 Xinxing Li
Xinxing Li Wendong Huang
Wendong Huang Xuan Xu
Xuan Xu Hong-Yu Zhang
Hong-Yu Zhang Qianqian Shi
Qianqian Shi- Hubei Key Laboratory of Agricultural Bioinformatics, College of Informatics, Huazhong Agricultural University, Wuhan, China
Spatially resolved transcriptomics (SRT) provides an unprecedented opportunity to investigate the complex and heterogeneous tissue organization. However, it is challenging for a single model to learn an effective representation within and across spatial contexts. To solve the issue, we develop a novel ensemble model, AE-GCN (autoencoder-assisted graph convolutional neural network), which combines the autoencoder (AE) and graph convolutional neural network (GCN), to identify accurate and fine-grained spatial domains. AE-GCN transfers the AE-specific representations to the corresponding GCN-specific layers and unifies these two types of deep neural networks for spatial clustering via the clustering-aware contrastive mechanism. In this way, AE-GCN accommodates the strengths of both AE and GCN for learning an effective representation. We validate the effectiveness of AE-GCN on spatial domain identification and data denoising using multiple SRT datasets generated from ST, 10x Visium, and Slide-seqV2 platforms. Particularly, in cancer datasets, AE-GCN identifies disease-related spatial domains, which reveal more heterogeneity than histological annotations, and facilitates the discovery of novel differentially expressed genes of high prognostic relevance. These results demonstrate the capacity of AE-GCN to unveil complex spatial patterns from SRT data.
Introduction
Spatially resolved transcriptomics (SRT) technologies, such as spatial transcriptomics (ST) (Ståhl et al., 2016), 10x Visium, and Slide-seqV2 (Stickels et al., 2021), can measure the transcript localization and abundance in the dissected tissue area, enabling novel insights into tissue development and tumor heterogeneity (Atta and Fan, 2021; Nasab et al., 2022). Their generated data (i.e., gene expression in tissue locations [spots] and spatial locational information) can be used to decipher the spatially functional regions and cellular architectures in tissues (Maniatis et al., 2021; Marx, 2021; Zeng et al., 2022). However, due to technical limitations (Xu et al., 2022), modeling and integrating the available SRT modalities for accurate spatial domain identification still remain challenging.
Currently, the spatial domain detection methods could be mainly divided into two categories: non-spatial and spatial clustering methods. Some non-spatial methods originally developed for single-cell RNA-sequencing (scRNA-seq) studies, e.g., Seurat (Butler et al., 2018) and Scanpy (Wolf et al., 2018), are also applied in SRT studies. They only utilize the expression profiles to cluster spots while often obtaining domains lacking in spatial continuity to some extent. To address such issues, spatial clustering approaches generally incorporate the additional spatial information into their models. For example, with the spatial prior, BayesSpace (Zhao et al., 2021) and HMRF (Dries et al., 2021) use the Markov random field model (or its variant) to encourage the spatially neighboring spots to belong to the same domain. SpaGCN (Hu et al., 2021) and SEDR (Fu et al., 2021) enable spatial clustering by learning the low-dimensional representation with graph constraints that represent the spatial dependency. STAGATE (Dong and Zhang, 2022) identifies spatial domains by adaptively learning the similarity of neighboring spots via attention mechanisms. Modeling the spatial dependency of gene expression fairly facilitates the discovery of spatial domains with spatial coherence.
Though these methods have provided useful information on the usage of expression profiles and spatial information, they usually depend on single models, which center on either expression data itself or spatially neighboring structure, thus probably resulting in the preferred usage of the focused data type. For example, the non-spatial clustering methods only models the gene expression itself, while the spatial clustering methods often take spatial neighbors prior as a hard constraint to ensure spatial clustering continuity, which may lead to over-smoothing of expression (Huang et al., 2018) and missing subtle spatial regions with a handful of spots. Thus, the rational combination of these different kinds of models can fairly generate more useful representations, enabling better spatial domain detection in SRT studies.
Here, we develop a novel combined model, AE-GCN (autoencoder-assisted graph convolutional neural network), which combines the autoencoder (AE) and graph convolutional neural network (GCN), for accurate and fine-grained spatial domain identification. Specifically, AE-GCN relies on AE for learning expression data-based representations and GCN for spatial graph-constrained learning. AE-GCN orderly transfers the AE-specific representations to GCN-specific layers and unifies these two types of neural networks for spatial clustering via a clustering-aware contrastive mechanism. In this way, AE-GCN combines the advantages of the two models and takes full integration of the expression data and spatial information during the representation learning process.
We demonstrate the effectiveness of AE-GCN on spatial domain identification and data denoising using SRT datasets generated from ST, 10x Visium, and Slide-seqV2 platforms. In particular, it is validated in two cancer samples that AE-GCN can refine the spatial functional regions and discover novel cancer-associated genes. These results show that AE-GCN is capable of unveiling complex tissue architecture from SRT data.
