 Yi Dong
Yi Dong Yi Zhang
Yi Zhang Yue Sheng1
Yue Sheng1 Fang Wang
Fang Wang Lv Liu
Lv Liu Liang-Liang Fan
Liang-Liang Fan- 1Department of Cell Biology, School of Life Sciences, Central South University, Changsha, China
- 2Medical Psychological Center, Medical Psychological Institute of Central South University, National Clinical Research Center for Mental Disorders, The Second Xiangya Hospital of Central South University, Changsha, China
- 3Department of Endocrinology, The Third Xiangya Hospital of Central South University, Changsha, Hunan, China
- 4Department of Respiratory Medicine, Clinical Center for Gene Diagnosis and Therapy, Diagnosis and Treatment Center of Respiratory Disease, Diagnosis and Treatment Center of Respiratory Disease, The Second Xiangya Hospital of Central South University, Changsha, China
Background: Heterozygous mutations in the dehydrodolichol diphosphate synthase (DHDDS) gene are one of the causes generating developmental and epileptic encephalopathies. So far, only eleven mutations in the DHDDS gene have been identified. The mutation spectrum of the DHDDS gene in the Chinese population remains unclear.
Methods: In this study, we enrolled a Chinese family with myoclonus and/or epilepsy and intellectual disability. The epilepsy and myoclonic tremor were improved after deep brain stimulation (DBS) of the subthalamic nucleus (STN) treatment. Whole exome sequencing and Sanger sequencing were employed to explore the genetic variations of the family.
Results: Subsequent to data filtering, we identified a recurrent pathogenic mutation (NM_001243564.1, c.113G>A/p.R38H) in the DHDDS gene in the proband. Sanger sequencing further validated that the presence of the mutation in his affected mother but absent in the health family members. Further bioinformatics analysis revealed that this mutation (p.R38H), located in an evolutionarily conserved region of DHDDS, was predicted to be deleterious.
Discussion: In this report, we present the first case of intractable epilepsy and/or myoclonus caused by p.R38H mutation of the DHDDS gene in the Chinese population. Furthermore, this study represents the third report of autosomal dominant familial inheritance of DHDDS mutation worldwide.
Introduction
Dehydrodolichol diphosphate synthase (DHDDS) is an enzyme crucial for the synthesis of dolichol monophosphate and for global N-linked glycosylation (Cunillera et al., 2000; Galosi et al., 2022). The gene encoding the DHDDS protein is located at 1p36.11 and was first identified as a disease-causing gene of retinitis pigmentosa, a recessive genetic disorder (Zelinger et al., 2011). However, in 2017, Hamdan et al. (2017) reported that heterozygous mutations in the DHDDS gene were responsible for developmental and epileptic encephalopathies (DEE). In addition, several other genes involved in the dolichol-dependent protein glycosylation pathway have also been identified, including NUS1 Dehydrodolichyl Diphosphate Synthase Subunit (NUS1) and ALG10 Alpha-1,2-Glucosyltransferase (ALG10), which are also disease-causing genes for epilepsy (Araki et al., 2020; Courage et al., 2021). At present, approximately 11 DHDDS mutations have been reported in 39 patients, resulting in various types of movement disorders, epilepsy, or other symptoms (Kim et al., 2021b; Galosi et al., 2022; Jiao et al., 2022). Most of these variants have been confirmed to be de novo events, except for two mutations (p.R205Q and p.R211Q) that have been reported in two families (Wood et al., 2021; Galosi et al., 2022). However, no family history-related DHDDS mutations have been reported in the Chinese population, and the relationship between different phenotypes and DHDDS mutations remains unclear.
