 Xiaoyan Xu1Jing Zhu1
Xiaoyan Xu1Jing Zhu1 Liwei Fang1Zhuo Zou2Jingjing Yuan1Min Peng3Guoliang Yu3
Liwei Fang1Zhuo Zou2Jingjing Yuan1Min Peng3Guoliang Yu3 De Wu1
De Wu1 Yun Liu2*Jiulai Tang1*
Yun Liu2*Jiulai Tang1*- 1Pediatric Neurorehabilitation Center, Pediatric Department, The First Affiliated Hospital of Anhui Medical University, Hefei, China
- 2Department of Rehabilitation, Kunming Children’s Hospital, Kunming Medical University, Kunming, Yunnan, China
- 3Chigene (Beijing) Translational Medical Research Center Co, Ltd, Beijing, China
5,10-methenyltetrahydrofolate synthetase (MTHFS) deficiency is a folate metabolism disorder known as a rare autosomal recessive neurodevelopmental disorder (MIM: #618367). With central nervous system involvements, it is mainly characterized by developmental delay, epilepsy, microcephaly, hypertonia, and cranial nerves involvement. Here, we report three new cases with MTHFS deficiency from two non-consanguineous Chinese families. All patients showed white matter dysplasia and global developmental delay, of which only patient 1 and 2 manifested tonic-clonic seizures. Moreover, patient 2 had severe eczema and patient 3 had recurrent diarrhea. Both phenotypic features are firstly found in MTHFS deficiency. Trio whole-exome sequencing and sanger sequencing were used to identify four novel variants, p.Y169Tfs*17, p.S53F, c.117+1delG, and p.E61G in the MTHFS gene. The identification of four novel pathogenic variants and varied clinical features in three affected patients expands the genotype and phenotype spectrum of MTHFS deficiency. We also reviewed all cases of MTHFS deficiency that had previously been reported. The experience of diagnosis and treatment from these cases provides us a more comprehensive understanding of this rare disease.
Introduction
The 5,10-methenyltetrahydrofolate synthetase (MTHFS), also known as 5-formyltetrahydrofolate (5-FTHF) cyclo-ligase, is encoded by the MTHFS gene (location: 15q25.1). MTHFS catalyzes the conversion of 5-FTHF to 5,10-MTHF in an ATP and Mg2+ dependent manner, which is subsequently reduced to 5-MTHF and other reduced folates required for neurotransmitter synthesis, amino acid metabolism, and purine/thymidylate biosynthesis (Dayan et al., 1995). MTHFS gene knockdown and overexpression in cell lines or mice indicate that MTHFS regulates cellular folate pools and determines folate levels in tissues, which is crucial for oligodendrocyte maturation and survival in the central nervous system (CNS) (Girgis et al., 1997; Field et al., 2011). MTHFS also potentially affect the translation of mitochondrial methionyl-tRNA by acting as an intra-mitochondrial folate pool regulator in human mitochondria, whose deficiency could contribute to secondary mitochondrial dysfunction (Bertrand et al., 1995; Krupenko et al., 2010). Since folate metabolism and mitochondrial energy metabolism are critical in neural cells and cerebrospinal fluid (CSF), disruption of MTHFS activity can cause a variety of neurological impairments (Zheng and Cantley, 2019).
In recent years, MTHFS deficiency (MIM: #618367) has only been reported in a few unrelated patients as a rare autosomal recessive neurodevelopmental disorder. A total of seven variants in the MTHFS gene were identified in six different cases till now, which results in a range of phenotypes associated with abnormal enzyme activity. The level of 5, 10-MTHF in cerebrospinal fluid is decreased due to MTHFS impairment, whereas peripheral folate levels are normal. The clinical features mainly include global developmental delay (GDD), hypotonia, epilepsy, microcephaly, delayed myelination or hypomyelination and other cerebral dysplasia (Rodan et al., 2018; Romero et al., 2019; Sakthivel et al., 2020; Cavusoglu et al., 2022; Vafaee-Shahi et al., 2022). However, as an extremely rare disorder, there is very limited understanding of this disease.
Here, we present clinical findings of three Chinese patients with MTHFS deficiency, identify four novel variants c.504del (p.Y169Tfs*17), c.158C>T (p.S53F), c.117+1del and c.182A>G (p.E61G) in the MTHFS gene, and review the literature on the clinical features and therapy responses of this disease.
Materials and methods
Subjects and study approval
We recruited two families with MTHFS deficiency. Family 1 has two affected siblings and experienced two pregnancies of embryonic demise. The two children were admitted with repetitive seizures to Pediatric Neurorehabilitation Center, The First Affiliated Hospital of Anhui Medical University. The proband of family 2 was referred to the Department of Pediatric Rehabilitation Medicine, Kunming Children’s Hospital due to delayed motor development. This study has been approved by the ethics committee of The First Affiliated Hospital of Anhui Medical University and Kunming Children’s Hospital. Written informed consent for genetic testing and publication of relative information has been obtained from the legal guardians of the children. All investigations followed the principles of the Declaration of Helsinki.
Genetic tests and analysis
After obtaining prior written consent of the parents, venous blood samples from all patients and their parents were collected and stored properly. Trio whole-exome sequencing (WES) was performed on the genomic DNA extracted from whole blood samples using xGen Exome Research Panel v1.0 (IDT, Iowa, United States) and paired-end sequenced (2 × 150 bp) on NovaSeq 6,000 (Illumina San Diego, CA). Raw sequence readouts were processed by fastp and paired-end sequence reads were aligned to the human GRCh37/hg19 genome using the Burrows-Wheeler Aligner (BWA) and the GATK software packages. Variants annotations were retrieved from the database-based online system independently developed by Chigene (https://www.chigene.org, Beijing, China), including public SNP databases, the Single Nucleotide Polymorphism database (dbSNP), 1000 Genomes Project, Exome Aggregation Consortium (ExAC), Genome Aggregation Database (gnomAD), and ESP, and Chigene in-house MAFs database. Genes and the associated clinical phenotypes in Mendelian disorders were annotated using data from UCSC, RefGene, GENCODE, and ENSEMBL transcripts, LOVD, SWISS, Clinvitae, HGMD, OMIM, and ClinVar databases. Functional and conservational predictions for the amino-acid changes were evaluated by the software packages Provean, SIFT, PolyPhen2, MutationTaster, M-CAP, REVEL, and CADD; MaxEntScan, dbscSNV, and GTAG; GERP, phyloP, and phastCons. Causative variants screening was done according to rare variants with minor allele frequency (MAF) < 0.05% and biological impact, gene functions and disease mechanism, recessive/dominant/de novo pattern of inheritance and segregation analysis, phenotypic relevance in Human Phenotype Ontology (HPO) terms and the scientific literature. Subsequently, variants were classified according to the ACMG clinical practice guidelines (Richards et al., 2015). Sanger sequencing was performed on the patients and the parents to validate the candidate variants. The crystal structure of human MTHFS with 10-formyltetrahydrofolate (PDB ID: 3HY3) and disease-associated residues were visualized by Chimera-X.
