 Young Hoon Jung1†
Young Hoon Jung1† Jay Jiyong Kwak2†Kwangsic Joo1†
Jay Jiyong Kwak2†Kwangsic Joo1† Hyuk Jun Lee3
Hyuk Jun Lee3 Kyu Hyung Park3
Kyu Hyung Park3 Min Seok Kim1
Min Seok Kim1 Eun Kyoung Lee3Suk Ho Byeon2Christopher Seungkyu Lee2
Eun Kyoung Lee3Suk Ho Byeon2Christopher Seungkyu Lee2 Jinu Han4
Jinu Han4 Junwon Lee4*
Junwon Lee4* Chang Ki Yoon3*
Chang Ki Yoon3* Se Joon Woo1*
Se Joon Woo1*- 1Department of Ophthalmology, Seoul National University College of Medicine, Seoul National University Bundang Hospital, Seongnam, Republic of Korea
- 2Institute of Vision Research, Department of Ophthalmology, Severance Eye Hospital, Yonsei University College of Medicine, Seoul, Republic of Korea
- 3Department of Ophthalmology, Seoul National University Hospital, Seoul, Republic of Korea
- 4Institute of Vision Research, Department of Ophthalmology, Gangnam Severance Hospital, Yonsei University College of Medicine, Seoul, Republic of Korea
Purpose: To investigate the clinical features, natural course, and genetic characteristics of Koreans with rhodopsin-associated retinitis pigmentosa (RHO-associated RP).
Design: We conducted a retrospective, multicenter, observational cohort study.
Participants: We reviewed the medical records of 42 patients with RHO-associated RP of 36 families who visited 4 hospitals in Korea.
Methods: Patients with molecular confirmation of pathogenic variants of the RHO gene were included. The patients were divided into two subgroups: the generalized and sector RP groups. A central visual field of the better-seeing eye of <10° or a best-corrected visual acuity of the better-seeing eye <20/40 indicated the progression to late-stage RP.
Results: The mean age at which symptoms first appeared was 26.3 ± 17.9 years (range: 8–78 years), and the mean follow-up period was 80.9 ± 68.7 months (range: 6–268 months). At the last follow-up visit, the generalized RP group showed a significantly higher rate of visual field impairment progression to late-stage RP than that of the sector RP group (22 of 35 [62.9%] vs. 0 of 7 [0.0%], p = 0.003). No cases in the sector RP group progressed to generalized RP. Best-corrected visual acuity deterioration to late-stage RP was observed only in the generalized RP group (13 of 35 patients; 37.1%), whereas no deterioration was observed in the sector RP group. We identified 16 known and three novel RHO mutations, including two missense mutations (p.T108P and p.G121R) and one deletion mutation (p.P347_A348del). The pathogenic variants were most frequently detected in exon 1 (14 of 36 [38.9%]). The most common pathogenic variants were p.P347L and T17M (5 of 36 [13.9%] families). Among 42 patients of 36 families, 35 patients of 29 families (80.6%) presented with the generalized RP phenotype, and seven patients of seven families (19.4%) presented with the sector RP phenotype. Three variants (p.T17M, p.G101E, and p.E181K) presented with both the generalized and sector RP phenotypes.
Conclusion: This multicenter cohort study provided information on the clinical and genetic features of RHO-associated RP in Koreans. It is clinically important to expand the genetic spectrum and understand genotype-phenotype correlations to ultimately facilitate the development of gene therapy.
1 Introduction
Retinitis pigmentosa (RP), the most common type of progressive inherited retinal dystrophy (IRD), is characterized by rod and cone photoreceptor degeneration (Hartong et al., 2006; Verbakel et al., 2018). RP occurs in approximately 1 of each 3,500 individuals with the following inheritance patterns: autosomal dominant (15%–20%), autosomal recessive (20%–25%), and X-linked (10%–15%) (Wang et al., 2005). The genetic and clinical features of RP are somehow similar to those of other IRDs, such as cone-rod dystrophy, macular dystrophies, Leber congenital amaurosis, and congenital stationary night blindness (Zhang, 2016). Therefore, an accurate diagnosis via gene sequencing is crucial.
Since a rhodopsin (RHO) gene mutation, p.P23H, was initially reported, >230 different mutations have been reported to be associated with RP, accounting for 25%–30% of all autosomal dominant RP cases (Liu et al., 2021). The common phenotypes in RHO mutations are generalized (classical) and sector RP. Clinical features and visual prognosis differ between patients with generalized and sector RP. Although sector RP progresses relatively slower than generalized RP, several reports stated that it eventually progresses to the generalized type (Ballios et al., 2021; Nguyen et al., 2021). However, the difference in genetic and clinical features between generalized and sector RP is unclear. Although there have been studies on RHO-associated RP in Caucasian, Japanese, and Chinese populations, no such studies have been conducted on Koreans.
Thus, our study aims to identify the clinical features, including natural course, pathogenic variants, and genotype-phenotype correlations, in Koreans with RHO-associated RP.
2 Materials and methods
2.1 Patients and inclusion criteria
This retrospective observational cohort study was conducted at four tertiary centers: Seoul National University Bundang Hospital, Seoul National University Hospital, Severance Eye Hospital, and Gangnam Severance Hospital. An institutional Review Board approval was obtained (IRB no: B-2206-762-101), and the study was conducted in accordance with the principles of the Declaration of Helsinki. Initially, 876 patients diagnosed with RP via gene sequencing were screened. We enrolled patients with the clinical features of RP and one or more pathogenic variants of RHO. Finally, 42 patients (42 of 876; 4.8%) from 36 families were included.
2.2 Genetic analyses
A comprehensive custom gene panel of 295 known and candidate genes or a 429 gene-targeted panel linked to IRDs was used for genetic analyses, as reported in previous studies (Seong et al., 2015; Kim et al., 2019; Kim et al., 2021). Targeted next-generation sequencing (Targeted NGS, Illumina NextSeq 550 system; San Diego, CA, USA) or whole exome sequencing (WES; Illumina NovaSeq 6000 system) were performed. Targeted NGS was performed using custom-designed RNA oligonucleotide probes and a target enrichment kit (Celemics, Seoul, South Korea). WES was performed using the xGen Exome Research Panel v1.0 (Integrated DNA Technologies, Inc., Coraville, IA, USA) and SureSelect Human All Exon v6 enrichment kit (Agilent Technologies, Santa Clara, CA, USA).
2.3 Clinical data collection and classification
We retrospectively reviewed the medical records, and the patients were divided into two subgroups: the generalized and sector RP groups. Ophthalmic examinations included best-corrected visual acuity (BCVA), refraction, intraocular pressure (IOP), wide-field color fundus photography (Optos PLC, Dunfermline, UK), spectral domain OCT (Heideiberg Engineering, Heidelberg, Germany; Carl Zeiss Meditec, Germany), full-field electroretinography (ERG), and Goldmann visual field. ERG was performed based on the standards of the International Society for Clinical Electrophysiology of Vision, available at www.iscev.org. Based on the criteria of the World Health Organization, the International Classification of Diseases 11 classifies vision impairment into five groups: no visual impairment (BCVA ≥20/40), mild visual impairment (20/70 ≤ BCVA <20/40), moderate visual impairment (20/200 ≤ BCVA <20/70), severe visual impairment (20/400 ≤ BCVA <20/200), and blindness (BCVA <20/400). As many patients with RP maintain relatively good visual acuity until the late stage, we considered a decline in BCVA to <20/40 in the better-seeing eye to be an indication of progression to late-stage RP. Moreover, late-stage RP was identified when the central VF of the better-seeing eye was <10°, as VF loss is relatively an early symptom of RP.
