 Hongwei Guo
Hongwei Guo Guoyan Zhang1,2
Guoyan Zhang1,2 Bo Zhu
Bo Zhu Zujun Yang
Zujun Yang Zixian Zeng
Zixian Zeng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 25 July 2023
Sec. Genomics of Plants and the Phytoecosystem
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1241201
Polyploidization plays important roles in the evolution and breeding of the common wheat. Aegilops tauschii, the D-genome progenitor of the common wheat, provides a valuable pool of resistance genes to multiple diseases. Extensive studies focus on the exploration of these genes for wheat improvement. However, few studies have unveiled alternations on genome-wide expression pattern and histone modifications induced by whole-genome doubling (WGD) process. In this study, we conducted transcriptome analysis for the diploid and tetraploid Ae. taushcii lines using the leaf and root tissues. Both lines tend to display similar tissue-specific pattern. Interestingly, we found that TEs located in genic regions were depleted of the repressive histone mark H3K27me3, whereas their adjacent chromatin was enriched with H3K27me3. The tetraploid line exhibited higher levels of H3K27me3 in those regions than the diploid line, particularly for genic regions associated with TEs of the long interspersed nuclear elements (LINEs), CACTA, PIF/Harbinger, Tc1/Mariner and unclassed DNA transposon. Surprisingly, the expression levels of these TEs cognate genes were negatively associated with the levels of H3K27me3 between the tetraploid and diploid lines, suggesting the five types of TEs located within genic regions might be involved in the regulation of the ploidy-related gene expression, possibly through differential enrichment of H3K27me3 in the genic regions. These findings will help to understand the potential role of specific types of TEs on transcription in response to WGD.
Autopolyploids and allopolyploids are considered to be arose from the WGD within a single species and through the merging of genomes from difference species followed by doubling, respectively (Parisod et al., 2010a; Van de Peer et al., 2017). A considerable number of allopolyploid plant species have been investigated, including hexaploid bread wheat (Zhang et al., 2014), Brassica juncea (Yang et al., 2016), cotton (Chen Z. J. et al., 2020) and strawberry (Edger et al., 2019). However, studies on autopolyploid plant species are limited to few species (Song et al., 2020; Wang et al., 2021; Guo et al., 2023).
WGD-induced gene expression changes were relatively mild, limited to a few hundred genes in tetraploid potato (Guo et al., 2023) and Arabidopsis thaliana (Yu et al., 2010), although the tetraploid derivatives from both species exhibited typical changes in phenotypes, including enlarged tissues. WGD-induced variation in gene expression pattern is affected by multiple factors, such as ploidy level (Auger et al., 2005), DNA sequence alternation (Chen Z. J. et al., 2020), histone modification (Guo et al., 2023), DNA methylation (Wang et al., 2021), as well as TE status (Gill et al., 2021). TEs take effect on the expression of their adjacent genes by various ways, including transposition and production of short interfering RNAs (siRNA) (Wang et al., 2013; Gill et al., 2021). Insertion of TE into genic region may result in aberrant or novel transcripts, while transposition of TE into promoter may disrupt the function of promoter or produce alternative promoter leading to new expression pattern (Hirsch and Springer, 2017). In addition, studies on epigenetic modifications associated with TEs mainly focused on DNA methylation (Madlung et al., 2005; Parisod et al., 2009; Zhang et al., 2015). However, the association of TEs in genic regions with WGD-induced enrichment of histone modifications in their cognate genes, as well as the resulting differential gene expression between ploidies remain unraveled.
Ae. tauschii (2n = 2x = 14, DD) is the D-genome progenitor of the hexaploid wheat (2n = 6x = 42, AABBDD) (Van Slageren, 1994) and its genome contains a large number of TEs (84.4%) (Luo et al., 2017). As a wild species, Ae. tauschii serves as an important germplasm for wheat improvement, since it contains a number of novel genes resistant to multiple diseases (Cox et al., 2017; Gaurav et al., 2022), such as rust (Lin et al., 2022) and power mildew (Xue et al., 2022). In addition, the synthetic tetraploid Ae. tauschii will also be useful as a bridge to generate hybrids or amphiploids by crossing with other tetraploid Triticeae species for wheat breeding (Wang et al., 2010; Mizuno et al., 2011). Thus, investigation of gene expression patterns associated with ploidy in Ae. tauschii may provide clues for breeding targets. In this study, we utilized previously developed diploid and tetraploid Ae. tauschii and conducted transcriptomes for both leaf and root tissues for each ploidy. Both diploid and tetraploid lines tend to display similar tissue-specific pattern. Interestingly, TEs located in the genic regions were depleted of H3K27me3, whereas their adjacent chromatin was enriched with H3K27me3. The tetraploid line displayed higher levels of H3K27me3 in those regions than the diploid line. In particular, we found that the genic regions associated with five types of TEs showed significantly higher levels of H3K27me3 in the tetraploid line compared with those in the diploid line. Surprisingly, the expression levels of these TEs cognate genes were negatively associated with the levels of H3K27me3 between the tetraploid and diploid lines, suggesting the five types of TEs located within genic regions might be involved in the regulation of the ploidy-related gene expression, possibly through differential enrichment of H3K27me3 in the genic regions.
The diploid (2n = 2x = 14) and tetraploid (2n = 4x = 28) Ae. Tauschii, provided by Professor Huaren Jiang, were grown in a walk-in growth chamber under the photoperiod of 16 h at 24°C daylight and 8 h at 22°C darkness. Both leaves and roots were harvested 60 days after germination, respectively, which were subject to RNA-seq and ChIP-seq experiments or immediately frozen in liquid nitrogen.
Both leaves and roots were used for RNA-seq and ChIP-seq, respectively. Each tissue from three individual plants were pooled together as a biological replicate for each line. Two biological replicates of RNA-seq libraries were developed and sequenced using an Illumina NovaSeq 6000 platform with the mode of 150 pair end (PE) sequencing. All RNA-seq libraries were developed and sequenced in Novagene company.
According to the published protocol (Zhang et al., 2012), the ChIP experiments were performed with the antibody against H3K27me3 (Millipore 07–449). Chromatin was digested into monomer nucleosome pattern (∼150 bp fragments) using MNase (Sigma N3755) to obtain the highest resolution of the histone modification signal. Chromatin in monomer nucleosome pattern carrying H3K27me3 were captured and precipitated using rProtein A Sepharose beads (GE 17-1279-01), followed by ChIP-DNA separation. The isolated ChIP-DNA was applied for library construction, which was subsequently sequenced using the same method as the RNA-seq libraries in Novagene company.