Results
Case description
Family 1
Patient 1 (P1) was a 16-month-old boy admitted to our department after having two seizures at the ages of 15 and 16 months in 2018. He had ocular hypertelorism, long nasolabial fold, and tent-shaped upper lip. Physical examination revealed involuntary movements and hypertonia when excited. He was born from the second pregnancy of a non-consanguineous couple, while the first pregnancy resulted in an embryonic demise, and he was the first child of family 1 (Figure 1A). His mother had hypothyroidism during pregnancy and was treated with levothyroxine. At 6 months of age, he could not actively grasp, and at 9 months of age, he presented an inability to sit without assistance, indicating developmental delay (DD). His cranial MRI at 9 months primarily showed abnormal myelination, widened bilateral frontotemporal subarachnoid, and widened cerebral sulci (Figure 1B).
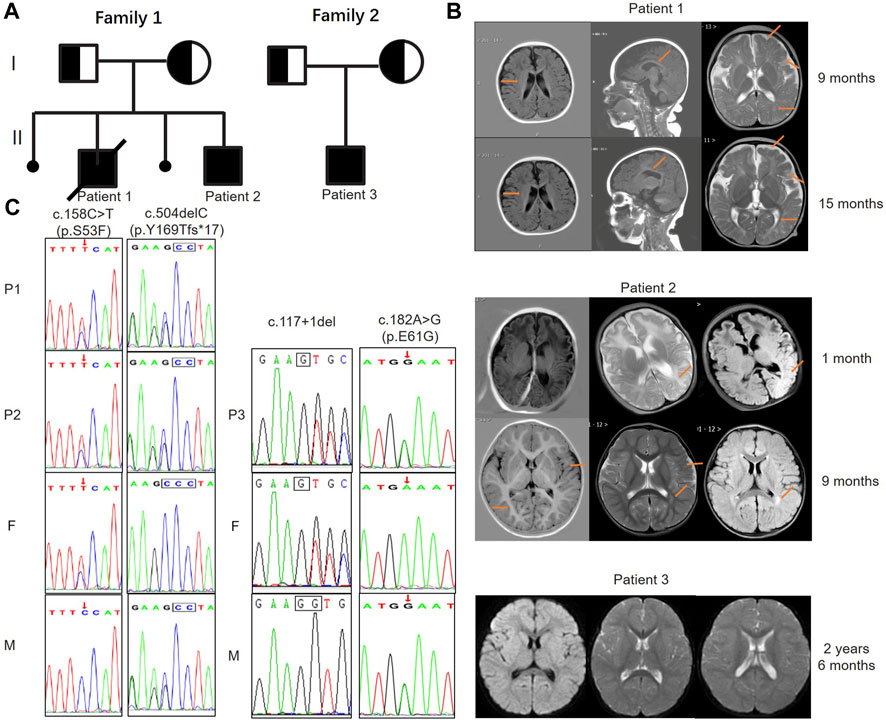
FIGURE 1. Pedigree diagrams, MRI images, and genetic findings of patients with MTHFS deficiency. (A) The pedigree diagrams of family 1 and 2 reveal a recessive inheritance pattern. Family 1 had two pregnancy loss and patient 1 died when he was 22 months old. Patient 3 is the only child in family 2. (B) Patient 1: images from left to right are axial T1-weighted, parasagittal T1-weighted, and axial T2-weighted. Widened bilateral frontotemporal subarachnoid, stripe hyperintensity around the lateral ventricle and centrum semiovale, decreased white matter volume, delayed myelination, thin corpus callosum, slightly enlarged lateral ventricles and widened cerebral sulci were observed (arrows) at 9 and 15 months. Patient 2: images from left to right are axial T1-weighted, axial T2-weighted and axial FLAIR. The images at 1 month revealed a slightly higher T2 and FLAIR signal in the left parieto-occipital lobe (arrow). The follow-up images at 9 months showed slightly widened bilateral sulcus fissure of the cortex, widened bilateral frontotemporal subdural space, decreased volume of white matter, and a patchy high T2 and FLAIR signal beside the left lateral ventricle (arrows). Patient 3: images from left to right are axial FLAIR and axial T2-weighted. The images at 2 years and 6 months of age only showed a decrease in the white matter volume of the brain, indicating hypomyelination. (C) Sanger sequencing confirms the pathogenic variants and both healthy parents are heterozygous carriers of the MTHFS mutations. Two variants of c.158C>T (p.S53F) and c.504del (p.Y169fs*17) in the MTHFS gene were identified in family 1; c.117+1del and c.182A>G (p.E61G) variants were identified in family 2. F: father; M: mother.
At 15 and 16 months of age, he respectively experienced frequent seizures after two fever episodes (including partial seizures, tonic seizures, tonic-clonic seizures, and atypical absence seizures), lasting between minutes to hours (status epilepticus). Hypertonia and athetosis were also observed during seizures. Phenobarbital was administered intravenously for sedation. His cranial MRI at 15 months re-demonstrated white matter dysplasia (Figure 1B). Blood routine examination showed mild anemia with low hemoglobin (Hb) of 104 g/L and low hematocrit (HCT) of 31.30%. Serum vitamin B12, lactate (LAC), peripheral folate, fasting blood glucose (FBG), thyroid function, and urine metabolism were normal. Diagnosed with epilepsy, cerebral palsy and GDD, he was treated with oxcarbazepine for anti-seizure therapy and had regular rehabilitation training.
After orally taken oxcarbazepine for 1 month, he developed multiple afebrile convulsions. Then the anti-seizure therapy regimen was replaced with sodium valproate and lamotrigine, but he still had petit mal seizures. At the age of 20 months, he had short stature, could not sit without assistance and pronounced unconsciously, indicating GDD. His cognitive, motor and speech functions did not improve after rehabilitation training. Unfortunately, the patient died from sudden unexpected death of epilepsy (SUDEP) at 22 months of age.
Patient 2 (P2), male, 3 months old, was admitted to our department due to recurrent seizures in 2022. He had craniofacial dysmorphisms including microcephaly, oxycephaly, long nasolabial fold, tent-shaped upper lip, and high arched palate. Tapered fingers were observed. Physical examination showed hypertonia, poor head control, poor eye contact and interaction, and severe eczematous dermatitis. He was the second child and was born from the fourth pregnancy of family 1. The third pregnancy of his mother was also ended by an embryonic demise (Figure 1A).
