 Xianda Wei1,2†
Xianda Wei1,2† Xu Zhou3†
Xu Zhou3† BoBo Xie1,2
BoBo Xie1,2 Meizhen Shi1,2Chunrong Gui1,2Bo Liu4Caiyan Li4Chi Zhang5Jiefeng Luo6*Cundong Mi4*
Meizhen Shi1,2Chunrong Gui1,2Bo Liu4Caiyan Li4Chi Zhang5Jiefeng Luo6*Cundong Mi4* Baoheng Gui1,2*
Baoheng Gui1,2*- 1Center for Medical Genetics and Genomics, The Second Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi, China
- 2The Guangxi Health Commission Key Laboratory of Medical Genetics and Genomics, The Second Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi, China
- 3The Second School of Medicine, Guangxi Medical University, Nanning, Guangxi, China
- 4Department of Rheumatology and Immunology, The Second Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi, China
- 5Department of Ultrasound Diagnosis, The Second Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi, China
- 6Department of Neurology, The Second Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi, China
Vascular Ehlers–Danlos syndrome (vEDS), the most severe type of Ehlers–Danlos syndrome, is caused by an autosomal-dominant defect in the COL3A1 gene. In this report, we describe the clinical history, specific phenotype, and genetic diagnosis of a man who died of vEDS. The precise diagnosis of this case using whole-exome sequencing provided solid evidence for the cause of death, demonstrating the practical value of genetic counseling and analysis. Early diagnosis for the proband’s son, who was also affected by vEDS, revealed initial complications of vEDS in early childhood, which have rarely been reported. We also reviewed the literature on COL3A1 missense mutations and related phenotypes. We identified an association between digestion tract events and non-glycine missense variants, which disproves a previous hypothesis regarding the genotype–phenotype correlation of vEDS. Our results demonstrate the necessity of offering comprehensive genetic testing for every patient suspected of having vEDS.
1 Introduction
Ehlers–Danlos syndrome (EDS), also known as congenital connective tissue hypoplasia syndrome, is associated with defects in collagen synthesis and metabolism. EDS represents a class of collagen disorders among the wider group of heritable connective tissue diseases. Vascular EDS (vEDS) is a specific form of EDS caused by autosomal-dominant mutations in COL3A1. Although vEDS is generally associated with mild skin lesions, severe cardiovascular lesions can occur in rare cases. These lesions can progress to cause aortic dissection and aneurysm, which are prone to spontaneous rupture, leading to death. As such, vEDS is the most dangerous type of EDS and has the worst prognosis.
In this report, we describe the clinical history, unique phenotype, and genetic cause of a man who died from vEDS; his son was also affected by vEDS, as confirmed by whole-exome sequencing (WES). In addition, we reviewed the literature on COL3A1 missense mutations and related phenotypes to provide more insight into the phenotype–genotype correlation in vEDS.
2 Clinical report
The proband was a 32-year-old man. In January 2021, he presented to a local hospital because of complaints of swelling of the left forearm and bulging of the blood vessels. He subsequently repeatedly visited doctors for systemic symptoms, including severe abdominal pain, lumbago, bulging vessels, sweating, a pale face, and transient amaurosis. Symptoms improved with a small dose of glucocorticoids and symptomatic treatment. Trauma caused by a violent impact was denied. In June 2021, he developed swelling of the right forearm; pain in the right forearm, back, and abdomen; and transient black spots in his vision. Ultrasound imaging revealed arterial abnormalities. Computed tomography (CT) angiography showed bilateral internal carotid and vertebral artery aneurysms, rupture of the left ulnar artery, and pseudoaneurysm.
In July 2021, the patient visited our hospital with abdominal pain and black stools. Physical examination revealed a short height (150 cm) and low body weight (36 kg). He exhibited aging skin of the extremities, subcutaneous venous exposure, and multiple ecchymoses (non-traumatic) of the chest, right waist, and upper extremities (Figure 1A). His fingers were slender, and the joints were abnormally flexible. His right hand had excessive dorsal flexion and the little finger was pressed against the back of the hand (Figure 1B). His feet rotated inward (Figure 1C). Gastroscopy revealed no bleeding in the upper digestive tract. Vascular and abdominal CT (Figure 1D) revealed multiple aneurysms and aneurysmal dilation of the celiac trunk, proper hepatic artery, left hepatic artery, splenic artery, and both kidneys, along with arterial dissection of the celiac trunk and splenic artery. The patient experienced pain on percussion in the right renal area. Color ultrasound of the urinary system indicated mild hydrops in the right kidney, excluding the presence of urinary stones. Distal occlusion of the upper pole branch of the right renal artery, decreased perfusion of the right renal parenchyma, and a high density of fat sacs around the right kidney suggested bleeding and infarction of the right kidney. Kidney stones were further excluded by ultrasonography. Blood biochemistry tests, immunoglobulin (Ig)A/IgM/IgG, rheumatoid factors, autoantibody spectra, and cardiac color Doppler ultrasonography all exhibited negative findings. The patient died of arterial rupture and hemorrhagic shock 2 weeks after admission.