RHO mutations are classified in HGMD (http://www.hgmd.cf.ac.uk/) based on the experimentally studied cellular and biochemical characteristics: class 1 (post Golgi trafficking and outer segments targeting), class 2 (misfolding and endoplasmic reticulum retention and instability), class 3 (disrupted vesicular traffic and endocytosis), class 4 (altered post-translational modifications and reduced stability), class 5 (altered transduction activation), class 6 (constitutive activation), class 7 (dimerization deficiency), and unclassified (Athanasiou et al., 2018).
We also ranked and scaled the combined annotation dependent depletion (CADD) score, an in silico prediction tool. A scaled CADD score greater than 20 indicating a variant in the top 1% of deleterious variants in the human genome, while a score greater than 30 indicating a variant in the top 0.1%.
2.4 Statistical analysis
Statistical analyses were performed using the SPSS version 27.0 software (SPSS Inc., Chicago, Illinois, USA). Either the χ2 test or Fisher’s exact test was used to compare the categorical variables between the two groups, and the Mann-Whitney U test was used to compare the numerical variables. The Pearson correlation test was performed to evaluate the correlation between CADD score and the progression to late-stage RP.
3 Results
The demographic and clinical characteristics of the 42 patients of 36 families are listed in Table 1. Among 42 patients of 36 families, 35 patients of 29 families (80.6%) presented with the generalized RP phenotype, and seven patients of seven families (19.4%) presented with the sector RP phenotype. The mean age at which symptoms first appeared was 24.0 ± 17.9 years (range: 8–78 years) for generalized RP and 35.0 ± 16.9 years (range: 24–78 years) for sector RP (p = 0.100). The mean follow-up period was 85.9 ± 73.3 months (range: 6–268 months) for generalized RP and 58.9 ± 40.0 months (range: 6–108 months) for sector RP (p = 0.536). No significant differences between the two phenotypes were detected in terms of sex, age, follow-up period, refraction, IOP, or BCVA.
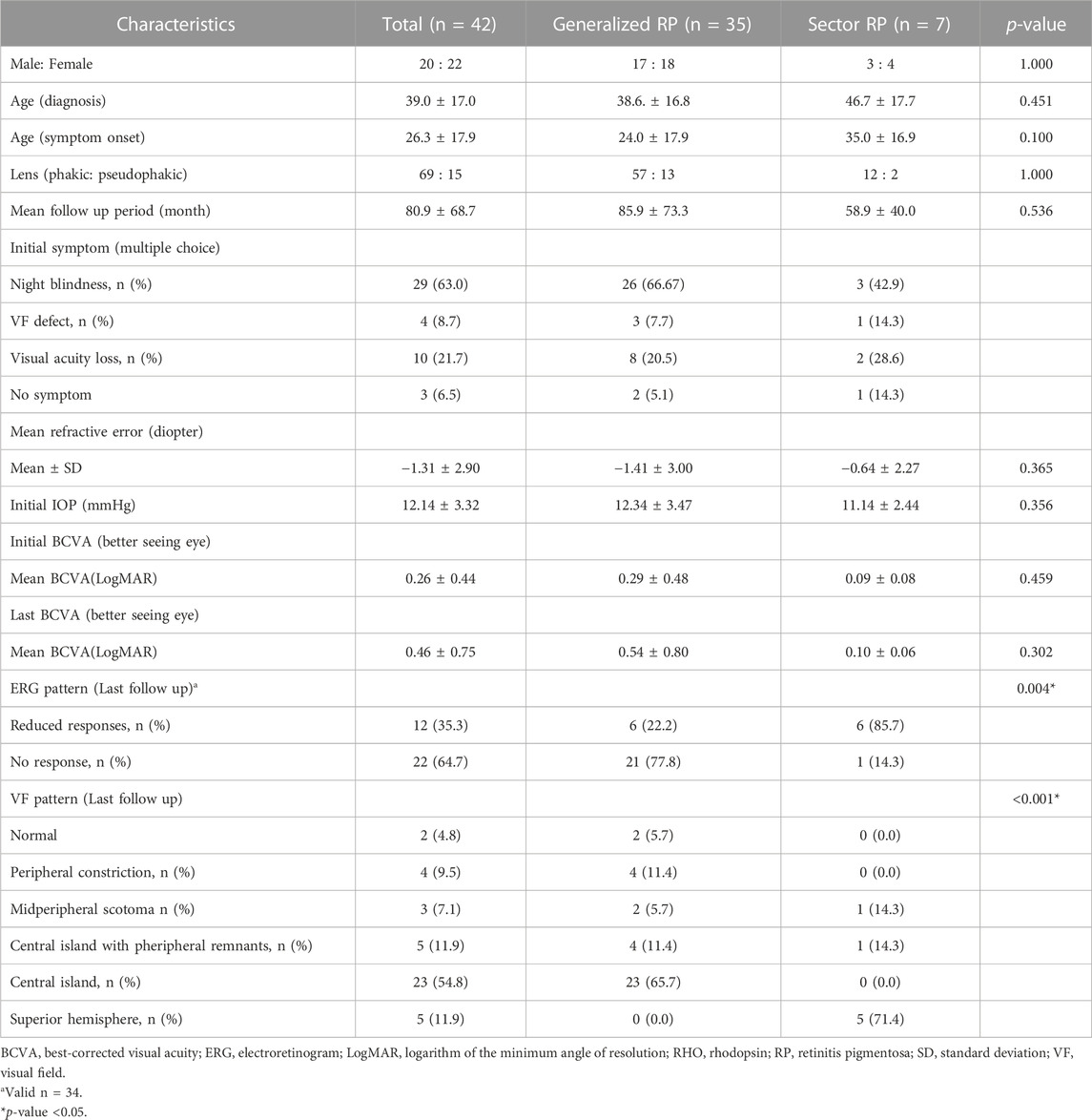
TABLE 1. Characteristics of patients with RHO-Associated RP.
ERG findings at both initial and last visit were documented in 34 patients. All patients with initially preserved central VF showed reduced or no response in photopic and scotopic ERG responses. Generalized RP showed a significantly higher rate of no response in ERG (21 of 27 [77.8%] vs. 1 of 7 [14.3%], p = 0.004) and central island patterns in VF (23 of 35 [65.7%] vs. 0 of 7 [0.0%], p < 0.001) than that of sector RP.
At the last follow-up visit, the generalized RP group showed a significantly higher rate of visual field impairment progression to late-stage RP than that of the sector RP group (22 of 35 [62.9%] vs. 0 of 7 [0.0%], p = 0.003) (Table 2). The generalized RP group showed a higher rate of visual acuity deterioration to late-stage RP than that of the sector RP group, although the p-value was not significant (13 of 35 [37.1%] vs. 0 of 7 [0.0%], p = 0.079).
