
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 15 May 2023
Sec. Genetics of Common and Rare Diseases
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1186882
 Mei-Rong Bai1,2,3†Hao-Yue Pei2,3†Ying Zhou1Huan-Lei Song2,3
Mei-Rong Bai1,2,3†Hao-Yue Pei2,3†Ying Zhou1Huan-Lei Song2,3 Wei-Hua Pan1Yi-Ming Gong1Wen-Jie Wu1
Wei-Hua Pan1Yi-Ming Gong1Wen-Jie Wu1 Wen-Wen Yu2,3Meng-Meng Cui2,3Bei-Lin Gu2,3
Wen-Wen Yu2,3Meng-Meng Cui2,3Bei-Lin Gu2,3 Xun Chu1,2,3*
Xun Chu1,2,3* Wei Cai1,2,3*
Wei Cai1,2,3*Background: Biliary atresia (BA) is a destructive, obliterative cholangiopathy characterized by progressive fibro-inflammatory disorder and obliteration of intra- and extrahepatic bile ducts. The Jagged1 (JAG1) gene mutations have been found in some isolated BA cases. We aim to explore the association of common variants in JAG1 with isolated BA risk in the Chinese Han population.
Methods: We genotyped 31 tag single nucleotide polymorphisms covering the JAG1 gene region in 333 BA patients and 1,665 healthy controls from the Chinese population, and performed case-control association analysis. The expression patterns of JAG1 homologs were investigated in zebrafish embryos, and the roles of jag1a and jag1b in biliary development were examined by morpholino knockdown in zebrafish.
Results: Single nucleotide polymorphisms rs6077861 [PAllelic = 1.74 × 10−4, odds ratio = 1.78, 95% confidence interval: 1.31–2.40] and rs3748478 (PAllelic = 5.77 × 10−4, odds ratio = 1.39, 95% confidence interval: 1.15–1.67) located in the intron region of JAG1 showed significant associations with BA susceptibility. The JAG1 homologs, jag1a and jag1b genes were expressed in the developing hepatobiliary duct of zebrafish, especially at 72 and 96 h postfertilization. Knockdown of both jag1a and jag1b led to poor biliary secretion, sparse intrahepatic bile duct network and smaller or no gallbladders compared with control embryos in the zebrafish model.
Conclusion: Common genetic variants of JAG1 were associated with BA susceptibility. Knockdown of JAG1 homologs led to defective intrahepatic and extrahepatic bile ducts in zebrafish. These results suggest that JAG1 might be implicated in the etiology of BA.
Biliary atresia (BA) is an inflammatory cholangiopathy of infancy characterized by progressive occlusion and fibrosclerosis of intra- and extrahepatic bile ducts (Hartley et al., 2009). The incidence of BA has significant geographic variation ranging from 1:5,000–1:10,000 in Asia (Wada et al., 2007; Chiu et al., 2013) to an estimated 1:15,000 in the United States and 1:19,000 in the Netherlands (Asai et al., 2015). BA occurs seasonally, with the highest incidence rate between December and March and has a female predominance of approximately 1.25 times that of males in syndromic BA (Asai et al., 2015). The definitive diagnosis of BA is usually made by surgery, liver biopsy and surgical cholangiography including collapsed gallbladder and invisible common bile duct or common hepatic duct (Li et al., 2008). The Kasai hepatic portoenterostomy is the standard therapy for BA to restore bile flow. However, even with a successful Kasai hepatic portoenterostomy, most infants have persisting liver dysfunction and more than 70% patients eventually develop cirrhosis and still need liver transplantation in the later stage (Sokol et al., 2007). Some BA patients are accompanied by a range of abnormalities including asplenia or polysplenia, vascular abnormalities, situs inversus and cardiac anomalies. Depending on the absence or presence of accompanied abnormalities, BA is usually classified as syndromic (about 15% of cases) and isolated (approximately 85% of cases) types (Lakshminarayanan and Davenport, 2016).
The pathogenesis of isolated BA is poorly understood, which is proposed to be caused by genetic proposition and environmental factors such as a virus and toxin (Asai et al., 2015; Bezerra et al., 2018). The most widely accepted hypothesis of initial hepatobiliary injury involves potential viral infections in the early perinatal period for non-syndromic or acquired BA (Malik et al., 2020). The viral infections and immune response might impair the integrity of the primary ciliary structure and the function on the top surface of bile duct cells, which was considered an important pathogenic factor for BA (Chu et al., 2012; Malik et al., 2020; Lam et al., 2021). Recent genome-wide association studies (GWASs) identified common variants in ADD3, GPC1, and ARF6 associated with isolated BA (Garcia-Barceló et al., 2010; Leyva-Vega et al., 2010; Cheng et al., 2013; Cui et al., 2013; Ningappa et al., 2015). Mutations in PKD1L1 and MAN1A2 have been linked to syndromic BA (Berauer et al., 2019; So et al., 2020). Mutations in EFEMP1 and three ciliary genes, including PCNT, KIF3B, and TTC17 have been found in both syndromic and non-syndromic BA patients (Chen et al., 2018; Lam et al., 2021). The lack of each of these genes impeded bile flow and intrahepatic bile duct development in zebrafish, which further emphasized the importance of variants in these gene in BA pathogenesis (Malik et al., 2020). Mutations in the Jagged1 (JAG1) gene have been found in non-syndromic BA (Kohsaka et al., 2002; Cheng et al., 2017; Sangkhathat et al., 2018). Kohsaka et al. (2002) explored the JAG1 gene mutations in 102 cases of the extrahepatic biliary atresia (EHBA), and found that the most JAG1 gene mutations were all missense and sporadic and were generally detected in severely ill patients who have received liver transplantation under 5 years old. However, none of the 9 EHBA patients with the JAG1 mutations showed any of the 5 major symptoms of Alagille syndrome (ALGS), nor any pathological findings similar to ALGS after 3 years of follow-up, which suggested the JAG1 gene abnormalities may be an aggravating factor in EHBA. Interestingly, approximately 95% of ALGS patients had mutations in JAG1 (Oda et al., 1997).
