 Huaming Wang
Huaming Wang Xi Lin2†
Xi Lin2† Guorong Lyu
Guorong Lyu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 06 September 2023
Sec. RNA
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1254829
This article is part of the Research Topic Expression and Variation in Non-coding RNAs for Diagnosis, Prevention and Cure - Volume 2 View all 4 articles
Background: Maternal body fluids contain abundant cell-free fetal RNAs which have the potential to serve as indicators of fetal development and pathophysiological conditions. In this context, this study aimed to explore the potential diagnostic value of maternal circulating long non-coding RNAs (lncRNAs) in ventricular septal defect (VSD).
Methods: The potential of lncRNAs as non-invasive prenatal biomarkers for VSD was evaluated using quantitative polymerase chain reaction (qPCR) and receiver operating characteristic (ROC) curve analysis. The biological processes and regulatory network of these lncRNAs were elucidated through bioinformatics analysis.
Results: Three lncRNAs (LINC00598, LINC01551, and GATA3-AS1) were found to be consistent in both maternal plasma and amniotic fluid. These lncRNAs exhibited strong diagnostic performance for VSD, with AUC values of 0.852, 0.957, and 0.864, respectively. The bioinformatics analysis revealed the involvement of these lncRNAs in heart morphogenesis, actin cytoskeleton organization, cell cycle regulation, and protein binding through a competitive endogenous RNA (ceRNA) network at the post-transcriptional level.
Conclusion: The cell-free lncRNAs present in the amniotic fluid have the potential to be released into the maternal circulation, making them promising candidates for investigating epigenetic regulation in VSD.
The development of the fetal heart is a complex and dynamic process; even a slight perturbation can be catastrophic and lead to a range of congenital heart defects (CHDs). Ventricular septal defect, one of the most common forms of CHD, has an occurrence rate of 4.3 in 1,000 births (Liu et al., 2019). With surgical intervention, infants with VSD have satisfactory long-term survival. However, they are still confronted with the risk of various short- and long-term complications (Ren et al., 2021). Therefore, novel insights into the underlying triggers of VSD are urgently needed to develop better diagnostic and therapeutic strategies for VSD. According to epidemiological investigations, abnormal heart development is associated with the accumulation of numerous molecular alterations (Deita et al., 2021). Although aberrant genetic alterations, such as aneuploidy, mutations, insertions, and deletions, are considered to be closely related to CHDs, they cannot be accounted for entirely by genetic inheritance (Gutierrez et al., 2022). In recent years, numerous studies have shown that epigenetic modifications play a significant role in regulating gene expression, which are also critical mediators for driving and maintaining heart development (Hou et al., 2020).
There are three main types of epigenetic modifications: non-coding RNAs (ncRNAs), histone modifications, and DNA methylation. Kan et al. (2022) revealed that long non-coding RNAs function as epigenetic regulators implicated in fetal heart gene expression and pathological processes of VSD. Several studies have found that differential expression of lncRNAs in CHD patients will participate in heart development by affecting cardiomyocyte proliferation. For instance, lncRNA SAP30-2:1 is downregulated in CHDs and regulates cell proliferation by targeting HAND2 (Ma et al., 2021); lncRNA-HA117 is upregulated in the myocardial tissue of Tetralogy of Fallot (ToF) children and negatively correlated with the prognosis (Wang et al., 2018). At the pre-transcriptional level, lncRNA can act as an enhancer or alter DNA methylation to inhibit cardiomyocyte differentiation and maintain cardiac stem cell pluripotency (Devaux et al., 2015; Zhang et al., 2019). At the post-transcriptional level, lncRNA-associated competitive endogenous RNA (ceRNA) is an essential epigenetic patterning of the genome. In ceRNA patterning, lncRNA binds to miRNAs to regulate the expression of downstream messenger RNA, thus causing CHDs to occur. For example, lncRNA FGD5-AS1 plays a repressive role in heart development through binding with has-miR-421 to regulate the expression of SMAD4 (Zhang et al., 2021). Given the adverse effects of VSD and the crucial role of lncRNAs in cardiac development, comprehending the regulatory mechanisms of these lncRNAs is central to the diagnosis and treatment of VSD.
Maternal body fluids are abundant in cell-free fetal DNAs and RNAs which can reflect fetal development and pathophysiological status (Wagner et al., 2019). Biomarkers from amniotic fluid and maternal plasma are considered powerful diagnostic tools for assessing fetal health and disease. Studies have shown that amniotic fluid ncRNA profiles play a significant role in clinical prediction in fetal infection and twin–twin transfusion syndrome recipients (Willner et al., 2021; Yoshikawa et al., 2021). In our previous publication, we found that some amniotic fluid lncRNAs (LINC00598, LINC01551, PWRN1, and GATA3-AS1) were differently expressed in VSD fetuses, which may be potential biomarkers for VSD fetuses (Wang et al., 2023). However, it remains to be investigated whether the expression of lncRNAs associated with VSD is consistent in maternal plasma and amniotic fluid. In addition, amniotic fluid biomarkers are not clinically appropriate as routine methods due to the complications of amniocentesis. Recently, research has shown that maternal circulating microRNAs can be used as non-invasive biomarkers for the prenatal diagnosis of CHDs (Gu et al., 2019). This motivates the possibility that circulating lncRNAs have the same potential.
Therefore, in the present study, we used qPCR to verify the expression of LINC00598, LINC01551, PWRN1, and GATA3-AS1 in maternal plasma. Subsequently, the integrated bioinformatics methods, including coding–non-coding gene co-expression network (CNC), weighted gene co-expression network analysis (WGCNA), and ceRNA regulation network, were used to predict the hypothetical functions and mechanisms of these lncRNAs in VSD.
All pregnant women were recruited from January 2021 to February 2023 at The Second Affiliated Hospital of Fujian Medical University. All pregnant women were assessed for prenatal ultrasound and echocardiography. Pregnant women who carried a VSD fetus were categorized into the VSD group, while those who carried a normal fetus were grouped into the control group. Exclusion standards included multiple pregnancies, pregnancies with placental anomalies, and fetuses with extracardiac anomalies. All fetuses were confirmed by postpartum ultrasound. Both groups of pregnant women did not report CHD risk factors, such as alcoholism, smoking, diabetes, hypertension, and pharmaceutical history. All pregnant women did not suffer from pregnancy complications, such as gestational diabetes mellitus and preeclampsia. This study was approved by the Ethics Committee of The Second Affiliated Hospital of Fujian Medical University (No. 2021-73) and conducted in accordance with the principles of the World Medical Association Declaration of Helsinki.