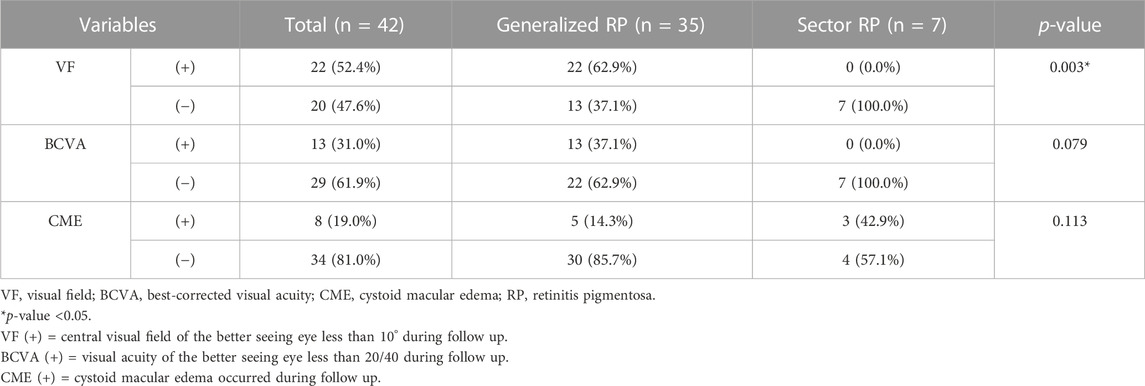
TABLE 2. Disease progression analysis of BCVA, VF, and CME.
The generalized RP group showed a lower rate of cystic macular edema (CME) than that of the sector RP group; however, the difference was not statistically significant (5 of 30 [14.3%] vs. 3 of 4 [42.9%], p = 0.113) (Table 2; Supplementary Figure S1). Furthermore, no association was observed between CME and any specific pathogenic variants.
We reported 14 (73.7%) missense mutations, 2 (10.5%) deletion mutations, 1 (5.3%) frameshift mutation, 1 (5.3%) nonsense mutation, and 1 (5.3%) splicing site mutation. Moreover, we identified three novel RHO mutations: two missense mutations (p.T108P and p.G121R) and one deletion mutation (p.P347_A348del) (Table 3; Figure 1). All patients harbored a single heterozygous variant of RHO. Pathogenic variants were most common in exon 1 (14 of 36 [38.9%]). Of the classified nucleotide changes, 6 of 10 (60%) were identified as either classification 2 or likely classification 2. The most common pathogenic variants were p.P347L (5 of 36 families; 13.8%) and p.T17M (5 of 36 families; 13.8%). p.G101E (4 of 36 families; 11.1%) and p.R135W (4 of 36 families; 11.1%) were the second most common pathogenic variants.
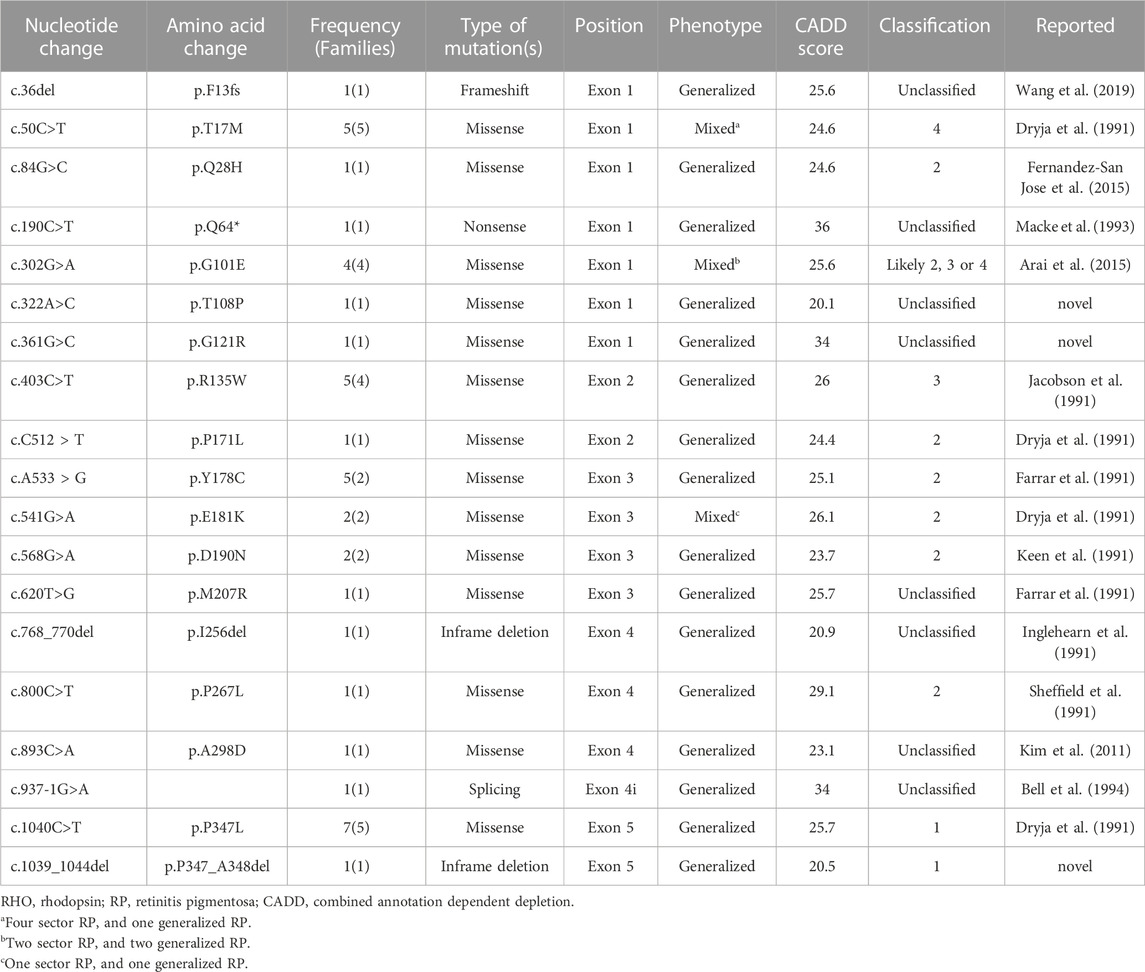
TABLE 3. Pathogenic variants included in this cohort of patients of RHO-assisted RP.
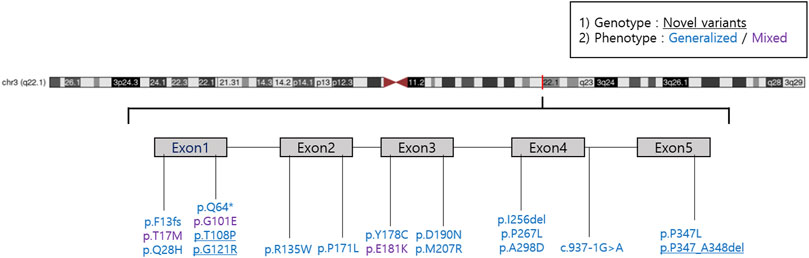
FIGURE 1. A lollipop diagram of the location of RHO mutations in the dataset.
Patients with the p.T17M variant presented with both the generalized (1 of 5) and sector RP (4 of 5) phenotypes Patients with the p.G101E variant presented with both the generalized (2 of 4) and sector RP (2 of 4) phenotypes (Figure 2). Patients with the p.E181K variant presented with both the generalized RP (1 of 2) and sector RP (1 of 2) phenotypes. During the follow-up period, all patients with sector RP (n = 7) showed bone-spicule pigmentation in the inferior retina, and none of these cases progressed to generalized RP. Three novel mutations (p.T108P, p.G121R, and p.P347_A348del) presented with bone-spicule pattern of pigmentary deposits in the mid-peripheral retina and were classified as generalized RP (Figure 3).