Raw reads generated from RNA-seq and ChIP-seq were first processed for quality control and adapter trimming using the program fsatp v0.32.2 with “-w 8” (Chen et al., 2018). Clean RNA-seq reads were mapped to the Ae. tauschii genome assembly (Ensembl v4.0) (Luo et al., 2017), using Hisat2 (Kim et al., 2019). Reads with mapping quality greater than 50 were retained for further analysis. The expression of all annotated genes was called using StringTie v2.1.5 (Pertea et al., 2015) and the differentially expression genes were identified using DESeq2 v1.32.0 (Love et al., 2014) with FDR <0.01, log2(Fold Change) > 1. The g:Profiler program (https://biit.cs.ut.ee/gprofiler/gost) was used for the gene ontology enrichment analysis. The statistical test for the reproducibility of RNA-seq data between biological replicates was conducted using Pearson correlation.
Clean ChIP-seq reads were mapped to the Ae. tauschii genome assembly, using the program of BWA “mem” with default parameters. The mapped reads were further filtered with mapping quality greater than 50. The uniquely mapped reads (mapped to a unique genomic position) were obtained and processed for downstream analysis. The histone modification signal was defined as the mid-point of the uniquely mapped paired reads. Quantification of the level of a histone modification within an interval was conducted by summarizing histone modification signals and normalizing to length of the interval, PE read number per million uniquely mapped reads and IgG. A histone modification enriched region was identified using the program MACS2 (Zhang et al., 2008) with -q 0.05. Statistical significance was tested using Wilcoxon signed-rank test with paired samples.
Repetitive sequences of the Ae. tauschii genome were downloaded from http://aegilops.wheat.ucdavis.edu/ATGSP/annotation/. Genomic distribution of TEs relative to the annotated genes was determined if more than half length of TEs is overlapped with a genomic feature.
All data were visualized using R program (https://www.r-project.org).
The tetraploid Ae. tauschii line (2n = 4x = 28, DDDD) was previously regenerated from the diploid Ae. tauschii (Zeng et al., 2012), one of the progenitors of the common wheat. The tetraploid line was associated with no visible structural variations on the chromosome level (Zeng et al., 2012), suggesting its genome tends to be homozygous at certain degree. However, typical phenotypic polymorphisms, which are frequently identified in other plant species (Miller et al., 2012; Saminathan et al., 2015), were found between two ploidies, including larger but less leaves (Supplementary Figure S1), as well as larger seeds (Zeng et al., 2012). Thus, we utilized the tetraploid derivative and its diploid progenitor for the transcriptome analysis.
We conducted RNA-seq for both root and leaf tissues of the tetraploid Ae. tauschii. The RNA-seq data between biological replicates were highly correlated (R2 = 0.97, p < 2.2e-16) (Supplementary Figure S2A; Supplementary Table S1). In comparison with leaves, a total of 6,136 genes were significantly upregulated in roots, while 5,660 genes were downregulated (FDR<0.01, log2 Fold Change (FC) > 1) (Figure 1A). The root upregulated genes were expressed at low levels in leaves (with median TPM = 0.4), which substantially differed in roots with median TPM of 16.1 (Figure 1B). In contrast, the differences in the expression levels of the root downregulated genes between leaves (with median TPM = 25.2) and roots (with median TPM of 6.2) were relatively minor (Figure 1B). This result indicates that the root upregulated genes might be associated with functions specific to root. Gene Ontology (GO) analysis revealed that the root upregulated genes were significantly enriched in terms related to hydrogen peroxide catabolic process, cell cycle and cell division (Figure 1C). The concentration of hydrogen peroxide in the root differentiation zone and the cell wall of root hairs affect the root elongation and root hair formation (Dunand et al., 2007). In addition, hydrogen peroxide was also found to be involved in response to abiotic stresses in roots (Hernandez et al., 2010). These functions are consistent with the root-specific upregulation of the genes. As expected, the root downregulated genes (that is leaf upregulated genes) were mainly associated with functions of photosynthesis and metabolic process (Figure 1D).
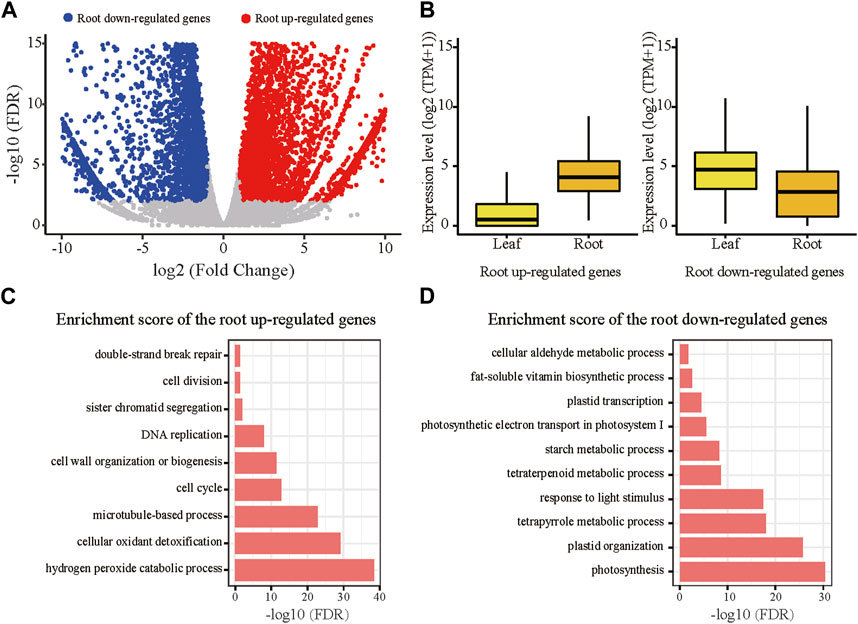
FIGURE 1. Differentially expressed genes (DEGs) between the root and leaf tissues in the tetraploid line. (A) Upregulated genes in root were marked with red color; downregulate genes in root were marked with blue color; non-DEGs were marked with gray color. DEGs were defined with the following criteria: FDR<0.01, fold change >1. (B) Expression levels of the root upregulated and downregulated genes in each tissue of the tetraploid line. (C) Significantly enriched GO terms of the root upregulated genes. (D) Significantly enriched GO terms of the root downregulated genes.