ALGS is an autosomal-dominant multisystem disorder defined clinically by at least three of the following five major features: Chronic cholestasis or intrahepatic bile duct paucity, congenital cardiac disease, skeletal anomalies (typically butterfly vertebrae), ocular abnormalities and characteristic facial feature (Mitchell et al., 2018). It was reported that about 89% of the ALGS patients had cholestasis and about 75% had bile duct paucity (Mitchell et al., 2018). Given that cholestasis and bile duct abnormality are commonly seen in both ALGS and BA (Verkade et al., 2016; Feldman and Sokol, 2019), some ALGS patients display a considerable overlap with BA.
The JAG1 gene influenced duct formation during the embryonal stage (Louis et al., 1999). The Notch signaling pathway regulated by JAG1 was critical for postnatal bile duct growth and bile duct branching morphology (Loomes et al., 2007; Roos et al., 2022). Early lack of bile ducts led to cholestasis, which subsequently stimulated the proliferation of bile ducts (Tharehalli et al., 2018). Considering JAG1 mutations found in BA patients and its critical roles in the development of liver and bile duct, we investigated whether common variants of JAG1 were associated with BA risk through a case-control association study and examined the role of jag1 in biliary tract development in zebrafish model.
We recruited 333 BA patients from 2008 to 2018 who were diagnosed with BA by intraoperative cholangiography and pathology of liver tissue after ultrasonography evaluation in Xinhua hospital affiliated to Shanghai Jiao Tong University School of Medicine as described in our previous study (Bai et al., 2020). All the patients were isolated BA cases without the presence of other malformations. The clinical data of BA patients, including age at surgery, gender and liver biochemical indicators were collected, which were provided in our previous study (Bai et al., 2020). Healthy individuals who visited Xinhua hospital for routine health check were recruited as controls. A group of 1,665 unrelated healthy individuals without BA, other congenital diseases, autoimmune, or liver disease were enrolled as described previously (Bai et al., 2020). All the case and control participants in the study were biologically unrelated individuals. Written informed consent for the collection of blood samples was obtained from participants or their legal guardians. Peripheral blood samples were collected in standard EDTA tubes. Genomic DNA was extracted using QIAamp DNA Blood Mini Kit according to the manufacturer’s protocol (Qiagen, Hilden, Germany). The study was conducted in accordance with the Declaration of Helsinki (version 2002) and was approved by the Institutional Review Board of Xinhua Hospital affiliated to Shanghai Jiaotong University School of Medicine (IRB: XHEC-H-2022-001-1).
Tag single nucleotide polymorphisms (SNPs) for JAG1 were selected using the SNPinfo Web Server with minor allele frequency ≥0.01 and r2 ≥ 0.8 as the selection criteria based on the HapMap Chinese Han in Beijing data. A total of 31 tag SNPs were selected (Supplementary Table S1) and genotyped in 333 cases and 1,665 controls using Fluidigm 96.96 Dynamic Array IFCs (Fluidigm, San Francisco, CA, United States) (Supplementary Table S2). The HaploRegv4.1 database was used to predict the function of the associated SNPs (Supplementary Table S3).
All zebrafish experiments were carried out on the AB line zebrafish. All procedures were conducted in full accordance with the institutional guidelines approved by the Animal care and Use Committee of Xinhua Hospital (XHEC-WSJSW-2018-029). Total RNA was extracted from pools of 25 zebrafish embryos at different developmental time points using TRIzol reagent (Invitrogen, Cat # 9109, United States). The Primescript RT Master Kit (Takara, Catalyst # RR036A, Japan) was used to synthesize cDNA. To quantify the relative expression of jag1a and jag1b, q-PCR was performed using SYBR Green Master Mix (Applied Biosystems, Catalyst # A25742, United States) with fluorescent tags located on QuantStudio Dx Real Time PCR Instrument (Applied Biosystems, CA, United States). The primers for q-PCR were listed in Supplementary Table S4. The 18s ribosomal RNA (18-s) gene was used for standardization (McCurley and Callard, 2008). The relative expression levels of each sample with three independent triplicates were calculated using the RQ formula (RQ = 2−ΔΔCT). To demonstrate the effectiveness of the splice-blocking morpholinos (SBMOs), RT-PCR was performed using Premix Taq™ (Takara, Cat#RR902A, Japan). The primers for RT-PCR were listed in Supplementary Table S5.
We performed PCR to obtain the specific fragments of jag1a and jag1b, cloned the PCR fragments into pGEM-T Easy Vector (Promega, WI, United States), and generated digoxigenin-labeled (Roche Applied Science, Penzberg, Germany) RNA probes by in vitro transcription. PCR primers used to generate riboprobes were shown in Supplementary Table S6. WISH was performed as previously described (Lu et al., 2021). We used jag1a and jag1b antisense RNA probes at the concentration of 3 ng/μl to hybridize with zebrafish embryos.
MOs were designed to act at the translational start site or splicing acceptor site of the jag1a or jag1b and ordered from Gene Tools, LLC (Philomath, OR, United States). Standard MO was used as negative control. MOs dissolved in distilled water were microinjected into the yolk of 1–4 cell stage embryos as previously described (Cui et al., 2013; Lu et al., 2021). The sequences of MOs were listed in Supplementary Table S7. We performed dose-dependent experiments on jag1a-MO and jag1b-MO, and found the dose with the best effect was 2 ng.
The feeding of PED-6 (Invitrogen, Cat#D23739, United States), a fluorescent phosphor-lipase A2 reporter, was performed on 5 days postfertilization (5dpf) embryos at a concentration of 0.1 mg/mL for no less than 2 hours (Cui et al., 2013). We determined the metabolism of fluorescent lipids or biliary secretion by observing the size of the gallbladder and the concentration of PED-6 in the gallbladder using a stereo fluorescence microscope (Nikon SMZ25, Japan). Each group of zebrafish embryos was scored as “normal”, “faint” and “no” based on the size of the gallbladder and the concentration of PED-6 therein (Cui et al., 2013).
