 Wenxia Su
Wenxia Su Xiaojun Qian
Xiaojun Qian Keli Yang
Keli Yang Hui Ding
Hui Ding Chengbing Huang
Chengbing Huang Zhaoyue Zhang
Zhaoyue Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 07 June 2023
Sec. Computational Genomics
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1211020
This article is part of the Research TopicMethods In Computational GenomicsView all 15 articles
Introduction: Outer membrane proteins are crucial in maintaining the structural stability and permeability of the outer membrane. Outer membrane proteins exhibit several functions such as antigenicity and strong immunogenicity, which have potential applications in clinical diagnosis and disease prevention. However, wet experiments for studying OMPs are time and capital-intensive, thereby necessitating the use of computational methods for their identification.
Methods: In this study, we developed a computational model to predict outer membrane proteins. The non-redundant dataset consists of a positive set of 208 outer membrane proteins and a negative set of 876 non-outer membrane proteins. In this study, we employed the pseudo amino acid composition method to extract feature vectors and subsequently utilized the support vector machine for prediction.
Results and Discussion: In the Jackknife cross-validation, the overall accuracy and the area under receiver operating characteristic curve were observed to be 93.19% and 0.966, respectively. These results demonstrate that our model can produce accurate predictions, and could serve as a valuable guide for experimental research on outer membrane proteins.
Outer membrane proteins (OMPs) are a special type of proteins that are found in the outermost membranes of Gram-negative bacteria, mitochondria, and chloroplasts (Rollauer et al., 2015; Qi et al., 2022). OMPs serve a wide range of functions, including acting as adhesion factors in virulence, channels for small hydrophilic molecules, enzymes in biochemical reactions, and antigens in immune responses. They also work in concert with other substances to enhance the bacteria pathogenicity. Recent research on OMPs has revealed their potential for clinical diagnosis and disease prevention. Several published studies have explored OMPs as potential vaccine candidates (Budiardjo et al., 2021; Fahie et al., 2021; Cheng et al., 2022; Yu et al., 2022). The functions are determined by the OMP’s structure and the way it interacts with other molecules. OMPs are typically composed of a transmembrane β-barrel architecture, providing permeability to the outer membrane and maintaining structural stability. Among different types of OMPs, β-buckets consist of varying even numbers of β-folding sheets, ranging from 8 to 26 (Rollauer et al., 2015). The specific composition of the β-barrel architecture is determined by the amino acid sequence of the OMPs. Mutations in sequences can impact the stability and function of the protein.
Distinguishing OMPs from non-OMPs can aid researchers in identifying promising vaccine targets, developing new antibiotics and therapeutics, and understanding the evolution of Gram-negative bacteria. Despite their distinctive β-barrel structure, OMPs are exposed to numerous charged and polar residues in the membrane, making it challenging to distinguish them from non-OMPs. This is a primary challenge and a significant obstacle in the research process, given the considerable time and capital costs associated with laboratory studies of OMPs. As a result, OMP prediction has tremendous significance for the scientific community. Currently, various machine learning methods have been used for the identification of OMPs, such as support vector machine (SVM) (Park et al., 2005; Gromiha et al., 2006; Hu et al., 2017; Zhang et al., 2021), k-nearest neighbor (K-NN) method (Yan et al., 2008), neural networks (NN) (Hu et al., 2017). These methods utilize the amino acid composition, and physical and chemical properties of the amino acid sequences to construct the prediction models. Gromiha and Suwa (2003); Gromiha and Suwa (2005) developed multiple OMP prediction methods based on amino acid composition, residue pair preference, and motif sequence. However, these methods only achieved prediction accuracies of 80%–90%. Subsequently, a machine learning algorithm was proposed with a higher accuracy ranging from 90% to 94% (Gromiha et al., 2005; Gromiha et al., 2006). Lin (2008) further improved the OMP prediction model by introducing the Incremental Diversity with Quality Distinctness analysis, which combines the Markov discriminant method and the pseudo amino acid composition (Pse-AAC). Despite the progress made in OMP predictions, there is still room for further improvement in prediction quality.