Materials and methods
Overview of AE-GCN
AE-GCN is an integrative scheme that incorporates the AE and GCN learning processes, enabling tasks of spatial domain detection and data denoising (Figure 1).
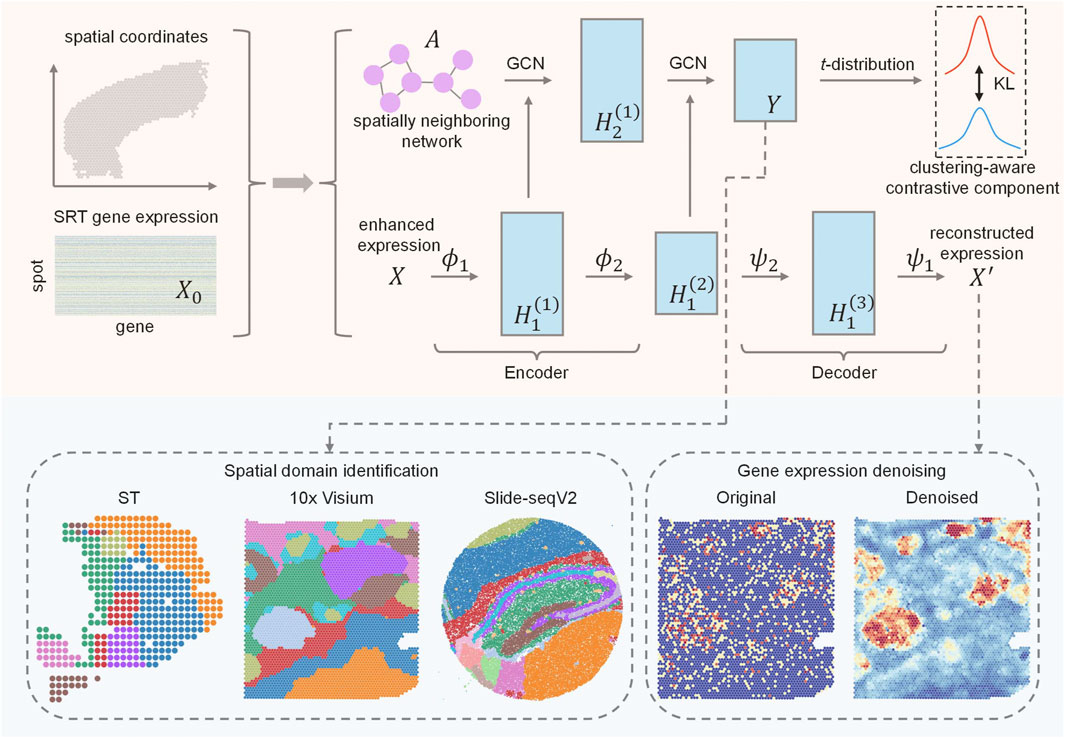
FIGURE 1. Schematic overview of AE-GCN and its potential applications. Given gene expression and spatial coordinates as input, AE-GCN first builds the spatially neighboring network
Given the original expression
When the learning process reaches convergence, the low-dimensional representation (i.e.,
Spatially neighboring network construction and expression augmentation
Spatially neighboring network construction
Assume that there are original expression matrix
Then, the weighted adjacency matrix
Spatial expression augmentation
Limited by the transcript capture rate of SRT technologies, expression data are often sparse and noisy. AE-GCN generates the enhanced expression data
where the tunable parameter
AE component
We employ AE to learn the useful representations from the expression data itself and assume that there are
where
The output (i.e.,
GCN component
AE-specific representations, e.g.,
where
Note that we denote the representation (i.e.,
Clustering-aware contrastive component
Although we have incorporated the encoder of AE into the neural network architecture of GCN to obtain the combined representation, this representation cannot be directly applied to the clustering problem. Herein, we propose a clustering-aware contrastive module to unify these two different deep learning models for effective spatial clustering.
Specifically, we use student’s t-distribution to measure the probability of assigning the spot
where
To optimize the AE-GCN-learnable representation from the high-confidence assignment, we make data representation closer to cluster centers for improving the cluster cohesion. Hence, we calculate the target distribution
where
This design is regarded as a clustering-aware contrastive mechanism, where the
where
Data collection and general preprocessing
The top 3,000 highly variable genes (HVGs) for 13 10x Visium datasets, one ST dataset, and one Slide-seqV2 dataset are selected using scanpy.pp.highly_variable_genes() from the Scanpy Python package. The log-transformation of the expression profiles is performed using scanpy.pp.log1p() on the original gene expression data.
Spatial domain detection and gene expression denoising
AE-GCN uses the combined latent representation
For the enhanced expression matrix
Performance evaluation
We use adjusted Rand index (ARI) (Hubert and Arabie, 1985) and cluster purity (i.e., Eq. 13) (Zhao et al., 2021) to quantify the accuracy of the identified spatial domain and the reference annotations from original publications.
where
Survival analysis
We use bulk expression data with patient survival information to evaluate the prognostic significance of genes via the Kaplan–Meier plotter (Zwyea et al., 2021) in the IDC and PDAC cancer studies.