Additionally, we conducted the transcriptome analysis for the diploid line (Supplementary Figure S2B; Supplementary Table S1) and revealed similar results as we found for the tetraploid line (Supplementary Figure S3). We also found 6,863 and 5,621 upregulated and downregulated genes in roots, respectively, compared with leaves. These results collectively suggest that both ploidies tend to be associated with similar tissue-specific expression pattern.
Comparison of the tissue-differentially expressed genes (DEGs) between the diploid and tetraploid lines revealed that 77.1% and 86.2% of the root upregulated genes (5,290) displayed the same direction of the tissue-specific differential expression in the diploid and tetraploid lines, respectively (Figure 2A). In contrast, only a small number of the root upregulated genes were specific to the diploid (1,570) and tetraploid lines (884) (Figure 2A). It is noticed that the diploid-specific root upregulated genes (1,570), which were expressed at substantially higher levels in roots than in leaves of the diploid line, also displayed higher expression levels in roots than in leaves of the tetraploid line (Figure 2B). A similar pattern was found for the tetraploid-specific root upregulated genes (884). However, the expression levels of the root downregulated genes were not consistently higher in leaves than in roots (Figure 2C). It is indicated that the root upregulated genes tend to transcribe constantly in both ploidies.
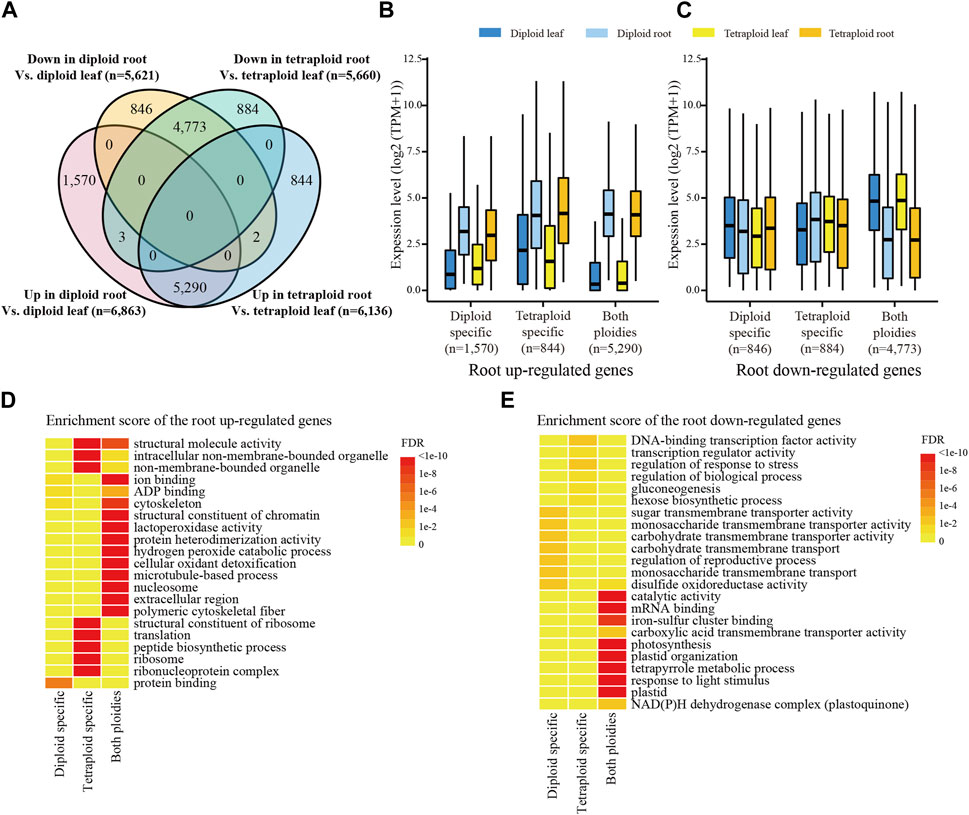
FIGURE 2. Ploidy-related expression in each tissue. (A) Venn diagram of the tissue-DEGs between two ploidies. Each ellipse represents the number of the DEGs between the root and leaf tissues of a given Ae. tauschii line. “Up” and “down” represents up- and downregulated genes, respectively. (B) The expression levels of the root upregulated genes in each tissue of the diploid and tetraploid lines. (C) The expression levels of the root downregulated genes in each tissue of the diploid and tetraploid lines. (D) Significantly enriched GO terms of the root upregulated genes. (E) Significant enriched GO terms of the root downregulated genes.
The GO analysis revealed that the majority of the root upregulated genes from both ploidies were enriched in terms associated with cytoskeleton and hydrogen peroxide catabolic process (Figure 2D), which are frequently involved in plant root growth (Dunand et al., 2007; Becker et al., 2014; Takatsuka and Ito, 2020) and response to salt stress (Wang et al., 2011; Chun et al., 2021). The tetraploid-specific root upregulated genes were associated with functions mainly related to ribosome and peptide biosynthetic process (Figure 2D). The peptides, such as the CLE family, are reported to have a role in root apical meristem maintenance, root hair development and lateral root development (Tavormina et al., 2015). Thus, upregulation of the genes related to protein synthesis and its apparatus in the tetraploid line indicates that the tetraploid line might be associated with vigorous roots. The root downregulated genes in both ploidies, however, were mainly associated with functions related to photosynthesis (Figure 2E). The tetraploid-specific root downregulated genes were mainly enriched in stress responses and sugar biosynthesis (Figure 2E). Enhanced stress responses were also found in other polyploid plant species (Dai et al., 2015; Guo et al., 2023), while upregulation of hexose synthesis and gluconeogenesis related genes was observed in sugarcane with high biomass (Wai et al., 2017), suggesting that the tetraploid line might be associated with better tolerance to abiotic stress and biomass production. The diploid-specific root downregulated genes were associated with carbohydrate transmembrane transport activity (Figure 2E), which is involved in enhancing photosynthesis (Ainsworth and Bush, 2011).