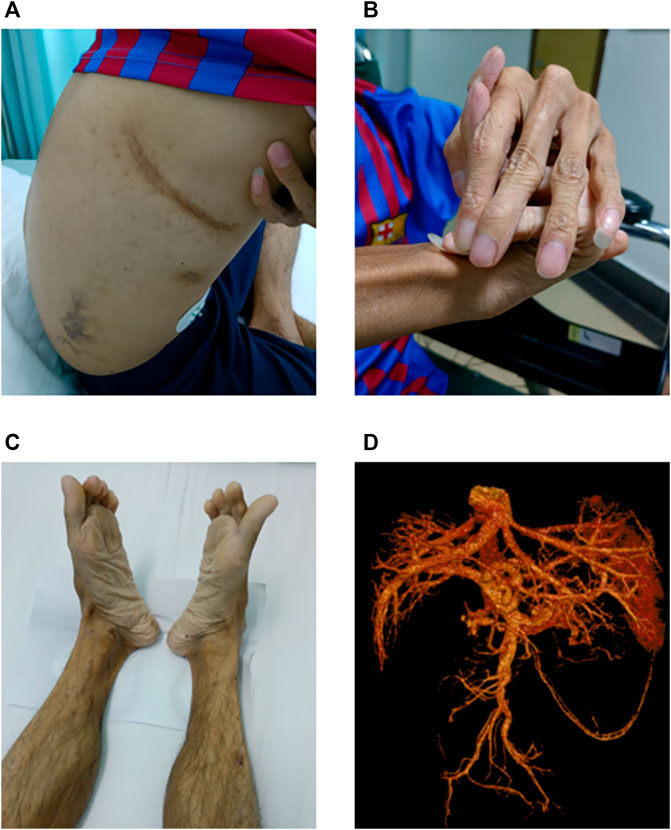
FIGURE 1. Main clinical findings of the proband. (A) Multiple ecchymoses (non-traumatic) in the anterior chest, right waist, and upper limbs. (B) Right finger with excessive dorsal flexion and the little finger pressed against the back of the hand. (C) Feet rotated inward. (D) Multiple aneurysms and aneurysmal dilation of the celiac trunk, proper hepatic artery, left hepatic artery, splenic artery, and both kidneys; arterial dissection of the celiac trunk and splenic artery is evident.
We collected basic information on the proband’s family members over three generations (Figure 2). The proband’s grandfather died of unknown cause, and the proband’s parents, sisters, and daughters did not show any abnormalities related to vEDS. However, the proband’s son, aged 3 years 4 months, had deep skin pigmentation, deep palm lines, poor wound healing of the skin, and occasional fresh blood in stools. Mosquito bites were also reported to be abnormally large at times. No surface hemangiomas were observed.
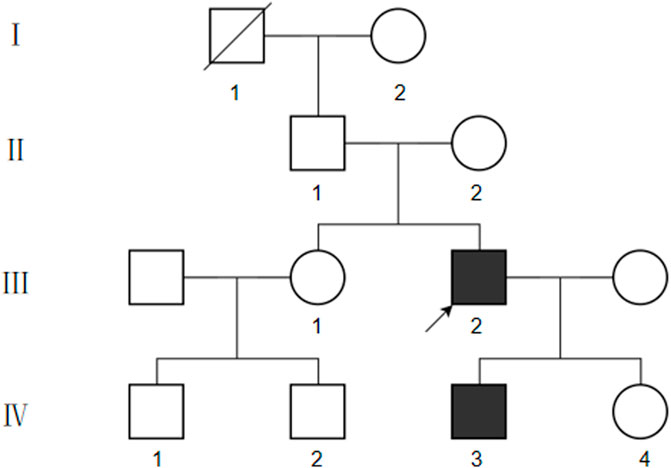
FIGURE 2. Pedigree chart of this case.
3 Genetic analysis
The rarity and complexity of the patient’s clinical manifestations implied the possibility of a genetic disease. To this end, we performed trio WES of the proband and his parents after genetic counseling; genetic testing was also performed on the proband’s son and proband’s older sister.
Genomic DNA was extracted from blood samples using a LabAid DNA kit (Zeesan Biotech Co., Ltd., Xiamen, China); target capture for WES (Human All Exon V5 Kit, Agilent Technologies, Foster City, CA, United States) and library sequencing (NextSeq CN500 platform, Illumina, San Diego, CA, United States) were conducted following the manufacturer protocols.
Data analysis and annotation were performed using the Genome Analysis Toolkit (GATK), version 3.4.0. The variants were identified and filtered using in-house protocols. The inclusion criteria for candidate variants were as follows: 1) heterozygous variants in an established causative gene of a dominant Mendelian disorder with a likely association to the patient’s phenotype; 2) variants with frequencies in the East Asian population in the gnomAD, NHLBI Exome Sequencing Project, 1000 Genomes Project, and in-house databases all <0.5%; and 3) computational evidence supports a deleterious effect of the variant. SpliceAI (Jaganathan et al., 2019), a deep-learning–based algorithm, was used to calculate the probability of splice-altering variants, providing a Δ score between 0 and 1. Candidate variants were verified by Sanger sequencing and true variants were validated following American College of Medical Genetics and Genomics/American Association of Molecular Pathology guidelines (Richards et al., 2015).