Case presentation
A two-generation Han Chinese pedigree, comprising six members, was recruited for this study (Figure 1A). The proband (III-1), a 28-year-old male, exhibited mild intellectual disability and was born to non-consanguineous Chinese parents following an uneventful full-term pregnancy. At the age of 3 years, he began to display mild intentional tremors in both hands. By the age of eight, he began experiencing absence seizures, and the myoclonus in his hands worsened with age, gradually extending to the lower extremities, head, and trunk. On neurological examination, generalized myoclonic tremors were detected at rest and were provoked by action (Supplementary Video S1, after deep brain stimulation (DBS) of the subthalamic nucleus (STN) treatment). Scalp electroencephalogram (EEG) monitoring conducted over a 24-h period revealed diffuse spike waves during the interictal period (Figure 1B). The myoclonic status was monitored during the ictal and awake phases, accumulating in the eyelids, neck muscles, bilateral upper extremities, and trunk. Spikes (deltoid and thenar muscles) were observed on synchronized surface electromyography (EMG) recordings (Figure 1C), and epileptiform discharges linked to EMG spikes were found on synchronized EEG recordings (Figure 1D). The patient was treated with sodium valproate, followed by lamotrigine, levetiracetam, clonazepam, and lorazepam; however, all had poor effects. Consequently, the patient underwent STN-DBS in 2020. The myoclonic tremor symptoms in this patient improved postoperatively, and the epilepsy was well controlled with the above-mentioned antiepileptic drugs (Figure 1E). Further investigation of family history revealed that the mother of the proband had a history of head tremors, whereas no other neurological disorders were observed in the other family members.
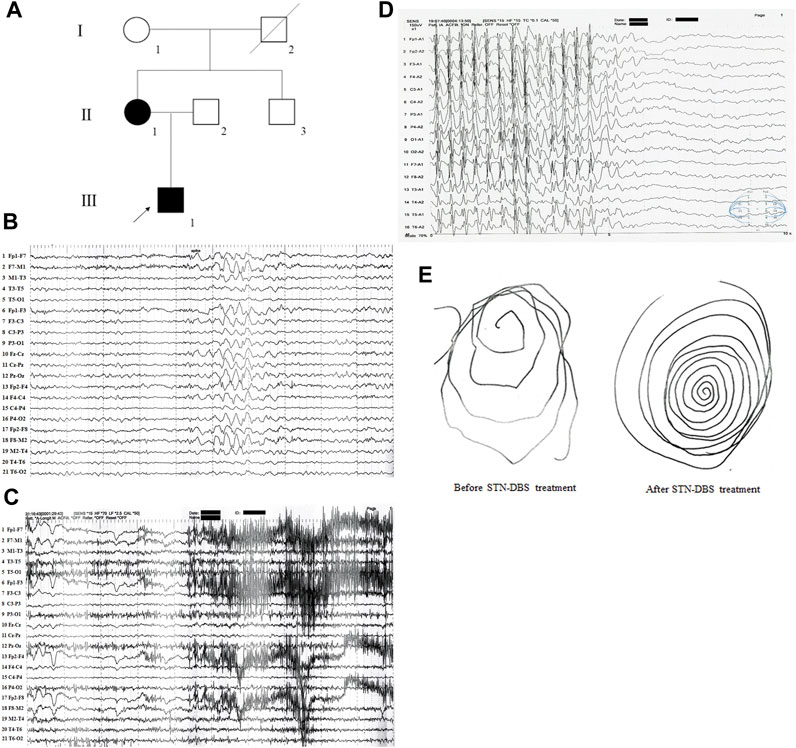
FIGURE 1. The clinical data of the family with epilepsy and/or myoclonus. (A) Pedigree figure. Family members are identified by generations and numbers. Squares indicate male family members; circles, female members; closed symbols, the affected members; open symbols, unaffected members; arrow, proband. (B) 24 h of video scalp EEG monitoring showed diffuse spike waves during the interictal period. (C) Synchronized surface EMG showed spikes when the patient has myoclonic seizures. (D) Synchronized surface EEG showed spikes when the patient has myoclonic seizures. (E) Patient’s handwriting showed tremor-like movement before and after STN-DBS treatment.
The proband was subjected to whole-exome sequencing, as previously described (Fan et al., 2018). The mean coverage of the target regions obtained for the proband was 99.8%, with an average sequencing depth of × 90.21. A total of 9,683 single nucleotide polymorphisms and 10,275 indels were identified. After data filtering (Figure 2A), 11 mutations were detected following public database filtering, bioinformatic program prediction, and family cosegregation analysis (Table 1). We then analyzed the evolutionary conservation (GERP) and the relationship between the candidate gene and phenotypes (OMIM) and found that the rare mutation (NM_001243564.1, c.113G>A/p. R38H) of DHDDS has a high possibility to be the genetic lesion of the family. Sanger sequencing confirmed that this mutation was present in the proband and his mother but absent in his father (Figure 2B). This rare mutation resulted in the substitution of arginine with histidine and was located at a highly evolutionarily conserved site (Figure 2C). This was not found in our local control cohort of 200 individuals and Genome Aggregation Database (gnomAD) database. Furthermore, a protein structure model constructed using the Swiss model revealed that the mutation (NM_001243564.1, c.113G>A/p. R38H) altered the polarity of the protein, which may further affect the protein function of DHDDS (Figure 2D). According to American College of Medical Genetics and Genomics guidelines (Richards et al., 2015), the mutation met the pathogenic criteria (PS1+PM1+PM2+PP1+PP2).