TEs have been reported to play a role in alternation of gene expression (Gill et al., 2021). To investigate if TEs and their underlying H3K27me3 are potentially associated with the difference in gene expression between two ploidies, we conducted ChIP-seq using antibody against H3K27me3 (Supplementary Table S2). A total of 3,452,972 TEs were identified in the genome. The majority of the TEs were distributed in the intergenic regions, while a noticeable proportion of TEs were located within genic regions (4.5% for Class I and 6.6% for Class II) (Supplementary Figure S4A). The integration of TEs in the genic regions indicates that they might potentially affect the expression of their cognate genes.
The analysis of H3K27me3 revealed that the diploid and tetraploid lines showed generally similar levels of H3K27me3 in TEs located in genic regions as well as the gene upstream regions (2 kb) in leaf tissue (Supplementary Figure S4B). In contrast, in root tissue, the tetraploid line displayed significantly lower levels of H3K27me3 in TEs in the corresponding regions compared with the diploid line (Supplementary Figure S4C). Interestingly, the genomic regions harboring TEs, including the genic regions, 2 kb upstream and 2 kb downstream regions, showed substantially higher levels of H3K27me3 in roots from the tetraploid line than those from the diploid line (Figures 3A–C), suggesting that the flanking regions of TEs within these genomic regions are enriched with H3K27me3 in the tetraploid line. In addition, these genome regions carrying TEs displayed higher levels of H3K27me3 than those without any TEs (Figures 3A–C), indicating TEs might play a role in the deposition of H3K27me3 to the flanking regions of their cognate genes, particularly for the tetraploid line. Dissection of TEs from the flanking regions confirmed that non-TE sequences within the genic regions as well as the upstream and downstream regions were enriched with H3K27me3 in the tetraploid line (Figures 3D–F).
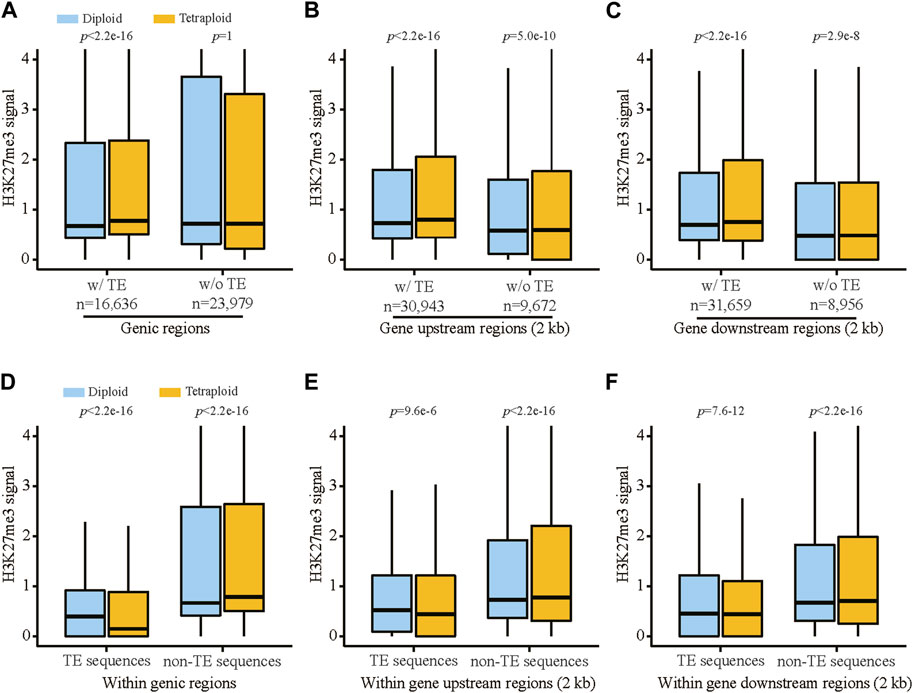
FIGURE 3. The levels of H3K27me3 associated with TEs and their adjacent non-TE sequences in the vicinity of genes in root tissue of the diploid and tetraploid lines. H3K27me3 signals in genic regions (A), gene 2 kb upstream regions (B) and gene 2 kb downstream regions (C) associated with and without TEs. H3K27me3 signals in TEs and non-TE sequences located within genic regions (D), gene 2 kb upstream regions (E) and gene 2 kb downstream regions (F). Statistical significance was tested using Wilcoxon signed-rank test with paired samples.
Further analysis for subclasses of TEs located in genic regions showed that all types of TEs were depleted of H3K27me3 compared with their flanking regions (Figure 4; Supplementary Figure S5). Interestingly, non-TE regions adjacent to LINEs, CACTA, PIF/Harbinger, Tc1/Mariner and unclassed DNA transposon in the tetraploid line were associated with significantly higher levels of H3K27me3 than those in the diploid line (Figure 4), which is coincident with the lower expression levels of their cognate genes in the tetraploid line compared with those in the diploid line (Supplementary Figure S6). The non-TE regions adjacent to the remaining types of TEs displayed similar levels of H3K27me3 between two ploidies (Figure 4), while their corresponding genes were generally expressed at similar levels (Supplementary Figure S6). It is indicated that LINE, CACTA, PIF/Harbinger, Tc1/Mariner and unclassed DNA transposon located within genic regions might be involved in the regulation of the ploidy-related gene expression, possibly through differential enrichment of H3K27me3 in the genic regions.