In this article, we proposed a novel method for predicting OMPs that combines Pse-AAC and SVM. To extract the features for amino acid composition and physical and chemical characteristics of amino acids, we used the Pse-AAC feature extraction method. Additionally, we introduced multi-level amino acid residue index correlation coefficients such as hydrophobic value, average polarity, and solvation-free energy to enhance the accuracy of our prediction model. To assess the effectiveness and reliability of our approach, we also conducted a comprehensive comparison and analysis of our proposed model with existing methods for predicting OMPs. Our developed approach will be useful for distinguishing OMPs from non-OMPs.
The construction of a reliable dataset is the basis for developing an accurate outer membrane protein prediction model (Su et al., 2021). A well-designed dataset is crucial for developing effective algorithms and an objective evaluation and prediction system. In this paper, membrane proteins were extracted from the PSORT-B database (https://www.psort.org/) (Gardy et al., 2003), and globular proteins were extracted from the PDB40D of SCOP_1.37 database (http://scop.mrc-lmb.cam.ac.uk/scop/) (Andreeva et al., 2020). As a result, a total of 208 OMPs were selected as the positive set, while 879 non-OMPs were chosen as the negative set. The negative set included 206 inner membrane proteins and 673 globular proteins. The globular protein dataset contained 154 complete
To construct a prediction model, it is necessary to represent the protein sequences as mathematical vectors. This conversion is commonly known as feature extraction (Basith et al., 2020; Dao et al., 2022b; Zhang Z.-Y. et al., 2022; Hunt et al., 2022; Karuna Nidhi et al., 2022; Sun et al., 2022; Tran and Nguyen, 2022; Wang et al., 2022; Yang et al., 2022). The amino acid composition (ACC) of the protein has a great impact on protein classification research (Awais et al., 2021; Shoombuatong et al., 2022b; Manavalan and Patra, 2022; Rout et al., 2022; Zhu et al., 2022). By using the ACC, a protein sequence can be represented as a 20-D (dimension) vector as follows:
In Eq. 1,
The type of amino acids is determined by their side chains, as the 20 types of amino acid side chains differ in shape, size, negativity, hydrophobicity, and acid-base properties. The distinct characteristics of the 20 amino acid side chains result in various combinations of amino acid sequences that exhibit different structures and functions. Therefore, algorithms based on the physicochemical properties of amino acids are another major category of feature extraction methods. Pse-AAC, originally proposed by Chou, is a feature extraction algorithm, that is, based on the physical and chemical properties of amino acids (Chou, 2005). By using Pse-AAC, a protein sample can be represented as follows:
where the first 20 numbers in Eq. 2 are the classic AAC features, and the next λ discrete numbers represent the position information of residues in amino acid sequences. For different problems, the optimal value of
SVM is a powerful supervised machine learning classification method based on statistical learning theory (Manavalan et al., 2019). It was originally designed based on the idea of the generalized linear classifier. First, features were mapped to high-dimensional space. Next, a separating hyperplane is constructed to separate the two categories in the high-dimensional feature space (Vapnik and Control, 2019). To avoid expensive computations, the mapping function only involves the relatively low-dimensional vector in the input space and the dot product in the feature space. The global optimization approach and avoidance of overfitting in SVM have made it a successful tool for addressing various bioinformatics problems (Zhang H. et al., 2022). In this paper, the support vector machine (SVM) was implemented using the widely used software LIBSVM (http://www.csie.ntu.edu.tw/∼cjlin/libsvm) (Chang and Lin, 2011). The radial basis function which is defined as
At present, k-fold cross-validation and jackknife cross-validation are widely used for prediction evaluation (Tabaie et al., 2021; Dao et al., 2022a; Xiao et al., 2022; Zhou et al., 2022). The jackknife test is a type of cross-validation that involves leaving one observation out of the dataset at a time and using the remaining observations to train a model. This process is repeated for each observation in the dataset, resulting in n different models, where n is the number of observations in the dataset. In this article, we used the Jackknife test to evaluate the prediction results. The sensitivity (Sn), specificity (Sp), average accuracy (AA), overall prediction accuracy (OA), and Matthew’s correlation coefficient (MCC), the area under ROC curve (auROC) were used to evaluate the prediction performance of the algorithm (Yang et al., 2021; Zhang Q. et al., 2022). The evaluation metrics are defined as follows:
where TP represents the number of the positive sample correctly identified, FN represents the positive sample wrongly identified as a negative sample, FP represents the negative sample wrongly identified as a positive sample, and TN represents the negative sample correctly identified. AuROC is an indicator that relates to the receiver operating characteristic (ROC) curve, which is a plot of a series of continuous (1-Sp) values on the horizontal axis against their corresponding Sn values on the vertical axis. The ROC curve is a useful tool for evaluating the sensitivity and specificity of a model (Hasan et al., 2022; Jeon et al., 2022). AuROC is calculated in this study as an indicator of classification ability and performance. A larger auROC value indicates better performance and classification ability of the model.