Results
Benchmarking AE-GCN against state-of-the-art methods
We evaluated the ability of AE-GCN to detect spatial domains using 12 human dorsolateral prefrontal cortex (DLPFC) slices generated using 10x Visium. The DLPFC dataset obtained from spatialLIBD (Pardo et al., 2022) is manually annotated as the layered regions by gene markers and cytoarchitecture. The annotations can be considered as the ground truth for benchmarking. Based on this dataset, we compared AE-GCN with the existing state-of-the-art methods, including six spatial clustering methods [i.e., BayesSpace (Zhao et al., 2021), Giotto (Dries et al., 2021), SEDR (Fu et al., 2021), SpaGCN (Hu et al., 2021), stLearn (Pham et al., 2020), and STAGATE (Dong and Zhang, 2022)] and three non-spatial algorithms [i.e., variational autoencoder (VAE) (Kingma and Welling, 2019), Leiden implemented in Scanpy (Wolf et al., 2018), and Louvain implemented in Seurat (Butler et al., 2018)]. The adjusted Rand index (ARI) is used to quantify the similarity between the manual labels and identified clusters, which ranges from 0 for poor consistency to 1 for identical clusters.
Generally, most of the spatial clustering methods performed better than non-spatial algorithms (Wilcoxon signed-rank test
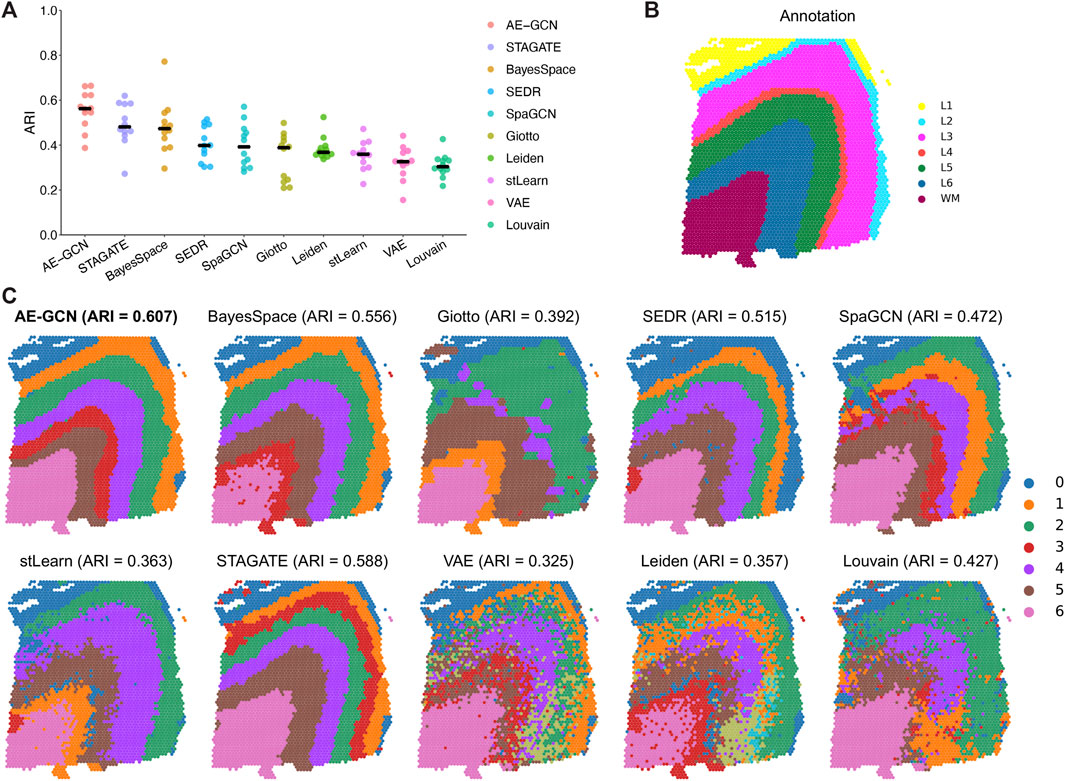
FIGURE 2. Benchmarking AE-GCN against state-of-the-art spatial domain detection methods. (A) Spatial clustering performance is compared using ARI on 12 manually annotated DLPFC datasets from spatialLIBD. The bold line represents the mean ARI value of each approach on all the datasets. (B) Slice 151673 with the manual annotation. (C) Comparative illustration of the identified spatial domain on slice 151673. The identified spatial domains of each method are distinguished by colors without strict correspondence.