P2 was delivered by cesarean section at term without asphyxia or hypoxia and his Apgar score was 10–10. On the second day after birth, he was hospitalized due to shivering. Electroencephalogram (EEG) was normal and symptomatic treatment such as hyperbaric oxygen therapy was given. At the age of 40 days, he had frequent seizures in 1 day (focal seizures with impaired awareness). Each seizure lasted for 1 minute and then stopped spontaneously. The 4-h video-EEG (VEEG) in the absence of seizures did not capture epileptiform discharge but revealed 2–4 Hz low amplitude slow wave activity in both the awake and sleep states. His cranial MRI showed slightly higher T2 and FLAIR signal in the left parieto-occipital lobe (Figure 1B). Cranial ultrasound revealed subependymal cysts in bilateral ventricles and mild enlargement of bilateral ventricles. Echocardiography detected the patent foramen oval.
At the age of two and a half months, the child suffered from frequent myoclonic seizures and tonic-clonic seizures, more than ten times a day. VEEG revealed the spike-and-slow-wave complex and sharp-wave in the left occipital and temporal regions, and the spike-and-slow-wave complex in the right occipital region during the sleep period. One epileptiform discharge in the left occipital region corresponds to the tonic seizures and tonic-clonic seizures observed in the video. Laboratory tests showed homocysteinemia (HCY) of 22.43 μmol/L, Hb of 98 g/L, mean corpuscular volume (MCV) of 108.1 fL, HCT of 30.50%, albumin of 37.1 g/L, total protein 52.1 g/L, and high neuron-specific enolase (NSE) level of 32.64 ng/mL, indicating hyperhomocysteinemia and macrocytic anemia. Serum vitamin B12, LAC, folate, FBG, thyroid function, and urine metabolism were normal. After treated with oral vigabatrin and pulse adrenocorticotropic hormone (ACTH) therapy, the frequency of seizures were significantly reduced. L-5-methyltetrahydrofolate (L-5-MTHF) and mecobalamin were orally taken regularly, and individualized rehabilitation therapy was carried out.
He still had focal seizures 1–2 times per day and recurrent eczematous dermatitis when he was 5.5 months of age. Topiramate were added orally, and no significant seizures were observed so far. The severe eczematous dermatitis was improved significantly after orally taken zinc gluconate (35 mg, bid) for 2 weeks. Repeat laboratory tests revealed a normal HCY level of 8.59 μmol/L, a slightly low Hb level of 120 g/L and a high NSE level of 39.87 ng/mL. VEEG at eight and a half months of age still showed the spike-and-slow-wave complex and sharp-wave in bilateral occipital and temporal regions. His cranial MRI at 9 months revealed decreased white matter volume, patchy high T2 and FLAIR signals beside the lateral ventricle, indicating cerebral dysplasia (Figure 1B). Other routine examinations and laboratory tests were normal.
At the follow-up of 13 months of age, he could pronounce “mom or dad”, interact with people, exchange objects from one hand to the other, sit without assistance, and stand for 2 min with support. The muscle tension of the extremities was lower than before. His Gesell assessment still revealed a moderate GDD: 57 in gross motor, 50 in fine motor, 46 in adaptability, 46 in language ability, and 57 in social ability.
Family 2
Patient 3 (P3) was a 30-month-old boy admitted to our department due to delayed motor milestone in 2022. He was the only child of a non-consanguineous couple (family 2) and was delivered at term by cesarean section with a normal birth weight (Figure 1A). After birth, he developed pathological jaundice, which was improved after exchange transfusion and phototherapy. He had feeding difficulties, slow growth rate, irritability, and recurrent diarrhea in the neonatal period without any drug or food allergies. The boy had normal motor milestones before 1 year old and could stand without assistance at 14 months of age. But he was unable to walk by himself until he was 30 months old, and even at that point he still showed bad stability and was prone to falling. Besides, he could only speak a few simple words and pronounce unclearly at 30 months. Griffith psychomotor developmental scale showed large motor developmental age of 20.5–21 months, individual-social developmental age of 19.5 months, language developmental age of 26–26.5 months, hand-eye coordinated developmental age of 18–8.5 months, and performance developmental age of 15–15.5 months, indicating GDD. Physical examination showed unstable muscle tone, and involuntary movements. He also had pigeon-toes and limited dorsiflexion of the feet. His EEG result was normal, and cranial MRI revealed partial cortical thickening in bilateral frontoparietal lobes, small bilateral white matter extent, and slightly higher white matter T2 and T2-Flair signals (Figure 1B). No abnormalities were detected in other routine examinations and laboratory tests of periphery blood.
After having regular rehabilitation for 3 months, his walking stability, cognitive level, speech and pronounce, hand function, and dorsiflexion limitation of both feet were improved. The involuntary movements of limbs were reduced. He could run, feet jump, climb 3 to 5 stairs, recognize size and color, distinguish up and down, and sing short nursery rhymes, but still had irritability and a slower response comparing with his peers.
Genetic and molecular analysis
Through trio-WES analysis and Sanger sequencing, four variants were identified in the MTHFS gene. The pedigree diagrams of family 1 and family 2 were presented in Figure 1A.
To be specific, P1 and P2 carried the same compound heterozygous variation in the MTHFS gene (NM_006441.4) consisting of c.158C>T (p.S53F) and c.504del (p.Y169fs*17), which were inherited from his father and mother, respectively (Figure 1C). The NM_006441.4 transcript encodes NP_006432.1 (isoform a). As a canonical sequence, it has 3 exons and encodes 203-amino-acid protein (Figure 2A). The variant of c.504del (p.Y169fs*17) is a detrimental frameshift variant at exon 3 that is unlikely to initiate the nonsense-mediated mRNA decay (NMD) and predicted to produce an abnormal 184aa product (Figure 2A). The other c.158C>T (p.S53F) variant is located at exon 2 and predicted to be deleterious by Provean, SIFT, PolyPhen-2, Mutation taster, and CADD. Moreover, S53 residue is relatively conserved in multiple species (Figure 2B). To evaluate the potential pathogenicity, we examined the residue change of S53F on the crystal structure of MTHFS protein (Figure 2C). The hydrogen bonds between S53 residue and multiple residues around stabilize the β-sheet structure. Replacement of S53 with phenylalanine would break the bond between S53 and M92 and affect the relative functions (Figure 2C). In addition, according to the ACMG guidelines, c.158C>T (p.S53F) is likely pathogenic (PM2+PM3+PP1+PP3+PP4) and c.504del (p.Y169fs*17) is pathogenic (PVS1+PM2+PP1+PP4). Neither of these variants has been observed or reported previously in any public genomic variants database (such as dbSNP, gnomAD, 1,000 Genomes, ESP6500, ExAC, Colinear, Clinvitae, SWISS, and HGMD).
