
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 29 August 2023
Sec. Human and Medical Genomics
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1240245
 Lana Salihefendić1,2
Lana Salihefendić1,2 Ivana Čeko1,2Larisa Bešić2Naida Mulahuseinović1Selma Durgut1Dino Pećar1Lejla Prnjavorac3Enis Kandić1
Ivana Čeko1,2Larisa Bešić2Naida Mulahuseinović1Selma Durgut1Dino Pećar1Lejla Prnjavorac3Enis Kandić1 Neven Meseldžić4
Neven Meseldžić4 Tamer Bego4
Tamer Bego4 Besim Prnjavorac3Damir Marjanović2,5Rijad Konjhodžić1,2
Besim Prnjavorac3Damir Marjanović2,5Rijad Konjhodžić1,2 Adna Ašić2*
Adna Ašić2*Introduction: COVID-19 has been a major focus of scientific research since early 2020. Due to its societal, economic, and clinical impact worldwide, research efforts aimed, among other questions, to address the effect of host genetics in susceptibility and severity of COVID-19.
Methods: We, therefore, performed next-generation sequencing of coding and regulatory regions of 16 human genes, involved in maintenance of the immune system or encoding receptors for viral entry into the host cells, in a subset of 60 COVID-19 patients from the General Hospital Tešanj, Bosnia and Herzegovina, classified into three groups of clinical conditions of different severity (“mild,” “moderate,” and “severe”).
Results: We confirmed that the male sex and older age are risk factors for severe clinical picture and identified 13 variants on seven genes (CD55, IL1B, IL4, IRF7, DDX58, TMPRSS2, and ACE2) with potential functional significance, either as genetic markers of modulated susceptibility to SARS-CoV-2 infection or modifiers of the infection severity. Our results include variants reported for the first time as potentially associated with COVID-19, but further research and larger patient cohorts are required to confirm their effect.
Discussion: Such studies, focused on candidate genes and/or variants, have a potential to answer the questions regarding the effect of human genetic makeup on the expected infection outcome. In addition, loci we identified here were previously reported to have clinical significance in other diseases and viral infections, thus confirming a general, broader significance of COVID-19-related research results following the end of the pandemic period.
COVID-19, caused by the SARS-CoV-2 virus outbreak in Wuhan, China, was declared a public health emergency of international concern on 30 January 2020, and a pandemic on 12 March 2020, by the World Health Organization (WHO) (Huang et al., 2020). The genome of SARS-CoV-2, at 29,881 nt and 9,860 amino acids, is a larger linear single-stranded RNA viral genome (Cheng et al., 2020). It encodes four structural proteins (spike S, envelope E, nucleocapsid N, and membrane M) and sixteen non-structural proteins (labeled Nsp1-16) (Naqvi et al., 2020; Wang et al., 2020).
COVID-19 represents an unprecedented challenge to governments all around the world due to virus transmissibility, symptom variability and severity, uncertainty regarding the immunity development following the course of infection, and the overall impact on healthcare systems and global economy (Fauci et al., 2020). For that reason, multiple parallel scientific approaches were used to address the issue as rapidly and efficiently as possible. In this context, scientists around the world sequenced the viral genome. For example, the Global Initiative on Sharing Avian Influenza Data (GISAID) has over 15,700,000 sequenced SARS-CoV-2 viruses, as of July 2023, and the number of submissions is growing daily (Elbe and Buckland-Merrett, 2017). On the other hand, huge variability in the severity of clinical picture of COVID-19 has also been investigated. It was accepted that environmental, demographic, and clinical factors all have an impact on severity of COVID-19, but that the host genetics may also have a significant role in the severity, as well as susceptibility to SARS-CoV-2 infection at the first place (Docherty et al., 2020). It is well-stablished that more severe symptoms and higher mortality rate are both observed in older patients (over 60 years of age), males, and people with other comorbidities, such as diabetes mellitus, cardiovascular diseases, and respiratory diseases, among others (Wang et al., 2020).
Therefore, the aim of our research was to perform the first study of COVID-19 host genetics in Bosnia and Herzegovina, and the Western Balkans region, by sequencing the coding and regulatory regions of 16 human genes in COVID-19 patients classified into three groups of mild, moderate, and severe clinical picture of the disease, in order to establish whether any of detected genetic variants can be associated with severity of COVID-19 and/or susceptibility to the infection. Study genes are mostly involved in maintenance and homeostasis of human immune system or are producing viral (co-) receptors expressed on the surface of human cells.
Ethical approvals for conducting this study were granted by the Joint Ethics Committee of the General Hospital Tešanj, Bosnia and Herzegovina, for patient DNA sample and clinical record use (11 January 2021, document number 01-4-17/21) and the Ethics Committee of the Faculty of Engineering and Natural Sciences, International Burch University Sarajevo, Bosnia and Herzegovina, for conducting the molecular analyses (23 March 2021, document number 04-51/21). Prior to sample collection, all patients signed an informed consent form, while physicians in charge filled in the patient questionnaire regarding general demographic characteristics, clinical presentation of COVID-19, and comorbidities. This research was carried out in accordance with the Declaration of Helsinki.
Whole blood samples were collected from RT-PCR-confirmed COVID-19 patients (n = 60) in the General Hospital Tešanj from March to October 2021, stored at −20°C, and delivered on ice to the ALEA Genetic Center (Sarajevo) laboratory. Based on the patients’ symptoms, overall condition, oxygen saturation levels, and laboratory and radiological testing results, samples were classified into three groups: mild (n = 20), moderate (n = 20), and severe symptoms group (n = 20), based on Baj et al., 2020.
Following sample delivery to the DNA laboratory, they were de-frosted, and DNA was extracted immediately using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). The original manufacturer’s protocol was modified only by using 100 µL of ATE buffer, instead of 200 μL, since DNA was not extracted right after sampling. Following extraction, DNA was quantified using Qubit™ 3.0 Fluorometer with dsDNA high-sensitivity (HS) kit (Thermo Fisher Scientific, Waltham, MA, United States).
Ion AmpliSeq Designer (Thermo Fisher Scientific) was used to create primers for 16 selected genes and their regulatory sequences, namely HLA-A, HLA-B, HLA-C, ACE2, IL-6, IL-4, TMPRSS2, IFITM3, IL-12, DDX58, IRF-7, IRF-9, IL-1B, IL-1A, CD55, and TNF-α. This custom-made primer panel was received frozen in two primer pools, whereby pool 1 contained 93 amplicons and pool 2 contained 92 amplicons. Candidate gene selection was performed in early 2020, meaning that we mainly relied on previously published literature regarding the effect of host genetics on SARS-CoV and MERS-CoV infections or the earliest reports related to biomarker changes that could potentially be indicative of variable immunological response to SARS-CoV-2 infection (Chapman and Hill, 2012; Trowsdale and Knight, 2013; Asselta et al., 2020; Ghafouri-Fard et al., 2020; Lingeswaran et al., 2020; Lipworth et al., 2020; Zhang et al., 2020; Zhou et al., 2020).
Library preparation was done using Ion AmpliSeq™ Library Kit 2.0 (Thermo Fisher Scientific) according to manufacturer’s instructions. The starting amount of DNA material was ranging from 30 to 100 ng, and the number of cycles was set to 24. Amplicon digestion, adapter ligation, and purification steps were performed according to manufacturer’s instructions. Product clean-up was done using Agencourt™ AMPure™ XP Reagent (Beckman Coulter, Brea, CA, United States). Following purification, libraries were quantified using real-time PCR and Ion Library TaqMan® Quantitation Kit (Thermo Fisher Scientific) according to manufacturer’s instructions. Libraries with concentration over 100 pM were diluted to 100 pM and pooled together before emulsion PCR and enrichment, which was done using Ion Chef System (Thermo Fisher Scientific). The chip was loaded automatically with Ion Chef System using Ion 510™ and Ion 520™ and Ion 530™ Kit (Thermo Fisher Scientific). Next-generation sequencing (NGS) was performed using Ion GeneStudio™ S5 System and data was analyzed using Torrent Browser Software (Thermo Fisher Scientific) through VCF (Variant Caller Files) format and Coverage Analysis.
Further modifications were made on the library preparation protocol, as it was concluded that primer pool 2 had lower coverage (more information was published in Salihefendić et al., 2022). Data analysis was ultimately done on 48 samples containing sequences from both primer pools and 12 samples containing only pool 1 amplicons.
Finally, clinical exome analysis was performed on three samples from the severe clinical symptoms group, using TruSight One Sequencing Panel (Illumina, San Diego, CA, United States), according to manufacturer’s instructions. Sequencing of these samples was performed on Illumina MiSeq platform.
All sequences can be accessed within the Sequence Read Archive (SRA) repository on the National Center for Biotechnology information (NCBI) website, as detailed below.
Genetic variants obtained through NGS were confirmed and the custom-made panel was validated via Sanger sequencing of five selected SNPs. Table 1 gives the position of these SNPs, their rs numbers, annealing temperatures, and designed primers.

TABLE 1. Designed primers for confirmatory Sanger sequencing and information about corresponding SNPs.