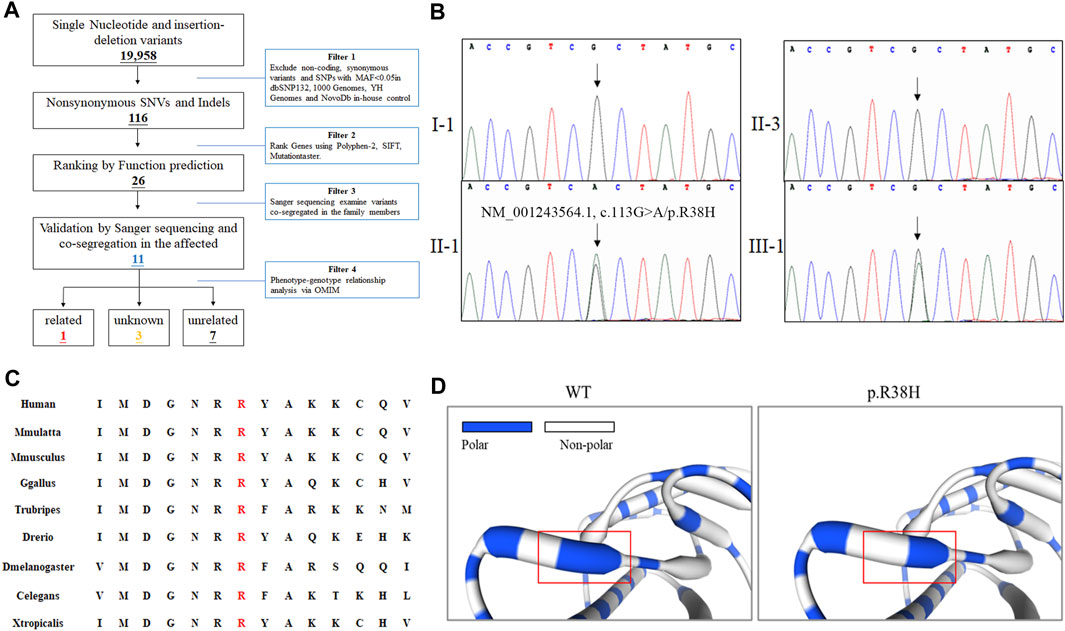
FIGURE 2. The genetic analysis of family. (A) Schematic representation of the filter strategies employed in our study. (B) Sanger sequencing of DHDDS confirmed the mutation (NM_001243564.1, c.113G>A/p. R38H) in II-1 and III-1. Arrow indicates the c.113G site. (C) Alignment of multiple DHDDS protein sequences across species. The R38H affected amino acid locates in the highly conserved amino acid region in different mammals (from Ensembl). Letters in red show the R38 site. (D) The part structure of WT and mutated DHDDS protein. Red squares represent R38 site.