After fixation in methanol/dimethyl sulfoxide, whole-mount cytokeratin immunostaining was performed on 5dpf larvae as previously described (Cui et al., 2013). Larvae were incubated with primary antibody against Keratin K18 (PROGEN, Cat#61028, Germany) at a dilution of 1:100 and Alexa Fluor 488 AffiniPure goat anti-mouse IgG (H + L) (YEASEN, Cat#33206ES60, China) diluted at 1:500 was used as secondary antibody. Immuno-stained larvae were examined using a laser scanning confocal microscope (Leica TCS SP8, Germany), and images were processed using Adobe Photoshop. ImageJ software was used to quantify the gallbladder area.
Hardy-Weinberg equilibrium tests for the genotype distribution of each SNP in the case and control groups was performed and case-control association analysis was conducted using PLINK 1.09. A study-wide level p-value <0.0016 (0.05/31) was considered statistically significant. All statistical analysis was performed by GraphPad Prism 8 software unless specified. The quantitative data was presented as the mean ± standard error of the mean (SEM). When the data conformed to the homogeneity of the variance, the two tailed Student’s t-test was used, otherwise the Mann-Whitney test was used. The uptake of PED-6 in larvae was analyzed by chi-square test. All p-values were bilateral, p < 0.05 was considered statistically significant.
All 31 tag SNPs conformed to Hardy-Weinsberg equilibrium and the minor allele frequencies were all above 0.01 in 1,665 controls. The allelic and genotypic distribution between cases and controls are shown in Supplementary Tables S1, S2, respectively. After multiple testing, we found that two SNPs reached the study-level significance, and the allele and genotype distribution of these two SNPs are shown in Table 1.
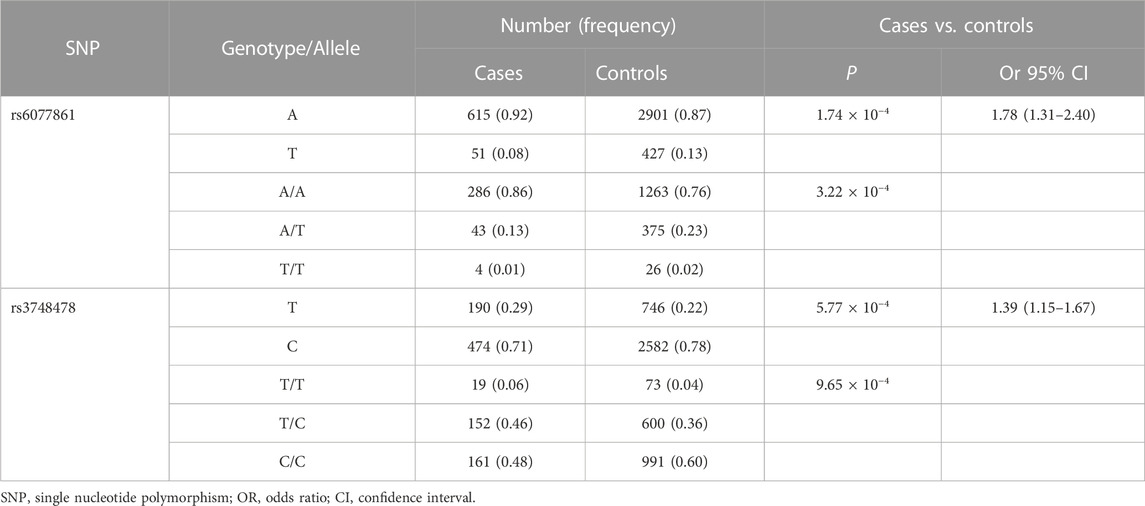
TABLE 1. Two tag SNPs showed association with sporadic BA risk in 333 patients and 1,665 controls.
Two tag SNPs located in the intron region of JAG1 showed significant association with BA susceptibility. Rs6077861 was the most associated SNP. The major allele A of rs6077861 was associated with BA risk [PAllelic = 1.74 × 10−4, odds ratio (OR) = 1.78, 95% confidence interval (95% CI): 1.31–2.40]. The frequency of rs6077861 allele A was 0.923 in cases and 0.8717 in controls (Supplementary Table S1). The second association signal was rs3748478. The minor allele T of rs3748478 was associated with BA risk (PAllelic = 5.77 × 10−4, OR = 1.39, 95% CI: 1.15–1.67). The frequency of rs3748478 allele T was 0.286 in cases and 0.224 in controls (Supplementary Table S1). The genotype distributions of the two SNPs in BA patients were also significantly different from controls (PGenotypic-rs6077861 = 3.22 × 10−4; PGenotypic-rs3748478 = 9.65 × 10−4; Supplementary Table S2). The two associated SNPs were in extreme low linkage disequilibrium (r2 < 0.01), which might represent an independent association signal.
Both rs6077861 and rs3748478 fell into strong enhancer activity regions and DNase I hypersensitivity sites and altered the sequences of regulatory motifs (Supplementary Table S3). Rs6077861 was an expression quantitative trait locus of JAG1 in the whole blood (p = 1.50 × 10−4) (Supplementary Table S3).
We first explored whether jag1a and jag1b were expressed in the developing hepatobiliary duct of zebrafish. The expression of jag1a was present at 12 h postfertilization (hpf), peaked at 24-48hpf, then decreased and maintained a stable expression level (Figure 1A). Similarly, the jag1b mRNA showed obvious expression at 12hpf, maintained high expression at 24-96hpf, and decreased expression at 5dpf (Figure 1C).