For every SNP, 10 samples were selected, taking care that both wild-type and variant allele-containing genotypes were selected. For genotypes with variant alleles, both heterozygous and homozygous individuals were selected, whenever possible. For PCR amplification, the final concentrations of 1x PCR Master Mix (Thermo Fisher Scientific) and 1 µM of both forward and reverse primers were used in a reaction of a total volume of 25 μL, including 10 ng of DNA. Initial denaturation was performed at 95°C for 3 min, followed by 40 cycles of denaturation at 95°C for 30 s, annealing for 30 s, and elongation at 72°C for 1 min. Final elongation was done at 72°C for 10 min.
Cycle sequencing was done using BigDye™ Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific) according to manufacturer’s instructions. Products were purified using Macherey-Nagel™ NucleoSpin™ Gel and PCR Clean-up columns (Macherey-Nagel, Düren, Germany) and Sanger sequencing was performed on SeqStudio™ Genetic Analyzer (Thermo Fisher Scientific).
Statistical analysis was performed using the chi-square test of goodness-of-fit to compare clinical severity categories between male and female participants, as well as among the patients with different comorbidities (cardiovascular, metabolic, respiratory, and other comorbidities). The same test was used to investigate the influence of detected genetic variants on the severity of COVID-19 presentation and infection susceptibility. We have used the exact test of goodness-of-fit to perform age-, sex-, and comorbidity-stratified analyses to determine the effect of these variables on the differences in the frequency of appearance of genetic variants detected previously. One-way ANOVA was used to compare the mean age among the different clinical severity groups. Kruskal-Wallis test was deployed as a non-parametric alternative to one-way ANOVA, whereby it assumes that mean ranks are the same among the three age groups. In all analyses, p-value of 0.05 was considered critical for detection of significant differences between the study groups.
In this study, we analyzed the effect of demographic characteristics, comorbidities, and genetic background of patients on severity of and susceptibility to COVID-19. The population age was ranging from 15 to 80, with median of 62.5, mean of 59.067, and standard deviation of 15.029, at a 95% confidence interval of 3.88 (upper 62.95, lower 55.18). Our results demonstrated that the male sex and older age are risk factors for severe symptoms of COVID-19 (Table 2).
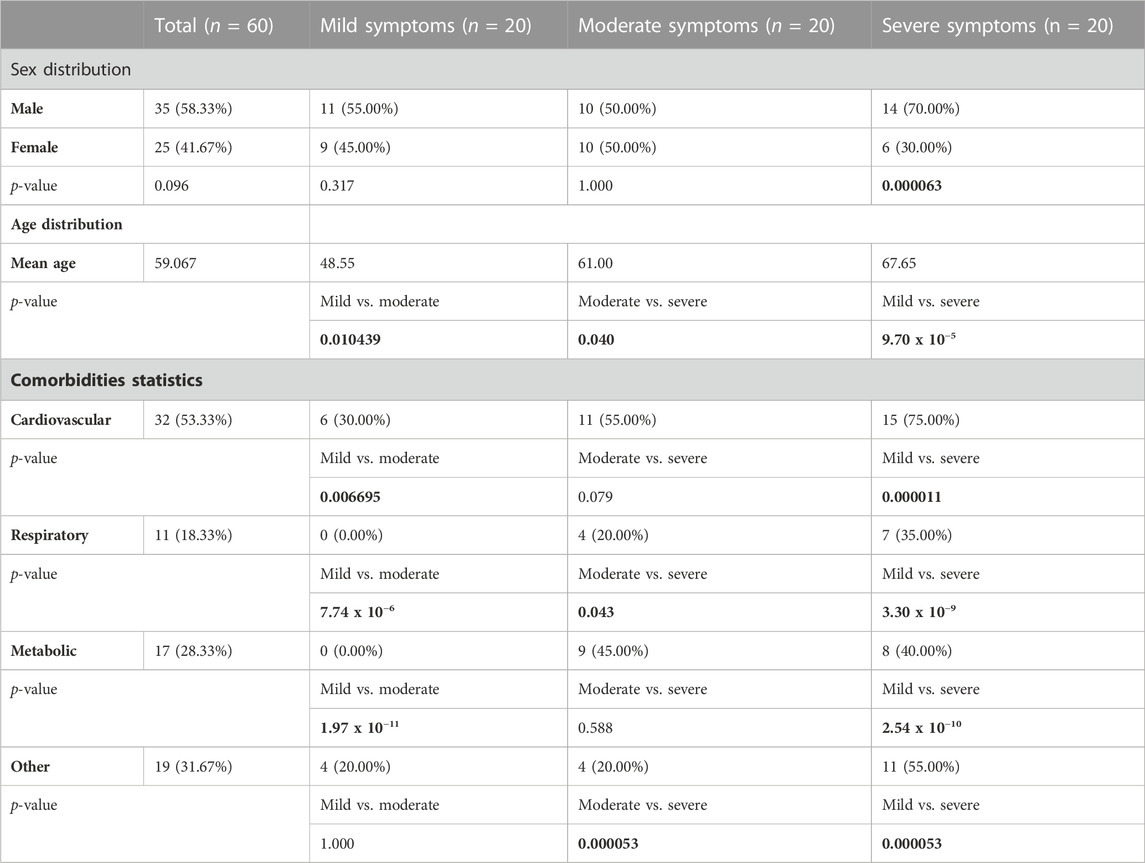
TABLE 2. Demographic characteristics and comorbidities compared between three study groups. p-values denoting statistically significant differences between the study groups are bolded.
By collecting patients’ clinical records, we were able to analyze the frequency of observed comorbidities in three clinical groups. Cardiovascular comorbidities were significantly more common in severe when compared to mild and moderate symptom groups, and include hypertension, chronic cardiomyopathy, brain stroke, angina pectoris, atrial fibrillation, aortic aneurysm, and myocardial infarction. Respiratory comorbidities, including bronchitis, asthma, and history of tuberculosis, were more common in moderate and severe groups, when compared to mild, but also in the moderate symptom group when compared to severe. We have also detected significantly less patients with metabolic comorbidities in mild symptom group when compared to both moderate and severe groups. These comorbidities include diabetes, hypothyroidism, glucose intolerance and chronic sideropenic anemia (iron insufficiency). Other comorbidities encompass rheumatoid arthritis, renal insufficiency, acute liver lesion, chronic gastritis, and hepatitis C infection, and were assessed together. We found that these comorbidities were significantly more common in severe symptom group when compared to either mild or moderate groups (Table 2).
In 11 of the study genes, we have observed genetic variants in our patients. Observed variants include single nucleotide polymorphisms, insertions, deletions, and complex variants (Table 3). We have detected variants that might be predisposing the patients towards milder or more severe symptoms of COVID-19, based on significant differences in the frequency of appearance of the study variants between the defined clinical groups. In our analyses, we grouped heterozygous and homozygous carriers of the variants together (Table 4; Figure 1). The full list of detected variants and accompanying statistics are given in Supplementary Table S1, while the results of clinical exome sequencing for three patients and observed allele frequencies of thus detected variants for European populations are given in Supplementary Table S2. We did not detect linkage disequilibrium or any other significant relationship between the viral-entry-associated genes (ACE2 and TMPRSS2) and the remaining, immunity-related genes, in terms of mutation distribution in individual patients and within the cohort (data not shown).
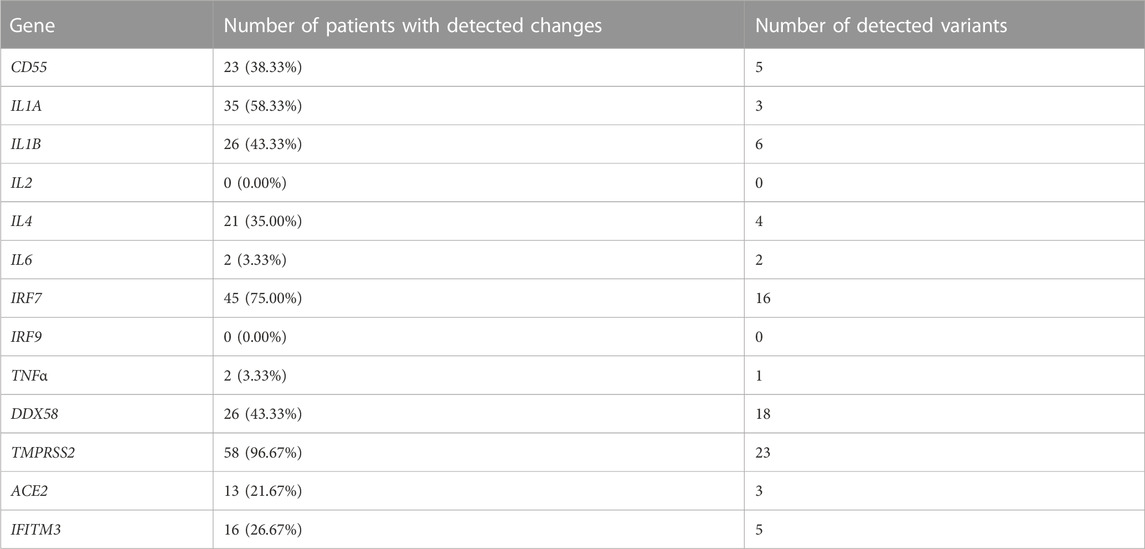
TABLE 3. The number of patients and percentage of the total patient population in which variants on 13 study genes were observed, regardless of the genotype, and the total number of variants observed in the custom-made panel.
