beta subunit gene in a patient with mucolipidosis III and a mild clinical phenotype. Am. J. Med. Genet. Part A 137 (3), 235–240. doi:10.1002/ajmg.a.30868
Tomatsu, S., Montaño, A. M., Nishioka, T., Gutierrez, M. A., Peña, O. M., Trandafirescu, G. G., et al. (2005). Mutation and polymorphism spectrum of the GALNS gene in mucopolysaccharidosis IVA (Morquio A). Hum. Mutat. 26 (6), 500–512. doi:10.1002/humu.20257
Tomatsu, S., Nishioka, T., Montaño, A. M., Gutierrez, M. A., Pena, O. S., Orii, K. O., et al. (2004). Mucopolysaccharidosis IVA: identification of mutations and methylation study in GALNS gene. J. Med. Genet. 41 (7), e98e98. doi:10.1136/jmg.2003.018010
Ullah, I., Nasir, A., Mehmood, S., Ahmed, S., Ullah, M. I., Ullah, A., et al. (2017). Identification and in silico analysis of GALNS mutations causing Morquio A syndrome in eight consanguineous families. Turkish J. Biol. 41 (3), 458–468. doi:10.3906/biy-1607-81
Valencia, M., Kim, S. R., Jang, Y., and Lee, S. H. (2021). Neuronal autophagy: characteristic features and roles in neuronal pathophysiology. Biomol. Ther. 29 (6), 605–614. doi:10.4062/biomolther.2021.012
Valstar, M. J., Neijs, S., Bruggenwirth, H. T., Olmer, R., Ruijter, G. J., Wevers, R. A., et al. (2010). Mucopolysaccharidosis type IIIA: clinical spectrum and genotype-phenotype correlations. Ann. neurology 68 (6), 876–887. doi:10.1002/ana.22092
Valstar, M. J., Ruijter, G. J. G., Van Diggelen, O. P., Poorthuis, B. J., and Wijburg, F. A. (2008). Sanfilippo syndrome: a mini-review. J. Inherit. metabolic Dis. 31 (2), 240–252. doi:10.1007/s10545-008-0838-5
Wolf, B., Grier, R. E., SecorMcVoy, J. R., and Heard, G. S. (1985). Biotinidase deficiency: a novel vitamin recycling defect. J. Inherit. metabolic Dis. 8 (1), 53–58. doi:10.1007/BF01800660
World Medical Association, (2013). World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. Jama 310 (20), 2191–2194. doi:10.1001/jama.2013.281053
Zhao, H. G., Li, H. H., Bach, G., Schmidtchen, A., and Neufeld, E. F. (1996). The molecular basis of Sanfilippo syndrome type B. Proc. Natl. Acad. Sci. U. S. A. 93, 6101–6105. doi:10.1073/pnas.93.12.6101
Zhao, H. G., Aronovich, E. L., and Whitley, C. B. (1998). Genotype-phenotype correspondence in Sanfilippo syndrome type B. Am. J. Hum. Genet. 62 (1), 53–63. doi:10.1086/301682
Keywords: Sanfilippo syndrome, Morquio syndrome, Pakistani families, whole genome sequencing, VWA3B
Citation: Gul R, Firasat S, Schubert M, Ullah A, Peña E, Thuesen ACB, Gjesing AP, Hussain M, Tufail M, Saqib M, Afshan K and Hansen T (2023) Identification of genetic variants associated with a wide spectrum of phenotypes clinically diagnosed as Sanfilippo and Morquio syndromes using whole genome sequencing. Front. Genet. 14:1254909. doi: 10.3389/fgene.2023.1254909
Received: 12 July 2023; Accepted: 17 August 2023;
Published: 11 September 2023.
Edited by:
Ammar Husami, Cincinnati Children’s Hospital Medical Center, United StatesReviewed by:
Abdul Nasir, Second Affiliated Hospital of Zhengzhou University, ChinaElsayed Abdelkreem, Sohag University, Egypt
Copyright © 2023 Gul, Firasat, Schubert, Ullah, Peña, Thuesen, Gjesing, Hussain, Tufail, Saqib, Afshan and Hansen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sabika Firasat, c2FiaWthLmZpcmFzYXRAcWF1LmVkdS5waw==; Torben Hansen, dG9yYmVuLmhhbnNlbkBTdW5kLmt1LmRr