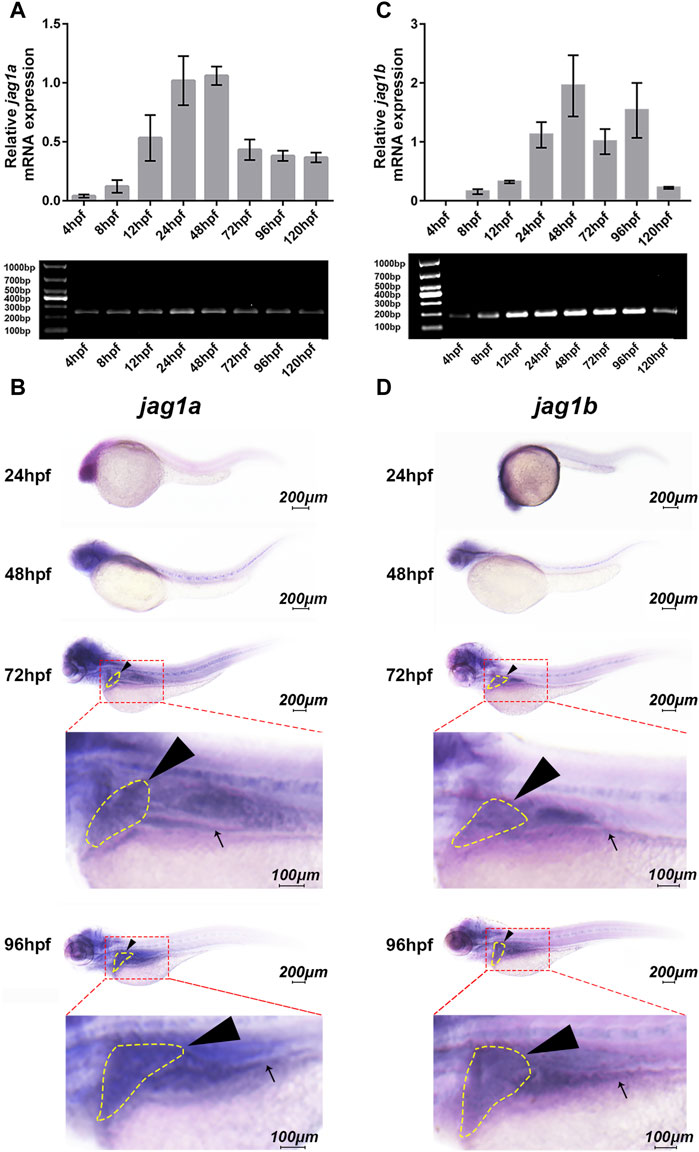
FIGURE 1. Spatiotemporal expression patterns of jag1a and jag1b in zebrafish larvae. (A, C) The relative transcript levels of jag1a and jag1b were analyzed by q-PCR during embryogenesis from 4hpf to 120hpf. (B, D) The spatial expression patterns of jag1a and jag1b were detected by WISH in 24hpf, 48hpf, 72hpf, and 96hpf zebrafish embryos. Black arrowheads and arrows mark liver and intestine respectively. Scale bar = 200 μm and 100 μm. All the data are shown as the mean ± SEM. q-PCR, quantitative polymerase chain reaction; hpf, hour postfertilization; WISH, whole-mount in situ hybridization; SEM, standard error of the mean.
To further explore the spatial expression patterns of jag1a and jag1b in the developing liver of zebrafish, WISH analysis was performed using antisense riboprobes. The results showed that jag1a was heavily expressed in the head at 24–96hpf, and moderate expression was observed in the liver and intestine, especially at 72hpf and 96hpf (Figure 1B). The expression pattern of jag1b was similar to that of jag1a, which was heavily expressed in the liver and intestine of zebrafish at 72hpf and 96hpf (Figure 1D), an important period in which bile duct growth is active, accompanied by rapid proliferation of cholangiocytes and formation of nascent ducts (Matthews et al., 2009). Therefore, we knocked down the expression of jag1a and jag1b to evaluate the influence on the biliary development.
We used two non-overlapping MOs for each gene: A translation-blocking MO (TBMO) and a SBMO. The SBMOs were complementary to the exon 6 splice acceptor site (intron5exon6, I5E6) of jag1a and the acceptor site of exon3 (intron2exon3, I2E3) of jag1b, respectively. Both TBMOs acted on the first exons (Figure 2A). To identify the effectiveness of SBMO, we performed RT-PCR and gel electrophoresis, which showed that injection of jag1a and jag1b SBMOs induced decreased expression of jag1 and alteration of transcription, as demonstrated by the appearance of abnormally spliced fragments marked by red arrows (Figures 2B, C).
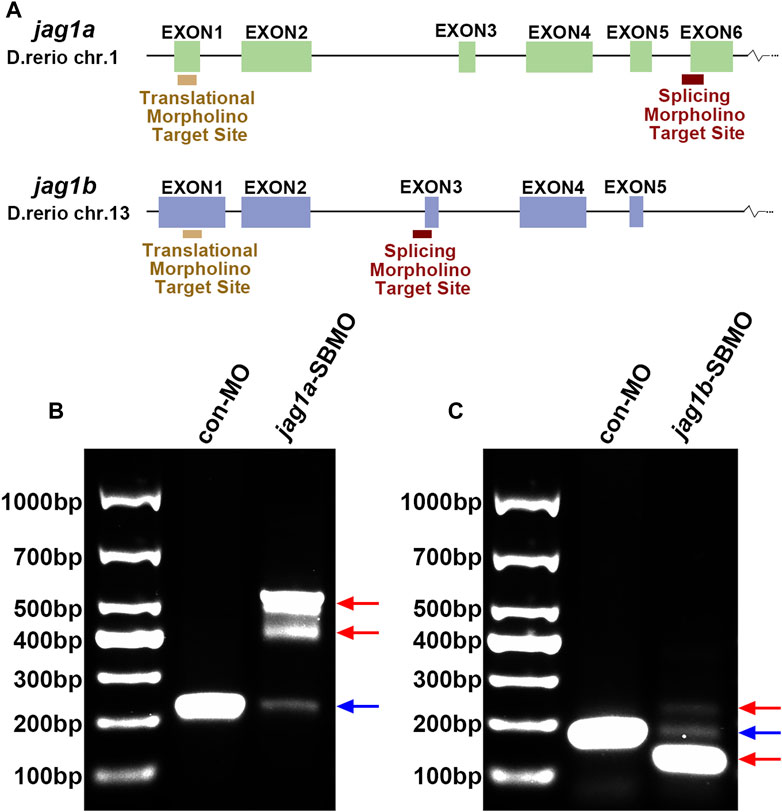
FIGURE 2. The expression of jag1a or jag1b was effectively knocked down with MO. (A) The target sites of jag1a-SBMO and jag1b-SBMO are at I5E6 and I2E3, respectively. The target sites of jag1a-TBMO and jag1b-TBMO are both located in E1. (B, C) RT-PCR confirmed the effectiveness of jag1a-SBMO and jag1b-SBMO, which demonstrated by the appearance of abnormally spliced fragments marked by red arrows. Abbreviations: SBMO, splice-blocking morpholino; TBMO, translation-blocking morpholino; E, exon; I, intron; RT-PCR, reverse transcription-polymerase chain reaction.