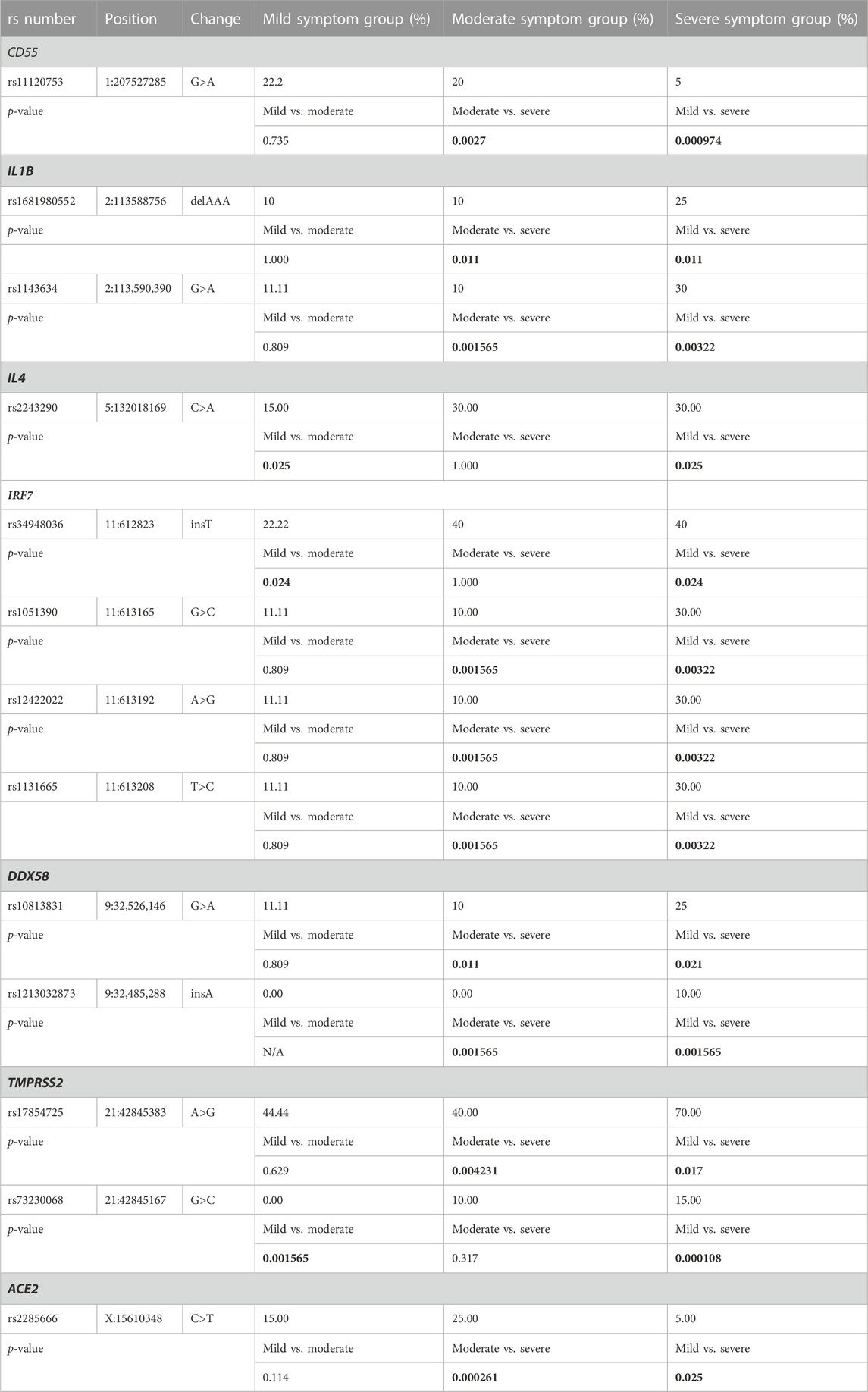
TABLE 4. Detected genetic polymorphisms with significant differences in frequency of appearance between the study groups. Frequencies are given as percentage of all tested patients per study group in which variant was detected, regardless of genotype. p-values denoting statistically significant differences between the study groups are bolded.
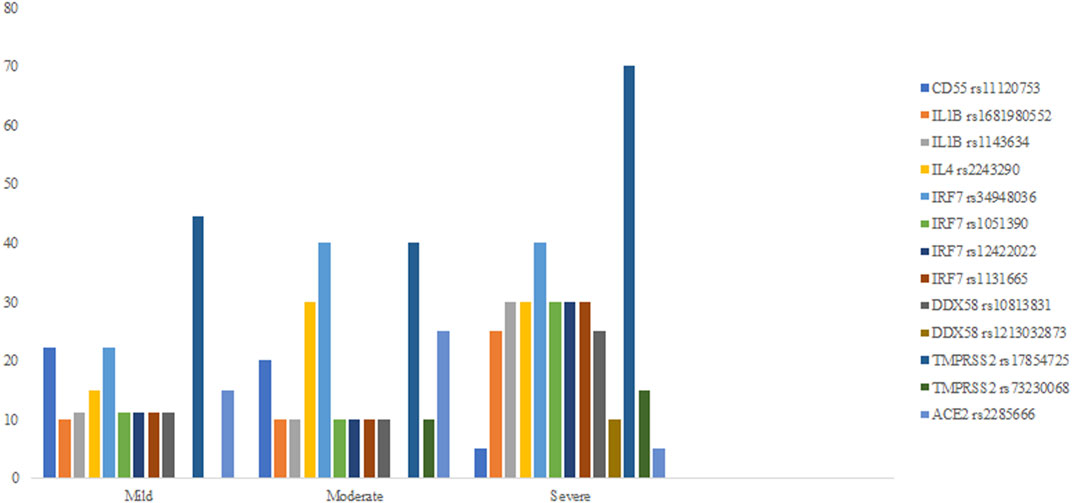
FIGURE 1. Percent fractions of detected genetic polymorphisms with significant differences in frequency between mild, moderate, and severe symptom groups.
In order to adjust our analysis for age, sex, and comorbidities and taking into account the size of the dataset, we divided our participants into 1) groups of patients younger than 60 vs. 60 years old or older, 2) females vs. males, and 3) with vs. without comorbidities. This way, we could analyze if the frequency of appearance of genetic variants within each clinical symptom group will depend on any of these confounding factors. Our results (Supplementary Table S3) show that age, sex, and comorbidity differences do cause statistically significant differences in genetic variant frequencies within the study groups in many reported variants and genes of interest.
When it comes to confirmatory Sanger sequencing, done with the goal of validating the results of the custom-made NGS panel, we have re-sequenced five SNPs, namely rs2243290 C>A (IL4 gene), rs370862493 G>A (IFITM3 gene), rs2285666 C>T (ACE2 gene), rs17854725 A>G, and rs12329760 C>T (both from TMPRSS2 gene) (Figure 2). We obtained 100% agreement for the variants rs370862493, rs2285666, and rs12329760. As for 90% agreement between the methods for rs2243290 polymorphism on IL4 gene, one sample from the severe symptom group was sequenced as homozygous variant using the Sanger method, while NGS reported it as a carrier of heterozygous genotype. When it comes to the TMPRSS2 variant rs178854725, the designed primers for this variant could not be optimized for Sanger sequencing, since the bands on gel electrophoresis were acquired during PCR protocol optimization, but the sequencing results did not give clear, readable electropherograms.
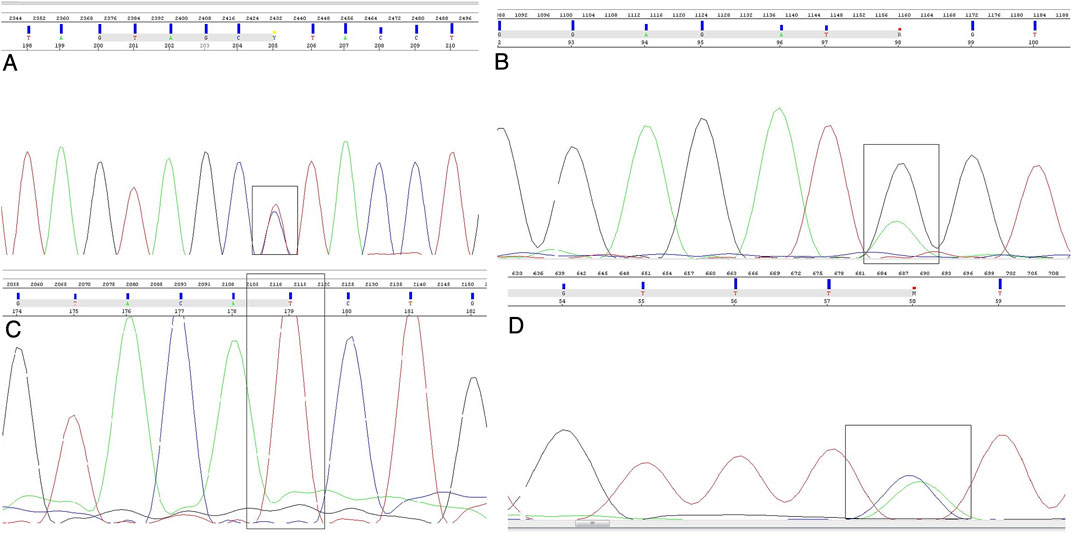
FIGURE 2. Sanger sequencing electropherograms. Nucleotide position of interest is shown in a black box. Nucleotides are stained as green—A, red—T, blue—C, and black—G. (A) Heterozygous genotype of ACE2 rs2285666 polymorphism, (B) heterozygous genotype of IFITM3 rs370862493 polymorphism, (C) homozygous mutant genotype of TMPRSS2 rs12329760 polymorphism, and (D) heterozygous genotype of IL4 rs2243290 polymorphism.