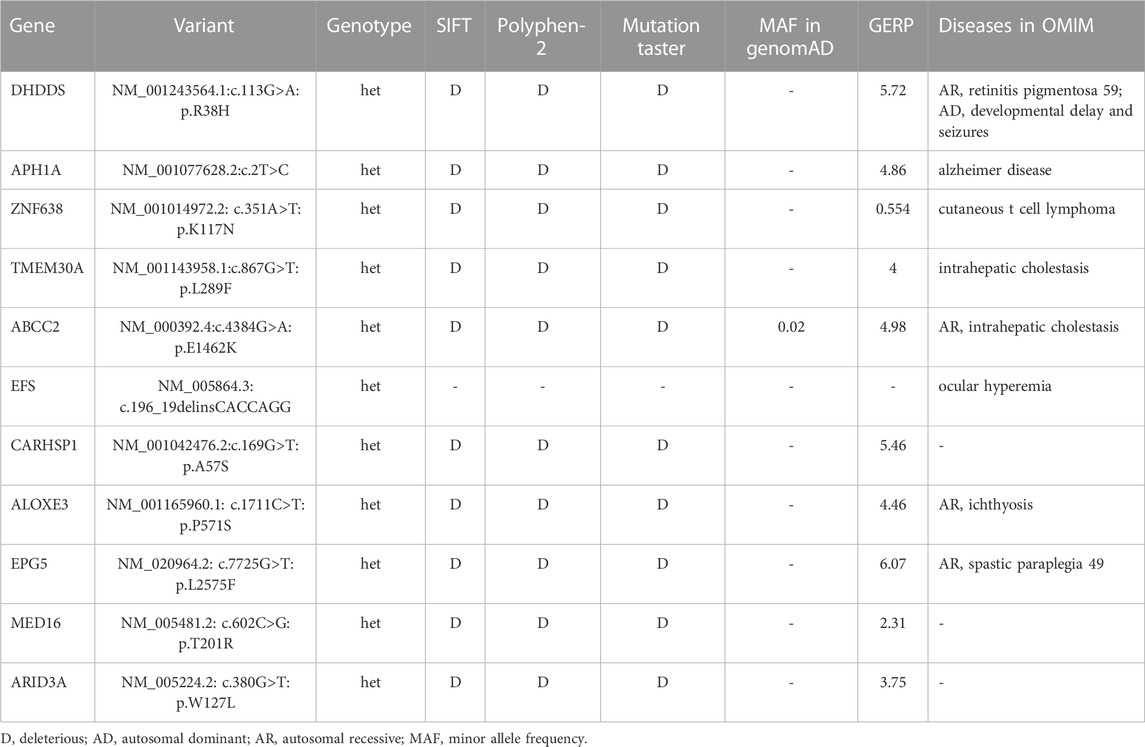
TABLE 1. The gene list of Sanger sequencing validation and co-segregation analysis.
Discussion
Currently, only 11 DHDDS mutations, identified in 39 patients, have been reported to result in DEE (Kim et al., 2021b; Galosi et al., 2022). Most patients were from Europe and North America, whereas 14 were from Eastern Asia (Togashi et al., 2020; Kim et al., 2021a; Jiao et al., 2022). In this study, we identified a rare mutation (NM_001243564.1, c.113G>A/p. R38H) of DHDDS in a Han Chinese family. This is the second report of DHDDS-related epilepsy in China and the third report of autosomal dominant familial inheritance of DHDDS mutation worldwide. Our findings further support that DHDDS play a crucial role in the occurrence and development of epilepsy.
Among the 39 patients with DHDDS mutations (Table 2), most presented with early onset epilepsy (0.5–8 years), and only nine showed late-onset epilepsy. Most of these patients exhibited different degrees of intellectual disability, generalized tonic-clonic seizures, and movement disorders, including tremor, dystonia, ataxia, and myoclonus. Interestingly, different phenotypes were associated with the DHDDS domain in which the mutations were located. The missense variant (NM_001243564.1, c.113G>A/p. R38H) in DHDDS was first reported in a patient with developmental and epileptic encephalopathy in an Epi4K study (Epi et al., 2013). Hamdan et al. subsequently identified de novo missense DHDDS mutations (NM_001243564.1, c.110G>A/p. R37H) that lie adjacent to p. R38H, and the defined DHDDS was found to be responsible for the DEE [4]. In our case, the proband also showed early onset epilepsy, intellectual disability, and myoclonus, consistent with most of the other reported cases (Kim et al., 2021b; Galosi et al., 2022).