The 5dpf embryos injected with jag1a-MOs and jag1b-MOs both exhibited significant morphological abnormalities, including pericardial edema, spinal and tail curvature and body shortening (Figures 3B, C), compared to embryos injected with control MO (Figure 3A). The statistics of malformation rate of jag1a and jag1b morphants were higher than that of control MO group. Except for jag1a-TBMO group, there was significant difference between them and control MO group (Figure 3D).
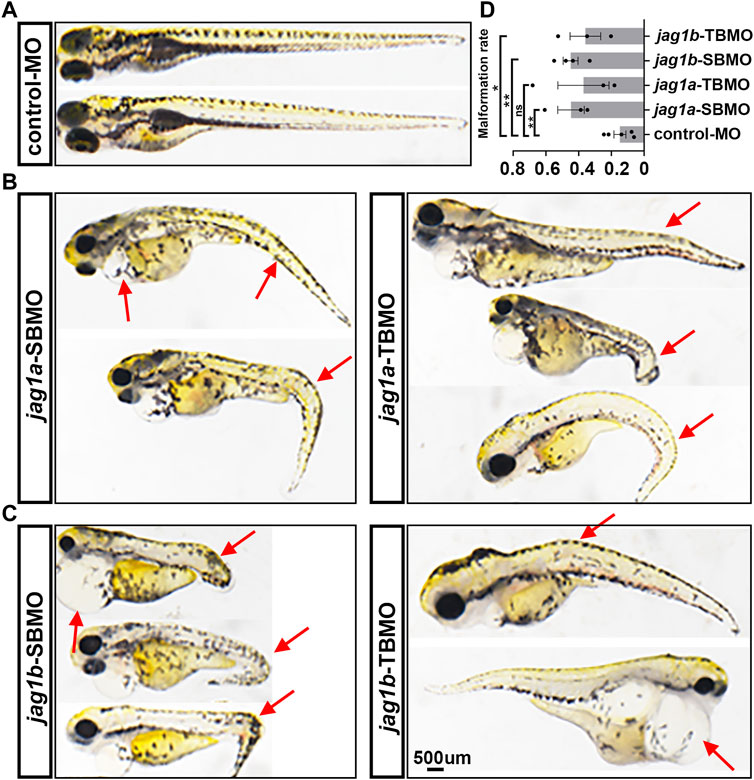
FIGURE 3. Morphological abnormality of 5dpf zebrafish injected with jag1a and jag1b MOs. (A) Presentative morphology of zebrafish injected with control MO; (B) The typical deformed zebrafish injected with jag1a MOs; (C) The typical deformed zebrafish injected with jag1b MOs; (D) The statistics of malformation rate of jag1a and jag1b morphants were higher than that of control MO group. Except for jag1a-TBMO group, there was significant difference between them and control MO group. Scale bar = 500 μm. All the data are shown as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. dpf, day postfertilization; MO, morpholino; SEM, standard error of the mean.
PED-6 is absorbed in the intestine after swallowing and enriches in the gallbladder after being processed by the liver and secreted with bile (Cui et al., 2013). The accumulation of PED-6 in the gallbladder is reduced if the intrahepatic bile ducts are abnormal (Cui et al., 2013). We conducted PED-6 swallowing to determine the effect of jag1 on bile secretion function. We found that jag1a and jag1b morphants showed poor bile drainage compared to embryos injected with control MO (Figure 4A) judged by gallbladder size and PED-6 enrichment within the gallbladder. There were statistically significant differences between the two morphants and the control (p < 0.0001, Figure 4B).
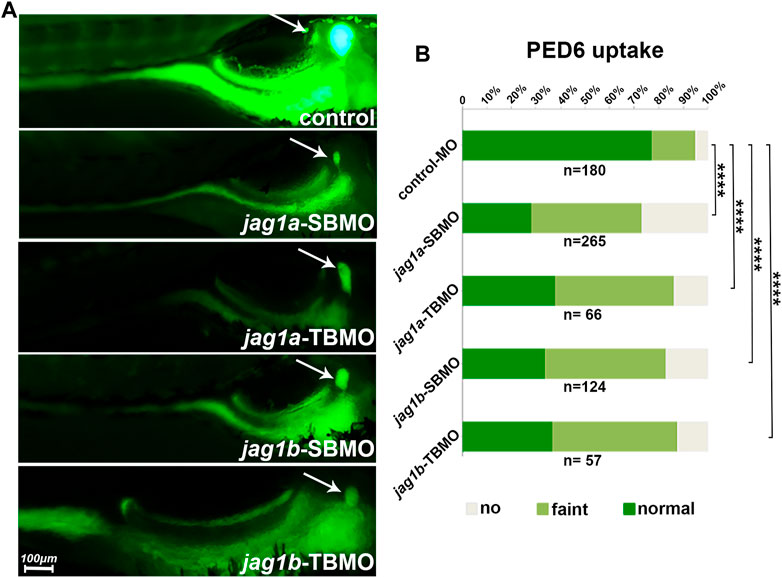
FIGURE 4. Knockdown of jag1a or jag1b led to biliary dysfunction in zebrafish. (A) The right lying view of 5dpf zebrafish larvae after swallowing PED-6 showed that the gallbladders of jag1a or jag1b morphants were weaker and smaller than those of embryos injected with control MO. (B) Quantitative analysis showed that the accumulation of PED-6 in the gallbladders of jag1a or jag1b morphants were significantly reduced (p < 0.0001, χ2 test). Scale bar = 100 μm *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Abbreviations: dpf, day post fertilization; PED-6, N-((6-(2,4-dinitrophenyl) amino) hexanoyl)-1-palmitoyl-2-BODIPY-FL-pentanoyl-sn-glycero-3-phosphoethanolamine.