In the present study, we identified male sex, older age, and different classes of comorbidities as significant predictors of severe COVID-19. These findings were confirmed on extremely large cohorts, based on data from the US and Chinese Centers for Disease Control and Prevention (CDCs), whereby male sex and, especially, age of 50 years of life or older, were confirmed to be significantly associated with severe COVID-19, defined as the infection which requires hospitalization, ICU, or results in patient’s death. In addition, comorbidities were identified as an additional contributing factor to severe COVID-19, whereby 69.2% of all US patients had comorbidities compared to only 26.7% of Chinese patients, which can be explained by different definitions of comorbidities in these two countries (Zheng and Song, 2021).
We also detected 13 variants of interest dispersed among seven human genes, playing different roles in the immune system maintenance and viral binding and entry into the host cells, with functional significance in COVID-19 and, potentially, other viral infections. A summary of variant annotation for eQTL status and possible association with other conditions is given in Table 5.
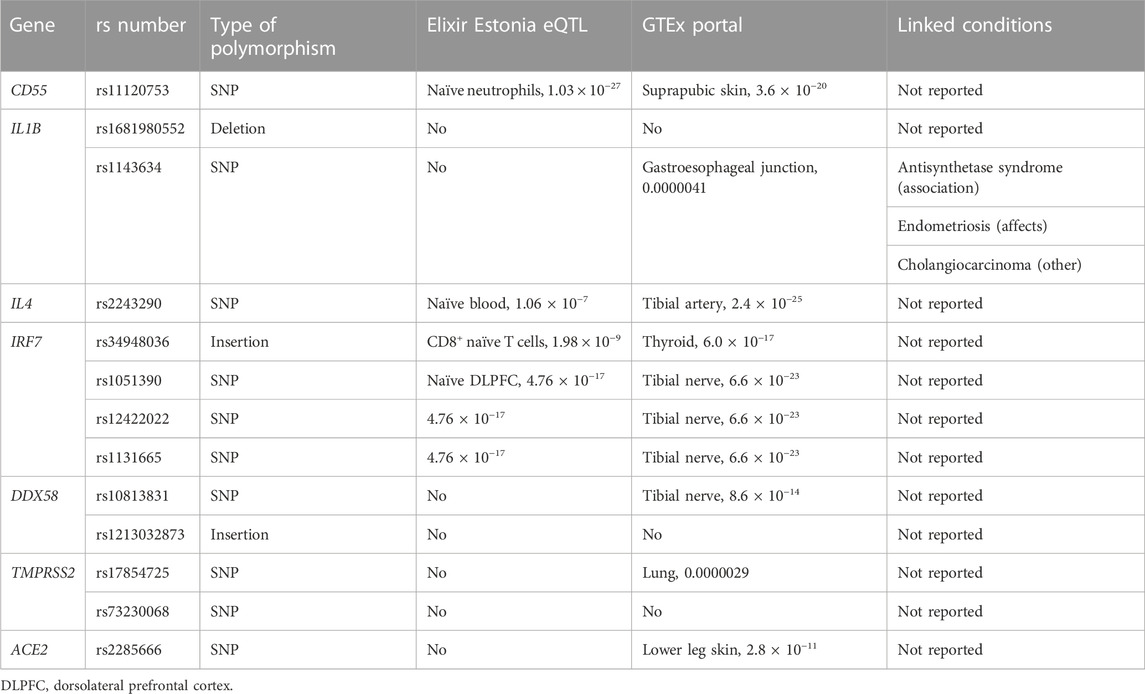
TABLE 5. Variant annotation was performed in order to detect the type of polymorphism, the significance of association of the locus with the eQTL character using two resources, namely ELIXIR Estonia eQTL Catalogue Browser (Kerimov et al., 2021) and GTEx Portal (Carithers et al., 2015), as well as whether these variants are associated with any other conditions using ClinVar database within NCBI (Landrum et al., 2018). For the eQTL identification, we are giving only the annotated association with the lowest p-value and sample identity in which association were identified for both resources.
TMPRSS2 and ACE2 gene products are necessary for viral invasion of the host cells, which is why their variants are heavily researched and expected to be associated with COVID-19 severity, as well as susceptibility (Hou et al., 2020; Paniri et al., 2021). TMPRSS2 is one of the main discoveries in understanding the mechanism of SARS-CoV-2 infection, as it codes for a cell-surface protein expressed by epithelial cells of different tissues, including the aerodigestive tract. SARS-CoV-2 entry into the host cells is dependent upon TMPRSS2 since viral S glycoprotein is cleaved by TMPRSS2, which helps with viral activation (Hoffmann et al., 2020). ACE2 is also crucial in SARS-CoV-2 infection, since the viral entry into the cell depends on ACE2 receptor, which can be found in respiratory tract, oral mucosa and heart cells (Aguiar et al., 2020). Kuba et al. (2005) found that expression of ACE2 gene is downregulated in cells infected by SARS-CoV (Kuba et al., 2005). It was speculated that the genetic variants and loss-of-function mutations in ACE2 might confer resistance to COVID-19, while hypomorphic variants of this gene could be protective against severe cases of COVID-19 disease (Casanova et al., 2020). There is evidence of sex-specific differences in the COVID-19 severity (Hou et al., 2020). For example, higher testosterone level increases the expression of TMPRSS2, which may cause higher susceptibility to COVID-19 in male patients (Bennani and Bennani-Baiti, 2020).
rs73230068 (G>C) is a single nucleotide change in the intronic region of TMPRSS2, which was present in five of our patients, whereby three of them belong to severe and two to moderate clinical symptoms group; all five individuals are heterozygous carriers. Our study, therefore, shows an increase in variant frequency in patients with more severe forms of COVID-19. While it was not a subject of previous research aiming to associate this variant with any diseases or clinical conditions, it has been a subject of a population study. Alternative allele frequency of 0.037 was recorded in 14,286 individuals of European ancestry (Phan et al., 2020), which is in good agreement with our allele frequency of 0.042 (p = 0.904) in a set of COVID-19 patients. rs17854725 A>G is a silent variant (c.879T>C, p.Ile293=) in the same gene, which is present in a significantly higher proportion of patients from the severe, when compared to mild and moderate symptom groups. The same variant is also present as a missense variant (c.879T>G, p.Ile293Met), which was not recorded in our study. This variant was previously investigated in terms of its association with COVID-19. Namely, rs17854725/rs75603675/rs12329760/rs4303795 polymorphisms have been associated with increased susceptibility to COVID-19 and more severe clinical symptoms (Rokni et al., 2022). In that study, mortality was more frequent in individuals who carried the rs17854725/AG genotype. They also showed that G allele of this SNP is related to increased susceptibility to COVID-19 infection. Combined haplotype rs17854725/AG, rs75603675/AC, rs12329760/TT, and rs4303795/AG was ruled as a risk factor for COVID-19 susceptibility, especially in the case of GATG and GCTG haplotypes. Most COVID-19 patients whose rs17854725 genotype was AG were affected by the severe form of the disease, while about 64% of the AA genotype carriers had mild clinical symptoms (Rokni et al., 2022). Additionally, in a bioinformatic prediction study (Paniri et al., 2021), rs75603675 was predicted to affect TMPRSS2 protein function according to PolyPen-2, but not according to SIFT (Paniri et al., 2021). Our study showed the presence of three polymorphisms together, that is, rs17854725/rs75603675/rs12329760 in two patients, whereby one patient belongs to mild and the other belongs to severe clinical symptoms group.
rs2285666 (C>T) intronic variant on X-chromosomal gene ACE2 was detected in nine samples in our study, including one male from the severe clinical symptoms group, five patients from the moderate group (three heterozygous females and two males), and three patients from the mild group (two heterozygous females and one male). rs2285666 polymorphism is located at the beginning of the intron 3, and it could theoretically affect gene expression with alternative splicing mechanisms (Yang et al., 2015). Srivastava et al. (2020) made a correlation between lower SARS-CoV-2 infection rate and the minor allele (T) in Indian population, therefore establishing a possibility of this polymorphism being associated with a protective role against infection (Srivastava et al., 2020). Möhlendick et al. (2021) have found a two-fold increased risk of SARS-CoV-2 infection and a three-fold increased risk for COVID-19-related fatality or severe form of COVID-19 in CC genotype (or C allele) carriers in German population (Möhlendick et al., 2021). Similarly, a meta-analysis reported GG carriers as individuals at risk of developing severe COVID-19 (Saengsiwaritt et al., 2022). Nonetheless, it is important to note that different studies report conflicting results regarding this polymorphism. A more recent meta-analysis of 11 studies reports rs2285666 as associated with more severe COVID-19 (Aziz and Islam, 2022). Also, T allele was identified as a risk factor for severe or fatal COVID-19, especially in males, regardless of age, hypertension, T2DM, and obesity (Martinez-Gomez et al., 2022). Our study shows that there is an increased number of alternative allele carriers in mild and moderate groups, when compared to the group of patients with severe form of COVID-19, meaning that our results corroborate the hypothesis of this SNP being more prevalent in mild and moderate clinical groups. However, this is not necessarily the case when considering the sex of the study participants. Males, carrying one alternative allele and having one copy of the gene, are distributed across three groups. Females are all heterozygous, which is important considering the fact that ACE2 escapes X chromosome inactivation (Gagliardi et al., 2020) and both copies of the gene remain transcriptionally active in all cells of female patients. This gives higher gene dosage to females, as well as evolutionary advantage in case of heterozygous carriers of harmful variants. This SNP, however, seems to be protective in females since it is found in moderate and mild groups only.