Maternal blood samples were collected from all pregnant women on an empty stomach. The fasting venous blood (5 mL) was collected from individuals with EDTA tubes. Blood samples were centrifuged at 3,000 g for 10 min. The isolated plasma was centrifuged at 13,500 g for 10 min to remove cell fragments, and then the supernatant samples were stored at −80°C until RNA extraction.
According to the manufacturer’s protocol, total RNA was extracted from plasma supernatant using TRIzol LS reagent (Invitrogen, Carlsbad, CA). Subsequently, NanoDrop ND-1000 was used to measure the RNA quantities and qualities. qPCR was performed according to PrimeScript™ RT Reagent Kit with gDNA Eraser (Takara, Dalian, China) and TB Green® Premix Ex Taq™ II (Takara, Dalian, China). Briefly, 1°μg of total RNA was used to synthesize cDNA. Then, the cDNA was amplified and quantified under the following cycling parameters: 95°C (30°s) at the first step; 95°C (5°s) and 60°C (34°s) for 40 cycles at the second step; and 95°C (15°s), 60°C (60°s), and 95°C (15°s) at the third step. The 2−ΔΔCT method was performed to calculate the gene expression levels. The sequences of the primers used are shown in Supplementary Table S1. The relative expression levels of lncRNAs were normalized to the expression levels of GAPDH and measured in triplicate for each sample.
It is known that lncRNAs can function by regulating mRNA expression. To further reveal the function of the aforementioned lncRNAs, co-expression analysis was used to unearth mRNAs associated with these lncRNAs. First, the mRNA expression data were obtained from our previous research (GEO: GSE204935), which contained 11 VSD fetuses and 11 normal fetuses (Wang et al., 2023). Second, based on the previously described co-expression analysis of lncRNAs and mRNAs, we constructed a CNC network by calculating Pearson’s correlation coefficient. Genes with Pearson’s correlation coefficients above 0.90 and p-values below 0.05 were selected for the network. These results were visualized using the Cytoscape (v2.8.2) program.
In the cell, active transcription and translation of mRNAs directly control the expression of localized proteins and subsequent signal regulation. To identify the potential role of lncRNAs, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were performed to reveal the key functional classifications, relationship networks, and disease information for mRNAs in the CNC network. The GO and KEGG categories were classified by Fisher’s exact test. The threshold of significance was defined by the false discovery rate (FDR) and p-value.
WGCNA, a systems biology approach, can be used to identify genetic modules of highly correlated genes and related modules to external sample traits. In order to reveal the VSD-related eigengene network, the WGCNA package of R was used to construct gene association patterns between different samples according to mRNA expression data (Langfelder et al., 2008). First, the top 75% of genes were screened according to median absolute deviation (MAD). Second, the weighted co-expression relationship among all genes was evaluated by calculating Pearson’s coefficient. Third, to ensure the construction of a scale-free network, we found a soft-threshold power β using the “pickSoftThreshold” function. Fourth, a topological overlap matrix separates all genes into multiple gene modules to represent network connectivity. The genes contained in each gene module are well correlated. The dynamic branch cut method was used to conduct hierarchical clustering dendrograms, and then each module was represented by a designated color (Langfelder and Horvath, 2008). Finally, key modules significantly associated with clinical traits were further identified by calculating module–trait relationships and gene significance. To further identify candidate genes of VSD, genes whose gene significance (GS) > 0.3 and module membership (MM) > 0.8 were defined as interesting genes in the key module.
One of the potential mechanisms of lncRNAs is working as miRNA sponges to affect mRNA’s function at the post-transcriptional level. Therefore, we constructed a ceRNA network to explore the potential role of lncRNAs in VSD. First, the positive lncRNA–mRNA pairs were identified based on the CNC network. Second, the miRcode and TargetScan databases were used to predict miRNA targets on lncRNAs and mRNAs, respectively. Finally, the intersection of lncRNA-predicted miRNAs and mRNA-predicted miRNAs was used to construct the lncRNA–miRNA–mRNA interaction network. The Cytoscape program was used to visualize the ceRNA network.
All data were analyzed using R software (4.1.3), SPSS (21.0), Stata (15.1), and GraphPad Prism (7.0). The quantitative data are expressed as the average ± standard deviation. The statistical analysis of age, gestational weeks, and gene expression levels was performed using the Student’s t-test. The receiver operating characteristic curve was constructed to evaluate the diagnostic value of lncRNAs. The significance of GO or KEGG enrichment was evaluated by Fisher’s exact test. p < 0.05 was considered to indicate a statistically significant difference.
In total, 22 pregnant women with VSD fetuses and 16 pregnant women with normal fetuses met the criteria and were included in the study. There were no differences in maternal age, the number of pregnancies, gestational age, and body mass index (BMI) between the two groups (Table 1; Supplementary Table S2). Maternal blood samples were collected from all pregnant women on an empty stomach.
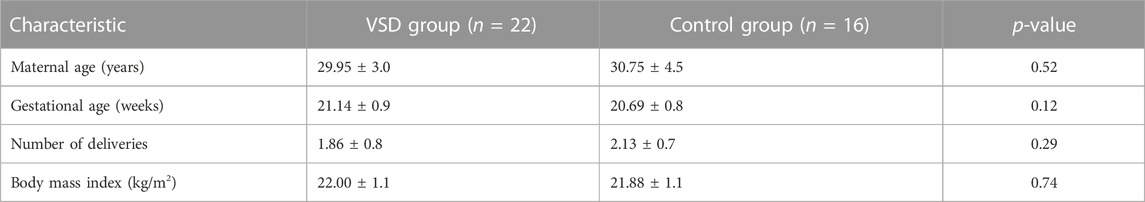
TABLE 1. Clinical characteristics of the VSD and control groups.