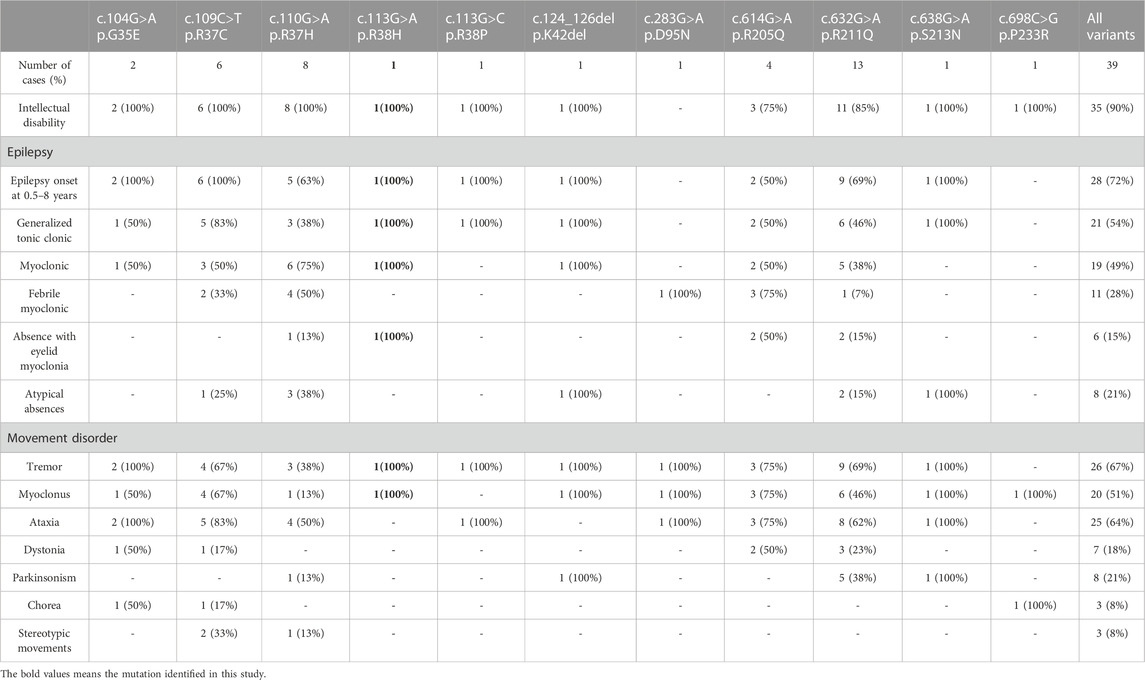
TABLE 2. Clinical features of DHDDS-related epilepsy.
DHDDS encodes a protein involved in dolichol biosynthesis on the cytoplasmic face of the endoplasmic reticulum (ER) (van Duijn et al., 1986). Previous studies have suggested that dolichol plays a crucial role in membrane trafficking, particularly between the ER and lysosomal-endosomal system, which is critical for brain development (Wu et al., 2017; Stojkovska et al., 2022). In addition, DHDDS can form a complex with the NUS1-encoded Nogo-B receptor (NgBR) (Bar-El et al., 2020), another key gene involved in neurodegeneration (Xue et al., 2022), which can further regulate protein glycosylation (Edani et al., 2020). In this study, the recurrent pathogenic mutation (NM_001243564.1, c.113G>A/p. R38H) of DHDDS altered the polarity of the protein, which may further disrupt the function of DHDDS protein.
In our study, the proband accepted STN-DBS treatment, which is usually intended for the appropriate treatment of a movement disorders. In recent years, STN-DBS has become a promising option for patients with epilepsy and appropriate indications for movement disorders, particularly for drug-refractory epilepsy (Valentin et al., 2013). Analogously, in our case, the patient’s epilepsy symptoms cannot be controlled after treatment with five different antiepileptic drugs. Hence, the patient accepted the STN-DBS treatment. After STN-DBS, the epilepsy of the patient was well controlled, and his myoclonic tremor symptoms improved. Our study further confirmed that STN-DBS surgery is an effective therapeutic regimen for epilepsy and an appropriate indication for movement disorders. Furthermore, our study also provided a viable treatment option for patients harboring DHDDS mutations.
Most patients (36/39) with DHDDS mutations have been validated for de novo events (Kim et al., 2021b; Galosi et al., 2022). Only two mutations (p.R205Q and p.R211Q) were found to have an autosomal-dominant familial inheritance pattern (Wood et al., 2021; Galosi et al., 2022). The mother of the proband carries a rare mutation (NM_001243564.1; c.113G>A/p. R38H) of DHDDS, presented only with tremors, and no other neurological disorders were detected, consistent with a previously reported family carrying the p.R205Q mutation, whereas the father of the proband, who carried the p.R211Q mutation, showed no similar symptoms. At present, the variable expression of epilepsy genes is known, and some cases could be carriers and show only EEG abnormalities. Mosaic carriers may explain this phenomenon (Robens et al., 2022). Here, we reported another DHDDS mutation which presented with autosomal dominant inheritance pattern and lead to intractable epilepsy and/or myoclonus.
Conclusion
In conclusion, by employing whole exome sequencing and Sanger sequencing, we identified a rare mutation (NM_001243564.1, c.113G>A/p. R38H) of DHDDS in a Chinese family. We may report the first case of intractable epilepsy and/or myoclonus caused by p.R38H mutation of DHDDS gene in the Chinese population. This study was also the third report of autosomal dominant familial inheritance of DHDDS mutation worldwide.