We identified a heterozygous missense variation, NM_000090.3:c.146C>T(p.Pro49Leu), in exon 2 of COL3A1, as the most likely genetic cause of the disease. The variant was heterozygous in the proband and his son and was absent in his parents and sister (Figure 3). The variant was classified as likely pathogenic according to the following criteria: 1) PS2: the variant was de novo, with both parental samples confirmed to be derived from the biological parents of the patient through single-nucleotide polymorphism analysis of trio-WES data; 2) PM2: this variant is not reported in the East Asian population database; and 3) PP2: the missense constraint Z-score of COL3A1 in the Gnomad database was 4.09, indicating that missense variations in this gene are a common cause of disease. According to the calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria (Pejaver et al., 2022), the prediction results obtained with a single prediction software tool can be used as a basis for PP3 evidence. The single prediction tool utilized in our laboratory to determine the usage and strength of PP3 is revel. In this case, the Revel score for the COL3A1 c.146C>T variant is only 0.586, which does not meet the criterion used for a PP3 level of supporting evidence (revel score ≥ 0.644). We also evaluated information about a previously reported variant c.145C>G p. (Pro49Ala) affecting the same residue as c.146C>T. Although homozygous c.145C>G variants have been detected in at least four patients with an autosomal recessive disorder caused by biallelic mutations in COL3A1, polymicrogyria with or without vascular-type EDS (OMIM 618343) (Vandervore, et al., 2017; Horn, et al., 2017), evidence supporting its pathogenicity in vEDS was insufficient. Thus, PM5 should not be applied to interpret c.146C>T(p.Pro49Leu) in our cases with vEDS.
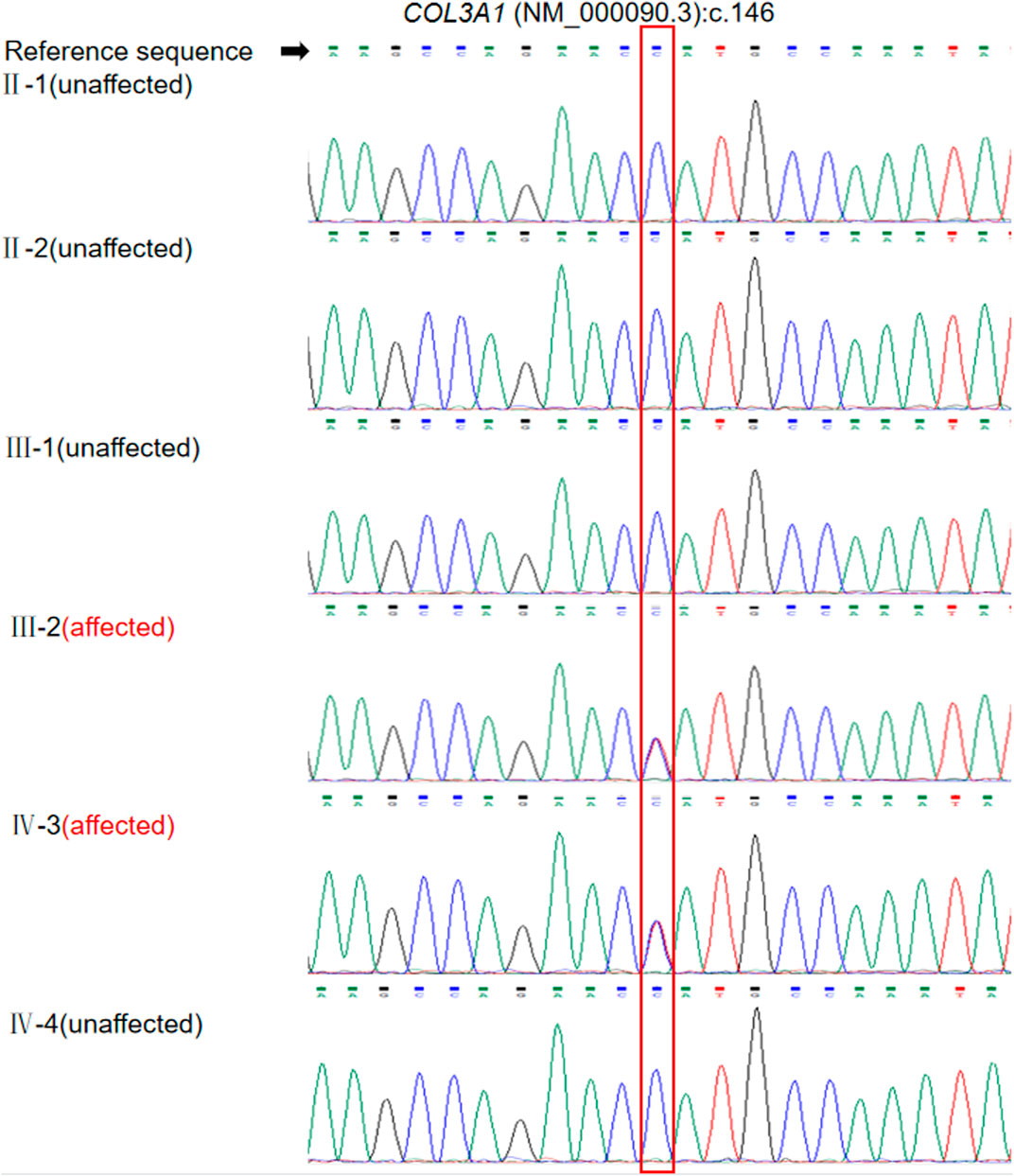
FIGURE 3. Validation of the COL3A1 mutation c.146C>T in the patient and family members by Sanger sequencing. Annotation in the red box indicates the location of the mutation.