To explore whether the expression of VSD-related genes is consistent in maternal plasma and amniotic fluid, four lncRNAs (PWRN1, GATA3-AS1, LINC00598, and LINC01551) were selected for qPCR. The qPCR results showed that three lncRNAs were consistent with amniotic fluid, including LINC00598, LINC01551, and GATA3-AS1. Subsequently, the ROC curve was drawn to evaluate the potential of these lncRNAs as biomarkers for VSD. The result showed that these lncRNAs have a good diagnostic performance on VSD with AUC values of 0.852, 0.957, and 0.864, respectively (Figure 1; Table 2). Furthermore, we assign an equal weight to each of the multiple lncRNAs to construct a combined model. The results show that the combined model with three lncRNAs has an AUC of 0.994, which is better than the combined model with only two lncRNAs. (Supplementary Figure S1). The RNA quantification and quality assurance are shown in Supplementary Table S3. In addition, the raw data of qPCR are shown in Supplementary Tables S4–S6.
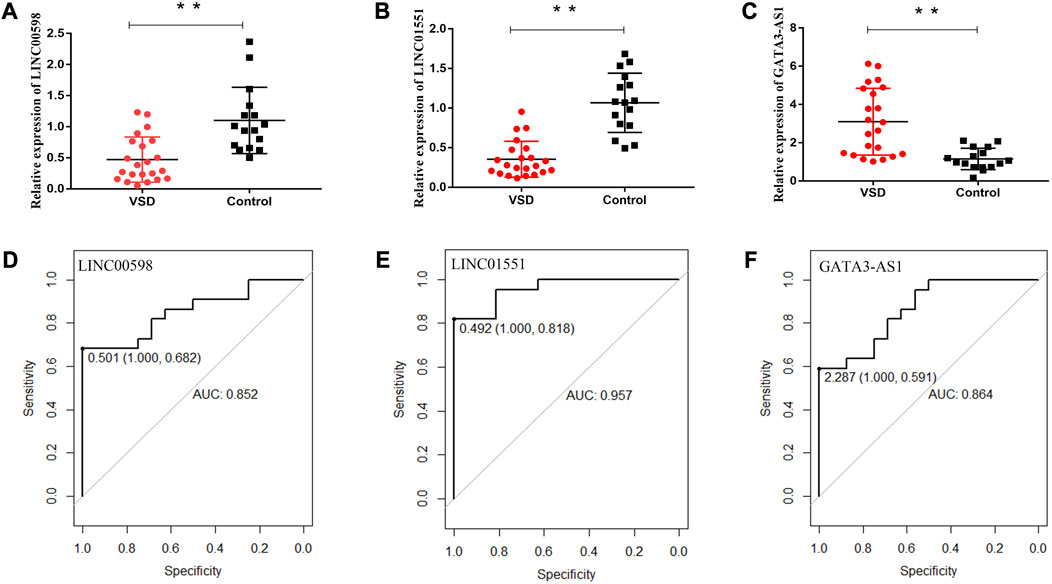
FIGURE 1. Potential of relevant RNAs as non-invasive prenatal biomarkers for VSD. Relative expression levels of lncRNAs in maternal plasma and the receiver operating characteristic (ROC) curves of LINC00598 (A, D), LINC01551 (B, E), and GATA3-AS1 (C, F). * indicates p < 0.05, and ** indicates p < 0.01. AUC, area under the ROC curve.
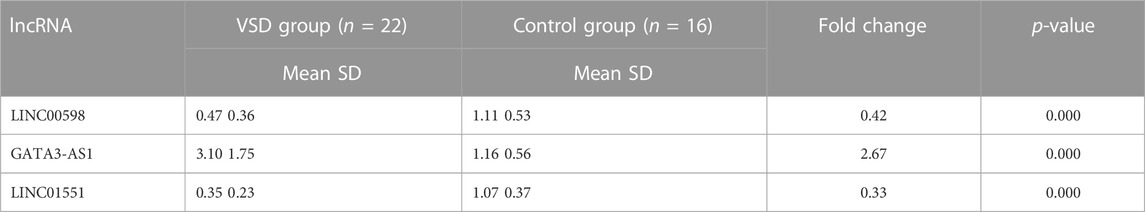
TABLE 2. Differential expression of lncRNAs in maternal plasma.
Among the co-expression, three lncRNAs and 125 mRNAs comprise the CNC network, which includes 178 network pairs of co-expressing lncRNAs and mRNAs. As shown in Figure 2, one hub lncRNA can be correlated with many mRNAs, and most of these relational pairs are presented as positive correlations.
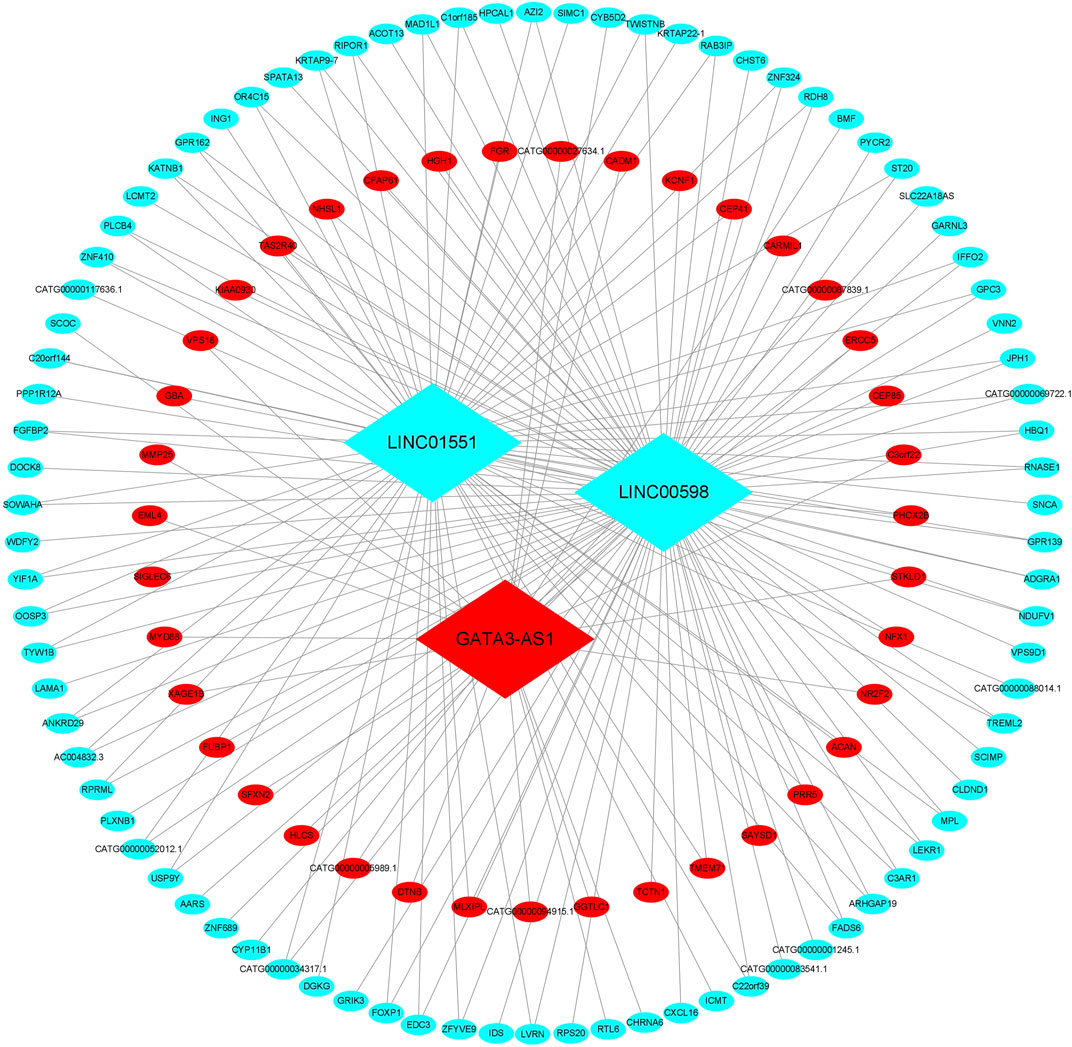
FIGURE 2. CNC network of the three lncRNAs. The ellipse represents mRNAs, and the diamond represents lncRNAs. Red and green colors represent up- and downregulated RNAs, respectively. CNC, coding–non-coding gene co-expression.