Data availability statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by the Second Xiangya Hospital of Central South University. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
YD and YZ performed the genetic analysis and wrote the draft of the manuscript. YS and WF performed the bioinformatics analysis. YZ, LL, and FW enrolled the samples. LL and L-LF designed the projected and supported the study. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Natural Science Foundation of Hunan Province (2021JJ40849 and 2023JJ20078), Research Project of the Hunan Provincial Health Commission (202203023480), the Scientific Research Launch Project for new employees of the Second Xiangya Hospital of Central South University (LL) and Hunan Provincial Innovation Foundation for Postgraduate (CX20210093).
Acknowledgments
The authors thank all the subjects for participating in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1208540/full#supplementary-material
References
Araki, K., Nakamura, R., Ito, D., Kato, K., Iguchi, Y., Sahashi, K., et al. (2020). NUS1 mutation in a family with epilepsy, cerebellar ataxia, and tremor. Epilepsy Res. 164, 106371. doi:10.1016/j.eplepsyres.2020.106371
Bar-El, M. L., Vankova, P., Yeheskel, A., Simhaev, L., Engel, H., Man, P., et al. (2020). Structural basis of heterotetrameric assembly and disease mutations in the human cis-prenyltransferase complex. Nat. Commun. 11, 5273. doi:10.1038/s41467-020-18970-z
Courage, C., Oliver, K. L., Park, E. J., Cameron, J. M., Grabinska, K. A., Muona, M., et al. (2021). Progressive myoclonus epilepsies-Residual unsolved cases have marked genetic heterogeneity including dolichol-dependent protein glycosylation pathway genes. Am. J. Hum. Genet. 108, 722–738. doi:10.1016/j.ajhg.2021.03.013
Cunillera, N., Arro, M., Fores, O., Manzano, D., and Ferrer, A. (2000). Characterization of dehydrodolichyl diphosphate synthase of Arabidopsis thaliana, a key enzyme in dolichol biosynthesis. FEBS Lett. 477, 170–174. doi:10.1016/s0014-5793(00)01798-1
Edani, B. H., Grabinska, K. A., Zhang, R., Park, E. J., Siciliano, B., Surmacz, L., et al. (2020). Structural elucidation of the cis-prenyltransferase NgBR/DHDDS complex reveals insights in regulation of protein glycosylation. Proc. Natl. Acad. Sci. U. S. A. 117, 20794–20802. doi:10.1073/pnas.2008381117
Epi, K. C., Epilepsy Phenome/Genome, P., Allen, A. S., Berkovic, S. F., Cossette, P., Delanty, N., et al. (2013). De novo mutations in epileptic encephalopathies. Nature 501, 217–221. doi:10.1038/nature12439
Fan, L. L., Huang, H., Jin, J. Y., Li, J. J., Chen, Y. Q., Zhao, S. P., et al. (2018). Whole exome sequencing identifies a novel mutation (c.333+2T>C) of TNNI3K in a Chinese family with dilated cardiomyopathy and cardiac conduction disease. Gene 648, 63–67. doi:10.1016/j.gene.2018.01.055
Galosi, S., Edani, B. H., Martinelli, S., Hansikova, H., Eklund, E. A., Caputi, C., et al. (2022). De novo DHDDS variants cause a neurodevelopmental and neurodegenerative disorder with myoclonus. Brain 145, 208–223. doi:10.1093/brain/awab299
Hamdan, F. F., Myers, C. T., Cossette, P., Lemay, P., Spiegelman, D., Laporte, A. D., et al. (2017). High rate of recurrent de novo mutations in developmental and epileptic encephalopathies. Am. J. Hum. Genet. 101, 664–685. doi:10.1016/j.ajhg.2017.09.008
Jiao, X., Xue, Y., Yang, S., Gong, P., Niu, Y., Wang, Q., et al. (2022). Phenotype of heterozygous variants of dehydrodolichol diphosphate synthase. Dev. Med. Child. Neurol. 64, 125–134. doi:10.1111/dmcn.14976
Kim, J., Kim, I., and Koh, S. B. (2021a). A novel variant of dehydrodolichol diphosphate synthase (DHDDS) mutation with adult-onset progressive myoclonus ataxia. Park. Relat. Disord. 87, 135–136. doi:10.1016/j.parkreldis.2021.05.010