In this study, the proteins were first obtained in FASTA format and then the PseAAC program (Shen and Chou, 2008) was used to extract the feature vectors of pseudo amino acid components. To achieve relatively optimal prediction results, different parameters were selected to extract pseudo amino acid component feature vectors of protein sequences. Specifically, feature vectors were extracted using different values of
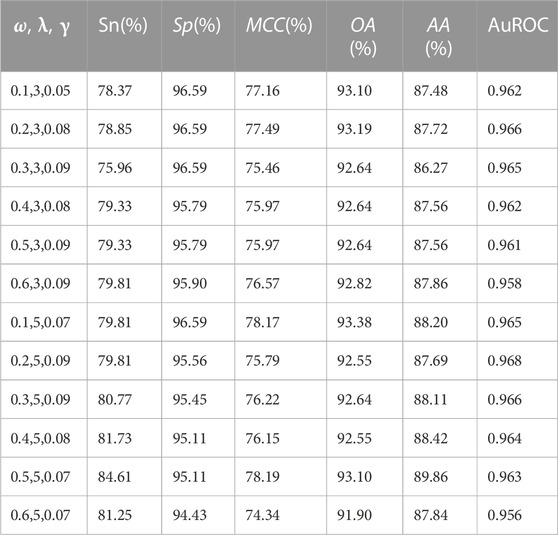
TABLE 1. The performance comparison of prediction models under different parameter conditions.
In this study, the benchmark dataset consisted of 208 OMPs and 879 non-OMPs. Due to this imbalanced dataset, using average accuracy as the sole evaluation criterion may lead to skewed results toward the negative sets. Thus, the paper used overall accuracy as the main criterion for model evaluation. By analyzing the data in Table 1, it was observed that high prediction sensitivity was achieved using Jackknife cross-validation with different parameters. And, with the best prediction result obtained with a weight factor of 0.5, the parameter
Various methods have been proposed by different researchers to predict and distinguish OMPs from other types of membrane proteins. Wu et al. (2007) proposed a prediction method that uses information differences to compare the distribution of subsequences and residual sequences, resulting in a prediction accuracy of 99.20%. Yan et al. (2008) proposed a method based on the K-nearest neighbor (KNN) method, which predicted the weighted Euclidean distance calculated by residual synthesis and achieved a recognition accuracy of 96.1%, sensitivity of 87.5%, specificity of 98.2% with 0.873 MCC. Gromiha et al. (2006) discriminate of OMPs and non-OMPs using different machine learning approaches, the best performance achieved sensitivity of 84.6%, specificity of 95.8% and accuracy of 93.7%. And the SVM-based model achieved sensitivity of 72.6%, specificity of 98.2% and accuracy of 93.3%. Park et al. (2005) proposed an SVM method that considers both amino acid composition and residue pair information, achieving sensitivity of 90.9 %, specificity of 94.7%, MCC 0.816 of and accuracy of 93.9%. Gao et al. (2010) developed a method that combined the structural and physicochemical characteristics of sequence-derived proteins with amino acid composition to distinguish OMPs and non-OMPs using SVM, with an overall accuracy of 97.8%, sensitivity of 91.8 %, specificity of 99.2% and MCC 0.928.
In this paper, the model constructed using the SVM algorithm achieved an overall accuracy of 93.10% and auROC of 0.963 under Jackknife cross-validation, respectively. Besides, the sensitivity, specificity, MCC, and average accuracy were found to be 84.61%, 95.11%, 78.19%, and 89.86%, respectively. Compared to previous SVM-based models, some progress has been made.