A total of 279 GO terms, including biological processes, molecular functions, and cellular components, were significantly enriched. The top ten terms of the GO results are shown in Figure 3A. Briefly, most genes were enriched in cardiac morphogenesis, cell proliferation, cell cycle, cell–cell junction, and protein binding. The KEGG pathway analysis showed that the mRNAs were enriched in the Hedgehog signaling (Shh) pathway, apoptosis, cardiovascular diseases, and regulation of the actin cytoskeleton. The top ten enriched KEGG pathways are shown in Figure 3B.
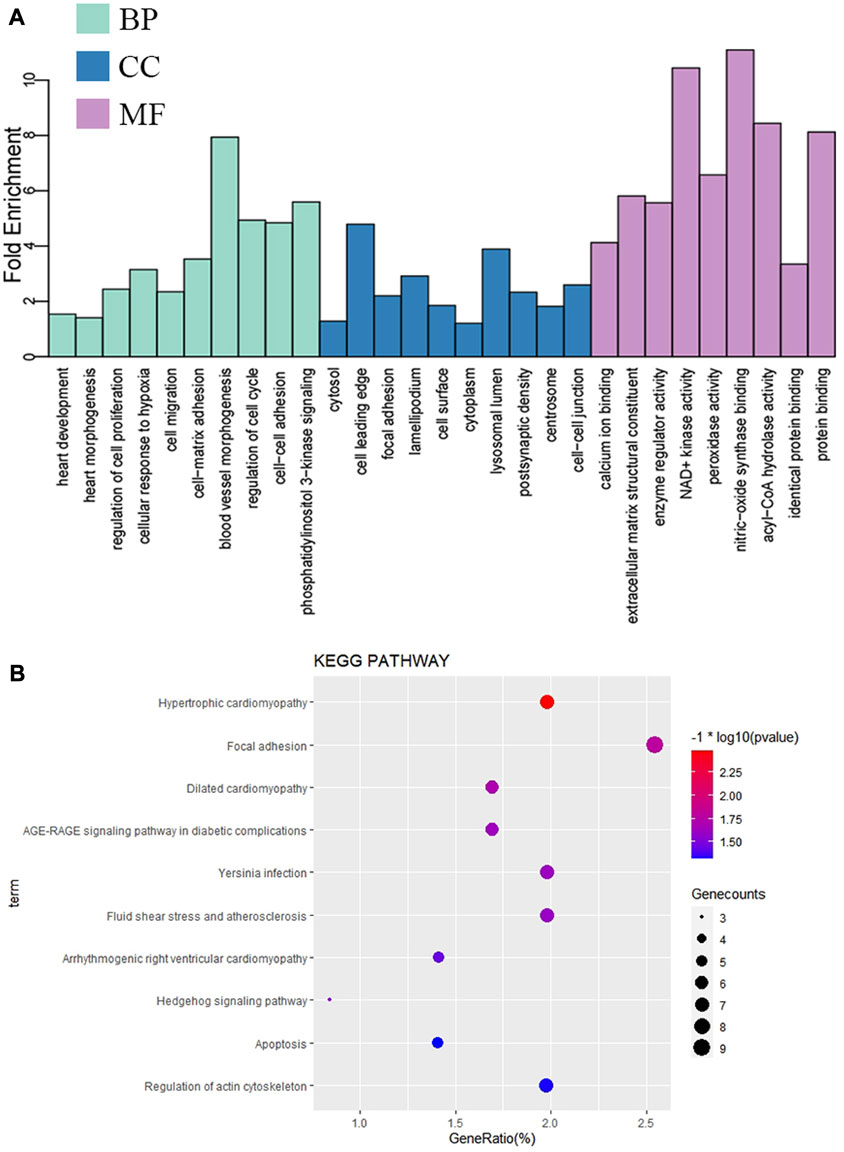
FIGURE 3. Enrichment analysis of significant DE-mRNAs in VSD. (A) Significantly enriched top ten GO terms. (B) Significantly enriched top ten KEGG pathways. GO, Gene Ontology; BP, biological process; CC, cellular component; MF, molecular function; KEGG, Kyoto Encyclopedia of Genes and Genomes.
To identify the relatively balanced mean connectivity and scale independence, soft-thresholding power β was set to 9, and the scale-free topology R2-value was set to 0.85 (Figure 4A). A total of 24 scale-free topology modules were constructed with the one-step network construction method (Figure 4B). The largest module was turquoise with 2,472 mRNAs, and the smallest module was dark gray with 32 mRNAs. The eigengene adjacency heatmap showed that modules were independent of each other (Figure 4C). The module–trait relationships are shown in Figure 4D. Among these modules, the red and purple modules were most positively associated with VSD. Therefore, we chose the red and purple modules as the key modules of VSD. Plots were charted to show the relationship between MM and GS in the red and purple modules (Figures 4E, F). Under the criteria of GS > 0.3 and MM > 0.8, 408 interesting mRNAs and 25 interesting mRNAs were singled out from the red and purple modules, respectively. After an integrated analysis of module genes and CNC network genes, 16 overlapping mRNAs were identified for further analysis (Supplementary Table S7).