CD55 variant rs11120753 (G>A) is reported in seven patients in our study, including one heterozygous carrier in severe symptom group, two homozygotes in moderate, and four patients from the mild symptom group, including three homozygous and one heterozygous carrier. According to the ALFA Project results (Phan et al., 2020), obtained from 17,796 individuals of European ancestry, alternate allele A is present with the frequency of 0.2697, which is higher when compared to our study population with allele frequency of 0.125, but the difference is not significant (p = 0.258). This is an intronic variant and there have been no previously published data on this variant regarding COVID-19 disease involvement. However, CD55 was found to be upregulated on the surface of monocytes in COVID-19 patients when compared to healthy controls (Lage et al., 2022), especially in lung tissue (Ge et al., 2023). In silico microarray data analysis from the ArrayExpress database revealed possible involvement of CD55 differential expression in COVID-19 (Vastrad et al., 2020). Since CD55 is a cell-surface-bound glycoprotein acting as a complement inhibitor, its overexpression is suggested to play a role in self-protection due to complement overactivation in case of viral infection and further prevention of host cell damage.
Intronic variant rs1681980552 (delAAA) in IL1B gene was detected in nine patients. Five patients belong to the severe clinical group (two homozygotes and three heterozygotes), two to the moderate (one homozygote and one heterozygote), and two to the mild clinical group (both heterozygous carriers). There are no reports on this variant for any disease association and there are no population studies on its frequency, but we are reporting it as a promising target for predicting possible severe symptoms of COVID-19. Polymorphism rs1143634 (G>A) from the same gene is detected in nine patients, including six patients from the severe, one patient from the moderate, and two patients from the mild clinical group, whereby all variant alleles were detected in heterozygous genotypes. This is a synonymous variant (p.Phe105=) reported in ClinVar (Landrum et al., 2018) as associated with antisynthetase syndrome and endometriosis. Jafrin, Aziz and Islam (2021) performed a meta-analysis which revealed that the presence of this polymorphism increases the risk of cancer development, more precisely gastric and breast cancers and multiple myeloma, especially in Asian populations (Jafrin et al., 2021). Several studies connect rs1143634 with chronic periodontitis, including a meta-analysis by da Silva et al. (2017), in which it was significantly associated with chronic periodontitis disease in Caucasian, Asian and mixed populations (da Silva et al., 2017). There is no reported data on this variant regarding its association with SARS-CoV-2 infection or the clinical course of the disease, prior to our study in which this variant seems to be overrepresented in the severe symptom group when compared to the other two. IL1B is an inflammatory cytokine involved in initiating the immunological response against viral infection and is therefore highly relevant for viral infections. Previous studies reported that IL1B deregulation could be among the causes of cytokine storm and critical and/or severe COVID-19 symptoms (Chua et al., 2020; Lee et al., 2020; Feng et al., 2022), as well as its high plasma levels in patients with post-acute sequelae of COVID-19 (PASC) (Schultheiss et al., 2022).
rs2243290 (C>T) is an intronic variant of IL4 gene and it has been detected in 15 patients in our study (six from severe, six from moderate and three form mild clinical group). Just like other variants on genes encoding for interleukins, this variant is enriched in the study groups with more pronounced COVID-19 symptoms and, therefore, might be associated with disease severity. This variant was not previously reported in relation to COVID-19; however, IL4 has been studied regarding susceptibility to SARS-CoV infection, and it was found that its protein product downregulates cell surface expression of ACE2, therefore inhibiting SARS-CoV replication (de Lang et al., 2006). This gene and its protein product were reported in relation to COVID-19, as IL4 is generally activated interleukin in bodily immune response to SARS-CoV-2 infection (Hasanvand, 2022). Increased IL4 serum levels were associated with patients with previous infection without signs of long COVID-19 (Queiroz et al., 2022), but also with lung tissue samples from COVID-19 patients who did not survive the infection (Vaz de Paula et al., 2021). Since individuals with asthma and allergic diseases, which are not commonly encountered comorbidities in COVID-19, experience overactivation of type 2 immune response, including IL4, it is also proposed that the overexpression of this protein might play a protective role against COVID-19 infection (Liu et al., 2020; Gao et al., 2022).
rs34948036 (insT) is an intronic variant from IRF7 gene detected in 16 patients, four of them belonging to mild, four to moderate, and eight to severe clinical symptom group. Its alternative allele (insT) frequency in European population is 0.259, based on the ALFA Project (Phan et al., 2020) on 24,292 individuals. Our results show, for the first time, that this single-nucleotide insertion could be clinically relevant and associated with severe clinical symptoms of COVID-19. rs1051390 (G>C), rs12422022 (A>G), and rs1131665 (T>C) variants, also on IRF7 gene, were detected in nine patients with identical distribution, as a haplotype. Six of these patients belong to the severe clinical symptoms group, one belongs to moderate, and two belong to mild symptoms group, whereby all nine participants presented with heterozygous genotype. While rs1051390 and rs12422022 are intronic, rs1131665 is a missense variant (g.613208T>C, p.Gln412Arg). Despite none of these variants being previously reported for COVID-19 association, IRF7 gene codes for protein necessary to produce IFN-I. Autosomal recessive IRF7 deficiency was reported in three patients with COVID-19 pneumonia symptoms, whereby IRF7-deficient patients are generally more prone to viral infection of the respiratory tract (Campbell et al., 2022). Additionally, type I interferon immunity deregulation due to IRF7 deficiency was suggested as a possible molecular mechanism of severe and life-threatening COVID-19 (Zhang et al., 2020).
rs10813831 (G>A) is a missense variant (g.5177C>T, p.Arg7Cys) in DDX58 gene, that was detected in eight patients in our study, including five from the severe clinical group (one heterozygous and four homozygous genotypes), one heterozygote from the moderate clinical group, and two heterozygotes from the mild clinical group. Our results point towards its involvement in progression of more severe COVID-19, especially in homozygous carriers of the variant, which was recently confirmed in an Iranian study of 182 patients with mild and 177 with severe COVID-19 were genotyped for this polymorphism, whereby AA genotype was significantly associated with severe COVID-19 when compared to GG, in a recessive model (Feizollahi et al., 2023). Previous research connected this variant with other conditions as well. For example, Wu et al. (2019) concluded that Chinese individuals carrying the rs10813831-G-allele-containing genotype were more liable to achieve spontaneous hepatitis C virus (HCV) clearance than the patients who were carriers of the alternate allele (Wu et al., 2019). Another interesting variant from DDX58 gene is rs1213032873 (insA), which is detected in only two heterozygous patients in the severe symptom group. It is an intronic variant with no clinical significance described in ClinVar (Landrum et al., 2018), including no reports on the variant association with COVID-19 susceptibility and/or severity. This insertion is extremely rare, with alternative allele frequency of 0.0002 in 8,676 individuals of European ancestry, according to the latest release of the ALFA Project (Phan et al., 2020), as compared to our allele frequency of 0.021 in the COVID-19 patient population, which is significantly different (p = 5.31 × 10−16). Since we are reporting this variant for the first time, to the best of our knowledge, as the variant potentially associated with severe COVID-19 manifestation and increased susceptibility to COVID-19, it should be researched on a large patient population and compared to the general population frequencies of the insertion allele. DDX58 gene, a carrier of these two variants, also known as RNA sensor RIG-I, is involved in viral double-stranded RNA recognition and antiviral immune response in host cells. It has been reported that DDX58 gene expression under SARS-CoV-2 infection is upregulated (Fricke-Galindo and Falfan-Valencia, 2021).