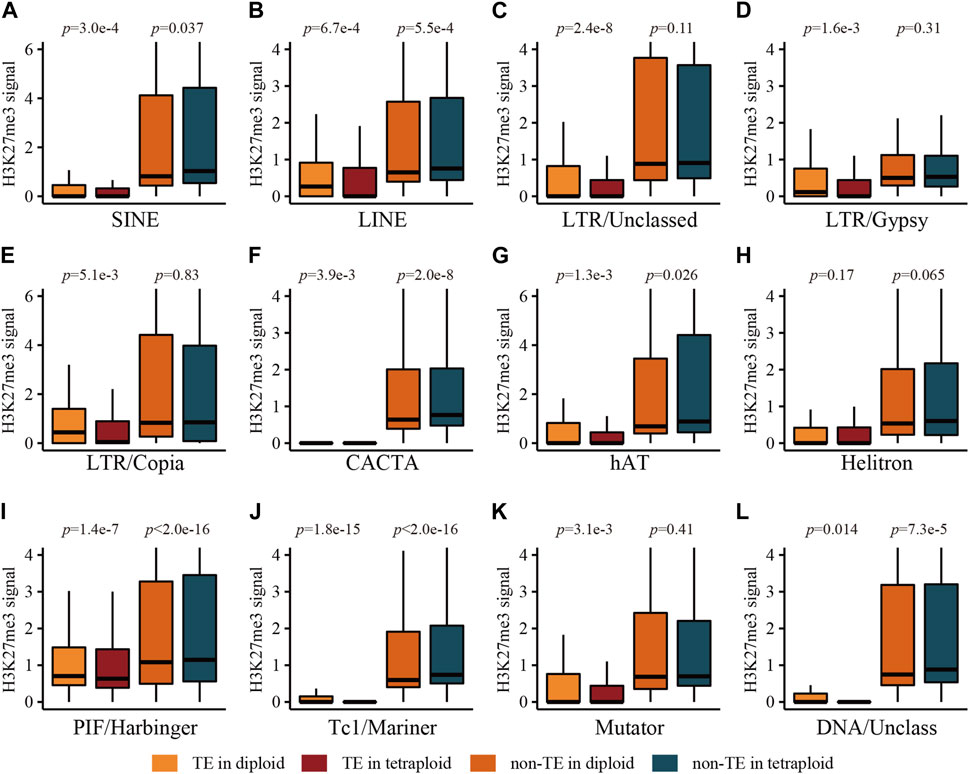
FIGURE 4. The levels of H3K27me3 associated with main types of TEs and their adjacent non-TE sequences in genic regions in root tissue of the diploid and tetraploid lines. (A) SINE, (B) LINE, (C) LTR/Unclassed, (D) LTR/Gypsy, (E) LTR/Copia, (F) CACTA, (G) hAT, (H) Helitron, (I) PIF/Harbinger, (J) Tc1/Mariner, (K) Mutator, (L) DNA/Unclassed. Statistical significance was tested using Wilcoxon signed-rank test with paired samples.
GO analysis revealed that these TE-associated genes were enriched in various terms of biological processes, including glycolysis, RNA phosphodiester bond hydrolysis, positive regulation of transcription and proteolysis (Supplementary Figure S7). Upregulation of genes associated with glycolysis has been found in roots from multiple genotypes of Banana (Musa spp.) (Zorrilla-Fontanesi et al., 2016), presumably involved in higher oxidative respiration. The GO result suggests that the tetraploid line might be associated dynamic energy shift and RNA/protein metabolism. In addition, we extracted the top10 differentially H3K27me3-enriched DEGs (tetraploid line Vs diploid line) carrying these five types of TEs, respectively (Supplementary Table S3). The majority of these genes were likely involved root growth and stress responses in roots, suggesting that the root- and stresses-related differential expression might be regulated with the involvement of H3K27me3 between the tetraploid and diploid lines.
WGD usually induces phenotypic changes, typically including enlarged leaves and seeds, increased stem diameter and changed plant architecture. Similar to other plant species (Chen, P. et al., 2020; Guo et al., 2023; Wang et al., 2021; Yu et al., 2010), the tetraploid Ae. tauschii line mainly displayed larger leaves and seeds than the diploid line (Zeng et al., 2012). However, the DEGs between two ploidies were limited to a few hundred or less. Interestingly, the ploidy-related DEGs in roots (n = 134, 0.33% of 40,615) were substantially less than those in leaves (n = 528, 1.3% of 40,165). Consistent trend was also reported in previous studies in potato, where the DEGs (n = 27–158) in tubers between the homozygous diploid and tetraploid Solanum phureja were only 0.07–0.4% of the total genes (39,400) (Guo et al., 2023), whereas the DEGs (n = 2,652–3,661) in leaves from the other homozygous diploid and tetraploid potato S. commersonii accounted for 6.7%–9.3% of the total genes (Fasano et al., 2016). GO analysis for the tetraploid Ae. tauschii line upregulated genes revealed that roots were associated with the terms related to conserved function compared with the leaves which were enriched in more specific processes (Supplementary Figure S8). Therefore, it is suggested that the gene expression in Ae. tauschii roots might not be affected by ploidy level as much as that in leaves. In addition, similar to potato tubers, roots are non-photosynthesis tissue (Henry et al., 2020), which may be associated with less influences from ambient environment compared with the up-ground tissues.
Immediate WGD from somatic cells likely results in very limited genomic changes in the current generation (Guo et al., 2023). Thus, alternation of gene expression pattern between different ploidies seems to be associated with epigenetic changes. Previous study in rice suggests that WGD increased methylation levels in class II transposable elements, suppressing the genome-wide expression levels of nearby genes (Zhang et al., 2015). Similar evidence was also documented in Spartina anglica genome, where TEs were associated with frequent methylation changes (Parisod et al., 2010a), likely leading to transcriptional changes in the vicinity of TEs following allopolyploidization (Parisod et al., 2010b; Parisod et al., 2009). However, studies on WGD-induced epigenetic changes on TEs and their potential effects on adjacent genes are relatively sporadic, while the majority of these studies only focused on DNA methylation (Madlung et al., 2005; Parisod et al., 2009; Zhang et al., 2015). In this study, we attempted to interrogate histone modification features associated with TEs between the diploid and tetraploid Ae. tauschii lines. We found genes containing TEs generally displayed higher levels of H3K27me3 in roots, compared with those without TEs, regardless of the ploidy level (Figures 3A–C), indicating that TEs might mediate the deposition of H3K27me3 to their adjacent regions. Interestingly, the TEs located in genic regions were associated with lower levels of H3K27me3 in the tetraploid line compared with those in the diploid line, while the adjacent regions of the TEs within genic regions were associated with higher levels of H3K27me3 in the tetraploid line than those in the diploid line (Figures 3D–F). H3K27me3 and DNA methylation are generally thought to be mutually exclusive (Rougee et al., 2021) and presumably TEs are associated with elevated DNA methylation levels in Ae. tauschii (Zhao et al., 2017). Thus, the genic TEs in the tetraploid line might be associated with higher level of DNA methylation than those in the diploid line. Hypermethylation of the TEs following WGD in rice not only suppress the expression of their nearby genes, but also stabilize the genomic structure (Zhang et al., 2015), implying that the regions containing the TEs in the tetraploid Ae. tauschii line might be less dynamic in root tissue.