AE-GCN reveals fine-grained anatomical regions on mouse hippocampus Slide-seqV2 data
To illustrate the effectiveness of AE-GCN on high-resolution SRT platforms, we applied AE-GCN to a mouse hippocampus Slide-seqV2 dataset (n = 41,786 spots). Slide-seqV2 can measure gene expression at near-cellular resolution (Stickels et al., 2021) but has lower number of transcripts per location/spot and higher dropouts than the 10x Visium platform. Thus, it poses more challenges for accurately distinguishing tissue structures from the data of high sparsity. To better validate the performance of AE-GCN, we also compared it with other domain detection methods and used the corresponding anatomical diagram from the Allen Mouse Brain Atlas (Sunkin et al., 2012) as the illustrative reference (Figure 3A).
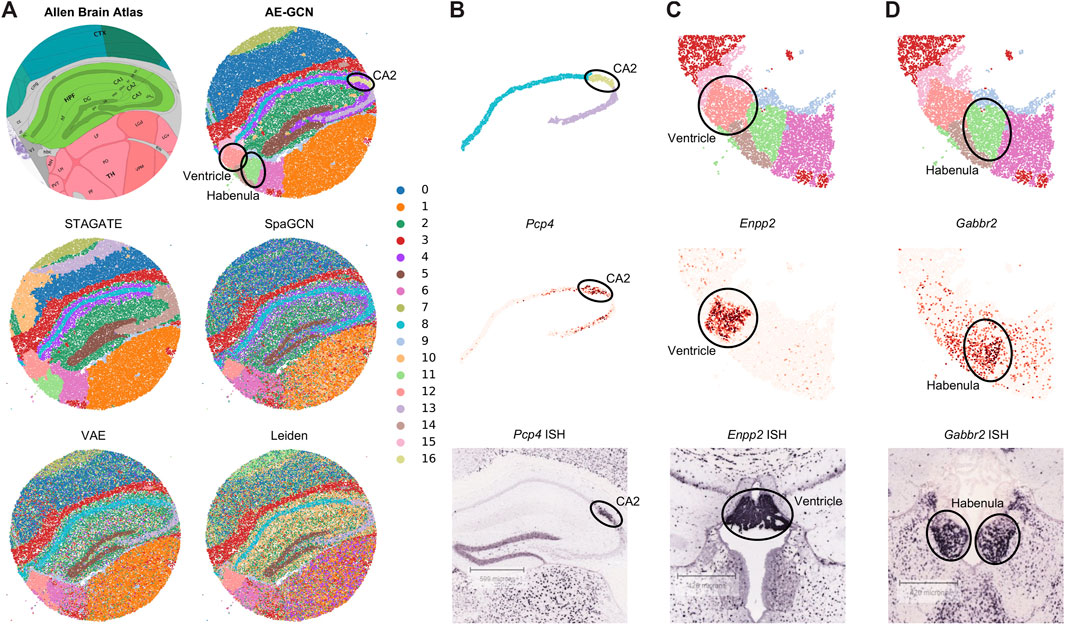
FIGURE 3. AE-GCN reveals the finer-grained anatomical regions on mouse hippocampus Slide-seqV2 data. (A) Corresponding anatomical diagram from the Allen Mouse Brain Atlas and spatial domains identified by each competed method. (B–D) CA2 and ventricle and habenula regions (at the top) from AE-GCN partitions are, respectively, validated by the known gene markers (i.e., Pcp4, Enpp2, and Gabbr2) from gene expression (at the middle) and ISH images (at the bottom). The ISH images of Pcp4, Enpp2, and Gabbr2 are also obtained from the Allen Mouse Brain Atlas.
Comparing with the reference, we found that AE-GCN and STAGATE can identify the spatially coherent domains compared to other involved methods. However, AE-GCN performed better to detect the fine-grained structures, such as the cornu ammonis 2 (CA2, AE-GCN domain 16), ventricle (AE-GCN domain 12), and habenula (AE-GCN domain 11) sections (Figure 3A). These sections are delineated with sharper boundaries and higher concordance with the anatomical annotation. We further isolated the focused regions and provided validations from other perspectives (Figures 3B–D). For the CA2 section, which is only detected by AE-GCN, the domain location showed good alignment with the marker gene expression (i.e., Pcp4 (San Antonio et al., 2014)) and independent in situ hybridization (ISH) image (Figure 3B). For ventricle and habenula sections, AE-GCN domains are closer to the shapes of their respective marker expression (Enpp2 for ventricle (Koike et al., 2006) and Gabbr2 for habenula (De Beaurepaire, 2018)) or stained regions and match the anatomical shape well (Figures 3C, D). Thus, for higher-resolution SRT data, AE-GCN is capable of effectively unveiling the fine-grained anatomical functional regions.
AE-GCN accurately discerns tumor regions on human pancreatic ductal adenocarcinoma data
To illustrate the effectiveness of AE-GCN on cancer tissue, we applied AE-GCN to the human pancreatic ductal adenocarcinoma (PDAC) ST dataset (n = 428 spots). The histopathological image and annotations were taken as references (Figures 4A, B). We assessed these spatial domain identification methods using cluster purity (see Methods) as the quantitative measure on cancer datasets with rough annotation information. AE-GCN achieved the highest cluster purity (purity = 0.756) and detected more spatially enriched functional regions in tumor tissue than other compared methods (Figure 4C).