Peripheral blood mononuclear cell (PBMC) immunophenotyping proved to be an additional highly informative tool of analysis when it comes to distinguishing between the severity of COVID-19 presentation, as well as viral persistence in infected individuals. A previous study performed PBMC immunophenotyping using single-cell RNA sequencing (scRNA-seq) technique on 11 healthy controls, five asymptomatic infected individuals and 33 symptomatic patients of different clinical presentations of the disease. They came up with 76 different immune cell subsets, some of which were found to be significantly more common in asymptomatic than symptomatic patients, while others were associated with more or less severe disease course, as well as capable of modulating the extent of viral presence in infected cells. For example, (TRAV1-2+CD8+) MAIT cells, (NCAM1hiCD160+) NK cells, (CD4loCSF1R−CD33−CD14+) classical monocytes, and (CD33− HLA-DMA-CD14+) classical monocytes were associated with asymptomatic infection. It was also shown that (CD68−CSF1R−IL1BhiCD14+) classical monocytes were positively associated with more severe COVID-19 presentation, but potentially also with the disease progression mechanism. Additionally, IL1B and IFITM3 were found to be upregulated in these cells in patients with severe COVID-19, when compared to healthy or asymptomatic individuals or patients with mild disease (Wang et al., 2022). Another study assessed 40 healthy individuals and 97 COVID-19 patients with different disease severity presentation and generated a dataset of 1,400 plasma proteins and 2,600 single-cell immune features to study the most commonly deregulated pathways during the progression of SARS-CoV-2 infection in the human body. It was found that JAK-STAT, MAPK-mTOR and NF-κB signaling pathways are deregulated in COVID-19 and might be used as early-stage predictors of COVID-19 severity. In addition, this study identified association of CD4 and CD8 T cell emergence in case of progression towards more severe COVID-19, as well as multiple proteome-level changes, such as RAS, lung homeostasis and hemostasis pathways enrichment, and cytokine storm elements, such as increased plasma levels of IL1B, IL-33, IL-6, and IFNγ. As an element of RAS system, increased ACE2 plasma levels were positively correlated with more severe COVID-19, which points towards its possible shedding from the cell surface and subsequent loss-of-function, which corroborates with expected increase in rate of cardiovascular damage and multi-organ injuries commonly seen in patients with severe COVID-19. An increased percentage of granulocytes in PBMC samples of patients with severe COVID-19 was also observed (Feyaerts et al., 2022). Another study compared immunophenotypic profiles of PBMCs and bronchoalveolar lavage fluid mononuclear cells (BALF-MCs) in 18 patients with COVID-19-associated acute respiratory distress syndrome (CARDS). The results showed similar profiles for these two types of cells in survivors and deceased patients. It was, however, reported that the frequency of classical monocytes and naïve CD4+ T cells in peripheral blood was higher in survivors than in non-survivors. Several differences were also detected between PBMCs and BALF-MCs, pointing towards specificities of disease progression in analyzed patients (Santinelli et al., 2023).
Genome-wide association study (GWAS) efforts are also an important source of information for COVID-19, especially since those studies are usually multicenter and based on large patient cohorts with matched controls. COVID Human Genetic Effort group studied 659 patients of different ancestries via whole-exome sequencing, as well as 534 controls with asymptomatic or mild clinical presentation. They detected 113 variants at 12 loci in the severely affected group, including nine predicted loss-of-function variants and 109 missense or in-frame indels, without copy-number variations. Significantly higher frequency of damaging loss-of-function or highly hypomorphic variants was identified in the severe group, including 24 rare nonsynonymous variants from eight genes, namely TLR3, IRF3, IRF7, IFNAR1, IFNAR2, TBK1, TICAM1 (TRIF), and UNC93B1 (Zhang et al., 2022). COVID-19 Host Genetics Initiative published their findings on 46 studies performed in 19 countries including a total of 49,562 COVID-19 cases and two million ancestry-matched controls, with the goal of identifying genomic loci associated with COVID-19 susceptibility and severity. Overall, 13 significant loci were detected, with four of these loci being associated with increased susceptibility, including ABO locus, 3p21.31 genomic region (rs2271616 from SLC6A20 gene), and PPP1R15A. Nine positions are linked to increased likelihood of more severe symptoms and worse clinical outcome, including variants in genes TYK2, DPP9, and CXCR6 (Niemi et al., 2021; COVID-19 Host Genetics Initiative, 2020). Genetics of Mortality in Critical Care (GenOMICC) conducted a study on 2,244 critically ill patients with COVID-19 infection hospitalized in 208 intensive care units throughout the United Kingdom, along with five ancestry-matched controls from the United Kingdom Biobank for each case. The strongest association was found on chromosome 3 (rs73064425, closest to LZTFL1), but several other loci were found significantly associated with critical illness in COVID-19, such as those in the proximity to genes OAS1, OAS2, OAS3, TYK2, DPP9, and IFNAR2. In addition, it was found that IFNAR2 underexpression, as well as TYK2 overexpression are associated with critical illness, just like the differential expression in lung tissue of CCR2, CCR3, CXCR6, and MTA2B in critical disease (Pairo-Castineira et al., 2021). The Severe COVID-19 GWAS group included 835 patients and 1,255 controls in Italy and 775 patients and 950 controls from Spain, to study the genetics of severe presentation of COVID-19. The strongest association with severe COVID-19 was observed for genomic locus 3p21.31 covering genes SLC6A20, LZTFL1, CCR9, FYCO1, CXCR6 and XCR1. Another signal was detected from 9q34.2, that overlaps with the ABO blood group locus. Subsequent HLA locus analysis did not reveal any SNPs associated with either susceptibility to COVID-19 or with disease severity (Ellinghaus et al., 2020).
Considering all previously presented findings and literature sources, it is important to identify the limitations of the present study. Firstly, our limited sample size does not permit us to draw any final conclusions related to the functional significance of detected variants. We are reporting these variants with recommendations for their future testing and analysis in order to fully understand their role, not only in COVID-19, but in other viral infections as well, in terms of both susceptibility and disease progression. Additional testing methods, including transcriptomics and proteomics analyses, flow cytometry for immunophenotyping, as well as WGS and WES sequencing would provide more comprehensive results and enable us to get a full picture of the molecular effect of detected variants and the mechanism of their action.
COVID-19 is probably the best representation of how significant discoveries can be made in short time periods, assuming that research teams are given adequate support. By studying COVID-19 from different perspectives, we accumulated a significant wealth of knowledge in less than 4 years since the beginning of the pandemic. Research into the host genetics of SARS-CoV-2 infection is still active and ongoing, with large consortia being formed with the goal of completing whole-genome or whole-exome studies on large patient and control groups. Despite being limited by small sample size and smaller portions of the genome being analyzed, this study also contributes to the present state of knowledge related to COVID-19, as it provides the only and unique look into the Western Balkans populations in relation to this infection. Apart from studying the influence of host genetics on COVID-19 susceptibility and severity, international research groups aim towards better understanding of critical COVID-19 and long-COVID, as well as how these findings can be used in future for better understanding and management of other viral infections in humans. Overall, given the current importance of this topic and availability of technical support, COVID-19 research promises to be one of the most fruitful areas of research in foreseeable future.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject/?term=912097.
The studies involving humans were approved by the Joint Ethics Committee of the General Hospital Tešanj, Bosnia and Herzegovina, for patient DNA sample and clinical record use (11 January 2021, document number 01-4-17/21) and the Ethics Committee of the Faculty of Engineering and Natural Sciences, International Burch University Sarajevo, Bosnia and Herzegovina, for conducting the molecular analyses (23 March 2021, document number 04-51/21). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Conceptualization: DM, RK, and AA; methodology: LS, IČ, NaM, SD, DP, LP, and NeM; software: LB; formal analysis: LB, EK, TB, and BP; investigation: LS, BP, RK, DM, and AA; resources: EK, TB, and BP; data curation: LS, NaM, and AA; writing—original draft preparation: LS and AA; writing—review and editing: all authors; supervision: RK and AA; project administration: AA; funding acquisition: AA. All authors contributed to the article and approved the submitted version.
This research is a part of the project titled “Personalized approach to COVID-19 infection through analysis of molecular genetic predisposition of the patients for a differential immune response,” that is co-financed by the Ministry of Science, Education, and Youth of the Sarajevo Canton (decision no. 11/05-34-12880-8/20). The authors would like to thank the International Burch University in Sarajevo for providing institutional support in covering a part of the article publication fee.
The authors are grateful to all volunteers for accepting to participate in the study.