Finally, we established a genetic diagnosis of vEDS for the patient and his son based on clinical and genetic findings.
4 Discussion
EDS is a heterogeneous group of connective tissue disorders that comprise a spectrum of monogenic conditions with multi-systematic and variable clinical manifestations (e.g., joint hypermotility, skin hyperextensibility, and tissue fragility) primarily affecting the skin, ligaments, joints, blood vessels, and internal organs. EDS is classified into 13 different subtypes, among which the vascular type (type IV) is the most severe and life-threatening, with arterial ruptures or dissections responsible for the majority of deaths. These events are unpredictable, and the fragility of the arterial walls often makes surgical repair difficult (Ruscitti et al., 2021).
The proband of this case had early symptoms, including peripheral vascular rupture events and skin abnormalities, which gradually evolved into thoracoabdominal vascular rupture. Although the clinical manifestations of the proband were in accordance with a diagnosis of vEDS, the rarity of this disease poses challenges for clinical diagnosis, especially for physicians in adult departments who may not have a strong awareness or sufficient knowledge of genetic disorders. We initially considered Marfan syndrome as a possible diagnosis, as the associated phenotypes overlap with those of vEDS, including aortic dissection, aortic aneurysm, and joint hypermotility. Following the advice of geneticists, we performed WES of the proband because of his multi-systematic and non-specific manifestations. The identification of a novel likely-pathogenic variation in COL3A1, c.146C>T, enabled a genetic diagnosis of vEDS for the patient. It should be noted that, though current evidences including the de novo occurrence and the absence in controls are sufficient to support the likely-pathogenicity according to the ACMG/AMP guidelines, functional evidences are still lacking. Further investigations of p.Pro49Leu mutant protein through in vitro or in vivo experiments will be vital to confirm the impact of this variant. In terms of this case, the diagnosis of vEDS with known prognosis eased the family’s concerns about the cause of death. Therefore, this case demonstrates the non-negligible practical value of genetic counseling and analysis.
Approximately two-thirds of published cases of genetically diagnosed vEDS are caused by point mutations in glycine residues (Ohyama et al., 2010). A single Gly substitution destabilizes the triple helix through a local disruption in hydrogen bonding and produces a discontinuity in the register of the helix (Brodsky and Persikov,2005).In COL1A1, COL2A1 and COL7A1, Gly substitutions have been reported to cause more severe disorders than any other single-aminoacid mutations (Marini et al., 2007; Xu et al., 2020; Gupta et al., 2023).According to a review article, all gastrointestinal events originate from glycine substitutions, splicing variants, and in-frame indels, whereas variants that cause haploinsufficiency and non-glycine missense variants are not associated with gastrointestinal events (Frank et al., 2015). To gather the latest evidence and re-analyze the association between the COL3A1 genotype and the vEDS phenotype, we conducted a literature review according to the following inclusion criteria: 1) reports of patients diagnosed with vEDS associated with only COL3A1 mutation and 2) the mutation type is consistent with non-glycine missense mutations.
This search retrieved reports of 35 cases of vEDS. In contrast to the review article of Frank et al. (2015), three of our reviewed cases involved non-glycine missense variants, c.1351G>A p.(Glu451Lys), c.2791G>A p.(Glu931Lys), and c.3511G>A p. (Glu1171Lys), that were indeed associated with digestive tract symptoms (Table 1). In the present case, both the proband and his son experienced digestive tract symptoms. Thus, the present case and previous three non-glycine missense variants reported put into question the currently established relationships between the genotype of COL3A1 and specific complications of vEDS. Accordingly, when performing genetic diagnosis for patients suspected as having vEDS, comprehensive testing of the COL3A1 gene covering all types of variants (e.g., through next-generation sequencing) is preferred over targeted analysis of limited sites or regions (e.g., Sanger sequencing), regardless of the presence or absence of any specific symptoms.