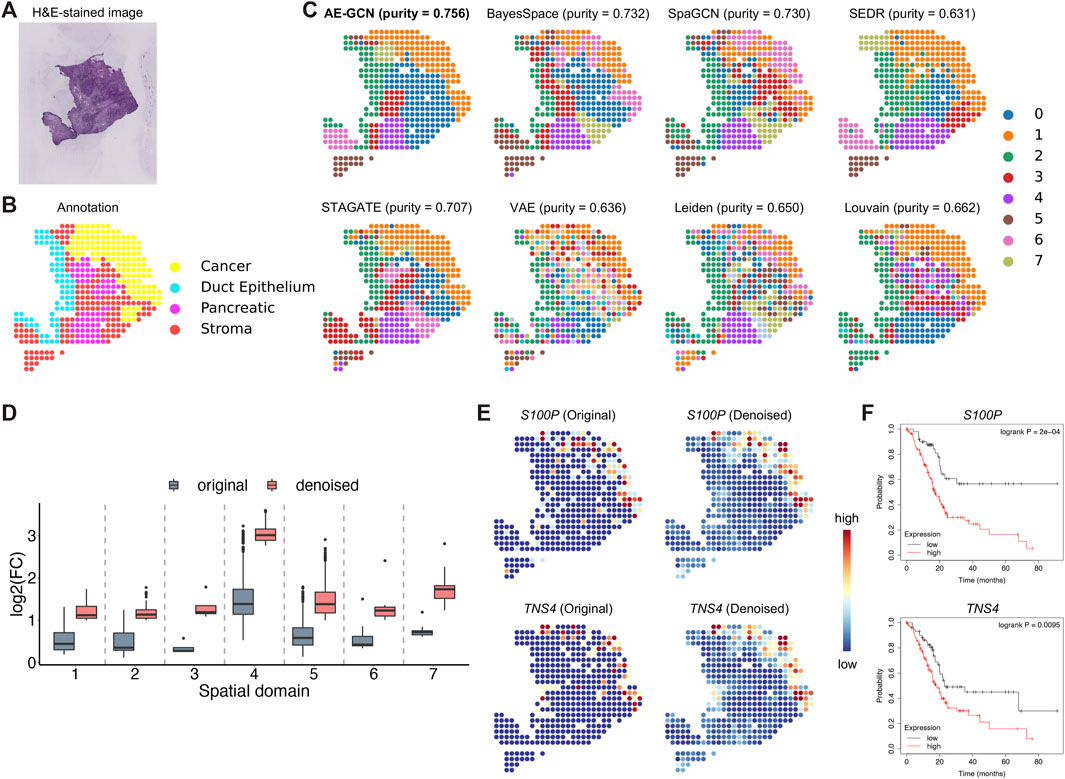
FIGURE 4. AE-GCN identifies tumor regions on human PDAC ST data. The H&E-stained image (A) and the corresponding manual annotation (B) are shown as references. (C) The identified spatial domains using all the compared methods are distinguished by different colors without strict correspondence. Cluster purity is used to compare the similarity between identified domains and the reference annotation. (D) The change in gene differential expression in each domain before and after data denoising. log2(FC): the logarithmic value of the gene expression fold change with base 2. (E) Spatial expression visualization of selected DEGs (i.e., S100P and TNS4) before and after data denoising. (F) Kaplan–Meier survival curves show the clinical relevance of the identified DEGs (i.e., S100P and TNS4).
Next, we examined whether AE-GCN could provide more insights into the underlying tumor heterogeneity, as data sparsity could hinder other downstream analytical tasks, for example, the identification of differentially expressed genes (DEGs). In this manner, we used the AE-GCN-reconstructed data to denoise the low-quality measurements and evaluated the effectiveness in recovering gene spatial expression patterns. Based on the denoised data, we selected the top 50 DEGs of each domain from the reconstructed data
AE-GCN reveals more intratumor heterogeneity on invasive ductal carcinoma data
To illustrate the generalization ability of AE-GCN on cancer tissues, we next tested AE-GCN using the invasive ductal carcinoma (IDC) Visium dataset (n = 4,727 spots). The histopathological annotations from the original paper (Zhao et al., 2021) were taken as the reference (Figures 5A, B). We found that the identified domains of AE-GCN were highly consistent with the manual annotations (purity = 0.865, Figure 5C). Compared with the domains captured by other methods, the clustering partitions from AE-GCN showed clear spatial separations with few scatter points and high regional continuity.