Kim, S., Kim, M. J., Son, H., Hwang, S., Kang, M. K., Chu, K., et al. (2021b). Adult-onset rapidly worsening progressive myoclonic epilepsy caused by a novel variant in DHDDS. Ann. Clin. Transl. Neurol. 8, 2319–2326. doi:10.1002/acn3.51483
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and Genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Robens, B. K., Yang, X., Mcgraw, C. M., Turner, L. H., Robens, C., Thyme, S., et al. (2022). Mosaic and non-mosaic protocadherin 19 mutation leads to neuronal hyperexcitability in zebrafish. Neurobiol. Dis. 169, 105738. doi:10.1016/j.nbd.2022.105738
Stojkovska, I., Wani, W. Y., Zunke, F., Belur, N. R., Pavlenko, E. A., Mwenda, N., et al. (2022). Rescue of α-synuclein aggregation in Parkinson's patient neurons by synergistic enhancement of ER proteostasis and protein trafficking. Neuron 110, 436–451.e11. doi:10.1016/j.neuron.2021.10.032
Togashi, N., Fujita, A., Shibuya, M., Uneoka, S., Miyabayashi, T., Sato, R., et al. (2020). Fifteen-year follow-up of a patient with a DHDDS variant with non-progressive early onset myoclonic tremor and rare generalized epilepsy. Brain Dev. 42, 696–699. doi:10.1016/j.braindev.2020.06.011
Valentin, A., Garcia Navarrete, E., Chelvarajah, R., Torres, C., Navas, M., Vico, L., et al. (2013). Deep brain stimulation of the centromedian thalamic nucleus for the treatment of generalized and frontal epilepsies. Epilepsia 54, 1823–1833. doi:10.1111/epi.12352
Van Duijn, G., Valtersson, C., Chojnacki, T., Verkleij, A. J., Dallner, G., and De Kruijff, B. (1986). Dolichyl phosphate induces non-bilayer structures, vesicle fusion and transbilayer movement of lipids: A model membrane study. Biochim. Biophys. Acta 861, 211–223. doi:10.1016/0005-2736(86)90423-2
Wood, K., Montgomery, T., and Devlin, A. M. (2021). DHDDS related epilepsy--Report of familial cases and review of the literature. Seizure 91, 189–191. doi:10.1016/j.seizure.2021.06.013
Wu, Y., Whiteus, C., Xu, C. S., Hayworth, K. J., Weinberg, R. J., Hess, H. F., et al. (2017). Contacts between the endoplasmic reticulum and other membranes in neurons. Proc. Natl. Acad. Sci. U. S. A. 114, E4859–E4867. doi:10.1073/pnas.1701078114
Xue, J., Zhu, Y., Wei, L., Huang, H., Li, G., Huang, W., et al. (2022). Loss of Drosophila NUS1 results in cholesterol accumulation and Parkinson's disease-related neurodegeneration. FASEB J. 36, e22411. doi:10.1096/fj.202200212R
Zelinger, L., Banin, E., Obolensky, A., Mizrahi-Meissonnier, L., Beryozkin, A., Bandah-Rozenfeld, D., et al. (2011). A missense mutation in DHDDS, encoding dehydrodolichyl diphosphate synthase, is associated with autosomal-recessive retinitis pigmentosa in Ashkenazi Jews. Am. J. Hum. Genet. 88, 207–215. doi:10.1016/j.ajhg.2011.01.002
Keywords: epilepsy, myoclonus, DHDDS, mutation, whole exome sequencing
Citation: Dong Y, Zhang Y, Sheng Y, Wang F, Liu L and Fan L-L (2023) Case report: Identification of a recurrent pathogenic DHDDS mutation in Chinese family with epilepsy, intellectual disability and myoclonus. Front. Genet. 14:1208540. doi: 10.3389/fgene.2023.1208540
Received: 19 April 2023; Accepted: 25 September 2023;
Published: 10 October 2023.
Edited by:
Jangsup Moon, Seoul National University Hospital, Republic of KoreaReviewed by:
Anita Devlin, Newcastle University, United KingdomMahmoud Koko, Wellcome Sanger Institute (WT), United Kingdom
Edoardo Monfrini, University of Milan, Italy
Copyright © 2023 Dong, Zhang, Sheng, Wang, Liu and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lv Liu, ZG9jbGl1bHZAY3N1LmVkdS5jbg==; Liang-Liang Fan, c3dmYW5saWFuZ2xpYW5nQGNzdS5lZHUuY24=
‡These authors have contributed equally to this work