This article focused on the prediction and recognition of OMPs using the method of combining Pse-AAC with SVM. The study achieved good results with the Pse-AAC method, which not only considers the content of 20 natural amino acids in each protein sequence but also includes the correlation between various amino acids, such as physical and chemical properties. This approach is more advanced than traditional methods that only consider amino acid composition, leading to more accurate prediction results. SVM is a widely used algorithm in bioinformatics (Hasan et al., 2020; Shoombuatong et al., 2022a; Bupi et al., 2023), and applying it to the prediction of OMPs is an inevitable trend in current research. The constructed model using the SVM algorithm achieved high performance with an overall accuracy of 93.10% and auROC of 0.963 under Jackknife cross-validation. The sensitivity, specificity, Matthew correlation coefficient, and average accuracy achieved 84.61%, 95.11%, 78.19%, and 89.86%, respectively. However, while feature extraction algorithms have been widely used in prediction methods and have achieved good performance, the relationship between the extracted information and protein structure and function needs to be further explored. This challenge will undoubtedly be the focus of our future research efforts aimed at identifying OMPs. The development of accurate prediction models for OMPs has the potential to significantly impact fields ranging from antibiotic discovery and vaccine development to biotechnology and bacterial diagnostics.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Conceptualization, HD and ZZ; data curation, WS and XQ; formal analysis, WS, XQ, and KY; funding acquisition, WS and ZZ; supervision, CH; writing—original draft, WS; writing—review and editing, CH and ZZ. All authors contributed to the article and approved the submitted version.
This research was funded by grants from the National Natural Science Foundation of China (Grant Nos. 62201299, 62102067), Natural Science Foundation of the Inner Mongolia of China (Grant No. 2021BS06003), Science and Technology Research Project of Colleges and Universities in Inner Mongolia of China (Grant No. NJZY21473), and Basic Scientific Research Foundation of Colleges and Universities directly under Inner Mongolia of China (Grant No. BR220505).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Andreeva, A., Kulesha, E., Gough, J., and Murzin, A. G. J. N. A. R. (2020). The SCOP database in 2020: Expanded classification of representative family and superfamily domains of known protein structures. Nucleic Acids Res. 48, D376–D382. doi:10.1093/nar/gkz1064
Awais, M., Hussain, W., Rasool, N., and Khan, Y. D. J. C. B. (2021). iTSP-PseAAC: identifying tumor suppressor proteins by using fully connected neural network and PseAAC. Curr. Bioinform. 16, 700–709. doi:10.2174/15748936mtezfmteby
Basith, S., Manavalan, B., Hwan Shin, T., and Lee, G. (2020). Machine intelligence in peptide therapeutics: A next-generation tool for rapid disease screening. Med. Res. Rev. 40, 1276–1314. doi:10.1002/med.21658
Budiardjo, S. J., Ikujuni, A. P., Firlar, E., Cordova, A., Kaelber, J. T., and Slusky, J. S. J. T. J. O. M. B. (2021). High-yield preparation of outer membrane protein efflux pumps by in vitro refolding is concentration dependent. J. Membr. Biol. 254, 41–50. doi:10.1007/s00232-020-00161-y
Bupi, N., Sangaraju, V. K., Phan, L. T., Lal, A., Vo, T. T. B., Ho, P. T., et al. (2023). An effective integrated machine learning framework for identifying severity of tomato yellow leaf curl virus and their experimental validation. Research 6, 0016. doi:10.34133/research.0016
Chang, C. C., and Lin, C. J. (2011). Libsvm: A library for support vector machines. ACM Trans. Intelligent Syst. Technol. 2, 1–27.