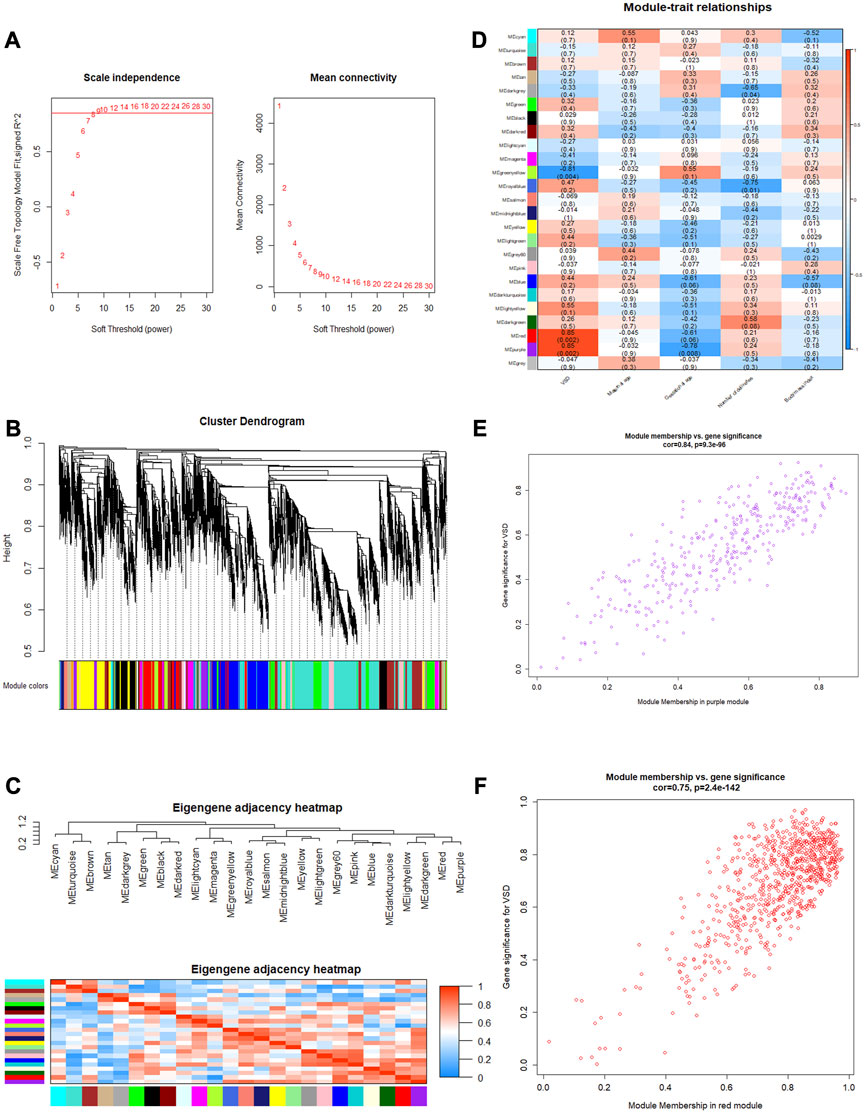
FIGURE 4. Weighted gene co-expression network analysis (WGCNA). (A) Determination of the soft threshold. Left: scale independence; right: mean connectivity. (B) Cluster dendrogram. The branches (at the top) represent genes, and each color represents one specific co-expression module. (C) Eigengene adjacency heatmap. The color blocks on the left and bottom of the panel correspond to modules. The red color indicates a remarkable correlation between modules. (D) Module–trait associations. The columns and rows correspond to trait and module eigengenes, respectively. The red color indicates a positive correlation, and the blue color indicates a negative correlation. (E, F) Scatterplots of MM vs. GS in the purple and red modules. GS, gene significance; MM, module membership.
To the best of our knowledge, lncRNAs are positively correlated with mRNAs in the ceRNA regulatory network. Based on the CNC network, 14 positive lncRNA–mRNA pairs, including three lncRNAs and nine mRNAs, were selected to construct the ceRNA. Subsequently, a total of 37 miRNAs were identified from the miRcode and TargetScan databases, exhibiting overlapping target sites on both lncRNAs and mRNAs. Eventually, the ceRNA regulatory network, which includes three lncRNAs, 38 miRNAs, and nine mRNAs, was constructed according to the pairs of lncRNA–mRNA, miRNA–mRNA, and lncRNA–mRNA (Figure 5).