Dissection of TE types revealed that non-TE regions adjacent to five types of TEs in the tetraploid line displayed higher levels of H3K27me3 than those in the diploid line (Figure 4). In addition, the genes containing those types of TEs were generally expressed at lower levels in the tetraploid lines than those in the diploid line (Supplementary Figure S6). The enrichment of H3K27me3 was evidenced in the flanking regions of TEs in Arabidopsis, possibly for the formation of heterochromatin and suppression of gene expression (Dong et al., 2012). The differential enrichment of H3K27me3 in these TE-associated genes between the diploid and tetraploid Ae. tauschii line may play a role in the ploidy-related gene expression. Collectively, the TEs, including LINE, CACTA, PIF/Harbinger, Tc1/Mariner and unclassed DNA transposon, might be involved in the deposition of H3K27me3 in their adjacent regions for the suppression of the cognate genes. However, this speculation requires further molecular evidence.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
ZZ and BZ conceived the idea; ZZ and DZ supervised for lab experiments; BZ and DZ provided funding for experiments; ZZ supervised the data analysis; HG and MZ performed the analysis; HG drew figures and wrote the first draft; GZ and MW performed lab experiments; ZY helped in manuscript preparation and discussion; ZZ, DZ, and ZY were involved in the final development and scientific proofreading of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Natural Science Foundation of China (31701060 to BZ), Sichuan Science and Technology Program (2021YFH0114 to DZ), State Key Laboratory of Crop Biology Open Fund (2020KF01 to BZ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1241201/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | The phenotypes of the diploid and tetraploid Ae. tauschii lines. (A) The diploid line. (B) The tetraploid line. Scale bar = 5 cm.
SUPPLEMENTARY FIGURE S2 | Correlation of expression levels between two biological replicates derived from the diploid line (A) and the tetraploid line (B), respectively. The Pearson correlation was measured using TPM values of 43,362 annotated Ae. tauschii genes.
SUPPLEMENTARY FIGURE S3 | DEGs between the root and leaf tissues in the diploid line. (A) Up-regulated genes in root were marked with red color; downregulate genes in root were marked with blue color; non-DEGs were marked with gray color. DEGs were defined with the following criteria: FDR < 0.01, fold change >1. (B) Expression levels of the root up-regulated and down-regulated genes in each tissue of the diploid line. (C) Significantly enriched GO terms of the root up-regulated genes. (D) Significantly enriched GO terms of the root down-regulated genes.
SUPPLEMENTARY FIGURE S4 | Genomic distribution of TEs identified in the Ae. tauschii genome. (A) Genomic distribution of TEs relative to the annotated genes. Class I and Class II represent retrotransposons and transposons, respectively. TE was assigned to a genomic feature if more than half length of TEs is overlapped with a genomic feature. Upstream indicates the region 2 kb upstream from a gene. Downstream indicates the region 2 kb downstream from a gene. Intergenic refers to the region 2 kb away from any genes. (B) The levels of H3K27me3 associated with TEs located in various genomic regions in leaf tissue. (C) The levels of H3K27me3 associated with TEs located in various genomic regions in root tissue. Statistical significance was tested using Wilcoxon signed-rank test with paired samples.
SUPPLEMENTARY FIGURE S5 | The profile of H3K27me3 associated with TEs located in genic regions. (A) SINE, (B) LINE, (C) LTR/Unclassed, (D) LTR/Gypsy, (E) LTR/Copia, (F) CACTA, (G) hAT, (H) Helitron, (I) PIF/Harbinger, (J) Tc1/Mariner, (K) Mutator, (L) DNA/Unclassed. Genes flanking regions (±1 kb) were analyzed in 50 bins.
SUPPLEMENTARY FIGURE S6 | Comparison of the expression levels of genes containing specific types of TEs between the diploid and tetraploid lines. The expression data was derived from the root tissue. Statistical significance was tested using Wilcoxon signed-rank test with paired samples.
SUPPLEMENTARY FIGURE S7 | Significantly enriched GO terms of the genes containing specific types of TEs.
SUPPLEMENTARY FIGURE S8 | Significantly enriched GO terms of the upregulated genes in the tetraploid line. Leaf represents the upregulated genes in leaf tissue of the tetraploid line in comparison with the leaf tissue of the diploid line. Root represents the upregulated genes in root tissue of the tetraploid line in comparison with the root tissue of the diploid line.
Ainsworth, E. A., and Bush, D. R. (2011). Carbohydrate export from the leaf: A highly regulated process and target to enhance photosynthesis and productivity. Plant Physiol. 155 (1), 64–69. doi:10.1104/pp.110.167684
Auger, D. L., Gray, A. D., Ream, T. S., Kato, A., Coe, E. H., and Birchler, J. A. (2005). Nonadditive gene expression in diploid and triploid hybrids of maize. Genetics 169 (1), 389–397. doi:10.1534/genetics.104.032987
Becker, J. D., Takeda, S., Borges, F., Dolan, L., and Feijo, J. A. (2014). Transcriptional profiling of Arabidopsis root hairs and pollen defines an apical cell growth signature. BMC Plant Biol. 14, 197. doi:10.1186/s12870-014-0197-3
Chen, P., Chen, J., Sun, M., Yan, H., Feng, G., Wu, B., et al. (2020). Comparative transcriptome study of switchgrass (Panicum virgatum L) homologous autopolyploid and its parental amphidiploid responding to consistent drought stress. Biotechnol. Biofuels 13, 170. doi:10.1186/s13068-020-01810-z
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34 (17), i884–i890. doi:10.1093/bioinformatics/bty560
Chen, Z. J., Sreedasyam, A., Ando, A., Song, Q., De Santiago, L. M., Hulse-Kemp, A. M., et al. (2020). Genomic diversifications of five Gossypium allopolyploid species and their impact on cotton improvement. Nat. Genet. 52 (5), 525–533. doi:10.1038/s41588-020-0614-5
Chun, H. J., Baek, D., Jin, B. J., Cho, H. M., Park, M. S., Lee, S. H., et al. (2021). Microtubule dynamics plays a vital role in plant adaptation and tolerance to salt stress. Int. J. Mol. Sci. 22 (11), 5957. doi:10.3390/ijms22115957
Cox, T. S., Wu, J., Wang, S., Cai, J., Zhong, Q., and Fu, B. (2017). Comparing two approaches for introgression of germplasm from Aegilops tauschii into common wheat. Crop J. 5 (5), 355–362. doi:10.1016/j.cj.2017.05.006