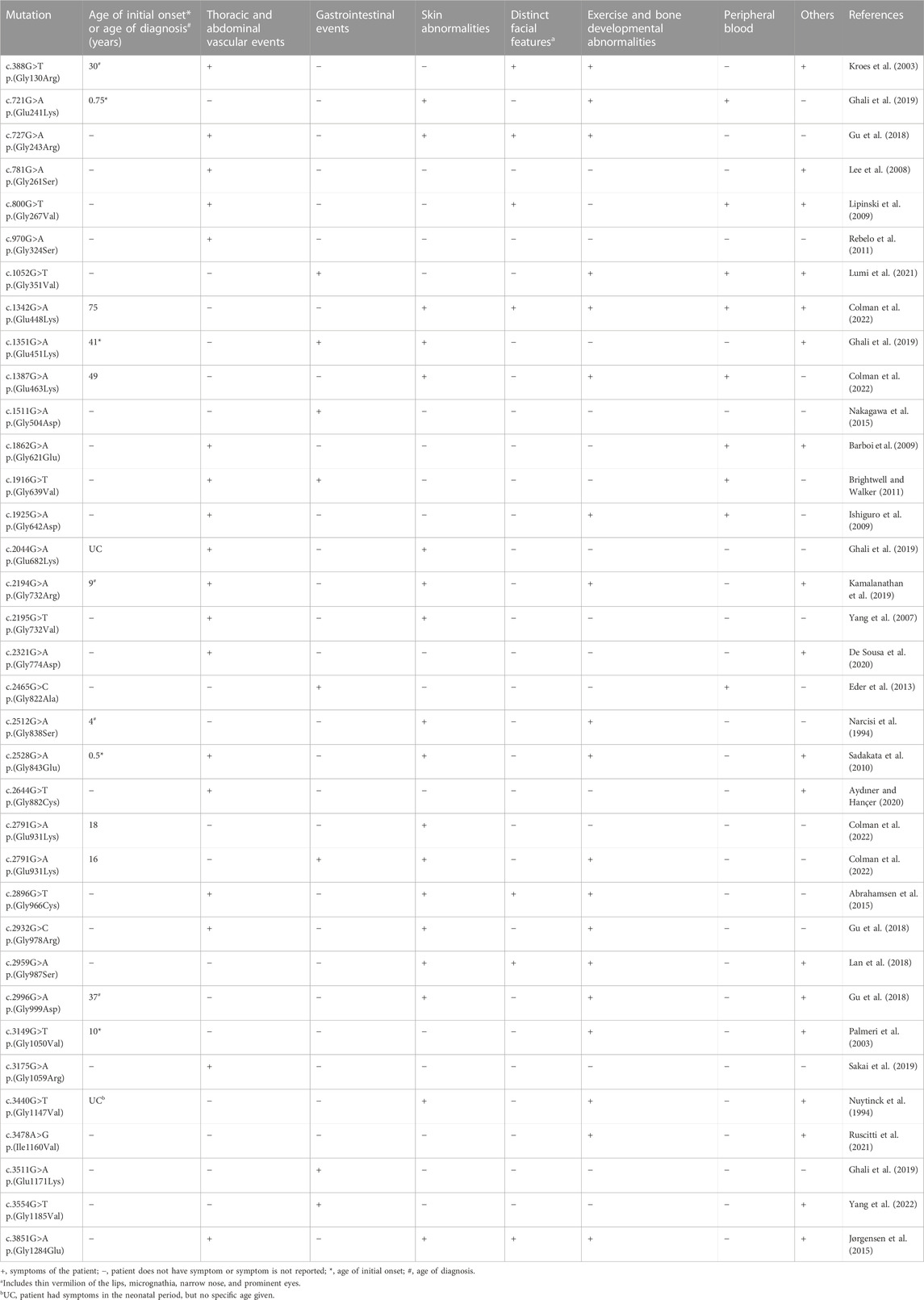
TABLE 1. Associations of vascular Ehlers–Danlos syndrome (vEDS) mutation sites with clinical phenotypes.
The proband’s son, who carried the same COL3A1 mutation, developed mild symptoms, including short stature, low body weight, skin pigmentation, deep palm prints, and poor skin healing. The boy also had black stool, indicating rupture of the blood vessels in the digestive tract. These observations suggest the initial complications of vEDS in early childhood but have rarely been reported in the published literature. In earlier reports, 25% of patients with vEDS had their first symptom by the age of 20 years and >80% had at least one symptom by the age of 40 years. Based on the high complication rates, the median age at the first major vascular event and at death for patients with vEDS are reported as 24.6 and 48 years, respectively (Pepin et al., 2000).
This early genetic diagnosis of the proband’s son (aged 3 years) presents an opportunity to assess the prognosis by referring to his father’s case, thereby enabling early interventions for management of the disease and prevention of further serious complications. For example, patients with vEDS require daily exercise and treatment. Care should be taken to avoid trauma (e.g., collision sports, heavy lifting, and extreme weight training). Arteriography should be discouraged and used only to identify life-threatening sources of bleeding before surgical intervention because of the risk of vascular injury (Byers, 1999).
In summary, this case highlights the importance of early and accurate diagnosis, genetic counseling, and avoiding high-risk activities and procedures in patients with vEDS (Frković et al., 2022). Thus, for late-onset genetic disorders such as vEDS, active utilization of genetic testing for younger relatives could create opportunities for early diagnosis of the disease, facilitating precise counseling and prevention of fatal disease progression. Moreover, the clinical findings and genetic analysis of this case could form the basis for the design of research programs, including further functional studies or modeling investigations.