Cheng, L., Qi, C., Yang, H., Lu, M., Cai, Y., Fu, T., et al. (2022). gutMGene: a comprehensive database for target genes of gut microbes and microbial metabolites. Nucleic Acids Res. 50, D795–D800. doi:10.1093/nar/gkab786
Chou, K.-C. J. B. (2005). Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics 21, 10–19. doi:10.1093/bioinformatics/bth466
Dao, F.-Y., Lv, H., Fullwood, M. J., and Lin, H. J. R. (2022a). Accurate identification of DNA replication origin by fusing epigenomics and chromatin interaction information. Res. (Wash D C) 2022, 9780293. doi:10.34133/2022/9780293
Dao, F.-Y., Lv, H., Zhang, Z.-Y., and Lin, H. J. C. B. (2022b). BDselect: A package for k-mer selection based on the binomial distribution. Curr. Bioinforma. 17, 238–244. doi:10.2174/1574893616666211007102747
Fahie, M. A., Yang, B., Chisholm, C. M., Chen, M. J. N. T. M., and Protocols, (2021). Protein analyte sensing with an outer membrane protein G (OmpG) nanopore. Methods Mol. Biol. 186, 77–94. doi:10.1007/978-1-0716-0806-7_7
Gao, Q.-B., Ye, X.-F., Jin, Z.-C., and He, J. J. A. B. (2010). Improving discrimination of outer membrane proteins by fusing different forms of pseudo amino acid composition. Anal. Biochem. 398, 52–59. doi:10.1016/j.ab.2009.10.040
Gardy, J. L., Spencer, C., Wang, K., Ester, M., Tusnady, G. E., Simon, I., et al. (2003). PSORT-B: Improving protein subcellular localization prediction for Gram-negative bacteria. Nucleic Acids Res. 31, 3613–3617. doi:10.1093/nar/gkg602
Gromiha, M. M., Ahmad, S., and Suwa, M. (2005). Application of residue distribution along the sequence for discriminating outer membrane proteins. Comput. Biol. Chem. 29, 135–142. doi:10.1016/j.compbiolchem.2005.02.006
Gromiha, M. M., and Suwa, M. J. B. (2005). A simple statistical method for discriminating outer membrane proteins with better accuracy. Bioinformatics 21, 961–968. doi:10.1093/bioinformatics/bti126
Gromiha, M. M., Suwa, M. J. B. E. B. A.-P., and Proteomics, (2006). Influence of amino acid properties for discriminating outer membrane proteins at better accuracy. Biochim. Biophys. Acta 1764, 1493–1497. doi:10.1016/j.bbapap.2006.07.005
Gromiha, M. M., and Suwa, M. J. I. J. O. B. M. (2003). Variation of amino acid properties in all-β globular and outer membrane protein structures. Int. J. Biol. Macromol. 32, 93–98. doi:10.1016/s0141-8130(03)00042-4
Gromiha, M. M., and Suwa, M. J. P. S. (2006). Discrimination of outer membrane proteins using machine learning algorithms. Funct. Bioinforma. Proteins 63, 1031–1037. doi:10.1002/prot.20929
Hasan, M. M., Schaduangrat, N., Basith, S., Lee, G., Shoombuatong, W., and Manavalan, B. (2020). HLPpred-fuse: Improved and robust prediction of hemolytic peptide and its activity by fusing multiple feature representation. Bioinformatics 36, 3350–3356. doi:10.1093/bioinformatics/btaa160
Hasan, M. M., Tsukiyama, S., Cho, J. Y., Kurata, H., Alam, M. A., Liu, X., et al. (2022). Deepm5C: A deep-learning-based hybrid framework for identifying human rna N5-methylcytosine sites using a stacking strategy. Mol. Ther. 30, 2856–2867. doi:10.1016/j.ymthe.2022.05.001
Hu, Y., Zhou, M., Shi, H., Ju, H., Jiang, Q., and Cheng, L. (2017). Measuring disease similarity and predicting disease-related ncRNAs by a novel method. Bmc Med. Genomics 10, 71. doi:10.1186/s12920-017-0315-9
Hunt, C., Montgomery, S., Berkenpas, J. W., Sigafoos, N., Oakley, J. C., Espinosa, J., et al. (2022). Recent progress of machine learning in gene therapy. Curr. Gene Ther. 22, 132–143. doi:10.2174/1566523221666210622164133
Jeon, Y. J., Hasan, M. M., Park, H. W., Lee, K. W., and Manavalan, B. (2022). Tacos: A novel approach for accurate prediction of cell-specific long noncoding RNAs subcellular localization. Brief. Bioinform 23, bbac243. doi:10.1093/bib/bbac243