Dai, F., Wang, Z., Luo, G., and Tang, C. (2015). Phenotypic and transcriptomic analyses of autotetraploid and diploid mulberry (morus alba L). Int. J. Mol. Sci. 16 (9), 22938–22956. doi:10.3390/ijms160922938
Dong, X., Reimer, J., Gobel, U., Engelhorn, J., He, F., Schoof, H., et al. (2012). Natural variation of H3K27me3 distribution between two Arabidopsis accessions and its association with flanking transposable elements. Genome Biol. 13 (12), R117. doi:10.1186/gb-2012-13-12-r117
Dunand, C., Crevecoeur, M., and Penel, C. (2007). Distribution of superoxide and hydrogen peroxide in Arabidopsis root and their influence on root development: Possible interaction with peroxidases. New Phytol. 174 (2), 332–341. doi:10.1111/j.1469-8137.2007.01995.x
Edger, P. P., Poorten, T. J., VanBuren, R., Hardigan, M. A., Colle, M., McKain, M. R., et al. (2019). Origin and evolution of the octoploid strawberry genome. Nat. Genet. 51 (3), 541–547. doi:10.1038/s41588-019-0356-4
Fasano, C., Diretto, G., Aversano, R., D'Agostino, N., Di Matteo, A., Frusciante, L., et al. (2016). Transcriptome and metabolome of synthetic Solanum autotetraploids reveal key genomic stress events following polyploidization. New Phytol. 210 (4), 1382–1394. doi:10.1111/nph.13878
Gaurav, K., Arora, S., Silva, P., Sanchez-Martin, J., Horsnell, R., Gao, L., et al. (2022). Population genomic analysis of Aegilops tauschii identifies targets for bread wheat improvement. Nat. Biotechnol. 40 (3), 422–431. doi:10.1038/s41587-021-01058-4
Gill, R. A., Scossa, F., King, G. J., Golicz, A. A., Tong, C., Snowdon, R. J., et al. (2021). On the role of transposable elements in the regulation of gene expression and subgenomic interactions in crop genomes. Crit. Rev. Plant Sci. 40 (2), 157–189. doi:10.1080/07352689.2021.1920731
Guo, H., Zhou, M., Zhang, G., He, L., Yan, C., Wan, M., et al. (2023). Development of homozygous tetraploid potato and whole genome doubling-induced the enrichment of H3K27ac and potentially enhanced resistance to cold-induced sweetening in tubers. Hortic. Res. 10 (3), uhad017. doi:10.1093/hr/uhad017
Henry, R. J., Furtado, A., and Rangan, P. (2020). Pathways of photosynthesis in non-leaf tissues. Biol. (Basel) 9 (12), 438. doi:10.3390/biology9120438
Hernandez, M., Fernandez-Garcia, N., Diaz-Vivancos, P., and Olmos, E. (2010). A different role for hydrogen peroxide and the antioxidative system under short and long salt stress in Brassica oleracea roots. J. Exp. Bot. 61 (2), 521–535. doi:10.1093/jxb/erp321
Hirsch, C. D., and Springer, N. M. (2017). Transposable element influences on gene expression in plants. Biochim. Biophys. Acta Gene Regul. Mech. 1860 (1), 157–165. doi:10.1016/j.bbagrm.2016.05.010
Kim, D., Paggi, J. M., Park, C., Bennett, C., and Salzberg, S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37 (8), 907–915. doi:10.1038/s41587-019-0201-4
Lin, G., Chen, H., Tian, B., Sehgal, S. K., Singh, L., Xie, J., et al. (2022). Cloning of the broadly effective wheat leaf rust resistance gene Lr42 transferred from Aegilops tauschii. Nat. Commun. 13 (1), 3044. doi:10.1038/s41467-022-30784-9
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 550. doi:10.1186/s13059-014-0550-8
Luo, M. C., Gu, Y. Q., Puiu, D., Wang, H., Twardziok, S. O., Deal, K. R., et al. (2017). Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 551 (7681), 498–502. doi:10.1038/nature24486
Madlung, A., Tyagi, A. P., Watson, B., Jiang, H., Kagochi, T., Doerge, R. W., et al. (2005). Genomic changes in synthetic Arabidopsis polyploids. Plant J. 41 (2), 221–230. doi:10.1111/j.1365-313X.2004.02297.x
Miller, M., Zhang, C., and Chen, Z. J. (2012). Ploidy and hybridity effects on growth vigor and gene expression in Arabidopsis thaliana hybrids and their parents. G3 (Bethesda) 2 (4), 505–513. doi:10.1534/g3.112.002162
Mizuno, N., Shitsukawa, N., Hosogi, N., Park, P., and Takumi, S. (2011). Autoimmune response and repression of mitotic cell division occur in inter-specific crosses between tetraploid wheat and Aegilops tauschii Coss. that show low temperature-induced hybrid necrosis. Plant J. 68 (1), 114–128. doi:10.1111/j.1365-313X.2011.04667.x
Parisod, C., Alix, K., Just, J., Petit, M., Sarilar, V., Mhiri, C., et al. (2010a). Impact of transposable elements on the organization and function of allopolyploid genomes. New Phytol. 186 (1), 37–45. doi:10.1111/j.1469-8137.2009.03096.x
Parisod, C., Holderegger, R., and Brochmann, C. (2010b). Evolutionary consequences of autopolyploidy. New phytol. 186 (1), 5–17. doi:10.1111/j.1469-8137.2009.03142.x
Parisod, C., Salmon, A., Zerjal, T., Tenaillon, M., Grandbastien, M. A., and Ainouche, M. (2009). Rapid structural and epigenetic reorganization near transposable elements in hybrid and allopolyploid genomes in Spartina. New Phytol. 184 (4), 1003–1015. doi:10.1111/j.1469-8137.2009.03029.x
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33 (3), 290–295. doi:10.1038/nbt.3122
Rougee, M., Quadrana, L., Zervudacki, J., Hure, V., Colot, V., Navarro, L., et al. (2021). Polycomb mutant partially suppresses DNA hypomethylation-associated phenotypes in Arabidopsis. Life Sci. Alliance 4 (2), e202000848. doi:10.26508/lsa.202000848
Saminathan, T., Nimmakayala, P., Manohar, S., Malkaram, S., Almeida, A., Cantrell, R., et al. (2015). Differential gene expression and alternative splicing between diploid and tetraploid watermelon. J. Exp. Bot. 66 (5), 1369–1385. doi:10.1093/jxb/eru486
Song, M. J., Potter, B. I., Doyle, J. J., and Coate, J. E. (2020). Gene balance predicts transcriptional responses immediately following ploidy change in Arabidopsis thaliana. Plant Cell. 32 (5), 1434–1448. doi:10.1105/tpc.19.00832
Takatsuka, H., and Ito, M. (2020). Cytoskeletal control of planar polarity in root hair development. Front. Plant Sci. 11, 580935. doi:10.3389/fpls.2020.580935
Tavormina, P., De Coninck, B., Nikonorova, N., De Smet, I., and Cammue, B. P. (2015). The plant peptidome: An expanding repertoire of structural features and biological functions. Plant Cell. 27 (8), 2095–2118. doi:10.1105/tpc.15.00440
Van de Peer, Y., Mizrachi, E., and Marchal, K. (2017). The evolutionary significance of polyploidy. Nat. Rev. Genet. 18 (7), 411–424. doi:10.1038/nrg.2017.26
Van Slageren, M. (1994). Wild wheats: A monograph of Aegilops L. And amblyopyrum (jaub. & spach) eig (poaceae). Wageningen: Agricultural University.