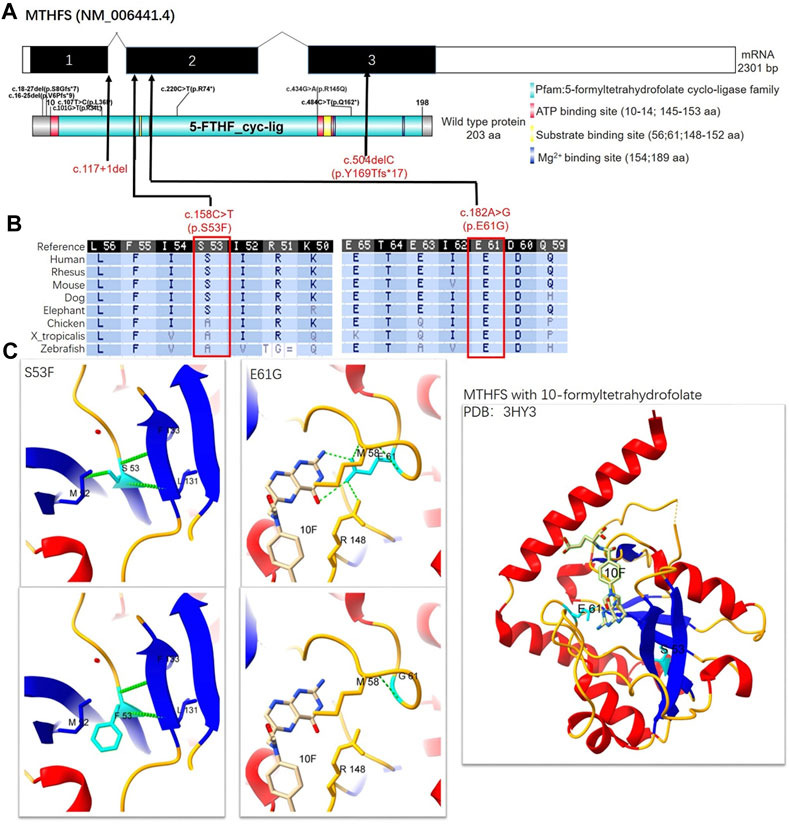
FIGURE 2. Schematic representation, conservation analysis, and structural analysis of the MTHFS protein. (A) The NM_006441.4 transcript encodes a longer isoform NP_006432.1 as the canonical sequence. It has 3 exons and encodes 203-amino-acid protein. MTHFS protein is also known as 5-formyltetrahydrofolate cyclo-ligase (10–198aa) with ATP binding sites (red box), substrate-binding sites (yellow box), and Mg2+ binding sites (dark blue box). Seven variants had been identified in previous published literature (small black text) and four novel variants were identified in this study (red text). (B) The conservation of MTHFS: S53 and G61 residues across multiple different species. (C) The crystal structure of the human MTHFS with 10-formyltetrahydrofolate is visualized using Protein Data Bank (PDB) structure 3HY3. β-sheet, α-helix, coil, and H-bond are shown in blue, red, orange, and green colors, respectively. S53 contacts with M92, F133, and L131 by hydrogen bonds, while F53 fails to form a hydrogen bond with M92. E61 contacts with M58 and R148 by hydrogen bonds and the carboxyl terminal of E61 also interacts with the NA2 and N3 of the substrate. G61 only contacts with M58. 10F: 10-formyltetrahydrofolate.
P3 from family 2 carried a different compound heterozygous variation consisting of c.117+1del and c.182A>G (p.E61G), which were inherited from his father and mother, respectively (Figure 1C). The canonical splice site variant c.117+1del is located at intron 1 (Figure 2A) and is predicted to cause improper splicing that alters the reading frame through exon skipping or intron retention, resulting in a possible NMD or encoding an aberrant protein. Regarding the other variant, multiple in silico analysis predictions show that c.182A>G (p.E61G) variant is deleterious. Homogeneous analysis demonstrated that E61 is highly conserved in different species (Figure 2B). In the MTHFS protein model, E61 localizes in the substrate binding site and contacts M58 and R148 by hydrogen bonds. The carboxyl terminal of E61 also interact with the NA2 and N3 of 10-FTHF substrate. The change of E61G would disrupt the hydrogen bonds with the M58 and R148 residues and break the interaction with substrate (Figure 2C). According to the ACMG guidelines, c.117+1del (PVS1+PM2+PP4) and c.182A>G (p.E61G) (PM2+PM3+PP3+PP4) are both likely pathogenic. Neither variant was reported in the public population databases such as dbSNP, 1,000 genomes, and gnomAD.
Combining the clinical characteristics and genetic results, all patients were diagnosed as MTHFS deficiency, and all four variants we identified in the MTHFS gene were novel.
Discussion
Folate metabolic network provides indispensable cofactors in nucleotide synthesis, neurotransmitter synthesis, methionine regeneration, and amino acid metabolism in several aspects of cell division, methylation, and homeostasis, especially in the CNS. Thus, folate metabolic deficiency can cause a variety of neurological impairments (Zheng and Cantley, 2019). MTHFS deficiency is a kind of inborn errors of folate metabolism caused by mutations in the MTHFS gene. As an extremely rare neurodevelopmental disorder with microcephaly, epilepsy, and hypomyelination (NEDMEHM), the phenotype was first created in OMIM (MIM: #618367) in 2019. The primary signs and symptoms include developmental delay (DD), microcephaly, hypertonia, feeding difficulties, exaggerated startle response or irritability, epilepsy, and cranial nerves affected in early infancy (Rodan et al., 2018; Romero et al., 2019; Sakthivel et al., 2020; Cavusoglu et al., 2022; Vafaee-Shahi et al., 2022).