Data availability statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by the Second Affiliated Hospital of Guangxi Medical University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
BG and JL organized the study. CM, BL, and CL conducted clinical evaluations. BX, MS, and CG analyzed the sequencing data. CZ conducted ultrasound testing. XW and XZ reviewed the literature and wrote this report. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by the National Natural Science Foundation of China (82001531 and 81860272 to BG), Guangxi Major Research Program (AB22035013 to BG), Guangxi Natural Science Foundation (2023GXNSFBA026124 to CG, 2018GXNSFAA281067 to BG), Shandong Natural Science Foundation (ZR2020QH050 to XW), Initial Scientific Research Fund for Advanced Talents from The Second Affiliated Hospital of Guangxi Medical University (2019112 to BG), Special Scientific Research Fund of Guangxi Ten-Hundred-Thousand Talents Project (2021186 to BG), Science Foundation for Young Scholars of Guangxi Medical University (GXMUYSF202115 to CG), Guangxi Natural Science Foundation (2021GXNSFAA196047 to BX), and Innovation Project of Guangxi Graduate Education (YCSW2023239 to XZ).
Acknowledgments
We thank the patient and his family members for their contributions to this medical research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abrahamsen, B. J. M., Kulseth, M. A. P., and Paus, B. M. P. (2015). A 19-year-old man with relapsing bilateral pneumothorax, hemoptysis, and intrapulmonary cavitary lesions diagnosed with vascular Ehlers-Danlos syndrome and a novel missense mutation in COL3A1. Chest 147, e166–e170. doi:10.1378/chest.13-3002
Aydıner, Ö., and Hançer, V. S. (2020). A novel COL3A1 c.2644G>T; p.(Gly882Cys) variant in a Turkish family with vascular Ehlers-Danlos syndrome. Mol. Syndromol. 11, 110–114. doi:10.1159/000506585
Barboi, A., Dennis, C., Timins, M., Peltier, W., Klotz, C. M., and Jaradeh, S. (2009). Neuromuscular manifestations in a patient with Ehlers-Danlos syndrome type IV. J. Clin. Neuromuscul. 11, 81–87. doi:10.1097/CND.0b013e3181bab6e3
Brightwell, R. E., and Walker, P. J. (2011). Lower limb arterio-venous fistula as a late complication of phlebectomy in a patient with Ehlers-Danlos type IV. Eur. J. Vasc. Endovasc. Surg. 42, 696–698. doi:10.1016/j.ejvs.2011.06.044
Brodsky, B., and Persikov, A. V. (2005). Molecular structure of the collagen triple helix. Adv. Protein Chem. 70, 301–339. doi:10.1016/S0065-3233(05)70009-7
Byers, P. H. (1999). “Vascular ehlers-danlos syndrome,” in GeneReviews (Seattle, WA: University of Washington).
Colman, M., Castori, M., Micale, L., Ritelli, M., Colombi, M., Ghali, N., et al. (2022). Atypical variants in COL1A1 and COL3A1 associated with classical and vascular Ehlers-Danlos syndrome overlap phenotypes: expanding the clinical phenotype based on additional case reports. Clin. Exp. Rheumatol. 40 (Suppl. 134), 46–62. doi:10.55563/clinexprheumatol/kzkq6y
De Sousa, F. D. R., Colucci, N., Dupuis, A., Toso, C., Buchs, N. C., and Abbassi, Z. (2020). Surgical management of vascular Ehlers-Danlos syndrome and its challenges: a case report. Swiss Med. Wkly. 150, w20379. doi:10.4414/smw.2020.20379
Eder, J., Laccone, F., Rohrbach, M., Giunta, C., Aumayr, K., Reichel, C., et al. (2013). A new COL3A1 mutation in Ehlers–Danlos syndrome type IV. Exp. Dermatol. 22, 231–234. doi:10.1111/exd.12105
Frank, M., Albuisson, J., Ranque, B., Golmard, L., Mazzella, J. M., Bal-Theoleyre, L., et al. (2015). The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers-Danlos syndrome. Eur. J. Hum. Genet. 23, 1657–1664. doi:10.1038/ejhg.2015.32
Frković, S. H., Marija Slišković, A., Toivonen, M., Crkvenac Gregorek, A., Šutalo, A., and Vrkić Kirhmajer, M. (2022). Vascular Ehlers-Danlos syndrome, an often unrecognized clinical entity: a case report of a novel mutation in the COL3A1 gene. Croat. Med. J. 63, 394–398. doi:10.3325/cmj.2022.63.394
Ghali, N., Baker, D., Brady, A. F., Burrows, N., Cervi, E., Cilliers, D., et al. (2019). Atypical COL3A1 variants (glutamic acid to lysine) cause vascular Ehlers–Danlos syndrome with a consistent phenotype of tissue fragility and skin hyperextensibility. Genet. Med. 21, 2081–2091. doi:10.1038/s41436-019-0470-9