We further examined the intra- and extrahepatic bile duct anatomy by cytokeratin immunostaining and found that both jag1a and jag1b morphants had sparse intrahepatic bile ducts and smaller or even no gallbladders compared to controls (Figures 5A, B). We counted the numbers of total ducts numbers, interconnecting ducts and terminal ductules, and found these ducts were significantly reduced in both jag1a and jag1b morphants compared with the embryos injected with control MO (p < 0.01, Figure 5C). In addition, we counted the number of gallbladder cells, and measured the area of gallbladder and the mean area of individual gallbladder cell. Compared to the embryos injected with control MO, both jag1a and jag1b morphants had smaller gallbladders and individual gallbladder cell (p < 0.01, Figure 5D). These results suggested that insufficiencies of jag1 led to developmental anomalies of intrahepatic and extrahepatic biliary, supporting a potential role of jag1 in BA etiology.
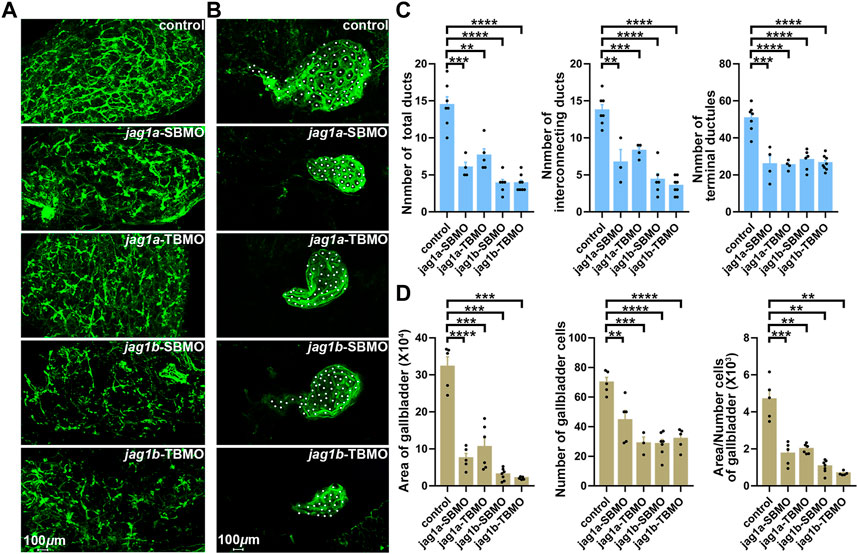
FIGURE 5. Knockdown of jag1a or jag1b led to biliary defects in zebrafish. (A) Confocal projection on 5dpf control larvae and jag1 morphants stained with cytokeratin 18 showed that the intrahepatic bile ducts of jag1a or jag1b morphants were sparse compared to controls. (B) Confocal projection on 5dpf control larvae and jag1 morphants stained with cytokeratin 18 showed that gallbladders of jag1a or jag1b morphants were smaller or even no compared to those of embryos injected with control MO. Gallbladder cells were noted by white dots. (C) Quantitative analysis of the numbers of total ducts, interconnecting ducts and terminal ductules showed that intrahepatic bile ducts were significantly reduced in both jag1a and jag1b morphants compared with the embryos injected with control MO. (D) Quantitative analysis of the number of gallbladder cells, the measured area of gallbladders and the mean area of individual gallbladder cell showed that both jag1a and jag1b morphants had smaller gallbladders and individual gallbladder cells compared to the embryos injected with control MO. Scale bar = 100 μm. All the data are shown as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. dpf, day post fertilization; SEM, standard error of the mean.
In this study, we performed an association analysis for common variation of JAG1 with BA susceptibility and found that two variants (rs6077861 and rs3748478) were associated with BA in the Chinese population. Knockdown of the expression of homologues of JAG1 in zebrafish led to biliary defects and small or even absent gallbladder. These results supported a role of JAG1 in the development of BA.
JAG1 rare mutations were repeatedly found in BA patients. Kohsaka et al. (2002) detected JAG1 missense mutations in 9 of 102 cases of EHBA, and JAG1 mutations were generally found in critically ill patients under 5 years of age who received liver transplantation. Moreover, none of the 9 EHBA patients with JAG1 mutations showed any of the five main symptoms of ALGS or any identical pathological features after 3 years of follow-up. These patients could be diagnosed as EHBA rather than atypical ALGS. Cheng et al. (2017) performed a cross-sectional observational study and discovered one de novo microdeletion of JAG1 in a BA patient diagnosed as type III BA by intraoperative cholangiography. Surasak et al. investigated rare nonsynonymous variants of 19 genes associated with infantile cholestasis using whole exome sequencing in 20 surgically and histopathologically diagnosed BA cases, and found JAG1 was the most frequent mutated gene (Sangkhathat et al., 2018). Our current study found that common SNPs in JAG1 gene region were associated with risk to sporadic BA, which reinforced the role of JAG1 in the etiology of BA development. Of note, for the number of cases was small, further independent replication was needed.