Wai, C. M., Zhang, J., Jones, T. C., Nagai, C., and Ming, R. (2017). Cell wall metabolism and hexose allocation contribute to biomass accumulation in high yielding extreme segregants of a Saccharum interspecific F2 population. BMC Genomics 18 (1), 773. doi:10.1186/s12864-017-4158-8
Wang, C.-J., Zhang, L.-Q., Dai, S.-F., Zheng, Y.-L., Zhang, H.-G., and Liu, D.-C. (2010). Formation of unreduced gametes is impeded by homologous chromosome pairing in tetraploid Triticum turgidum× Aegilops tauschii hybrids. Euphytica 175, 323–329. doi:10.1007/s10681-010-0173-4
Wang, C., Zhang, L. J., and Huang, R. D. (2011). Cytoskeleton and plant salt stress tolerance. Plant Signal Behav. 6 (1), 29–31. doi:10.4161/psb.6.1.14202
Wang, L., Cao, S., Wang, P., Lu, K., Song, Q., Zhao, F. J., et al. (2021). DNA hypomethylation in tetraploid rice potentiates stress-responsive gene expression for salt tolerance. Proc. Natl. Acad. Sci. U. S. A. 118 (13), e2023981118. doi:10.1073/pnas.2023981118
Wang, X., Weigel, D., and Smith, L. M. (2013). Transposon variants and their effects on gene expression in Arabidopsis. PLoS Genet. 9 (2), e1003255. doi:10.1371/journal.pgen.1003255
Xue, S., Hu, S., Chen, X., Ma, Y., Lu, M., Bai, S., et al. (2022). Fine mapping of Pm58 from Aegilops tauschii conferring powdery mildew resistance. Theor. Appl. Genet. 135 (5), 1657–1669. doi:10.1007/s00122-022-04061-8
Yang, J., Liu, D., Wang, X., Ji, C., Cheng, F., Liu, B., et al. (2016). The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat. Genet. 48 (10), 1225–1232. doi:10.1038/ng.3657
Yu, Z., Haberer, G., Matthes, M., Rattei, T., Mayer, K. F., Gierl, A., et al. (2010). Impact of natural genetic variation on the transcriptome of autotetraploid Arabidopsis thaliana. Proc. Natl. Acad. Sci. U. S. A. 107 (41), 17809–17814. doi:10.1073/pnas.1000852107
Zeng, Z., Zhang, T., Li, G., Liu, C., and Yang, Z. (2012). Phenotypic and epigenetic changes occurred during the autopolyploidization of Aegilops tauschii. Cereal Res. Commun. 40 (4), 476–485. doi:10.1556/crc.40.2012.0014
Zhang, H., Zhu, B., Qi, B., Gou, X., Dong, Y., Xu, C., et al. (2014). Evolution of the BBAA component of bread wheat during its history at the allohexaploid level. Plant Cell. 26 (7), 2761–2776. doi:10.1105/tpc.114.128439
Zhang, J., Liu, Y., Xia, E. H., Yao, Q. Y., Liu, X. D., and Gao, L. Z. (2015). Autotetraploid rice methylome analysis reveals methylation variation of transposable elements and their effects on gene expression. Proc. Natl. Acad. Sci. U. S. A. 112 (50), E7022–E7029. doi:10.1073/pnas.1515170112
Zhang, W., Wu, Y., Schnable, J. C., Zeng, Z., Freeling, M., Crawford, G. E., et al. (2012). High-resolution mapping of open chromatin in the rice genome. Genome Res. 22 (1), 151–162. doi:10.1101/gr.131342.111
Zhang, Y., Liu, T., Meyer, C. A., Eeckhoute, J., Johnson, D. S., Bernstein, B. E., et al. (2008). Model-based analysis of ChIP-seq (MACS). Genome Biol. 9 (9), R137. doi:10.1186/gb-2008-9-9-r137
Zhao, G., Zou, C., Li, K., Wang, K., Li, T., Gao, L., et al. (2017). The Aegilops tauschii genome reveals multiple impacts of transposons. Nat. Plants 3 (12), 946–955. doi:10.1038/s41477-017-0067-8
Keywords: whole-genome doubling, Aegilops tauschii, transcriptome, transposable elements, H3K27me3
Citation: Guo H, Zhang G, Zhou M, Wan M, Zhu B, Yang Z, Zeng D and Zeng Z (2023) Whole genome doubling-induced the enrichment of H3K27me3 in genes carrying specific TEs in Aegilops tauschii. Front. Genet. 14:1241201. doi: 10.3389/fgene.2023.1241201
Received: 16 June 2023; Accepted: 17 July 2023;
Published: 25 July 2023.
Edited by:
Pengtao Ma, Yantai University, ChinaReviewed by:
Ying Wu, Northeast Normal University, ChinaCopyright © 2023 Guo, Zhang, Zhou, Wan, Zhu, Yang, Zeng and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zixian Zeng, emVuZ3ppeGlhbkBzaWNudS5lZHUuY24=; Deying Zeng, ZGV5aW5nLnplbmdAb3V0bG9vay5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.