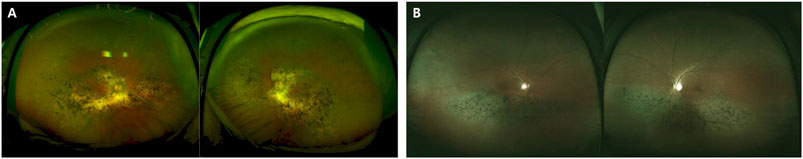
FIGURE 2. Images of different phenotypes of patients with RP with p.G101E mutations. (A) A 78-year-old man carrying the missense mutation (p.G101E). A wide field color fundus photo showing bone-spicule pattern of pigmentary deposits. (B) A 61-year-old man carrying the same missense mutation (p.G101E). A wide field color fundus photo showing inferior bone-spicule degeneration fundus pattern of pigmentary deposits.
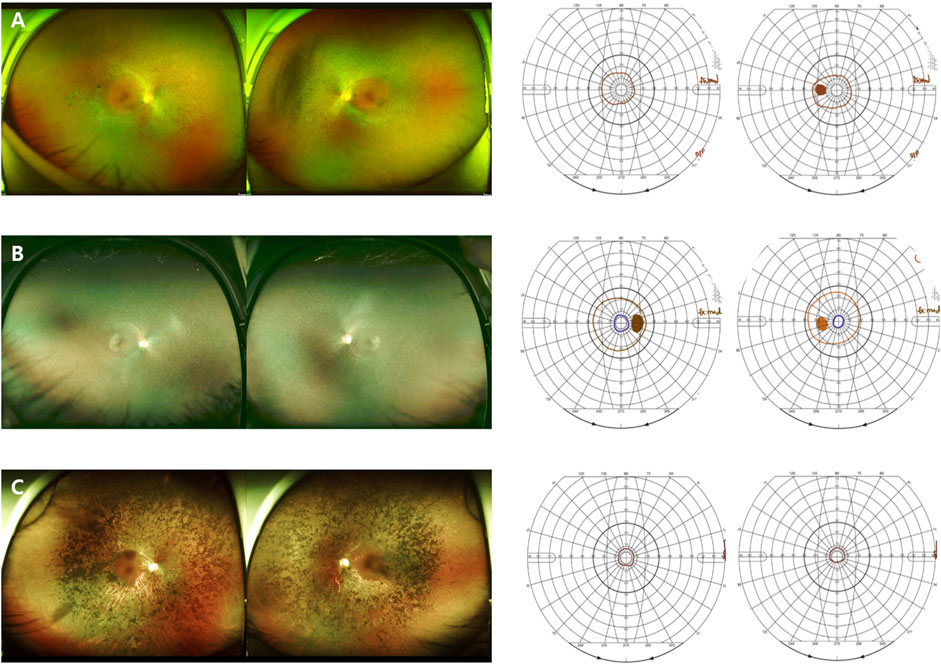
FIGURE 3. Images of the RP patients with novel RHO mutations. (A) A 50-year-old female carrying the missense mutation (p.T108P). A wide field color fundus photo showing bone-spicule pattern of pigmentary deposits in the mid-peripheral retina. The Goldmann visual field test showing a central island on both eyes. (B) A 21-year-old female with the missense mutation (p.G121R). A wide field color fundus photo showing some bone-spicule pattern of pigmentary deposits in the mid-peripheral retina. The Goldmann visual field test showing a peripheral constriction field defect. (C) A 45-year-old male with the deletion mutation (p.P347_A348del). A wide field color fundus photo showing diffuse bone-spicule pattern of pigmentary deposits. The Goldmann visual field test showing a central island on both eyes.
There were no significant differences in the progression to late-stage RP between class A and class B RHO-associated adRP phenotypes for visual field or visual acuity (p = 1.000, respectively).
No correlation was observed between the CADD score and the progression of visual field impairment to late-stage RP (Pearson correlation coefficient r = 0.106, p = 0.505), as well as the deterioration of visual acuity to late-stage RP (Pearson correlation coefficient r = −0.069, p = 0.664).
4 Discussion
In this study, we analyzed RHO-associated RP cases from multiple centers in Korea and compared the two distinctive clinical patterns: generalized and sector RP. Mutations in the RHO gene are associated with autosomal dominant RP (adRP), autosomal dominant congenital stationary night blindness, and autosomal recessive RP (Dryja et al., 1991). Patients with RHO-associated adRP experience distinct patterns of retinal dysfunction, either generalized or sector RP, depending on the specific mutation. Generalized RP is characterized by a decrease in rod function across the entire retina, leading to night blindness, peripheral visual field loss, and eventual central vision loss (Figure 4). Sector RP affects one or two quadrants of the retina, primarily the inferior retina (Figure 5). (Berson and Howard, 1971; Berson et al., 1991; Kranich et al., 1993; Van Woerkom and Ferrucci, 2005).
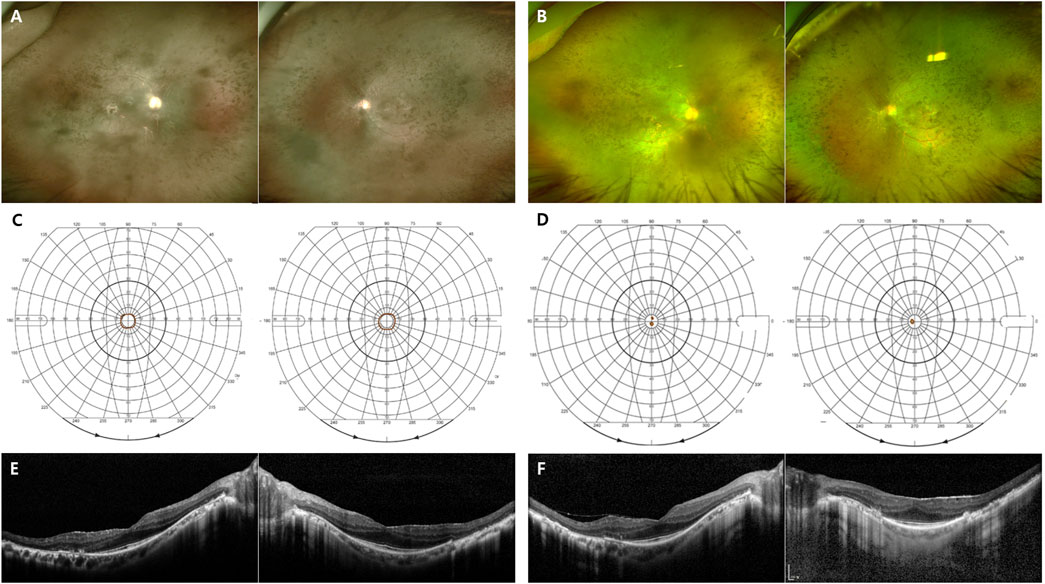
FIGURE 4. Representative images of the generalized RHO-associated RP. (A–D) A 48-year-old man carrying the missense mutation (p.R135W). (A, B) A wide field color fundus photo at the initial and last visits (11 years gap), respectively. (C, D) The Goldmann visual field test at the initial and last visits (11 years gap), respectively. (E, F) A 45-year-old man carrying the missense mutation (p.R135W). Horizontal and vertical scans of spectral domain OCT at the initial and last visits (7 years gap), respectively.