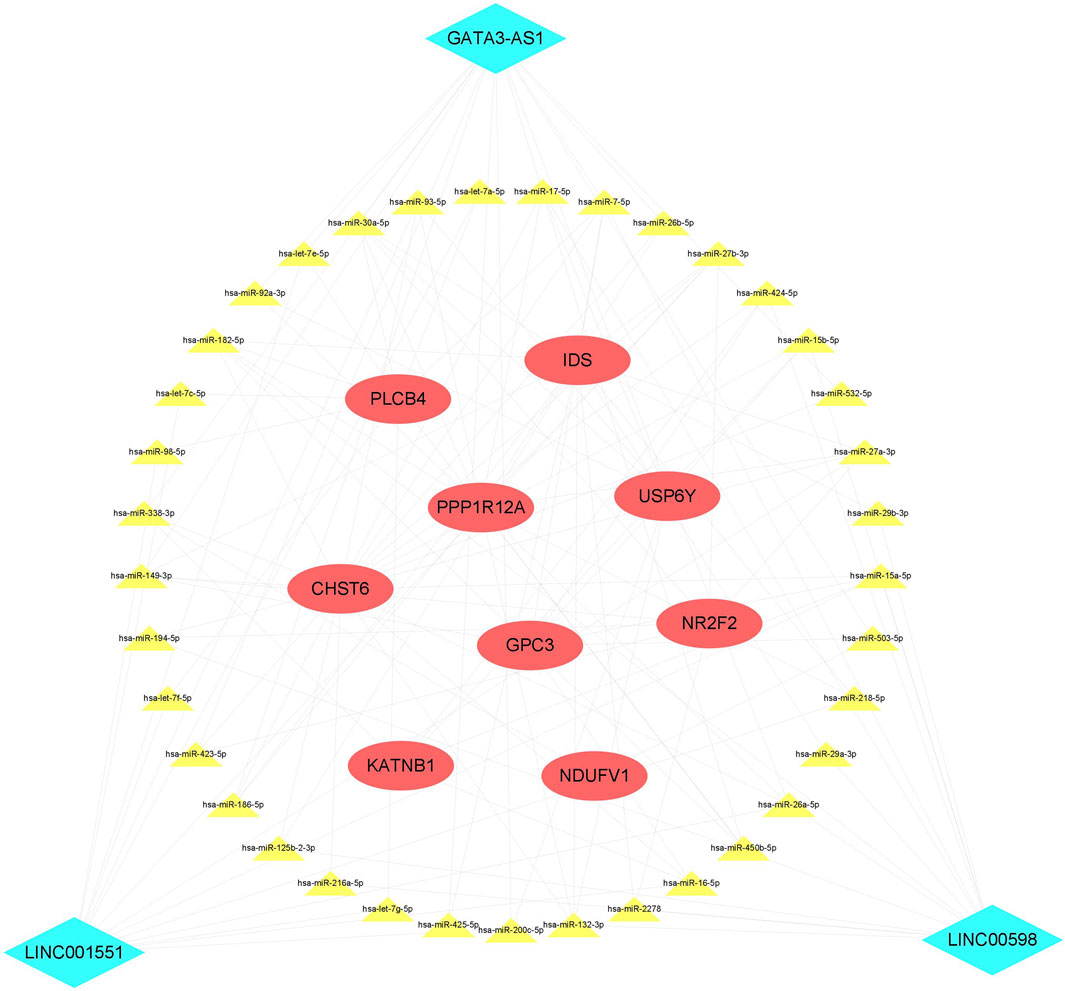
FIGURE 5. ceRNA network. The red ellipse represents mRNAs, the green diamond represents lncRNAs, and the yellow triangle represents miRNAs. ceRNA, competitive endogenous RNA.
As we all know, cardiac development involves many signaling pathways and is regulated by ncRNAs and mRNAs both temporally and spatially. It has been shown that amniotic fluid remains in constant exchange with maternal blood by the placenta (Rosner et al., 2021), and cell-free fetal RNAs derived from fetal tissues were remarkably stable in peripheral blood (Liu et al., 2022). To explore whether the expression of VSD-related lncRNAs is consistent in maternal plasma and amniotic fluid, we selected four lncRNAs validated in our previous study and performed qPCR in plasma samples. The result showed that three lncRNAs (LINC01551, LINC00598, and GATA3-AS1) were consistent with amniotic fluid. Subsequently, the ROC curves showed that the three lncRNAs were able to significantly differentiate VSD patients from controls. These data indicate that cell-free fetal lncRNAs are capable of bidirectional transport between amniotic fluid and maternal plasma and are expected to be useful biomarkers for VSD diagnosis.
How lncRNAs coordinate to control cell fate remains largely elusive. One possibility is that lncRNAs regulate specific gene expression programs to influence cell function. In the combined analysis of the CNC network and functional enrichment, we found that mRNAs associated with these three lncRNAs were enriched in heart morphogenesis, actin cytoskeleton regulation, cell cycle, and protein binding. Abnormal heart morphogenesis is at the root of CHDs, and the potential CHD may arise at each heart development stage (Baban et al., 2022). For example, the secondary heart field is very important for the outflow tract and chamber formation and, when disrupted, will lead to conotruncal defects (Houyel and Meilhac, 2021). The actin cytoskeleton is pivotal to the proper function of cardiac muscle and the deployment process of second heart field progenitor cells (Wu et al., 2017). The correct cardiogenic lineage cell cycle directs cardiomyocyte reprogramming and stem cell differentiation (Hubert et al., 2018).
As previously discussed, the biological activity of these mRNAs contributes to the differentiation of specific cell types and morphogenesis of critical layers in the developing heart, but the key mRNA for VSD remained unclear. Through WGCNA, the red and purple modules were identified as positively correlated with VSD, and then 16 mRNAs were identified as associated with VSD pathogenesis. Several studies have reported that these mRNAs may cause perturbations in the cardiac structure and function by interfering with cell-type differentiation and specification. In mammals, NR2F2 is abundantly expressed in the developing fetal heart and is considered a candidate gene contributing to CHD (Qiao et al., 2018; Wang et al., 2019). KATNB1 is a critical regulator of heart development, and mutations in KATNB1 will cause heart left–right asymmetry defects (Furtado et al., 2017). GPC3 is widely expressed in embryonic mesodermal tissues and plays an important role in regulating cell proliferation and apoptosis in the embryonic heart (Liu et al., 2021). IDS contributes to cardiomyocyte differentiation disorder and aberrant heart development by affecting the catabolism of GAGs (Matsuhisa and Imaizumi, 2021).
The sponging effect is a particular feature of lncRNAs, which can regulate mRNA by competitively binding to shared miRNAs (Marques and Gama-carvalho, 2022). Therefore, knowledge of the lncRNA-associated ceRNA regulation network and its patterning is central to the understanding of CHD diagnosis and etiology. To reveal the underlying regulatory network between lncRNAs and mRNAs, we constructed a ceRNA network with three lncRNAs, 38 miRNAs, and nine mRNAs. In the ceRNA network, many miRNAs have been confirmed to be involved in the development of CHDs. For example, previous research has reported that has-let-7e (Li et al., 2014), has-miR-27b (Long et al., 2020), and has-miR-218 (Ramachandran et al., 2022) were dysregulated in CHD patients. In addition, Yang et al. (2020) found that has-miR-29b was significantly overexpressed in the right ventricular outflow tract of CHD patients and inhibited cardiomyocyte proliferation by targeting NOTCH2. Liu et al. (2016) found that has-miR-30c was upregulated in the heart tissues with VSD and confirmed that miR-30c regulated P19 cell differentiation, apoptosis, and proliferation via the Hedgehog signaling pathway. The aforementioned studies show that an lncRNA can sponge off one or more miRNAs to affect downstream target gene expression and participate in the formation of VSD.