MTHFS acts as the only enzyme in the folate-mediated one-carbon pathway (FOCM) known to catalyze both endogenous 5-FTHF and exogenous folic acid into 5,10-MTHF, which is subsequently reduced to 5-MTHF and other reduced folates (Dayan et al., 1995). Therefore, the lack of MTHFS (homozygous mutations) results in the increased level of 5-FTHF and the low-normal 5-MTHF level in CSF (Romero et al., 2019; Sakthivel et al., 2020). In some patients with MTHFS deficiency (heterozygous mutations), there is a low folate CSF level (Rodan et al., 2018; Cavusoglu et al., 2022). Several mechanisms could be involved to explain why cerebral folate deficiency contributes to the possible pathophysiology of this neurodevelopmental disorder. First, the downstream low-level 5-MTHF in MTHFS deficiency could lead to a lack of S-adenosylmethionine (SAM)—a methyl donor of the myelin basic protein and then decrease the stability of myelin. Besides, the substituted generation of SAM through choline oxidation pathway leads to CNS choline deficiency, which may trigger aberrant myelination due to a decline in sphingomyelin and phosphatidylcholine levels (Romero et al., 2019). Second, 5-MTHF is the largest contributor to serum folate and participates in various 5-MTHF-dependent biological processes, including neurotransmitter syntheses such as dopamine, serotonin, and norepinephrine (Miller, 2008). Third, the increased 5-FTHF level may hinder serine hydroxy methyltransferase and 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase activity, inhibit thymidylate synthesis and de novo purine metabolism, and impair DNA synthesis in cell growth and division of processes such as red blood cell production, therefore contribute to hyperhomocysteinemia and anemia (Field et al., 2006). High homocysteine level would attenuate the integrity and increase permeability of blood-brain barrier (Kamath et al., 2006; Beard et al., 2011), lead to toxin-mediated brain injury-induced seizures (Mattson and Shea, 2003; Vitvitsky et al., 2004). Fourth, MTHFS deficiency also exhibits an intramitochondrial folate deficiency, which would shunt intramitochondrial translation of mitochondrial methionyl-tRNA and lead to secondary mitochondrial dysfunction, resulting in deficient energy for brain development (Bertrand et al., 1995; Fox and Stover, 2008; Wang et al., 2021).
We summarized clinical features of the three new cases with MTHFS deficiency and previously published ones (Table 1). First described in 2018, Rodan et al. reported two unrelated patients, aged 8 and 11 years, who both carried biallelic variants in the MTHFS gene and showed similar phenotypes. Microcephaly, feeding difficulties, exaggerated startle response, short stature, severe GDD, progressive spasticity and hypomyelination were initially found in early infancy. Laboratory test detected low-normal levels of 5-MTHF in the CSF. The onset of epilepsy occurs when the patient was 2–3 years of age (Rodan et al., 2018). Next in 2019, Romero et al. reported the third patient of MTHFS deficiency with the identical presentation, such as microcephaly, short stature, spasticity, hypertonia, DD, cerebral hypomyelination and seizures. Novel findings of high-arched palate, macrocytic anemia, and elevated neopterin were recorded (Romero et al., 2019). Then, Sakthivel et al. described the fourth patient with homozygous mutation in the MTHFS gene in 2020. The patient suffered from seizures since she was 3 years old, and she had vocal fold paralysis at 9 years old. Dysphagia with feeding difficulties, hypertonia, DD, decreased level of CSF 5-MTHF were shown. Unfortunately, the patient passed away at the age of 18 due to hepatic failure. However, this study did not present the neuroimaging results (Sakthivel et al., 2020). The fifth patient with MTHFS deficiency is a girl characterized by similar features, including microcephaly, GDD, hypertonia, seizure, and cerebral hypomyelination, while she was initially diagnosed with homocystinuria. Genetic analysis helped to confirm the diagnosis (Cavusoglu et al., 2022). The sixth case was mainly characterized by microcephaly, DD, seizures, high signal intensity in the bilateral parietal and occipital lobes, macrocytic anemia, increased homocysteine, and low CSF folate level. Her unusual manifestations included nystagmus, hypotonia, and bilateral deep brain calcification. Genetic tests identified novel variants in the MTHFS gene and confirmed the diagnosis of MTHFS deficiency (Vafaee-Shahi et al., 2022).
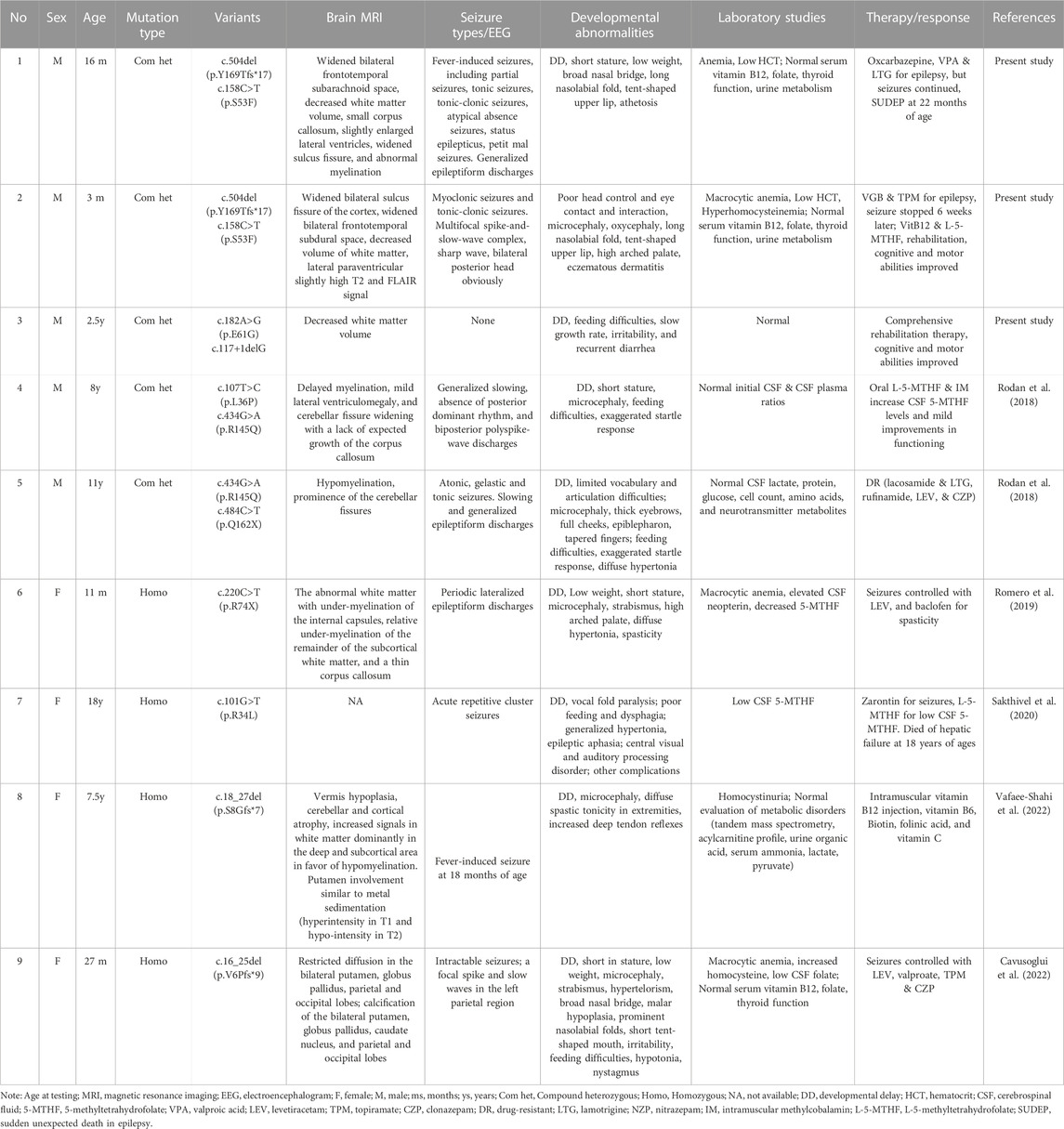
TABLE 1. Clinical features and therapy response of the patients with MTHFS variants.