Karuna Nidhi, M. B., Ganapathy, R., Subbiah, P., Suvaiyarasan, S., and Karuppasamy, M. P. J. C. B. (2022). GenNBPSeq: Online web server to generate never born protein sequences using toeplitz matrix approach with structure analysis. Curr. Bioinform. 17, 565–577. doi:10.2174/1574893617666220519110154
Lin, H. J. J. O. T. B. (2008). The modified Mahalanobis discriminant for predicting outer membrane proteins by using Chou's pseudo amino acid composition. J. Theor. Biol. 252, 350–356. doi:10.1016/j.jtbi.2008.02.004
Manavalan, B., Basith, S., Shin, T. H., Wei, L., and Lee, G. J. B. (2019). mAHTPred: a sequence-based meta-predictor for improving the prediction of anti-hypertensive peptides using effective feature representation. Bioinformatics 35, 2757–2765. doi:10.1093/bioinformatics/bty1047
Manavalan, B., and Patra, M. C. J. J. O. M. B. (2022). Mlcpp 2.0: An updated cell-penetrating peptides and their uptake efficiency predictor. J. Mol. Biol. 434, 167604. doi:10.1016/j.jmb.2022.167604
Park, K.-J., Gromiha, M. M., Horton, P., and Suwa, M. J. B. (2005). Discrimination of outer membrane proteins using support vector machines. Bioinformatics 21, 4223–4229. doi:10.1093/bioinformatics/bti697
Qi, C., Cai, Y., Qian, K., Li, X., Ren, J., Wang, P., et al. (2022). SCovid: Single-cell atlases for exposing molecular characteristics of COVID-19 across 10 human tissues. Nucleic acids Res. 50, D867–D874. doi:10.1093/nar/gkab881
Rollauer, S. E., Sooreshjani, M. A., Noinaj, N., and Buchanan, S. K. J. P. T. O. T. R. S. B. B. S. (2015). Outer membrane protein biogenesis in Gram-negative bacteria. Philos. Trans. R. Soc. Lond B Biol. Sci. 370, 20150023.
Rout, R. K., Hassan, S. S., Sheikh, S., Umer, S., Sahoo, K. S., and Gandomi, A. H. (2022). Feature-extraction and analysis based on spatial distribution of amino acids for SARS-CoV-2 Protein sequences. Comput. Biol. Med. 141, 105024. doi:10.1016/j.compbiomed.2021.105024
Shen, H.-B., and Chou, K.-C. J. a. B. (2008). PseAAC: A flexible web server for generating various kinds of protein pseudo amino acid composition. Anal. Biochem. 373, 386–388. doi:10.1016/j.ab.2007.10.012
Shoombuatong, W., Basith, S., Pitti, T., Lee, G., and Manavalan, B. J. J. O. M. B. (2022b). Throne: A new approach for accurate prediction of human RNA N7-methylguanosine sites. J. Mol. Biol. 434, 167549. doi:10.1016/j.jmb.2022.167549
Shoombuatong, W., Basith, S., Pitti, T., Lee, G., and Manavalan, B. (2022a). Throne: A new approach for accurate prediction of human rna N7-methylguanosine sites. J. Mol. Biol. 434, 167549. doi:10.1016/j.jmb.2022.167549
Su, W., Liu, M.-L., Yang, Y.-H., Wang, J.-S., Li, S.-H., Lv, H., et al. (2021). Ppd: A manually curated database for experimentally verified prokaryotic promoters. J. Mol. Biol. 433, 166860. doi:10.1016/j.jmb.2021.166860
Sun, Z., Huang, Q., Yang, Y., Li, S., Lv, H., Zhang, Y., et al. (2022). PSnoD: Identifying potential snoRNA-disease associations based on bounded nuclear norm regularization. Brief. Bioinform. 23, bbac240. doi:10.1093/bib/bbac240
Tabaie, A., Orenstein, E. W., Nemati, S., Basu, R. K., Kandaswamy, S., Clifford, G. D., et al. (2021). Predicting presumed serious infection among hospitalized children on central venous lines with machine learning. Comput. Biol. Med. 132, 104289. doi:10.1016/j.compbiomed.2021.104289
Tran, H. V., and Nguyen, Q. H. J. C. B. (2022). iAnt: combination of convolutional neural network and random Forest models using PSSM and BERT features to identify antioxidant proteins. Curr. Bioinform. 17, 184–195. doi:10.2174/1574893616666210820095144
Vapnik, V. N. J. A., and Control, R. (2019). Complete statistical theory of learning. Inf. Fusion 80, 1949–1975.