In the present study, we confirmed that several VSD-related lncRNAs (GATA3-AS1, LINC01551, and LINC00598) were consistent in both maternal plasma and amniotic fluid, suggesting that the amniotic fluid cell-free lncRNAs could be released into the maternal circulation and investigated as potential markers for CHDs. The effects of these lncRNAs on VSD may be involved in cardiac morphogenesis, actin cytoskeleton, cell cycle, and protein binding by the ceRNA network at the post-transcriptional level. Nevertheless, our study has several limitations. First, the sample size enrolled in the present study is insufficient. In the future, larger samples with VSD are needed to validate those candidate lncRNAs. Second, we have only predicted the potential role of lncRNAs in the development of VSD through bioinformatics analysis, but the targets and specific mechanisms of these lncRNAs have not been validated. Therefore, the corresponding functional and mechanistic experiments need to be initiated urgently in vivo and in vitro.
Publicly available datasets were analyzed in this study. These data can be found at: Gene Expression Omnibus (GEO) site (https://www.ncbi.nlm.nih.gov/geo/) (GEO: GSE204935).
The studies involving humans were approved by the Ethics Committee of The Second Affiliated Hospital of Fujian Medical University. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
HW: formal analysis, data curation, methodology, validation, visualization, and writing—original draft. XLn: data curation, validation, writing—original draft, and investigation. XW: validation, writing—original draft, methodology, and visualization. XLu: writing—original draft, investigation, and software. SH: writing—original draft, formal analysis, and project administration. GL: formal analysis, conceptualization, funding acquisition, and writing—review and editing.
This work was supported by the Qihang Fund of Fujian Medical University (grant number 2020QH2039) and Quanzhou City Science and Technology Program of China (grant number 2021C059R).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1254829/full#supplementary-material
Baban, A., Lodato, V., Parlapiano, G., and Drago, F. (2022). Genetics in congenital heart diseases: unraveling the link between cardiac morphogenesis, heart muscle disease, and electrical disorders. Heart Fail. Clin. 18, 139–153. doi:10.1016/j.hfc.2021.07.016
Deita, M., Cisneros, B., and Rosas-Vargas, H. (2021). Genetics of transposition of great arteries: between laterality abnormality and outflow tract defect. J. Cardiovasc. Transl. Res. 14, 390–399. doi:10.1007/s12265-020-10064-x
Devaux, Y., Zangrando, J., Schroen, B., Creemers, E. E., Pedrazzini, T., Chang, C. P., et al. (2015). Long noncoding RNAs in cardiac development and ageing. Nat. Rev. Cardiol. 12, 415–425. doi:10.1038/nrcardio.2015.55
Furtado, M. B., Merriner, D. J., Berger, S., Rhodes, D., Jamsai, D., and O'Bryan, M. K. (2017). Mutations in the Katnb1 gene cause left-right asymmetry and heart defects. Dev. Dyn. official Publ. Am. Assoc. Anatomists 246, 1027–1035. doi:10.1002/dvdy.24564
Gu, H., Chen, L., Xue, J., Huang, T., Wei, X., Liu, D., et al. (2019). Expression profile of maternal circulating microRNAs as non-invasive biomarkers for prenatal diagnosis of congenital heart defects. Biomed. Pharmacother. = Biomedecine Pharmacother. 109, 823–830. doi:10.1016/j.biopha.2018.10.110
Gutierrez, J., Davis, B. A., Nevonen, K. A., Ward, S., Carbone, L., and Maslen, C. L. (2022). DNA methylation analysis of turner syndrome BAV. Front. Genet. 13, 872750. doi:10.3389/fgene.2022.872750
Hou, Y. S., Wang, J. Z., Shi, S., Han, Y., Zhang, Y., Zhi, J. X., et al. (2020). Identification of epigenetic factor KAT2B gene variants for possible roles in congenital heart diseases. Biosci. Rep. 40, BSR20191779. doi:10.1042/BSR20191779
Houyel, L., and Meilhac, S. M. (2021). Heart development and congenital structural heart defects. Annu. Rev. genomics Hum. Genet. 22, 257–284. doi:10.1146/annurev-genom-083118-015012
Hubert, F., Payan, S. M., and Rochais, F. (2018). FGF10 signaling in heart development, homeostasis, disease and repair. Front. Genet. 9, 599. doi:10.3389/fgene.2018.00599
Kan, R. L., Chen, J., and Sallam, T. (2022). Crosstalk between epitranscriptomic and epigenetic mechanisms in gene regulation. Trends Genet. TIG 38, 182–193. doi:10.1016/j.tig.2021.06.014
Langfelder, P., and Horvath, S. (2008). Wgcna: an R package for weighted correlation network analysis. BMC Bioinforma. 9, 559. doi:10.1186/1471-2105-9-559
Langfelder, P., Zhang, B., and Horvath, S. (2008). Defining clusters from a hierarchical cluster tree: the dynamic tree cut package for R. Bioinforma. Oxf. Engl. 24, 719–720. doi:10.1093/bioinformatics/btm563
Li, D., Ji, L., Liu, L., Liu, Y., Hou, H., Yu, K., et al. (2014). Characterization of circulating microRNA expression in patients with a ventricular septal defect. PloS one 9, e106318. doi:10.1371/journal.pone.0106318
Liu, J., Liu, Q., Yang, S., Ma, N., Pang, J., Peng, Y., et al. (2021). Prenatal case of Simpson-Golabi-Behmel syndrome with a de novo 370Kb-sized microdeletion of Xq26.2 compassing partial GPC3 gene and review. Mol. Genet. genomic Med. 9, e1750. doi:10.1002/mgg3.1750
Liu, X., Li, M., Peng, Y., Hu, X., Xu, J., Zhu, S., et al. (2016). miR-30c regulates proliferation, apoptosis and differentiation via the Shh signaling pathway in P19 cells. Exp. Mol. Med. 48, e248. doi:10.1038/emm.2016.57