Gu, G., Yang, H., Cui, L., Fu, Y., Li, F., Zhou, Z., et al. (2018). Vascular Ehlers-Danlos syndrome with a novel missense COL3A1 mutation present with pulmonary complications and iliac arterial dissection. Vasc. Endovasc. Surg. 52, 138–142. doi:10.1177/1538574417745432
Gupta, D., Jayashankar, C., Srinivas, M., Baraka Vishwanathan, G., Reddy, K. R., Kubba, A., et al. (2023). Clinical and allelic heterogeneity in dystrophic epidermolysis bullosa-lessons from an Indian cohort. PLoS One 18, e0289558. doi:10.1371/journal.pone.0289558
Horn, D., Siebert, E., Seidel, U., Rost, I., Mayer, K., Abou Jamra, R., et al. (2017). Biallelic COL3A1 mutations result in a clinical spectrum of specific structural brain anomalies and connective tissue abnormalities. Am. J. Med. Genet. A. Part A 173, 2534–2538. doi:10.1002/ajmg.a.38345
Ishiguro, T., Takayanagi, N., Kawabata, Y., Matsushima, H., Yoshii, Y., Harasawa, K., et al. (2009). Ehlers-Danlos syndrome with recurrent spontaneous pneumothoraces and cavitary lesion on chest X-ray as the initial complications. Intern. Med. 48, 717–722. doi:10.2169/internalmedicine.48.1818
Jaganathan, K., Kyriazopoulou Panagiotopoulou, S., McRae, J. F., Darbandi, S. F., Knowles, D., Li, Y. I., et al. (2019). Predicting splicing from primary sequence with deep learning. Cell 176, 535–548. doi:10.1016/j.cell.2018.12.015
Jørgensen, A., Fagerheim, T., Rand-Hendriksen, S., Lunde, P. I., Vorren, T. O., Pepin, M. G., et al. (2015). Vascular Ehlers-Danlos syndrome in siblings with biallelic COL3A1 sequence variants and marked clinical variability in the extended family. Eur. J. Hum. Genet. 23, 796–802. doi:10.1038/ejhg.2014.181
Kamalanathan, K. C., Barnacle, A. M., Holbrook, C., and Rees, C. (2019). Splenic rupture secondary to vascular Ehlers–Danlos syndrome managed by coil embolization of the splenic artery. Eur. J. Pediatr. Surg. Rep. 7, e83–e85. doi:10.1055/s-0039-3399555
Kroes, H. Y., Pals, G., and van Essen, A. J. (2003). Ehlers–Danlos syndrome type IV: unusual congenital anomalies in a mother and son with a COL3A1 mutation and a normal collagen III protein profile. Clin. Genet. 63, 224–227. doi:10.1034/j.1399-0004.2003.00047.x
Lan, N. S. R., Fietz, M., Pachter, N., Paul, V., and Playford, D. (2018). A case of vascular Ehlers-Danlos Syndrome with a cardiomyopathy and multi-system involvement. Cardiovasc. Pathol. 35, 48–51. doi:10.1016/j.carpath.2018.04.006
Lee, S. T., Kim, J. A., Jang, S. Y., Kim, D. K., Kim, J. W., and Ki, C. S. (2008). A novel COL3A1 gene mutation in patient with aortic dissected aneurysm and cervical artery dissections. Heart Vessels 23, 144–148. doi:10.1007/s00380-007-1027-4
Lipinski, M. J., Lipinski, S. E., Kripalani, S., Friesen, L. D., Uthlaut, B. S., and Braddock, S. R. (2009). An unusual presentation of Ehlers-Danlos syndrome vascular type with deep vein thrombosis: a case for multidisciplinary management. Am. J. Med. Genet. A 149A, 698–701. doi:10.1002/ajmg.a.32687
Lumi, X., Bergant, G., Lumi, A., and Mahnic, M. (2021). Outcomes of vitrectomy for retinal detachment in a patient with Ehlers–Danlos syndrome type IV: a case report. J. Med. Case Rep. 15, 249. doi:10.1186/s13256-021-02855-w
Marini, J. C., Forlino, A., Cabral, W. A., Barnes, A. M., San Antonio, J. D., Milgrom, S., et al. (2007). Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum. Mutat. 28, 209–221. doi:10.1002/humu.20429
Nakagawa, H., Wada, H., Hajiro, T., Nagao, T., Ogawa, E., Hatamochi, A., et al. (2015). Ehlers-Danlos syndrome type IV with bilateral pneumothorax. Intern. Med. 54, 3181–3184. doi:10.2169/internalmedicine.54.4947
Narcisi, P., Richards, A. J., Ferguson, S. D., and Pope, F. M. (1994). A family with Ehlers-Danlos syndrome type III/articular hypermobility syndrome has a glycine 637 to serine substitution in type III collagen. Hum. Mol. Genet. 3, 1617–1620. doi:10.1093/hmg/3.9.1617
Nuytinck, L., De Paepe, A., Renard, J. P., Adriaens, F., and Leroy, J. (1994). Single-strand conformation polymorphism (SSCP) analysis of the COL3A1 gene detects a mutation that results in the substitution of glycine 1009 to valine and causes severe Ehlers–Danlos syndrome type IV. Hum. Mutat. 3, 268–274. doi:10.1002/humu.1380030315