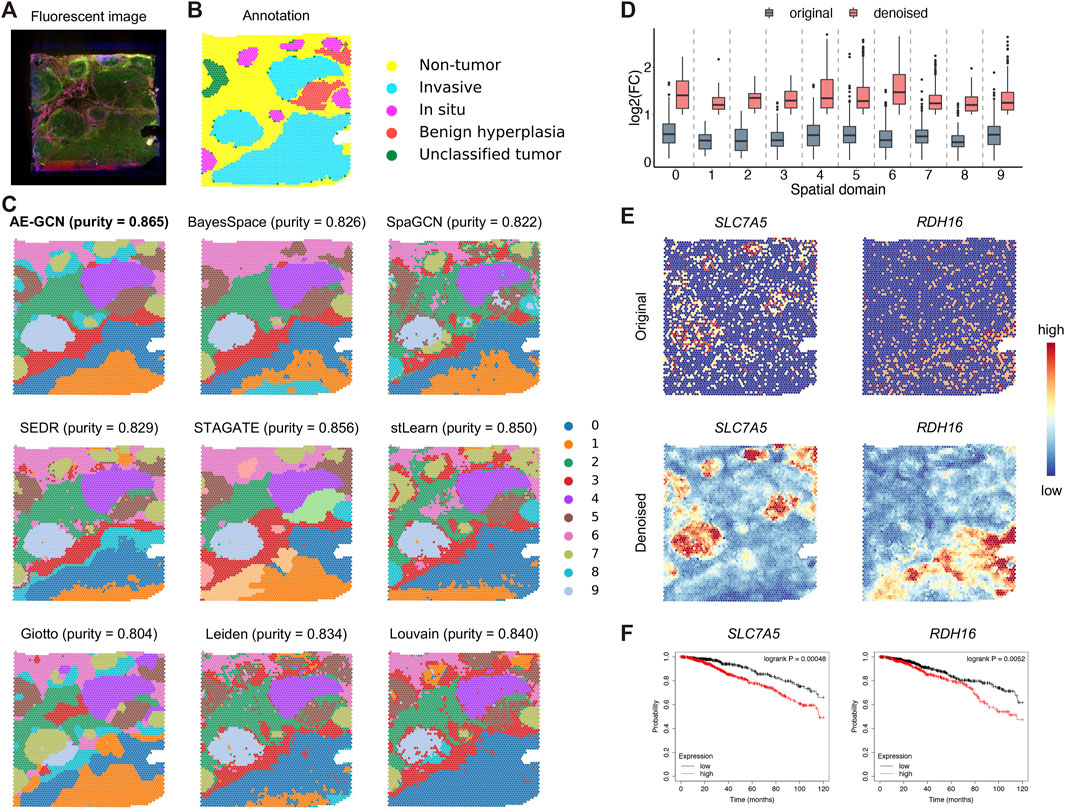
FIGURE 5. AE-GCN provides more biological insights into intratumor heterogeneity on the IDC 10x Visium dataset. The fluorescent image (A) and the corresponding manual annotation (B) are shown as references. Each spot is colored due to the annotation label in (B). (C) The spatial domains obtained by all involved methods are distinguished using different colors without strict correspondence. Cluster purity is used to compare the similarities between identified outcomes and reference annotation. (D) The change gene FC before and after data denoising. (E) Spatial expression visualization of the selected domain-specific genes (i.e., SLC7A5 and RDH16) before and after data denoising. (F) Kaplan–Meier survival curves show the clinical relevance of the newly identified DEGs (i.e., SLC7A5 and RDH16).
Then, for functional gene identification, we identified the top 50 DEGs of each cluster from the denoised data
Discussion
Spatially resolved transcriptomics technologies measure gene expression on each spot while preserving spatial context, which can support computational methods to identify functional regions of tissue and further resolve organizational heterogeneity. The combined modeling of gene expression and spatial information enables the improved identification accuracy of spatial domains, especially for complex spatial architecture, e.g., tumor microenvironments. In this paper, AE-GCN combines the autoencoder and graph convolutional neural network to achieve effective latent representations from expression data itself and spot neighboring structure. The superiority of AE-GCN is shown not only on the accurate and fine-grained identification of spatial domains for multiple SRT platforms but also on the recovery or identification of gene spatial expression patterns. In particular, the application on cancer slices (i.e., IDC and PDAC) demonstrates that AE-GCN reveals more functional regions and novel cancer prognostic genes for interpreting cancer heterogeneity, suggesting that AE-GCN has great capability of unveiling tissue heterogeneity from SRT data.
The effectively combined modeling is key to the superiority of AE-GCN in the SRT study. Generally, AE models learn the representations from expression data itself, while GCN models learn the structured representations from the sample graph structure by providing an approximate second-order graph regularization, which may suffer from over-smoothing issues. AE-GCN combines the characteristics of these two deep learning methods and integrates them to learn effective representations so that AE is used to weaken the problem of overfitting while simultaneously learning the structured representations in GCN. Additionally, the proposed clustering-aware contrastive module in AE-GCN further promotes the combined model from processes independent of clustering targets to the model that achieves effective spatial clustering. Thus, AE-GCN can not only effectively use the information of the expression data itself but also reasonably regularize the learned information from expression data by spatial structure between spots, which has better advantages than the spatial domain detection methods based on a single-model design in SRT studies.