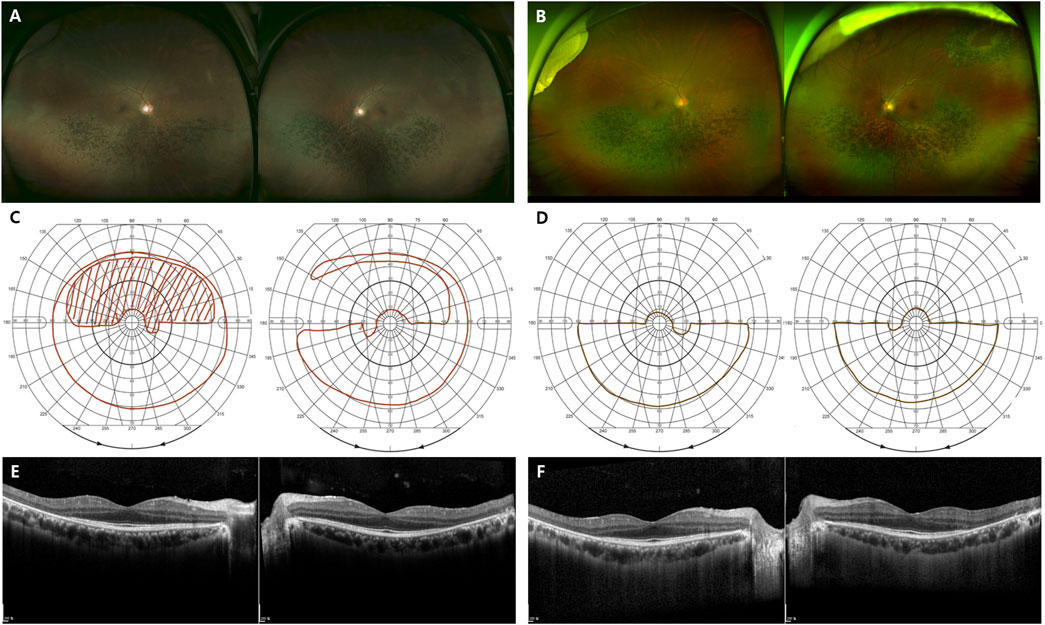
FIGURE 5. Representative images of the sector RHO-associated RP. (A–F) A 48-year-old man carrying the missense mutation (p.T17M). (A, B) A wide field color fundus photo at the initial and last visits (7 years gap), respectively. (B) A barrier laser scar showing at superotemporal area on left eye. (C, D) The Goldmann visual field test at the initial and last visits (7 years gap), respectively. (E, F) Horizontal and vertical scans of spectral domain OCT at the initial and last visits (7 years gap), respectively.
The clinical features and visual prognosis are better in sector RP than in generalized RP. It has been reported that 24 of 26 patients (83.3%) with sector RP retained a BCVA better than 0.3 logarithm of the minimal angle of resolution (LogMAR) (Georgiou et al., 2021). Consistent with this finding, our study showed a mean final logMAR visual acuity of the sector RP group of 0.10, which was better than the 0.54 of the generalized group (Table 2). In addition, a significant difference in the last follow-up visit reported VF pattern was detected between the two groups (p < 0.001). No statistically significant difference in visual acuity deterioration during follow-up was observed between the two groups; however, a significant difference in the rate of VF impairment was detected. The significantly higher rate of VF impairment progression to late-stage RP in the generalized RP group was consistent with that of another study (Hartong et al., 2006). However, Nguyen et al. reported a significant difference in visual acuity between the two groups, at the last examination. This discrepancy might have occurred because our study had a relatively small sample size and a short follow-up period. Thus, a few patients had advanced photoreceptor degeneration involving the foveal center.
All patients with sector RP (n = 7) showed bone-spicule pigmentation in the inferior retina, and several studies may explain this result. Generally, the inferior retina is more prone to light exposure, including sunlight and ceiling light, because multiple light sources are oriented upwards. Schwartz et al. (Schwartz et al., 2003) reported that the highest doses of ultraviolet and blue light are located at the superior VF. Tam and Naash et al. (Naash et al., 1996; Tam and Moritz, 2007) reported that the density of apoptotic cells was the highest in the inferior retina and that an increased exposure to a darkened environment delayed retinal degeneration progression in P23H rats. All our study patients with sector RP (seven patients) showed inferior degeneration, which is consistent with the findings of previous studies.
The presence of a central VF does not guarantee normal photopic ERG responses. Even when the central VF is preserved, the photopic ERG responses typically show reduced or no response. Similarly, in our study and other studies of RHO-associated RP, there were no cases of normal photopic ERG responses, regardless of visual field preservation.
Consistent with the findings of the current study (4.8%), the expected frequency of RHO mutations in RP has been reported to range from 4% to 7% in previous studies (Chan et al., 2001; Gao et al., 2019). We identified 16 pathogenic variants of RHO, including three novel variants. Frequently reported mutations are summarized in Table 4. In the US population, the RHO p.P23H mutation is the most common, with a frequency of up to 15% in families with adRP and 61.5% (24 of 39 families) in patients with RHO-associated RP (Dryja et al., 1990b; Sung et al., 1991). Moreover, RHO p.P347L is a widely reported mutation in the Japanese, Lithuanian, South African, and Spanish populations (Fujiki et al., 1992; Kucinskas et al., 1999; Roberts et al., 2000; Fernandez-San Jose et al., 2015). RHO p.R135W is a common mutation in the Han-Chinese family (Wu et al., 2019), and RHO p. E181K and p. D190Y are common mutations in the Dutch and Belgian populations (Nguyen et al., 2021). Consistent with previous reports in literature, our study identified p.P347L as one of the two most common pathogenic variants (5 of 36 families; 13.8%). Moreover, we frequently observed the p.T17M variant (5 of 36 families; 13.8%). p.G101E (4 of 36 families; 11.1%) and p.R135W (4 of 36 families; 11.1%) were the second most common variants. These results showed that the variant frequencies largely overlap between different ethnicities, whereas there are several ethnicity-dependent variations such as p.P23H. Pathogenic variants in RHO were most frequently detected in exon 1, in 14 of 36 patients (38.9%).