Ohyama, Y., Iso, T., Niño, A. C. V., Obokata, M., Takahashi, R., Okumura, W., et al. (2010). Multiple spontaneous coronary artery ruptures and cardiac tamponade in vascular Ehlers-Danlos syndrome. J. Cardiol. Cases 3, e29–e32. doi:10.1016/j.jccase.2010.09.002
Palmeri, S., Mari, F., Meloni, I., Malandrini, A., Ariani, F., Villanova, M., et al. (2003). Neurological presentation of Ehlers-Danlos syndrome type IV in a family with parental mosaicism. Clin. Genet. 63, 510–515. doi:10.1034/j.1399-0004.2003.00075.x
Pejaver, V., Byrne, A. B., Feng, B. J., Pagel, K. A., Mooney, S. D., Karchin, R., et al. (2022). Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am. J. Hum. Genet. 109, 2163–2177. doi:10.1016/j.ajhg.2022.10.013
Pepin, M., Schwarze, U., Superti-Furga, A., and Byers, P. H. (2000). Clinical and genetic features of Ehlers–Danlos syndrome type IV, the vascular type. N. Engl. J. Med. 342, 673–680. doi:10.1056/NEJM200003093421001
Rebelo, M., Ramos, L., Lima, J., Vieira, J. D., Tavares, P., Teixeira, L., et al. (2011). Ehlers-Danlos syndrome type IV in association with a (c.970G>A) mutation in the COL3A1 gene. Acta Medica Port. 24, 1079–1086. doi:10.20344/amp.1405
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and Genomics and the association for molecular Pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Ruscitti, F., Trevisan, L., Rosti, G., Gotta, F., Cianflone, A., Geroldi, A., et al. (2021). A novel mutation in COL3A1 associates to vascular Ehlers–Danlos syndrome with predominant musculoskeletal involvement. Mol. Genet. Genom. Med. 9, e1753. doi:10.1002/mgg3.1753
Sadakata, R., Hatamochi, A., Kodama, K., Kaga, A., Yamaguchi, T., Soma, T., et al. (2010). Ehlers-Danlos syndrome type IV, vascular type, which demonstrated a novel point mutation in the COL3A1 gene. Intern. Med. 49, 1797–1800. doi:10.2169/internalmedicine.49.3435
Sakai, K., Toda, M., Kyoyama, H., Nishimura, H., Kojima, A., Kuwabara, Y., et al. (2019). Vascular Ehlers-Danlos syndrome with a novel missense mutation in COL3A1: a man in his 50s with aortic dissection after interventional treatment for hemothorax as the first manifestation. Intern. Med. 58, 3441–3447. doi:10.2169/internalmedicine.2983-19
Vandervore, L., Stouffs, K., Tanyalçin, I., Vanderhasselt, T., Roelens, F., Holder-Espinasse, M., et al. (2017). Bi-allelic variants in COL3A1 encoding the ligand to GPR56 are associated with cobblestone-like cortical malformation, white matter changes and cerebellar cysts. J. Med. Genet. 54, 432–440. doi:10.1136/jmedgenet-2016-104421
Xu, Y., Li, L., Wang, C., Yue, H., Zhang, H., Gu, J., et al. (2020). Clinical and molecular characterization and discovery of novel genetic mutations of Chinese patients with col2a1-related dysplasia. Int. J. Biol. Sci. 16, 859–868. doi:10.7150/ijbs.38811
Yang, F., Yang, R. J., Li, Q., Zhang, J., Meng, Y. X., Liu, X. J., et al. (2022). Whole-exome sequencing facilitates the differential diagnosis of Ehlers-Danlos syndrome (EDS). Mol. Genet. Genom. Med. 10, e1885. doi:10.1002/mgg3.1885
Keywords: vascular Ehlers–Danlos syndrome, vascular rupture, COL3A1, novel mutation, genotype–phenotype correlations
Citation: Wei X, Zhou X, Xie B, Shi M, Gui C, Liu B, Li C, Zhang C, Luo J, Mi C and Gui B (2023) Importance of comprehensive genetic testing for patients with suspected vascular Ehlers–Danlos syndrome: a family case report and literature review. Front. Genet. 14:1246712. doi: 10.3389/fgene.2023.1246712
Received: 24 June 2023; Accepted: 04 December 2023;
Published: 20 December 2023.
Edited by:
Tomoki Kosho, Shinshu University, JapanReviewed by:
Samira Kalayinia, Shaheed Rajaei Cardiovascular Medical and Research Center, IranTomomi Yamaguchi, Shinshu University Hospital, Japan
Copyright © 2023 Wei, Zhou, Xie, Shi, Gui, Liu, Li, Zhang, Luo, Mi and Gui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Baoheng Gui, QmFvaGVuZ0d1aUB5ZWFoLm5ldA==; Cundong Mi, bWQtMzkyQDEyNi5jb20=; Jiefeng Luo, ZHJsamY5OEAxNjMuY29t
†These authors have contributed equally to this work