Currently, AE-GCN only models gene expression and spatial information from SRT data and cannot utilize histological images which are also provided by several SRT technologies, e.g., 10x Visium. Although some methods have used histological images in spatial domain detection, histological images are mainly used to enhance the quality of expression data and lack of modeling image data separately, e.g., stLearn (Pham et al., 2020). Compared with expression data and spatial information, histological image data are one type of modalities more suitable for deep learning modeling. The future work to extend AE-GCN is to integrate deep learning models for each multi-modal data characteristic (i.e., gene expression, histological images, and spatial information) to improve the performance of current methods in SRT research.
Data availability statement
The datasets presented in this study can be found in online repositories. The available web resources include human DLPFC datasets (available in spatialLIBD package), mouse hippocampus Slide-seqV2 dataset (https://singlecell.broadinstitute.org), human IDC 10x Visium dataset (https://www.10xgenomics.com/resources/datasets) and human PDAC ST dataset (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE111672). Python source code of AE-GCN is available at https://github.com/zccqq/AE-GCN.
Author contributions
QS conceived and designed the framework and the experiments. XL and WH performed the experiments. WH and XX developed the Python package and documentation website of the framework. QS analyzed the data and wrote the paper. QS and HZ revised the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 61802141).
Acknowledgments
The authors thank the reviewers for useful suggestions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Atta, L., and Fan, J. (2021). Computational challenges and opportunities in spatially resolved transcriptomic data analysis. Nat. Commun. 12 (1), 5283. doi:10.1038/s41467-021-25557-9
Butler, A., Hoffman, P., Smibert, P., Papalexi, E., and Satija, R. (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36 (5), 411–420. doi:10.1038/nbt.4096
De Beaurepaire, R. (2018). A review of the potential mechanisms of action of baclofen in alcohol use disorder. Front. Psychiatry 9, 506. doi:10.3389/fpsyt.2018.00506
Dong, K., and Zhang, S. (2022). Deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto-encoder. Nat. Commun. 13 (1), 1739–1812. doi:10.1038/s41467-022-29439-6
Dries, R., Zhu, Q., Dong, R., Eng, C.-H. .L., Li, H., Liu, K., et al. (2021). Giotto: A toolbox for integrative analysis and visualization of spatial expression data. Genome Biol. 22 (1), 78–31. doi:10.1186/s13059-021-02286-2
El Ansari, R., Craze, M. .L., Miligy, I., Diez-Rodriguez, M., Nolan, C. .C., Ellis, I. .O., et al. (2018). The amino acid transporter SLC7A5 confers a poor prognosis in the highly proliferative breast cancer subtypes and is a key therapeutic target in luminal B tumours. Breast Cancer Res. 20, 21–17. doi:10.1186/s13058-018-0946-6
Fu, H., Hang, X. .U., and Chen, J. (2021). Unsupervised spatial embedded deep representation of spatial transcriptomics. bioRxiv.
Gao, C., Zhuang, J., Li, H., Liu, C., Zhou, C., Liu, L., et al. (2020). Development of a risk scoring system for evaluating the prognosis of patients with Her2-positive breast cancer. Cancer Cell Int. 20 (1), 121–212. doi:10.1186/s12935-020-01175-1
Hu, J., Li, X., Coleman, K., Schroeder, A., Ma, N., Irwin, D. .J., et al. (2021). SpaGCN: Integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat. methods 18 (11), 1342–1351. doi:10.1038/s41592-021-01255-8
Huang, M., Wang, J., Torre, E., Dueck, H., Shaffer, S., Bonasio, R., et al. (2018). Saver: Gene expression recovery for single-cell RNA sequencing. Nat. methods 15 (7), 539–542. doi:10.1038/s41592-018-0033-z
Hubert, L., and Arabie, P. (1985). Comparing partitions. J. Classif. 2 (1), 193–218. doi:10.1007/bf01908075
Kingma, D. .P., and Welling, M. (2019). An introduction to variational autoencoders. Found. Trends® Mach. Learn. 12 (4), 307–392. doi:10.1561/2200000056
Koike, S., Keino-Masu, K., Ohto, T., and Masu, M. (2006). The N-terminal hydrophobic sequence of autotaxin (ENPP2) functions as a signal peptide. Genes cells 11 (2), 133–142. doi:10.1111/j.1365-2443.2006.00924.x
Maniatis, S., Petrescu, J., and Phatnani, H. (2021). Spatially resolved transcriptomics and its applications in cancer. Curr. Opin. Genet. Dev. 66, 70–77. doi:10.1016/j.gde.2020.12.002
Marx, V. (2021). Method of the year: Spatially resolved transcriptomics. Nat. methods 18 (1), 9–14. doi:10.1038/s41592-020-01033-y
Nasab, R. .Z., Ghamsari, M. .R. .E., Argha, A., Macphillamy, C., Beheshti, A., Alizadehsani, R., et al. (2022). Deep learning in spatially resolved transcriptomics: A comprehensive technical view. arXiv preprint arXiv:221004453 2022.