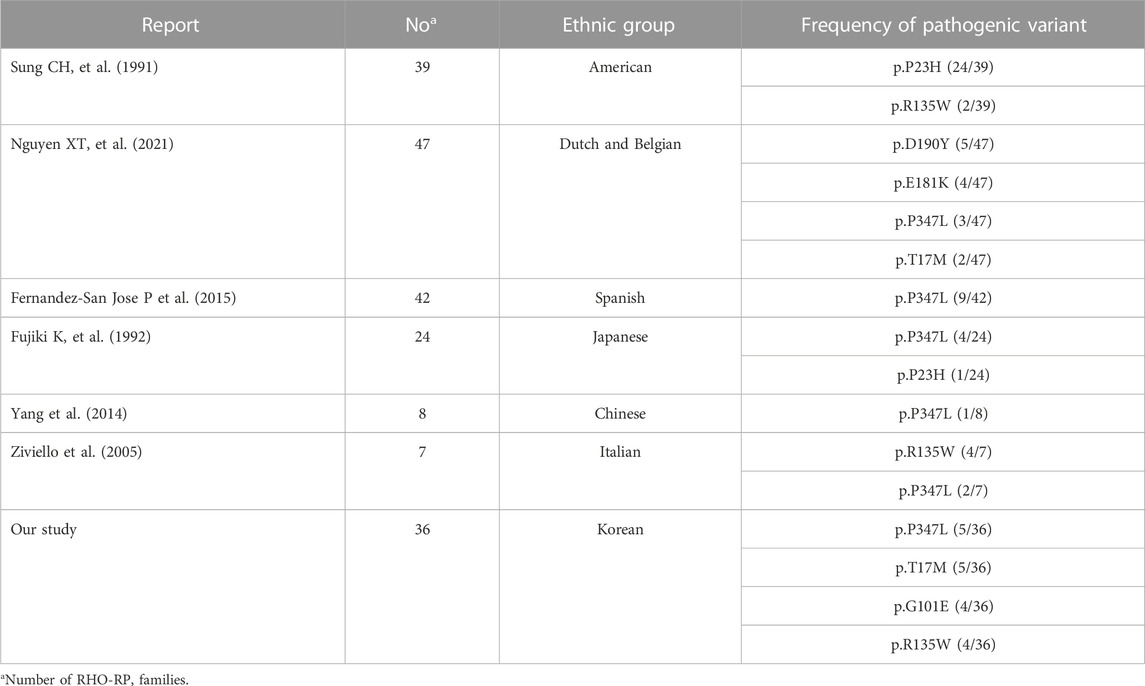
TABLE 4. Reports of the frequency of pathogenic variants among RHO-RP families.
We identified three novel RHO-associated RP mutations: p.T108P, p.G121R, and p.P347_A348del; their phenotypes were generalized RP, and for p.T108P and p.P347_A348del mutations, the central VF was <10° at the initial and last visits. For the p.G121R mutation case, the central VF was 20–30° at the last visit, possibly because the patient aged 21 years at the last visit, i.e., an early stage of RP.
In our study, the p.T17M, p.G101E, and p.E181K variants showed both generalized and sector RP phenotypes, and none of the variants presented sector RP alone. One possible explanation for this phenomenon is that sector RP eventually progressed to generalized RP in the later stages. Although none of the patients that were initially diagnosed with sector RP progressed to generalized RP, some patients that were initially diagnosed with generalized RP might have started with sector RP. In a previous study, the mutant allele (p.M39R; heterozygous) was transmitted from the affected father to the son, and the phenotype was generalized RP for the father and sector RP for the son (Ramon et al., 2014). Another possibility is that the phenotype could be influenced by other environmental factors such as light exposure. Additional long-term follow-up studies are required to confirm these findings.
RHO, comprising 348 amino acids, has a structure divided into three regions: the cytoplasmic, transmembrane, and intradiscal domains (Dryja et al., 1990a; Macke et al., 1993). In this study, seven of seven families (p.T17M, p.G101E, and p.E181K) were diagnosed with sector RP, and these pathogenic variants were located in the intradiscal domain. Consistent with our findings, several studies have reported that RHO-associated sector RP is caused by missense mutations; most of which are located in the intradiscal domain (Kranich et al., 1993; Napier et al., 2015; Xiao et al., 2019). Additionally, some sector RP rhodopsin mutations, p.M39R and p.N55K, can also be found in the transmembrane domain (Ramon et al., 2014).
Another approach to the genotype-phenotype correlation of adRP RHO mutations is based on the classification into two major classes in clinical settings (Cideciyan et al., 1998). Class A exhibits early onset severe rod dysfunction, while class B is associated with a later onset and less severe phenotype with slower progression. However, other factors, such as genetic modifiers and the environment, can also influence the disease presentation and may lead to interfamilial variability (Iannaccone et al., 2006). In this study, among eight patients with mutations corresponding to class A (p.P347L or p.P347_A348del), three demonstrated progression to late-stage RP. However, five patients did not show progression to late-stage RP until the last follow-up, suggesting the possible influence of genetic modifiers or environmental factors on disease progression. A patient with p.Q64* corresponding to class B was diagnosed with RP at the age of 13, indicating early onset RP, but only exhibited progression to late-stage RP at the age of 56. Therefore, prospective future studies are necessary to further elucidate the impact of genetic and environmental factors on disease progression.
Many ongoing clinical trials using gene supplementation therapies have been conducted on autosomal recessive and X-linked IRD since the US Food and Drug Administration approved the gene therapy voretigene neparvovec-rzyl (Luxturna). In contrast, autosomal dominant diseases, such as RHO-associated RP, are caused by gain-of-function mutations and require a substantially different approach. Targeted gene therapy is promising for RHO-associated RP; however, it requires the inhibition of mutant RHO protein expression with simultaneously increasing the wild-type-to-mutant RHO ratio to effectively reduce the rate of retinal degeneration. Several treatments are currently being developed, including stem cell or retinal tissue transplantation, nutritional supplements, retinal implants, and targeted and non-targeted gene therapies (Meng et al., 2020). Although no universally effective treatment has yet been established for RHO-associated RP, understanding and analyzing its genotype, phenotype, and underlying mechanisms can help develop future therapies.
This study had two limitations. First, the sample size was too small to perform a survival analysis. Second, the details of the follow-up protocol were not standardized because of the retrospective design of the study. However, this is the first Korean multicenter study conducted on RHO-associated RP that included the largest number of Korean patients with RHO-associated RP.
This multicenter cohort study provided information on the clinical and genetic features of RHO-associated RP in Koreans. One of the two most commonly reported pathogenic variants was p.P347L, consistent with the findings of previous reports of non-American populations. Furthermore, we identified p.T17M as a common pathogenic variant. Generalized and sector RP showed different courses of VF and BCVA progression. It is clinically important to expand the genetic spectrum and understand genotype-phenotype correlations to ultimately facilitate the development of gene therapy.
Data availability statement
The datasets presented in this article are not readily available because data obtained from targeted next-generation sequencing or whole exome sequencing are sensitive, and our ethics committee does not authorize the sharing of these data. Requests to access the datasets should be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by Institutional Review Board of Seoul National University Bundang Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
Conceptualization, SW; methodology, YJ, JK, and KJ; validation, YJ and SW; formal analysis, YJ; data curation, YJ, HL, JK, and EL; writing—original draft, YJ; writing—review and editing, KJ, KP, MK, EL, SB, CL, JH, JL, CY, and SW; visualization, YJ; supervision, JL, CY, and SW; project administration, SW. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by grants from the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education (grant number: 2021R1I1A1A01045648). This study was supported by the National Research Foundation (NRF) grant RS-2023-00248480 funded by the Korean government (MSIT).
Acknowledgments
The authors alone are responsible for the content and writing of this paper.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1240067/full#supplementary-material
Abbreviations
BCVA, best-corrected visual acuity; CME, cystic macular edema; D, diopters; ERG, electroretinography; GVF, Goldmann visual field; IOP, intraocular pressure; IRD, inherited retinal dystrophies; LogMAR, logarithm of the minimal angle of resolution; RHO, rhodopsin; RP, retinitis pigmentosa; SD, standard deviation; VF, visual field.