The occurrence of BA has regional and racial differences (Asai et al., 2015; Bezerra et al., 2018). The frequencies of the risk allele A for rs6077861 and the risk allele T for rs3748478 in the Asian population are higher than that of other populations, which might have a role in higher prevalence of BA in the Asian population compared to other populations. Both rs6077861 and rs3748478 are located in the enhancer activity regions of JAG1 gene. The enhancer is a key gene regulatory element that controls the spatiotemporal gene expression program specific to cell type and its function is the basis of the regulatory process of establishing gene expression patterns in cells (Ong and Corces, 2011; Schoenfelder and Fraser, 2019). It usually enhances gene transcription by combining with specific transcription factors, and enhances the expression of target genes through long-range chromosome interaction (Schoenfelder and Fraser, 2019). The sequence variation and genome rearrangement of enhancer may be the basis of disease susceptibility and developmental abnormalities, though altering the transcription level of target genes (Schoenfelder and Fraser, 2019). Therefore, it could be speculated that rs6077861 and rs3748478 may lead to a decreased expression of JAG1, underlying the susceptibility of BA.
Zebrafish emerged as a useful model in studying the pathogenesis of BA. PED-6, a fluorescent lipid reporter, is swallowed, absorbed by the intestine, processed through the liver, and excreted into bile, concentrating in the gallbladder. It is often used for examination of biliary function of zebrafish, and decreased gallbladder uptake of PED-6 would be caused by the maldevelopment of gallbladder (Cheng et al., 2013). BODIPY C5 (4,4-Difluoro-5,7-Dimethyl-4-Bora-3a,4a-Diaza-s-Indacene-3-Pentanoic Acid) is another fluorescent lipid reporter, which is often used to investigate bile canaliculi and intrahepatic bile conduits of zebrafish in addition to the gallbladder (So et al., 2018). The roles of susceptibility genes from recent GWASs in the pathogenesis of BA were evaluated in zebrafish model. In 2010, Leyva Vega et al. identified GPC1 as a BA susceptibility gene for deletions in 2q37.3 containing GPC1 in BA patients (Leyva-Vega et al., 2010). Knockdown of gpc1 in zebrafish led to developmental biliary defects and activation of the Hedgehog pathway (Cui et al., 2013). ADD3 was identified in a GWAS in the Chinese population, and knockdown of add3 in zebrafish resulted biliary trees defects (Garcia-Barceló et al., 2010; Cheng et al., 2013; Tang et al., 2016). In 2015, Ningappa et al. conducted genotyping in the American population and found a significant association between ARF6 and BA (Ningappa et al., 2015). Knockdown of arf6 in zebrafish resulted in sparse intrahepatic bile duct network, defects in bile duct epithelial cells and poor bile excretion, suggesting that ARF6 might be a susceptibility gene for BA (Ningappa et al., 2015). In 2020, a GWAS study for BA requiring liver transplantation identified the susceptibility gene MAN1A2. Knockdown of man1a2 in zebrafish impaired the development of biliary network (So et al., 2020). In addition, man1a2 morphant zebrafish displayed ciliary dysgenesis in Kupffer’s vesicle, and cardiac and liver heterotaxy, indicating a novel molecular mechanism for multiple system defects in BA (So et al., 2020).
Knockdown of jag1 in zebrafish caused the poor formation of intrahepatic bile duct network and the dysfunction of bile excretion, manifested by the lack of PED-6 accumulation in the gallbladder. In addition, jag1 knockdown also impaired the development of extrahepatic bile ducts including the reduction of area, cell numbers and unicellular area of gallbladder. The jag1 morphants mimic syndromes found in some BA patients including failure to excrete radionuclides into extrahepatic bile duct during hepatobiliary scanning, failed surgical drainage, extrahepatic bile duct occlusion and small or absent gallbladders, and lack of bile duct (Li et al., 2008). The intrahepatic bile ducts could not be examined before the onset of jaundice or before the occurrence of extrahepatic biliary tract obstruction in BA patients. Therefore, it was now unclear whether the alterations in the intrahepatic bile ducts of BA children were primary lesions or caused by the obstruction of extrahepatic bile duct. Continuous biopsies from four children diagnosed with EHBA showed that the intrahepatic bile duct deficiency without fibrosis preceded bile duct hyperplasia and cirrhosis (Azar et al., 2002). Knockdown of homologs of JAG1 and other BA risk genes (GPC1, ARF6, ADD3, and MAN1A2) (Cui et al., 2013; Ningappa et al., 2015; Tang et al., 2016; So et al., 2020) showed the paucity of intrahepatic bile ducts. These lines of evidence supported that the alteration of intrahepatic bile ducts was a primary lesion in BA infants.
JAG1 encodes the jagged 1 protein, which is the ligand for the receptor notch 1 involved in signaling processes. Notch signaling is an evolutionarily conserved intercellular signaling pathway which regulates the fate and differentiation of cells (Fortini, 2009), and the development and repair of bile ducts (Sparks et al., 2010). JAG1-mediated Notch signaling pathway is critical for postnatal bile duct growth and bile duct branching (Loomes et al., 2007; Roos et al., 2022). The target gene Hes1 of Notch signaling contributes to the maturation and cytoarchitectonic organization of biliary epithelial cells (Zhang et al., 2018). Therefore, it is reasonable that JAG1 leads to or aggravates the development of BA by affecting the primitive development of hepatobiliary ducts. JAG1 is also involved in the inflammatory processes. JAG1-mediated Notch signaling regulates IL-8 production and inflammatory injury by HSF1/Snail and NLRP3 inflammasome activation in the liver (Kohsaka et al., 2002; Jin et al., 2020). NLRP3 regulates the bile duct obstruction in experimental BA (Yang et al., 2018). The interaction between Notch and NF- κB plays an important role in immune system development and the immune system response to inflammation and viral infection (Wang et al., 2001). Jag1 is expressed in the biliary epithelium (Loomes et al., 2002). Thus, it could be speculated that JAG1 regulates inflammatory damage in the hepatic bile ducts, thereby causing or exacerbating BA. Since the primary disorder of bile duct development (Zani et al., 2015) and the continued activation of immune inflammatory processes (Bezerra et al., 2002; Wang et al., 2020) were the main features of BA, JAG1 might contribute to BA by regulating the development of bile duct and inflammation in liver and bile duct.