Pardo, B., Spangler, A., Weber, L. .M., Page, S. .C., Hicks, S. .C., Jaffe, A. .E., et al. (2022). spatialLIBD: an R/Bioconductor package to visualize spatially-resolved transcriptomics data. BMC genomics 23 (1), 434. doi:10.1186/s12864-022-08601-w
Pham, D., Tan, X., Xu, J., Grice, L. .F., Lam, P. .Y., Raghubar, A., et al. (2020). stLearn: integrating spatial location, tissue morphology and gene expression to find cell types, cell-cell interactions and spatial trajectories within undissociated tissues. bioRxiv.
Sakashita, K., Mimori, K., Tanaka, F., Kamohara, Y., Inoue, H., Sawada, T., et al. (2008). Prognostic relevance of Tensin4 expression in human gastric cancer. Ann. Surg. Oncol. 15, 2606–2613. doi:10.1245/s10434-008-9989-8
San Antonio, A., Liban, K., Ikrar, T., Tsyganovskiy, E., and Xu, X. (2014). Distinct physiological and developmental properties of hippocampal CA2 subfield revealed by using anti-Purkinje cell protein 4 (PCP4) immunostaining. J. Comp. Neurology 522 (6), 1333–1354. doi:10.1002/cne.23486
Ståhl, P. .L., Salmén, F., Vickovic, S., Lundmark, A., Navarro, J. .F., Magnusson, J., et al. (2016). Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353 (6294), 78–82. doi:10.1126/science.aaf2403
Stickels, R. .R., Murray, E., Kumar, P., Li, J., Marshall, J. .L., Di Bella, D. .J., et al. (2021). Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat. Biotechnol. 39 (3), 313–319. doi:10.1038/s41587-020-0739-1
Sunkin, S. .M., Ng, L., Lau, C., Dolbeare, T., Gilbert, T. .L., Thompson, C. .L., et al. (2012). Allen Brain Atlas: An integrated spatio-temporal portal for exploring the central nervous system. Nucleic acids Res. 41 (D1), D996-D1008–D1008. doi:10.1093/nar/gks1042
Traag, V. .A., Waltman, L., and Van Eck, N. .J. (2019). From Louvain to leiden: Guaranteeing well-connected communities. Sci. Rep. 9 (1), 5233–5312. doi:10.1038/s41598-019-41695-z
Wang, Q., Zhang, Y.-N., Lin, G.-L., Qiu, H.-Z., Wu, B., Wu, H.-Y., et al. (2012). S100P, a potential novel prognostic marker in colorectal cancer. Oncol. Rep. 28 (1), 303–310. doi:10.3892/or.2012.1794
Wolf, F. .A., Angerer, P., and Theis, F. .J. (2018). Scanpy: Large-scale single-cell gene expression data analysis. Genome Biol. 19 (1), 15–5. doi:10.1186/s13059-017-1382-0
Xu, C., Jin, X., Wei, S., Wang, P., Luo, M., Xu, Z., et al. (2022). DeepST: Identifying spatial domains in spatial transcriptomics by deep learning. Nucleic Acids Res. 50 (22), e131–e. doi:10.1093/nar/gkac901
Zeng, Z., Li, Y., Li, Y., and Luo, Y. (2022). Statistical and machine learning methods for spatially resolved transcriptomics data analysis. Genome Biol. 23 (1), 83–23. doi:10.1186/s13059-022-02653-7
Zhao, E., Stone, M. .R., Ren, X., Guenthoer, J., Smythe, K. .S., Pulliam, T., et al. (2021). Spatial transcriptomics at subspot resolution with BayesSpace. Nat. Biotechnol. 39 (11), 1375–1384. doi:10.1038/s41587-021-00935-2
Keywords: spatially resolved transcriptomics, spatial domain identification, spatial information, graph convolutional neural network, autoencoder
Citation: Li X, Huang W, Xu X, Zhang H-Y and Shi Q (2023) Deciphering tissue heterogeneity from spatially resolved transcriptomics by the autoencoder-assisted graph convolutional neural network. Front. Genet. 14:1202409. doi: 10.3389/fgene.2023.1202409
Received: 08 April 2023; Accepted: 09 May 2023;
Published: 25 May 2023.
Edited by:
Tao Huang, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Chengming Zhang, The University of Tokyo, JapanXiaoping Liu, University of Chinese Academy of Science, China
Copyright © 2023 Li, Huang, Xu, Zhang and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qianqian Shi, cXFzaGlAbWFpbC5oemF1LmVkdS5jbg==