References
Athanasiou, D., Aguila, M., Bellingham, J., Li, W., Mcculley, C., Reeves, P. J., et al. (2018). The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog. Retin Eye Res. 62, 1–23. doi:10.1016/j.preteyeres.2017.10.002
Ballios, B. G., Place, E. M., Martinez-Velazquez, L., Pierce, E. A., Comander, J. I., and Huckfeldt, R. M. (2021). Beyond sector retinitis pigmentosa: expanding the phenotype and natural history of the rhodopsin gene codon 106 mutation (Gly-to-Arg) in autosomal dominant retinitis pigmentosa. Genes (Basel) 12, 1853. doi:10.3390/genes12121853
Bell, C., Converse, C. A., Hammer, H. M., Osborne, A., and Haites, N. E. (1994). Rhodopsin mutations in a Scottish retinitis pigmentosa population, including a novel splice site mutation in intron four. Br. J. Ophthalmol. 78, 933–938. doi:10.1136/bjo.78.12.933
Berson, E. L., and Howard, J. (1971). Temporal aspects of the electroretinogram in sector retinitis pigmentosa. Arch. Ophthalmol. 86, 653–665. doi:10.1001/archopht.1971.01000010655008
Berson, E. L., Rosner, B., Sandberg, M. A., and Dryja, T. P. (1991). Ocular findings in patients with autosomal dominant retinitis pigmentosa and a rhodopsin gene defect (Pro-23-His). Arch. Ophthalmol. 109, 92–101. doi:10.1001/archopht.1991.01080010094039
Chan, W. M., Yeung, K. Y., Pang, C. P., Baum, L., Lau, T. C., Kwok, A. K., et al. (2001). Rhodopsin mutations in Chinese patients with retinitis pigmentosa. Br. J. Ophthalmol. 85, 1046–1048. doi:10.1136/bjo.85.9.1046
Cideciyan, A. V., Hood, D. C., Huang, Y., Banin, E., Li, Z. Y., Stone, E. M., et al. (1998). Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proc. Natl. Acad. Sci. U. S. A. 95, 7103–7108. doi:10.1073/pnas.95.12.7103
Dryja, T. P., Hahn, L. B., Cowley, G. S., Mcgee, T. L., and Berson, E. L. (1991). Mutation spectrum of the rhodopsin gene among patients with autosomal dominant retinitis pigmentosa. Proc. Natl. Acad. Sci. U. S. A. 88, 9370–9374. doi:10.1073/pnas.88.20.9370
Dryja, T. P., Mcgee, T. L., Hahn, L. B., Cowley, G. S., Olsson, J. E., Reichel, E., et al. (1990a). Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N. Engl. J. Med. 323, 1302–1307. doi:10.1056/NEJM199011083231903
Dryja, T. P., Mcgee, T. L., Reichel, E., Hahn, L. B., Cowley, G. S., Yandell, D. W., et al. (1990b). A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature 343, 364–366. doi:10.1038/343364a0
Farrar, G. J., Kenna, P., Redmond, R., Shiels, D., Mcwilliam, P., Humphries, M. M., et al. (1991). Autosomal dominant retinitis pigmentosa: a mutation in codon 178 of the rhodopsin gene in two families of Celtic origin. Genomics. 11, 1170–1171.
Fernandez-San Jose, P., Blanco-Kelly, F., Corton, M., Trujillo-Tiebas, M. J., Gimenez, A., Avila-Fernandez, A., et al. (2015). Prevalence of rhodopsin mutations in autosomal dominant retinitis pigmentosa in Spain: clinical and analytical review in 200 families. Acta Ophthalmol. 93, e38–e44. doi:10.1111/aos.12486
Fujiki, K., Hotta, Y., Hayakawa, M., Sakuma, H., Shiono, T., Noro, M., et al. (1992). Point mutations of rhodopsin gene found in Japanese families with autosomal dominant retinitis pigmentosa (ADRP). Jpn. J. Hum. Genet. 37, 125–132. doi:10.1007/BF01899733
Gao, F. J., Li, J. K., Chen, H., Hu, F. Y., Zhang, S. H., Qi, Y. H., et al. (2019). Genetic and clinical findings in a large cohort of Chinese patients with suspected retinitis pigmentosa. Ophthalmology 126, 1549–1556. doi:10.1016/j.ophtha.2019.04.038
Georgiou, M., Grewal, P. S., Narayan, A., Alser, M., Ali, N., Fujinami, K., et al. (2021). Sector retinitis pigmentosa: extending the molecular genetics basis and elucidating the natural history. Am. J. Ophthalmol. 221, 299–310. doi:10.1016/j.ajo.2020.08.004
Hartong, D. T., Berson, E. L., and Dryja, T. P. (2006). Retinitis pigmentosa. Lancet 368, 1795–1809. doi:10.1016/S0140-6736(06)69740-7
Iannaccone, A., Man, D., Waseem, N., Jennings, B. J., Ganapathiraju, M., Gallaher, K., et al. (2006). Retinitis pigmentosa associated with rhodopsin mutations: correlation between phenotypic variability and molecular effects. Vis. Res. 46, 4556–4567. doi:10.1016/j.visres.2006.08.018
Inglehearn, C. F., Bashir, R., Lester, D. H., Jay, M., Bird, A. C., Bhattacharya, S. S., et al. (1991). A 3-bp deletion in the rhodopsin gene in a family with autosomal dominant retinitis pigmentosa. Am. J. Hum. Genet. 48, 26–30.
Jacobson, S. G., Kemp, C. M., Sung, C. H., and Nathans, J. (1991). Retinal function and rhodopsin levels in autosomal dominant retinitis pigmentosa with rhodopsin mutations. Am. J. Ophthalmol. 112, 256–271. doi:10.1016/s0002-9394(14)76726-1
Keen, T. J., Inglehearn, C. F., Lester, D. H., Bashir, R., Jay, M., Bird, A. C., et al. (1991). Autosomal dominant retinitis pigmentosa: four new mutations in rhodopsin, one of them in the retinal attachment site. Genomics 11, 199–205. doi:10.1016/0888-7543(91)90119-y
Kim, K. J., Kim, C., Bok, J., Kim, K. S., Lee, E. J., Park, S. P., et al. (2011). Spectrum of rhodopsin mutations in Korean patients with retinitis pigmentosa. Mol. Vis. 17, 844–853.
Kim, H. M., Joo, K., Han, J., and Woo, S. J. (2021). Clinical and genetic characteristics of Korean congenital stationary night blindness patients. Genes (Basel) 12, 789. doi:10.3390/genes12060789
Kim, M. S., Joo, K., Seong, M. W., Kim, M. J., Park, K. H., Park, S. S., et al. (2019). Genetic mutation profiles in Korean patients with inherited retinal diseases. J. Korean Med. Sci. 34, e161. doi:10.3346/jkms.2019.34.e161
Kranich, H., Bartkowski, S., Denton, M. J., Krey, S., Dickinson, P., Duvigneau, C., et al. (1993). Autosomal dominant 'sector' retinitis pigmentosa due to a point mutation predicting an Asn-15-Ser substitution of rhodopsin. Hum. Mol. Genet. 2, 813–814. doi:10.1093/hmg/2.6.813
Kucinskas, V., Payne, A. M., Ambrasiene, D., Jurgelevicius, V., Steponaviciute, D., Arciuliene, J. V., et al. (1999). Molecular genetic study of autosomal dominant retinitis pigmentosa in Lithuanian patients. Hum. Hered. 49, 71–74. doi:10.1159/000022847
Liu, W., Guo, R., Hao, H., and Ji, J. (2021). Identification of a novel RHO heterozygous nonsense mutation in a Chinese family with autosomal dominant retinitis pigmentosa. BMC Ophthalmol. 21, 360. doi:10.1186/s12886-021-02125-9
Macke, J. P., Davenport, C. M., Jacobson, S. G., Hennessey, J. C., Gonzalez-Fernandez, F., Conway, B. P., et al. (1993). Identification of novel rhodopsin mutations responsible for retinitis pigmentosa: implications for the structure and function of rhodopsin. Am. J. Hum. Genet. 53, 80–89.