In this study, we present three more cases with MTHFS deficiency from two unrelated families, with phenotypes varying from mild to severe. Similarly, three patients all showed DD and cerebral hypomyelination. P1 and P2 were siblings both had some craniofacial dysmorphisms including ocular hypertelorism, broad and flat nasal bridge, long nasolabial groove, and tent-shaped upper lip. Apart from GDD, they also suffered from seizures and anemia. P1 was the first case of SUDEP and P2 had severe seizures at birth, indicating a more severe phenotype than other cases. While P3 had feeding difficulty, irritability, and mild DD without seizures. Unlike the previously recorded cases, P3 had poor muscle coordination and more involuntary movements, which was easily misdiagnosed in clinical practice as dyskinetic cerebral palsy. Due to the mild disease presentation of P3, we suppose that the c.117+1delG variant only generates partial splicing errors, resulting in an incomplete loss of MTHFS activity. Although previous research found that the amino acid change of E61A almost completely abolished catalytic activity of MTHFS (Wu et al., 2009), the outcome of c.182A>G (p.E61G) remains unknown. Further experiments are warranted to interpret the pathogenic variants in order to elucidate the genetic heterogeneity of MTHFS deficiency.
Moreover, the athetosis of P1, the eczematous dermatitis of P2, and the recurrent diarrhea of P3 were novel features reported in MTHFS deficiency. Athetosis might be related to neurological impairments induced by folate metabolic deficiency, as mentioned above. In terms of the eczema, studies have shown that a lower methyl donor pool (e.g., lower serum folate levels, impaired folate metabolism) is associated with a higher risk of atopy (Friso et al., 2002; Husemoen et al., 2006). Since the folate metabolic deficiency might lead to epigenetic changes that tilt the immunophenotypic balance toward allergic diseases (Matsui and Matsui, 2009). Considering the mechanism of the link between MTHFS deficiency and recurrent diarrhea, regeneration of the intestinal surface may depend on adequate folate status on the one hand, and may be related to the role of folate in innate and/or adaptive antigen-specific immune responses on the other (Dhur et al., 1991; Manger et al., 2011). Disturbance of folate metabolism disrupts the normal renewal of intestinal epithelial cells or interferes with the immune response, prolonging recovery from the initial infection and leading to recurring episodes of diarrhea. The above manifestations were only seen in a few individuals; thus, more research is still needed to explore the specific mechanisms.
So far, there is no definitive treatment protocol for MTHFS deficiency (Table 1). Treatment with folinic acid and folic acid in a patient with low-normal CSF 5-MTHF level was unsuccessful, indicating that the non-reduced form of folate has no improvement in the management of cerebral folate deficiency (Rodan et al., 2018). When treated with L-5-MTHF, a useable reduced form of folate, and mecobalamin, a methylated form of cobalamin to utilize 5-MTHF, the motor functions and CSF 5-MTHF level improved (Rodan et al., 2018; Ramaekers and Quadros, 2022). Since most of the patients had seizures, anti-epilepsy therapy is also the main treatment. Previous study showed that one MTHFS deficiency patient with refractory seizures and recurrent episodes of hyperthermia responded to lamotrigine (Rodan et al., 2018). However, in our cases, P1 had fever-induced, refractory, status seizures, and his seizures still continued with the treatment of oxcarbazepine, sodium valproate, and lamotrigine.
Fortunately, P2, the younger brother of P1, had myoclonic seizures and tonic-clonic seizures stopped, and his HCY level returned to normal with a combination of anti-epilepsy drugs (vigabatrin and topiramate), L-5-MTHF, and mecobalamin. The severe eczema was also improved by taking zinc gluconate orally. Combined with goal-targeted rehabilitation, P2’s cognitive and motor abilities had become better than before. Till now, P3 has not shown any signs of seizures, but a longer-term follow-up with repeat MRI and EEG is required. It is worth noting that severe complications are the leading cause of death in this disease. One case was reported to die at the age of 18 due to hepatic failure (Sakthivel et al., 2020), and P1 died at 22 months old because of SUDEP. So, regular follow-up and meticulous care are essential in the management of such disease.
MTHFS deficiency can present with varying degrees of neurodevelopmental abnormalities, ranging from mild GDD to refractory epilepsy to SUDEP. A broad range of differential diagnosis may exist between it and other diseases, such as cerebral folate transport deficiency (FOLR1), dihydrofolate reductase (DHFR) deficiency, methylenetetrahydrofolate reductase (MTHFR) deficiency, hereditary folate malabsorption (SLC46A1), etc. Thus, genetic analysis is required to confirm the diagnosis of MTHFS deficiency. Furthermore, the mother of P1 and P2 had recurrent pregnancy loss (RPL), which could be linked to a folate metabolism disorder during embryonic development (Hong Li and Marren, 2018). There is mounting evidence that impaired FOCM and elevated homocysteine levels are associated with adverse pregnancy outcomes (Rady, 2018; Mo et al., 2019; Zhang et al., 2019; Liu et al., 2020; Nwogu et al., 2020; Bala et al., 2021). As folate levels fall, dTMP synthesis and DNA methylation levels were decreased, resulting in unstable genomes and aneuploid chromosomal abnormalities (Wang et al., 2004). The elevated HCY may lead to placental arterial embolization, impaired vascular endothelium and coagulation, and cause placental insufficiency with direct toxic effects on the embryo (Eskes, 2001; Jakubowski, 2019; Zaric et al., 2019). Therefore, RPL is introduced as a danger sign in MTHFS deficiency, and prenatal genetic screening on the MTHFS gene in such families would avoid inborn errors of metabolism.