Liu, Y., Chen, S., Zühlke, L., Black, G. C., Choy, M. K., Li, N., et al. (2019). Global birth prevalence of congenital heart defects 1970-2017: updated systematic review and meta-analysis of 260 studies. Int. J. Epidemiol. 48, 455–463. doi:10.1093/ije/dyz009
Liu, Z., Wang, T., Yang, X., Zhou, Q., Zhu, S., Zeng, J., et al. (2022). Polyadenylation ligation-mediated sequencing (PALM-Seq) characterizes cell-free coding and non-coding RNAs in human biofluids. Clin. Transl. Med. 12, e987. doi:10.1002/ctm2.987
Long, L., Xiao, Y., Yin, X., Gao, S., Zhou, L., and Liu, H. (2020). Expression of serum miR-27b and miR-451 in patients with congenital heart disease associated pulmonary artery hypertension and risk factor analysis. Exp. Ther. Med. 20, 3196–3202. doi:10.3892/etm.2020.9042
Ma, J., Chen, S., Hao, L., Sheng, W., Chen, W., Ma, X., et al. (2021). Long non-coding RNA SAP30-2:1 is downregulated in congenital heart disease and regulates cell proliferation by targeting HAND2. Front. Med. 15, 91–100. doi:10.1007/s11684-020-0778-5
Marques, T. M., and Gama-Carvalho, M. (2022). Network approaches to study endogenous RNA competition and its impact on tissue-specific microRNA functions. Biomolecules 12 (2), 332. doi:10.3390/biom12020332
Matsuhisa, K., and Imaizumi, K. (2021). Loss of function of mutant IDS due to endoplasmic reticulum-associated degradation: new therapeutic opportunities for mucopolysaccharidosis type II. Int. J. Mol. Sci. 22, 12227. doi:10.3390/ijms222212227
Qiao, X. H., Wang, Q., Wang, J., Liu, X. Y., Xu, Y. J., Huang, R. T., et al. (2018). A novel NR2F2 loss-of-function mutation predisposes to congenital heart defect. Eur. J. Med. Genet. 61, 197–203. doi:10.1016/j.ejmg.2017.12.003
Ramachandran, V., Bhagavatheeswaran, S., Shanmugam, S., Vasudevan, M., Ragunathan, M., Cherian, K. M., et al. (2022). Deep sequencing unveils altered cardiac miRNome in congenital heart disease. Mol. Genet. genomics MGG 297, 1123–1139. doi:10.1007/s00438-022-01908-z
Ren, C., Wu, C., Pan, Z., and Li, Y. (2021). Minimally invasive closure of transthoracic ventricular septal defect: postoperative complications and risk factors. J. Cardiothorac. Surg. 16, 30. doi:10.1186/s13019-021-01415-z
Rosner, M., Kolbe, T., and Hengstschläger, M. (2021). Fetomaternal microchimerism and genetic diagnosis: on the origins of fetal cells and cell-free fetal DNA in the pregnant woman. Rev. Mutat. Res. 788, 108399. doi:10.1016/j.mrrev.2021.108399
Wagner, R., Tse, W. H., Gosemann, J. H., Lacher, M., and Keijzer, R. (2019). Prenatal maternal biomarkers for the early diagnosis of congenital malformations: A review. Pediatr. Res. 86, 560–566. doi:10.1038/s41390-019-0429-1
Wang, H., Lin, X., Wang, Z., He, S., Dong, B., and Lyu, G. (2023). Differential lncRNA/mRNA expression profiling and ceRNA network analyses in amniotic fluid from foetuses with ventricular septal defects. PeerJ 11, e14962. doi:10.7717/peerj.14962
Wang, J., Abhinav, P., Xu, Y. J., Li, R. G., Zhang, M., Qiu, X. B., et al. (2019). NR2F2 loss-of-function mutation is responsible for congenital bicuspid aortic valve. Int. J. Mol. Med. 43, 1839–1846. doi:10.3892/ijmm.2019.4087
Wang, Q., Wang, Z., Wu, C., Pan, Z., Xiang, L., Liu, H., et al. (2018). Potential association of long noncoding RNA HA117 with tetralogy of Fallot. Genes & Dis. 5, 185–190. doi:10.1016/j.gendis.2018.03.002
Willner, E. C., Galan, H. L., Cuneo, B. F., Hoffman, H. A., Neltner, B., Schuchardt, E. L., et al. (2021). Amniotic fluid microRNA profiles in twin-twin transfusion syndrome with and without severe recipient cardiomyopathy. Am. J. obstetrics Gynecol. 225, 439.e1–439439.e10. doi:10.1016/j.ajog.2021.06.066
Wu, T., Mu, Y., Bogomolovas, J., Fang, X., Veevers, J., Nowak, R. B., et al. (2017). HSPB7 is indispensable for heart development by modulating actin filament assembly. Proc. Natl. Acad. Sci. U. S. A. 114, 11956–11961. doi:10.1073/pnas.1713763114
Yang, Q., Wu, F., Mi, Y., Wang, F., Cai, K., Yang, X., et al. (2020). Aberrant expression of miR-29b-3p influences heart development and cardiomyocyte proliferation by targeting NOTCH2. Cell Prolif. 53, e12764. doi:10.1111/cpr.12764
Yoshikawa, K., Kiyoshima, C., Hirakawa, T., Urushiyama, D., Fukagawa, S., Izuchi, D., et al. (2021). Diagnostic predictability of miR-4535 and miR-1915-5p expression in amniotic fluid for foetal morbidity of infection. Placenta 114, 68–75. doi:10.1016/j.placenta.2021.08.059
Zhang, X., Gao, Y., Zhang, X., Zhang, X., Xiang, Y., Fu, Q., et al. (2021). FGD5-AS1 is a hub lncRNA ceRNA in hearts with tetralogy of Fallot which regulates congenital heart disease genes transcriptionally and epigenetically. Front. Cell Dev. Biol. 9, 630634. doi:10.3389/fcell.2021.630634
Keywords: long non-coding RNA, ventricular septal defect, maternal body fluids, prenatal diagnosis, epigenetic
Citation: Wang H, Lin X, Wang X, Liu X, He S and Lyu G (2023) Maternal body fluid lncRNAs serve as biomarkers to diagnose ventricular septal defect: from amniotic fluid to plasma. Front. Genet. 14:1254829. doi: 10.3389/fgene.2023.1254829
Received: 07 July 2023; Accepted: 21 August 2023;
Published: 06 September 2023.
Edited by:
Subhashree Nayak, National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIH), United StatesReviewed by:
Nir Pillar, University of California, Los Angeles, United StatesCopyright © 2023 Wang, Lin, Wang, Liu, He and Lyu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guorong Lyu, bGdyX2ZldXNAc2luYS5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.