Wang, P., Zhang, S., He, G., Du, M., Qi, C., Liu, R., et al. (2022). microbioTA: an atlas of the microbiome in multiple disease tissues of Homo sapiens and Mus musculus. Nucleic acids Res. 51, D1345–D1352. doi:10.1093/nar/gkac851
Wu, Z., Feng, E., Wang, Y., Chen, L. J. P., and Letters, P. (2007). Discrimination of outer membrane proteins by a new measure of information discrepancy. Protein Pept. Lett. 14, 37–44. doi:10.2174/092986607779117254
Xiao, J., Liu, M., Huang, Q., Sun, Z., Ning, L., Duan, J., et al. (2022). Analysis and modeling of myopia-related factors based on questionnaire survey. Comput. Biol. Med. 150, 106162. doi:10.1016/j.compbiomed.2022.106162
Yan, C., Hu, J., and Wang, Y. J. a. A. (2008). Discrimination of outer membrane proteins using a K-nearest neighbor method. Amino Acids 35, 65–73. doi:10.1007/s00726-007-0628-7
Yang, H., Luo, Y., Ren, X., Wu, M., He, X., Peng, B., et al. (2021). Risk prediction of diabetes: Big data mining with fusion of multifarious physical examination indicators. Inf. Fusion 75, 140–149. doi:10.1016/j.inffus.2021.02.015
Yang, Y., Gao, D., Xie, X., Qin, J., Li, J., Lin, H., et al. (2022). DeepIDC: A prediction framework of injectable drug combination based on heterogeneous information and deep learning. Clin. Pharmacokinet. 61, 1–11. doi:10.1007/s40262-022-01180-9
Yu, H., Shen, Z.-A., Zhou, Y.-K., and Du, P.-F. (2022). Recent advances in predicting protein-lncRNA interactions using machine learning methods. Curr. Gene Ther. 22, 228–244. doi:10.2174/1566523221666210712190718
Zhang, H., Wang, S., and Huang, T. (2021). Identification of chronic hypersensitivity pneumonitis biomarkers with machine learning and differential Co-expression analysis. Curr. Gene Ther. 21, 299–303. doi:10.2174/1566523220666201208093325
Zhang, H., Zou, Q., Ju, Y., Song, C., and Chen, D. J. C. B. (2022a). Distance-based support vector machine to predict DNA N6-methyladenine modification. Curr. Bioinform. 17, 473–482. doi:10.2174/1574893617666220404145517
Zhang, Q., Li, H., Liu, Y., Li, J., Wu, C., and Tang, H. J. C. O. (2022b). Exosomal non-coding RNAs: New insights into the biology of hepatocellular carcinoma. Curr. Oncol. 29, 5383–5406. doi:10.3390/curroncol29080427
Zhang, Z.-Y., Ning, L., Ye, X., Yang, Y.-H., Futamura, Y., Sakurai, T., et al. (2022c). iLoc-miRNA: extracellular/intracellular miRNA prediction using deep BiLSTM with attention mechanism. Brief. Bioinform. 23, bbac395. doi:10.1093/bib/bbac395
Zhou, H., Wang, H., Ding, Y., and Tang, J. J. C. B. (2022). Multivariate information fusion for identifying antifungal peptides with Hilbert-Schmidt Independence Criterion. Curr. Bioinform. 17, 89–100. doi:10.2174/1574893616666210727161003
Keywords: outer membrane protein, pseudo amino acid composition, support vector machine, jackknife test, prediction model
Citation: Su W, Qian X, Yang K, Ding H, Huang C and Zhang Z (2023) Recognition of outer membrane proteins using multiple feature fusion. Front. Genet. 14:1211020. doi: 10.3389/fgene.2023.1211020
Received: 24 April 2023; Accepted: 24 May 2023;
Published: 07 June 2023.
Edited by:
Lei Chen, Shanghai Maritime University, ChinaReviewed by:
Yongqiang Xing, Inner Mongolia University of Science and Technology, ChinaCopyright © 2023 Su, Qian, Yang, Ding, Huang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chengbing Huang, YWJ0Y2hjYkBxcS5jb20=; Zhaoyue Zhang, enl6aGFuZ0B1ZXN0Yy5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.