In conclusion, we reported three new cases of MTHFS deficiency and the process of diagnosis and treatment, which expands the phenotype spectrum with the novel features of athetosis, eczema, and recurrent diarrhea. Meanwhile, we identified four novel variants in the MTHFS gene and expanded the genotype spectrum of this rare disease. Further studies on exploring the genotype-phenotype correlation of MTHFS deficiency are warranted.
Data availability statement
All data generated or analyzed during this study are included in this published article. The original contributions presented in this study are publicly available. The MTHFS variants NM_006441.4: c.158C>T (p.S53F), c.504del (p.Y169Tfs*17), c.117+1del and c.182A>G (p.E61G) were submitted to the LOVD database (https://databases.lovd.nl/shared/variants/MTHFS/), with the LOVD Variant ID: https://databases.lovd.nl/shared/variants/0000933017#00014020, https://databases.lovd.nl/shared/variants/0000933018#00014020, https://databases.lovd.nl/shared/variants/0000933019#00014020, https://databases.lovd.nl/shared/variants/0000933020#00014020.
Ethics statement
The studies involving humans were approved by The ethics committee of The First Affiliated Hospital of Anhui Medical University and the ethics committee of Kunming Children’s and Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants and legal guardians/next of kin. Written informed consent was obtained from the minor(s) and legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
XX, DW, YL, and JT diagnosed the patients and designed this study. XX, JZ, LF, ZZ and JY gathered clinical information of the patients, drafted the manuscript. MP, GY performed the genetic analysis. All authors contributed to the article and approved the submitted version.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We are indebted to the patients and the parents who participated in this study.
Conflict of interest
Authors MP and GY were employed by Chigene (Beijing) Translational Medical Research Center Co, Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Bala, R., Verma, R., Verma, P., Singh, V., Yadav, N., Rajender, S., et al. (2021). Hyperhomocysteinemia and low vitamin B12 are associated with the risk of early pregnancy loss: A clinical study and meta-analyses. Nutr. Res. 91, 57–66. doi:10.1016/j.nutres.2021.05.002
Beard, R. S., Reynolds, J. J., and Bearden, S. E. (2011). Hyperhomocysteinemia increases permeability of the blood-brain barrier by NMDA receptor-dependent regulation of adherens and tight junctions. Blood 118 (7), 2007–2014. doi:10.1182/blood-2011-02-338269
Bertrand, R., Beauchemin, M., Dayan, A., Ouimet, M., and Jolivet, J. (1995). Identification and characterization of human mitochondrial methenyltetrahydrofolate synthetase activity. Biochim. Biophys. Acta 1266 (3), 245–249. doi:10.1016/0167-4889(95)00020-s
Cavusoglu, D., Kose, M., Er, E., Elmas, M., Gencpinar, P., Dundar, N. O., et al. (2022). A female case of 5,10-methenyltetrahydrofolate synthetase deficiency with novel neuro-imaging abnormalities. Brain Dev. 44 (9), 640–644. doi:10.1016/j.braindev.2022.05.008
Dayan, A., Bertrand, R., Beauchemin, M., Chahla, D., Mamo, A., Filion, M., et al. (1995). Cloning and characterization of the human 5,10-methenyltetrahydrofolate synthetase-encoding cDNA. Gene 165 (2), 307–311. doi:10.1016/0378-1119(95)00321-v
Dhur, A., Galan, P., and Hercberg, S. (1991). Folate status and the immune system. Prog. Food Nutr. Sci. 15 (1-2), 43–60.
Eskes, T. K. (2001). Clotting disorders and placental abruption: homocysteine-a new risk factor. Eur. J. Obstet. Gynecol. Reprod. Biol. 95 (2), 206–212. doi:10.1016/s0301-2115(00)00492-9
Field, M. S., Anderson, D. D., and Stover, P. J. (2011). Mthfs is an essential gene in mice and a component of the purinosome. Front. Genet. 2, 36. doi:10.3389/fgene.2011.00036
Field, M. S., Szebenyi, D. M., and Stover, P. J. (2006). Regulation of de novo purine biosynthesis by methenyltetrahydrofolate synthetase in neuroblastoma. J. Biol. Chem. 281 (7), 4215–4221. doi:10.1074/jbc.M510624200
Fox, J. T., and Stover, P. J. (2008). Folate-mediated one-carbon metabolism. Vitam. Horm. 79, 1–44. doi:10.1016/s0083-6729(08)00401-9
Friso, S., Choi, S. W., Girelli, D., Mason, J. B., Dolnikowski, G. G., Bagley, P. J., et al. (2002). A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proc. Natl. Acad. Sci. U. S. A. 99 (8), 5606–5611. doi:10.1073/pnas.062066299
Girgis, S., Suh, J. R., Jolivet, J., and Stover, P. J. (1997). 5-Formyltetrahydrofolate regulates homocysteine remethylation in human neuroblastoma. J. Biol. Chem. 272 (8), 4729–4734. doi:10.1074/jbc.272.8.4729
Hong Li, Y., and Marren, A. (2018). Recurrent pregnancy loss: A summary of international evidence-based guidelines and practice. Aust. J. Gen. Pract. 47 (7), 432–436. doi:10.31128/ajgp-01-18-4459
Husemoen, L. L., Toft, U., Fenger, M., Jørgensen, T., Johansen, N., and Linneberg, A. (2006). The association between atopy and factors influencing folate metabolism: is low folate status causally related to the development of atopy? Int. J. Epidemiol. 35 (4), 954–961. doi:10.1093/ije/dyl094
Jakubowski, H. (2019). Homocysteine modification in protein structure/function and human disease. Physiol. Rev. 99 (1), 555–604. doi:10.1152/physrev.00003.2018
Kamath, A. F., Chauhan, A. K., Kisucka, J., Dole, V. S., Loscalzo, J., Handy, D. E., et al. (2006). Elevated levels of homocysteine compromise blood-brain barrier integrity in mice. Blood 107 (2), 591–593. doi:10.1182/blood-2005-06-2506
Krupenko, N. I., Dubard, M. E., Strickland, K. C., Moxley, K. M., Oleinik, N. V., and Krupenko, S. A. (2010). ALDH1L2 is the mitochondrial homolog of 10-formyltetrahydrofolate dehydrogenase. J. Biol. Chem. 285 (30), 23056–23063. doi:10.1074/jbc.M110.128843
Liu, C., Luo, D., Wang, Q., Ma, Y., Ping, L., Wu, T., et al. (2020). Serum homocysteine and folate concentrations in early pregnancy and subsequent events of adverse pregnancy outcome: the Sichuan homocysteine study. BMC Pregnancy Childbirth 20 (1), 176. doi:10.1186/s12884-020-02860-9
Manger, M. S., Taneja, S., Strand, T. A., Ueland, P. M., Refsum, H., Schneede, J., et al. (2011). Poor folate status predicts persistent diarrhea in 6- to 30-month-old north Indian children. J. Nutr. 141 (12), 2226–2232. doi:10.3945/jn.111.144220
Matsui, E. C., and Matsui, W. (2009). Higher serum folate levels are associated with a lower risk of atopy and wheeze. J. Allergy Clin. Immunol. 123 (6), 1253–1259. doi:10.1016/j.jaci.2009.03.007
Mattson, M. P., and Shea, T. B. (2003). Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci. 26 (3), 137–146. doi:10.1016/s0166-2236(03)00032-8
Miller, A. L. (2008). The methylation, neurotransmitter, and antioxidant connections between folate and depression. Altern. Med. Rev. 13 (3), 216–226.