Parts of this research have been presented at the 12th ISABS Conference on Forensic and Anthropological Genetics and Mayo Clinic Lectures in Individualized Medicine held in June 2022 in Dubrovnik, Croatia. The manuscript is deposited as a pre-print on the Research Square, under the doi number https://doi.org/10.21203/rs.3.rs-2397519/v1.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1240245/full#supplementary-material
Aguiar, J. A., Tremblay, B. J., Mansfield, M. J., Woody, O., Lobb, B., Banerjee, A., et al. (2020). Gene expression and in situ protein profiling of candidate SARS-CoV-2 receptors in human airway epithelial cells and lung tissue. Eur. Respir. J. 56 (3), 2001123. doi:10.1183/13993003.01123-2020
Asselta, R., Paraboschi, E. M., Mantovani, A., and Duga, S. (2020). ACE2 and TMPRSS2 variants and expression as candidates to sex and country differences in COVID-19 severity in Italy. Aging 12 (11), 10087–10098. doi:10.18632/aging.103415
Aziz, M. A., and Islam, M. S. (2022). Association of ACE1 I/D rs1799752 and ACE2 rs2285666 polymorphisms with the infection and severity of COVID-19: A meta-analysis. Mol. Genet. Genom. Med. 10, e2063. doi:10.1002/mgg3.2063
Baj, J., Karakuła-Juchnowicz, H., Teresiński, G., Buszewicz, G., Ciesielka, M., Sitarz, R., et al. (2020). COVID-19: specific and non-specific clinical manifestations and symptoms: the current state of knowledge. J. Clin. Med. 9 (6), 1753. doi:10.3390/jcm9061753
Bennani, N. N., and Bennani-Baiti, I. M. (2020). Androgen deprivation therapy may constitute a more effective COVID-19 prophylactic than therapeutic strategy. Ann. Oncol. 31 (11), 1585–1586. doi:10.1016/j.annonc.2020.08.2095
Campbell, T. M., Liu, Z., Zhang, Q., Moncada-Velez, M., Covill, L. E., Zhang, P., et al. (2022). Respiratory viral infections in otherwise healthy humans with inherited IRF7 deficiency. J. Exp. Med. 219 (7), e20220202. doi:10.1084/jem.20220202
Carithers, L. J., Ardlie, K., Barcus, M., Branton, P. A., Britton, A., Buia, S. A., et al. (2015). A novel approach to high-quality postmortem tissue procurement: the GTEx project. Biopreserv. Biobank. 13 (5), 311–319. doi:10.1089/bio.2015.0032
Casanova, J. L., Su, H. C., Abel, L., Aiuti, A., Almuhsen, S., Arias, A. A., et al. (2020). A global effort to define the human genetics of protective immunity to SARS-CoV-2 infection. Cell. 181 (6), 1194–1199. doi:10.1016/j.cell.2020.05.016
Chapman, S. J., and Hill, A. V. (2012). Human genetic susceptibility to infectious disease. Nat. Rev. Genet. 13 (3), 175–188. doi:10.1038/nrg3114
Cheng, C. Y., Lee, Y. L., Chen, C. P., Lin, Y. C., Liu, C. E., Liao, C. H., et al. (2020). Lopinavir/ritonavir did not shorten the duration of SARS CoV-2 shedding in patients with mild pneumonia in Taiwan. J. Microbiol. Immunol. Infect. 53 (3), 488–492. doi:10.1016/j.jmii.2020.03.032
Chua, R. L., Lukassen, S., Trump, S., Hennig, B. P., Wendisch, D., Pott, F., et al. (2020). COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 38 (8), 970–979. doi:10.1038/s41587-020-0602-4
COVID-19 Host Genetics Initiative (2020). The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur. J. Hum. Genet. 28 (6), 715–718. doi:10.1038/s41431-020-0636-6
da Silva, M. K., de Carvalho, A. C. G., Alves, E. H. P., da Silva, F. R. P., Pessoa, L. D. S., and Vasconcelos, D. F. P. (2017). Genetic factors and the risk of periodontitis development: findings from a systematic review composed of 13 studies of meta-analysis with 71,531 participants. Int. J. Dent. 2017, 1914073. doi:10.1155/2017/1914073
de Lang, A., Osterhaus, A. D., and Haagmans, B. L. (2006). Interferon-gamma and interleukin-4 downregulate expression of the SARS coronavirus receptor ACE2 in Vero E6 cells. Virology 353 (2), 474–481. doi:10.1016/j.virol.2006.06.011
Docherty, A. B., Harrison, E. M., Green, C. A., Hardwick, H. E., Pius, R., Norman, L., et al. (2020). Features of 20 133 UK patients in hospital with COVID-19 using the ISARIC WHO clinical characterisation protocol: prospective bservational cohort study. BMJ 369, m1985. doi:10.1136/bmj.m1985
Elbe, S., and Buckland-Merrett, G. (2017). Data, disease and diplomacy: GISAID's innovative contribution to global health. Glob. Chall. 1 (1), 33–46. doi:10.1002/gch2.1018
Ellinghaus, D., Degenhardt, F., Bujanda, L., Buti, M., Albillos, A., Invernizzi, P., et al. (2020). Genomewide association study of severe covid-19 with respiratory failure. N. Engl. J. Med. 383 (16), 1522–1534. doi:10.1056/NEJMoa2020283
Fauci, A. S., Lane, H. C., and Redfield, R. R. (2020). Covid-19 - navigating the uncharted. N. Engl. J. Med. 382 (13), 1268–1269. doi:10.1056/NEJMe2002387
Feizollahi, P., Zamanian, M. H., Falahi, S., Salari, F., Mahmoudi, Z., Faryadi, E., et al. (2023). Association of IFIH1 and DDX58 genes polymorphism with susceptibility to COVID-19. Med. Microbiol. Immun. 212 (3), 221–229. doi:10.1007/s00430-023-00764-x
Feng, S., Song, F., Guo, W., Tan, J., Zhang, X., Qiao, F., et al. (2022). Potential genes associated with COVID-19 and comorbidity. Int. J. Med. Sci. 19 (2), 402–415. doi:10.7150/ijms.67815
Feyaerts, D., Hédou, J., Gillard, J., Chen, H., Tsai, E. S., Peterson, L. S., et al. (2022). Integrated plasma proteomic and single-cell immune signaling network signatures demarcate mild, moderate, and severe COVID-19. Cell. Rep. Med. 3, 100680. doi:10.1016/j.xcrm.2022.100680
Fricke-Galindo, I., and Falfán-Valencia, R. (2021). Genetics insight for COVID-19 susceptibility and severity: A review. Front. Immunol. 12, 622176. doi:10.3389/fimmu.2021.622176
Gagliardi, M. C., Tieri, P., Ortona, E., and Ruggieri, A. (2020). ACE2 expression and sex disparity in COVID-19. Cell. Death Discov. 6, 37. doi:10.1038/s41420-020-0276-1
Gao, Y. D., Agache, I., Akdis, M., Nadeau, K., Klimek, L., Jutel, M., et al. (2022). The effect of allergy and asthma as a comorbidity on the susceptibility and outcomes of COVID-19. Int. Immunol. 34 (4), 177–188. doi:10.1093/intimm/dxab107
Ge, X., Yu, Z., Guo, X., Li, L., Ye, L., Ye, M., et al. (2023). Complement and complement regulatory proteins are upregulated in lungs of COVID-19 patients. Pathol. Res. Pract. 247, 154519. doi:10.1016/j.prp.2023.154519
Ghafouri-Fard, S., Noroozi, R., Vafaee, R., Branicki, W., Poṡpiech, E., Pyrc, K., et al. (2020). Effects of host genetic variations on response to, susceptibility and severity of respiratory infections. Biomed. Pharmacother. 128, 110296. doi:10.1016/j.biopha.2020.110296
Hasanvand, A. (2022). COVID-19 and the role of cytokines in this disease. Inflammopharmacology 30 (3), 789–798. doi:10.1007/s10787-022-00992-2
Hoffmann, M., Kleine-Weber, H., Schroeder, S., Krüger, N., Herrler, T., Erichsen, S., et al. (2020). SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 181 (2), 271–280. doi:10.1016/j.cell.2020.02.052
Hou, Y., Zhao, J., Martin, W., Kallianpur, A., Chung, M. K., Jehi, L., et al. (2020). New insights into genetic susceptibility of COVID-19: an ACE2 and TMPRSS2 polymorphism analysis. BMC Med. 18 (1), 216. doi:10.1186/s12916-020-01673-z
Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., et al. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet (London, Engl. 395 (10223), 497–506. doi:10.1016/S0140-6736(20)30183-5
Jafrin, S., Aziz, M. A., and Islam, M. S. (2021). Role of IL-1β rs1143634 (+3954C>T) polymorphism in cancer risk: an updated meta-analysis and trial sequential analysis. J. Int. Med. Res. 49 (12), 3000605211060144. doi:10.1177/03000605211060144
Kerimov, N., Hayhurst, J. D., Peikova, K., Manning, J. R., Walter, P., Kolberg, L., et al. (2021). A compendium of uniformly processed human gene expression and splicing quantitative trait loci. Nat. Genet. 53 (9), 1290–1299. doi:10.1038/s41588-021-00924-w
Kuba, K., Imai, Y., Rao, S., Gao, H., Guo, F., Guan, B., et al. (2005). A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 11 (8), 875–879. doi:10.1038/nm1267
Lage, S. L., Rocco, J. M., Laidlaw, E., Rupert, A., Galindo, F., Kellogg, A., et al. (2022). Activation of complement components on circulating blood monocytes from COVID-19 patients. Front. Immunol. 13, 815833. doi:10.3389/fimmu.2022.815833
Landrum, M. J., Lee, J. M., Benson, M., Brown, G. R., Chao, C., Chitipiralla, S., et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46 (D1), D1062–D1067. doi:10.1093/nar/gkx1153
Lee, J. S., Park, S., Jeong, H. W., Ahn, J. Y., Choi, S. J., Lee, H., et al. (2020). Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci. Immunol 5, eabd1554. doi:10.1126/sciimmunol.abd1554
Lingeswaran, M., Goyal, T., Ghosh, R., Suri, S., Mitra, P., Misra, S., et al. (2020). Inflammation, immunity and immunogenetics in COVID-19: A narrative review. Indian J. Clin. biochem. 35 (3), 260–273. doi:10.1007/s12291-020-00897-3
Lipworth, B., Chan, R., and Kuo, C. R. (2020). Predicting severe outcomes in COVID-19. J. Aller. Cl. Imm.-Pract. 8, 2582–2584. doi:10.1016/j.jaip.2020.06.039
Liu, S., Zhi, Y., and Ying, S. (2020). COVID-19 and asthma: reflection during the pandemic. Clin. Rev. Allerg. Immu. 59 (1), 78–88. doi:10.1007/s12016-020-08797-3
Martínez-Gómez, L. E., Herrera-López, B., Martinez-Armenta, C., Ortega-Peña, S., Camacho-Rea, M. D. C., Suarez-Ahedo, C., et al. (2022). ACE and ACE2 gene variants are associated with severe outcomes of COVID-19 in men. Front. Immunol. 13, 812940. doi:10.3389/fimmu.2022.812940
Möhlendick, B., Schönfelder, K., Breuckmann, K., Elsner, C., Babel, N., Balfanz, P., et al. (2021). ACE2 polymorphism and susceptibility for SARS-CoV-2 infection and severity of COVID-19. Genomics 31 (8), 165–171. doi:10.1097/FPC.0000000000000436
Niemi, M. E. K., Karjalainen, J., Liao, R. G., Neale, B. M., Daly, M., Ganna, A., et al. (2021). Mapping the human genetic architecture of COVID-19. Nature 600 (7889), 472–477. doi:10.1038/s41586-021-03767-x
Naqvi, A. A. T., Fatima, K., Mohammad, T., Fatima, U., Singh, I. K., Singh, A., et al. (2020). Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: structural genomics approach. Biochim. Biophys. Acta. Mol. Basis Dis. 1866 (10), 165878. doi:10.1016/j.bbadis.2020.165878
Pairo-Castineira, E., Clohisey, S., Klaric, L., Bretherick, A. D., Rawlik, K., Pasko, D., et al. (2021). Genetic mechanisms of critical illness in COVID-19. Nature 591 (7848), 92–98. doi:10.1038/s41586-020-03065-y
Paniri, A., Hosseini, M. M., and Akhavan-Niaki, H. (2021). First comprehensive computational analysis of functional consequences of TMPRSS2 SNPs in susceptibility to SARS-CoV-2 among different populations. J. Biomol. Struct. Dyn. 39 (10), 3576–3593. doi:10.1080/07391102.2020.1767690
Phan, L., Jin, Y., Zhang, H., Qiang, W., Shekhtma, E., Shao, D., et al. Alfa: allele frequency aggregator. (2020) Available at: www.ncbi.nlm.nih.gov/snp/docs/gsr/alfa/.