Meng, D., Ragi, S. D., and Tsang, S. H. (2020). Therapy in rhodopsin-mediated autosomal dominant retinitis pigmentosa. Mol. Ther. 28, 2139–2149. doi:10.1016/j.ymthe.2020.08.012
Naash, M. L., Peachey, N. S., Li, Z. Y., Gryczan, C. C., Goto, Y., Blanks, J., et al. (1996). Light-induced acceleration of photoreceptor degeneration in transgenic mice expressing mutant rhodopsin. Invest. Ophthalmol. Vis. Sci. 37, 775–782.
Napier, M. L., Durga, D., Wolsley, C. J., Chamney, S., Alexander, S., Brennan, R., et al. (2015). Mutational analysis of the rhodopsin gene in sector retinitis pigmentosa. Ophthalmic Genet. 36, 239–243. doi:10.3109/13816810.2014.958862
Nguyen, X. T., Talib, M., Van Cauwenbergh, C., Van Schooneveld, M. J., Fiocco, M., Wijnholds, J., et al. (2021). Clinical characteristics and natural history of rho-associated retinitis pigmentosa: a long-term follow-up study. Retina 41, 213–223. doi:10.1097/IAE.0000000000002808
Ramon, E., Cordomi, A., Aguila, M., Srinivasan, S., Dong, X., Moore, A. T., et al. (2014). Differential light-induced responses in sectorial inherited retinal degeneration. J. Biol. Chem. 289, 35918–35928. doi:10.1074/jbc.M114.609958
Roberts, L., Ramesar, R., and Greenberg, J. (2000). Low frequency of rhodopsin mutations in South African patients with autosomal dominant retinitis pigmentosa. Clin. Genet. 58, 77–78. doi:10.1034/j.1399-0004.2000.580114.x
Schwartz, L., Boëlle, P. Y., D'hermies, F., Ledanois, G., and Virmont, J. (2003). Blue light dose distribution and retinitis pigmentosa visual field defects: an hypothesis. Med. Hypotheses 60, 644–649. doi:10.1016/s0306-9877(02)00391-2
Seong, M. W., Seo, S. H., Yu, Y. S., Hwang, J. M., Cho, S. I., Ra, E. K., et al. (2015). Diagnostic application of an extensive gene panel for Leber congenital amaurosis with severe genetic heterogeneity. J. Mol. Diagn 17, 100–105. doi:10.1016/j.jmoldx.2014.09.003
Sung, C. H., Davenport, C. M., Hennessey, J. C., Maumenee, I. H., Jacobson, S. G., Heckenlively, J. R., et al. (1991). Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proc. Natl. Acad. Sci. U. S. A. 88, 6481–6485. doi:10.1073/pnas.88.15.6481
Tam, B. M., and Moritz, O. L. (2007). Dark rearing rescues P23H rhodopsin-induced retinal degeneration in a transgenic Xenopus laevis model of retinitis pigmentosa: a chromophore-dependent mechanism characterized by production of N-terminally truncated mutant rhodopsin. J. Neurosci. 27, 9043–9053. doi:10.1523/JNEUROSCI.2245-07.2007
Van Woerkom, C., and Ferrucci, S. (2005). Sector retinitis pigmentosa. Optometry 76, 309–317. doi:10.1016/s1529-1839(05)70314-6
Verbakel, S. K., Van Huet, R. a. C., Boon, C. J. F., Den Hollander, A. I., Collin, R. W. J., Klaver, C. C. W., et al. (2018). Non-syndromic retinitis pigmentosa. Prog. Retin Eye Res. 66, 157–186. doi:10.1016/j.preteyeres.2018.03.005
Wang, D. Y., Chan, W. M., Tam, P. O., Chiang, S. W., Lam, D. S., Chong, K. K., et al. (2005). Genetic markers for retinitis pigmentosa. Hong Kong Med. J. 11, 281–288.
Wang, J., Xu, D., Zhu, T., Zhou, Y., Chen, X., Wang, F., et al. (2019). Identification of two novel RHO mutations in Chinese retinitis pigmentosa patients. Exp. Eye. Res. 188, 107726. doi:10.1016/j.exer.2019.107726
Wu, Y., Guo, Y., Yi, J., Xu, H., Yuan, L., Yang, Z., et al. (2019). Heterozygous RHO p.R135W missense mutation in a large Han-Chinese family with retinitis pigmentosa and different refractive errors. Biosci. Rep. 39. doi:10.1042/BSR20182198
Xiao, T., Xu, K., Zhang, X., Xie, Y., and Li, Y. (2019). Sector Retinitis Pigmentosa caused by mutations of the RHO gene. Eye (Lond) 33, 592–599. doi:10.1038/s41433-018-0264-3
Yang, G., Xie, S., Feng, N., Yuan, Z., Zhang, M., Zhao, J., et al. (2014). Spectrum of rhodopsin gene mutations in Chinese patients with retinitis pigmentosa. Mol. Vis. 20, 1132–1136.
Zhang, Q. (2016). Retinitis pigmentosa: progress and perspective. Asia Pac J. Ophthalmol. (Phila) 5, 265–271. doi:10.1097/APO.0000000000000227
Keywords: retinitis pigmentosa, rhodopsin, generalized retinitis pigmentosa, sector retinitis pigmentosa, Koreans
Citation: Jung YH, Kwak JJ, Joo K, Lee HJ, Park KH, Kim MS, Lee EK, Byeon SH, Lee CS, Han J, Lee J, Yoon CK and Woo SJ (2023) Clinical and genetic features of Koreans with retinitis pigmentosa associated with mutations in rhodopsin. Front. Genet. 14:1240067. doi: 10.3389/fgene.2023.1240067
Received: 14 June 2023; Accepted: 14 August 2023;
Published: 29 August 2023.
Edited by:
Ekim Zihni Taskiran, Hacettepe University, TürkiyeReviewed by:
Pere Garriga, Universitat Politecnica de Catalunya, SpainYuanyuan Chen, University of Pittsburgh, United States
Bo Lei, Henan Provincial People’s Hospital, China
Copyright © 2023 Jung, Kwak, Joo, Lee, Park, Kim, Lee, Byeon, Lee, Han, Lee, Yoon and Woo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Se Joon Woo, c2Vqb29uMUBzbnUuYWMua3I=; Chang Ki Yoon, c3lzdDE4QGdtYWlsLmNvbQ==; Junwon Lee, YnJhdmV3b25AeXVocy5hYw==
†These authors have contributed equally to this work and shared first authorship