Mo, H., Rao, M., Wang, G., Long, Y. X., Wang, H. W., and Tang, L. (2019). Polymorphism of MTHFR 1298A>C in relation to adverse pregnancy outcomes in Chinese populations. Mol. Genet. Genomic Med. 7 (5), e642. doi:10.1002/mgg3.642
Nwogu, C. M., Okunade, K. S., Adenekan, M. A., Sekumade, A. I., John-Olabode, S., and Oluwole, A. A. (2020). Association between maternal serum homocysteine concentrations in early pregnancy and adverse pregnancy outcomes. Ann. Afr. Med. 19 (2), 113–118. doi:10.4103/aam.aam_41_19
Rady, H. A. (2018). Maternal methyltetrahydrofolate reductase gene mutation in patients with missed abortions. J. Taibah Univ. Med. Sci. 13 (1), 93–96. doi:10.1016/j.jtumed.2017.04.004
Ramaekers, V. T., and Quadros, E. V. (2022). Cerebral folate deficiency syndrome: early diagnosis, intervention and treatment strategies. Nutrients 14 (15), 3096. doi:10.3390/nu14153096
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Rodan, L. H., Qi, W., Ducker, G. S., Demirbas, D., Laine, R., Yang, E., et al. (2018). 5,10-methenyltetrahydrofolate synthetase deficiency causes a neurometabolic disorder associated with microcephaly, epilepsy, and cerebral hypomyelination. Mol. Genet. Metab. 125 (1-2), 118–126. doi:10.1016/j.ymgme.2018.06.006
Romero, J. A., Abdelmoumen, I., Hasbani, D., Khurana, D. S., and Schneider, M. C. (2019). A case of 5,10-methenyltetrahydrofolate synthetase deficiency due to biallelic null mutations with novel findings of elevated neopterin and macrocytic anemia. Mol. Genet. Metab. Rep. 21, 100545. doi:10.1016/j.ymgmr.2019.100545
Sakthivel, D., Lei, Y., Cao, X., and Finnell, R. H. (2020). Homozygous mutation in theMTHFSGene may contribute to the development of cerebral folate deficiency syndrome. Med. Nexus 4 (2), 72–80. doi:10.4103/2096-2924.288022
Vafaee-Shahi, M., Amirkashani, D., Ashrafi, M. R., Tahernia, L., and Riahi, A. (2022). Methenyltetrahydrofolate synthease deficiency (MTHFS deficiency): novel mutation and brain MRI findings: A case report and glance to other cases. Clin. Neurol. Neurosurg. 215, 107153. doi:10.1016/j.clineuro.2022.107153
Vitvitsky, V., Dayal, S., Stabler, S., Zhou, Y., Wang, H., Lentz, S. R., et al. (2004). Perturbations in homocysteine-linked redox homeostasis in a murine model for hyperhomocysteinemia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 287 (1), R39–R46. doi:10.1152/ajpregu.00036.2004
Wang, F., Zhang, D., Zhang, D., Li, P., and Gao, Y. (2021). Mitochondrial protein translation: emerging roles and clinical significance in disease. Front. Cell Dev. Biol. 9, 675465. doi:10.3389/fcell.2021.675465
Wang, X., Thomas, P., Xue, J., and Fenech, M. (2004). Folate deficiency induces aneuploidy in human lymphocytes in vitro-evidence using cytokinesis-blocked cells and probes specific for chromosomes 17 and 21. Mutat. Res. 551 (1-2), 167–180. doi:10.1016/j.mrfmmm.2004.03.008
Wu, D., Li, Y., Song, G., Cheng, C., Zhang, R., Joachimiak, A., et al. (2009). Structural basis for the inhibition of human 5,10-methenyltetrahydrofolate synthetase by N10-substituted folate analogues. Cancer Res. 69 (18), 7294–7301. doi:10.1158/0008-5472.Can-09-1927
Zaric, B. L., Obradovic, M., Bajic, V., Haidara, M. A., Jovanovic, M., and Isenovic, E. R. (2019). Homocysteine and hyperhomocysteinaemia. Curr. Med. Chem. 26 (16), 2948–2961. doi:10.2174/0929867325666180313105949
Zhang, Y., He, X., Xiong, X., Chuan, J., Zhong, L., Chen, G., et al. (2019). The association between maternal methylenetetrahydrofolate reductase C677T and A1298C polymorphism and birth defects and adverse pregnancy outcomes. Prenat. Diagn 39 (1), 3–9. doi:10.1002/pd.5396
Keywords: 5, 10-methenyltetrahydrofolate synthetase, MTHFS deficiency, folate metabolism, seizure, developmental delay
Citation: Xu X, Zhu J, Fang L, Zou Z, Yuan J, Peng M, Yu G, Wu D, Liu Y and Tang J (2023) Exome sequencing identified novel variants in three Chinese patients with 5,10-methenyltetrahydrofolate synthetase deficiency. Front. Genet. 14:1236849. doi: 10.3389/fgene.2023.1236849
Received: 11 July 2023; Accepted: 25 August 2023;
Published: 18 September 2023.
Edited by:
Augusto Rojas-Martinez, Escuela de Medicina y Ciencias de la Salud Tec Salud, Tecnológico de Monterrey, MexicoReviewed by:
Jorge Román Corona-Rivera, University of Guadalajara, MexicoHui Xi, Hunan Provincial Maternal and Child Health Care Hospital, China
Copyright © 2023 Xu, Zhu, Fang, Zou, Yuan, Peng, Yu, Wu, Liu and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yun Liu, bGl1eXVuQGV0eXkuY24=; Jiulai Tang, dGFuaml1bGFpODg4OEAxNjMuY29t