Queiroz, M. A. F., Neves, P. F. M. D., Lima, S. S., Lopes, J. D. C., Torres, M. K. D. S., Vallinoto, I. M. V. C., et al. (2022). Cytokine profiles associated with acute COVID-19 and long COVID-19 syndrome. Front. Cell. Infect. Mi. 12, 922422. doi:10.3389/fcimb.2022.922422
Rokni, M., Heidari Nia, M., Sarhadi, M., Mirinejad, S., Sargazi, S., Moudi, M., et al. (2022). Association of TMPRSS2 gene polymorphisms with COVID-19 severity and mortality: A case-control study with computational analyses. Appl. Biochem. Biotechnol. 194 (8), 3507–3526. doi:10.1007/s12010-022-03885-w
Saengsiwaritt, W., Jittikoon, J., Chaikledkaew, U., and Udomsinprasert, W. (2022). Genetic polymorphisms of ACE1, ACE2, and TMPRSS2 associated with COVID-19 severity: A systematic review with meta-analysis. Rev. Med. Virol. 32, 4, e2323. doi:10.1002/rmv.2323
Salihefendić, L., Ašić, A., Čeko, I., Pećar, D., Bešić, L., Mulahuseinović, N., et al. (2022). Challenges in obtaining high-quality data from a custom-made panel for the next generation sequencing (NGS) using Ion Torrent GeneStudio™ S5 platform. J. Bioanthropol. 2 (2), 56–61. doi:10.54062/jb.2.1.1
Santinelli, L., Lazzaro, A., Sciarra, F., Maddaloni, L., Frasca, F., Fracella, M., et al. (2023). Cellular immune profiling of lung and blood compartments in patients with SARS-CoV-2 infection. Pathog.-Basel 12 (3), 442. doi:10.3390/pathogens12030442
Schultheiß, C., Willscher, E., Paschold, L., Gottschick, C., Klee, B., Henkes, S. S., et al. (2022). The IL-1β, IL-6, and TNF cytokine triad is associated with post-acute sequelae of COVID-19. Cell. Rep. Med. 3 (6), 100663. doi:10.1016/j.xcrm.2022.100663
Srivastava, A., Bandopadhyay, A., Das, D., Pandey, R. K., Singh, V., Khanam, N., et al. (2020). Genetic association of ACE2 rs2285666 polymorphism with COVID-19 spatial distribution in India. Front. Genet. 11, 564741. doi:10.3389/fgene.2020.564741
Trowsdale, J., and Knight, J. C. (2013). Major histocompatibility complex genomics and human disease. Ann. Rev. Genom. Hum. G. 14, 301–323. doi:10.1146/annurev-genom-091212-153455
Vastrad, B., Vastrad, C., and Tengli, A. (2020). Identification of potential mRNA panels for severe acute respiratory syndrome coronavirus 2 (COVID-19) diagnosis and treatment using microarray dataset and bioinformatics methods. 3 Biotech. 10, 422. doi:10.1007/s13205-020-02406-y
Vaz de Paula, C. B., Nagashima, S., Liberalesso, V., Collete, M., da Silva, F. P. G., Oricil, A. G. G., et al. (2021). COVID-19: immunohistochemical analysis of TGF-β signaling pathways in pulmonary fibrosis. Int. J. Mol. Sci. 23 (1), 168. doi:10.3390/ijms23010168
Wang, D., Hu, B., Hu, C., Zhu, F., Liu, X., Zhang, J., et al. (2020a). Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in wuhan, China. JAMA 323 (11), 1061–1069. doi:10.1001/jama.2020.1585
Wang, M. Y., Zhao, R., Gao, L. J., Gao, X. F., Wang, D. P., and Cao, J. M. (2020b). SARS-CoV-2: structure, biology, and structure-based therapeutics development. Front. Cell. Infect. Microbiol. 10, 587269. doi:10.3389/fcimb.2020.587269
Wang, X., Bai, H., Ma, J., Qin, H., Zeng, Q., Hu, F., et al. (2022). Identification of distinct immune cell subsets associated with asymptomatic infection, disease severity, and viral persistence in COVID-19 patients. Front. Immunol. 13, 812514. doi:10.3389/fimmu.2022.812514
Wu, X., Zang, F., Liu, M., Zhuo, L., Wu, J., Xia, X., et al. (2019). Genetic variants in RIG-I-like receptor influences HCV clearance in Chinese Han population. Epidemiol. Infect. 147, e195. doi:10.1017/S0950268819000827
Yang, M., Zhao, J., Xing, L., and Shi, L. (2015). The association between angiotensin-converting enzyme 2 polymorphisms and essential hypertension risk: A meta-analysis involving 14,122 patients. J. Renin Angiotensin Aldosterone Syst. 16 (4), 1240–1244. doi:10.1177/1470320314549221
Zhang, Q., Bastard, P., Cobat, A., and Casanova, J. L. (2022). Human genetic and immunological determinants of critical COVID-19 pneumonia. Nature 603 (7902), 587–598. doi:10.1038/s41586-022-04447-0
Zhang, Q., Bastard, P., Liu, Z., Le Pen, J., Moncada-Velez, M., Chen, J., et al. (2020a). Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Sciences 370 (6515), eabd4570. doi:10.1126/science.abd4570
Zhang, Y., Qin, L., Zhao, Y., Zhang, P., Xu, B., Li, K., et al. (2020b). Interferon-Induced transmembrane protein 3 genetic variant rs12252-C associated with disease severity in coronavirus disease 2019. J. Infect. Dis. 222 (1), 34–37. doi:10.1093/infdis/jiaa224
Zheng, M., and Song, L. (2021). Shift in the distributions of pre-existing medical condition, gender and age across different COVID-19 outcomes. Aging Dis. 12 (2), 327–329. doi:10.14336/AD.2020.1222
Keywords: ACE2, COVID-19, host genetics, IRF7, SARS-CoV-2, TMPRSS2
Citation: Salihefendić L, Čeko I, Bešić L, Mulahuseinović N, Durgut S, Pećar D, Prnjavorac L, Kandić E, Meseldžić N, Bego T, Prnjavorac B, Marjanović D, Konjhodžić R and Ašić A (2023) Identification of human genetic variants modulating the course of COVID-19 infection with importance in other viral infections. Front. Genet. 14:1240245. doi: 10.3389/fgene.2023.1240245
Received: 14 June 2023; Accepted: 11 August 2023;
Published: 29 August 2023.
Edited by:
Manuel Corpas, University of Westminster, United KingdomReviewed by:
Karthikeyan Subbarayan, Martin Luther University of Halle-Wittenberg, GermanyCopyright © 2023 Salihefendić, Čeko, Bešić, Mulahuseinović, Durgut, Pećar, Prnjavorac, Kandić, Meseldžić, Bego, Prnjavorac, Marjanović, Konjhodžić and Ašić. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adna Ašić, YWRuYS5hc2ljQGlidS5lZHUuYmE=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.