ALGS and BA have some overlapping clinical features. The pathologic characteristics of ALGS differed from those of BA cases with intrahepatic biliary hyperplasia and obstructed extrahepatic bile ducts (Kohsaka et al., 2002; Han et al., 2022). Germline mutations of JAG1 cause ALGS (Oda et al., 1997; Mitchell et al., 2018); however, mutations of JAG1 were also found in BA and common SNPs were associated with sporadic BA in the current result. There are several explanations for these results. First, the decreased expression of JAG1 caused by associated SNPs or BA mutations results in intrahepatic duct paucity. The paucity of bile ducts was likely to lead to cholestasis, which stimulated the proliferation of intrahepatic bile ducts (Tharehalli et al., 2018). The proliferation of intrahepatic bile ductular epithelium was increased in livers of BA patients (Kinugasa et al., 1999). The increased number of intrahepatic bile duct epithelial cells was found to be a poor prognosis factor for BA, and the density of proliferating cells was higher in patients with poor bile drainage than those of good bile drainage (Kinugasa et al., 1999). Therefore, it could be reasoned that common variation or mutations might work as an aggravation factor of BA, but as a casual factor of AGS. Second, ALGS mutations and BA mutations domains distributed in different functional domains of JAG1 (Kohsaka et al., 2002). The mutation with less harmful effects might impair the expression of JAG1 and contribute to the development of BA. However, the deleterious jag1 mutations could lead to failure of liver regeneration and prevent bile duct proliferation (Zhao et al., 2022), which causes ALGS.
In summary, our study found that common variation of JAG1 was associated with susceptibility of BA in the Chinese Han population. Knockdown of jag1 led to defective bile duct formation and dysfunction of bile ducts in zebrafish model, which indicated that JAG1 implicated in the etiology of BA. The current findings revealed new etiologic factors for BA development.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by the Institutional Review Board of Xinhua Hospital affiliated to Shanghai Jiaotong University School of Medicine. Written informed consent to participate in this study was provided by the participants and legal guardian/next of kin. The animal study was reviewed and approved by the Animal care and Use Committee of Xinhua Hospital.
WC and XC conceived the study. XC and M-RB wrote the manuscript. M-RB and XC performed the statistical analysis. M-RB, H-YP, H-LS, W-JW, W-WY, and M-MC performed the experiments. YZ, W-HP, Y-MG, W-JW, and B-LG collected the samples and clinical data. All authors critically reviewed the article and approved the final manuscript. All authors contribution to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (82170527, XC); Natural Science Foundation of Shanghai (21ZR1452600, XC); Foundation for Shanghai Key Laboratory of Pediatric Gastroenterology and Nutrition (17DZ2272000, WC).
We sincerely thank Professor Michael Pack and Dr. Kristin Lorent from University of Pennsylvania School of Medicine for the kind help and detailed experimental protocols of zebrafish experiments.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1186882/full#supplementary-material
Asai, A., Miethke, A., and Bezerra, J. A. (2015). Pathogenesis of biliary atresia: Defining biology to understand clinical phenotypes. Nat. Rev. Gastroenterol. Hepatol. 12 (6), 342–352. doi:10.1038/nrgastro.2015.74
Azar, G., Beneck, D., Lane, B., Markowitz, J., Daum, F., and Kahn, E. (2002). Atypical morphologic presentation of biliary atresia and value of serial liver biopsies. J. Pediatr. Gastroenterol. Nutr. 34 (2), 212–215. doi:10.1097/00005176-200202000-00020
Bai, M .R., Niu, W. B., Zhou, Y., Gong, Y. M., Lu, Y. J., Yu, X. X., et al. (2020). Association of common variation in ADD3 and GPC1 with biliary atresia susceptibility. Aging (Albany NY) 12 (8), 7163–7182. doi:10.18632/aging.103067
Berauer, J. P., Mezina, A. I., Okou, D. T., Sabo, A., Muzny, D. M., Gibbs, R. A., et al. (2019). Identification of polycystic kidney disease 1 like 1 gene variants in children with biliary atresia splenic malformation syndrome. Hepatology 70 (3), 899–910. doi:10.1002/hep.30515
Bezerra, J. A., Tiao, G., Ryckman, F. C., Alonso, M., Sabla, G. E., Shneider, B., et al. (2002). Genetic induction of proinflammatory immunity in children with biliary atresia. Lancet 360 (9346), 1653–1659. doi:10.1016/S0140-6736(02)11603-5
Bezerra, J. A., Wells, R. G., Mack, C. L., Karpen, S. J., Hoofnagle, J. H., Doo, E., et al. (2018). Biliary atresia: Clinical and research challenges for the twenty-first century. Hepatology 68 (3), 1163–1173. doi:10.1002/hep.29905
Chen, Y., Gilbert, M. A., Grochowski, C. M., McEldrew, D., Llewellyn, J., Waisbourd-Zinman, O., et al. (2018). A genome-wide association study identifies a susceptibility locus for biliary atresia on 2p16.1 within the gene EFEMP1. PLoS Genet. 14 (8), e1007532. doi:10.1371/journal.pgen.1007532
Cheng, G., Tang, C. S., Wong, E. H., Cheng, W. W. C., So, M. T., Miao, X., et al. (2013). Common genetic variants regulating ADD3 gene expression alter biliary atresia risk. J. Hepatol. 59 (6), 1285–1291. doi:10.1016/j.jhep.2013.07.021
Cheng, G., Chung, P. H., Chan, E. K., So, M. T., Sham, P. C., Cherny, S. S., et al. (2017). Patient complexity and genotype-phenotype correlations in biliary atresia: A cross-sectional analysis. BMC Med. Genomics 10 (1), 22. doi:10.1186/s12920-017-0259-0
Chiu, C. Y., Chen, P. H., Chan, C. F., Chang, M. H., and Wu, T. C.Taiwan Infant Stool Color Card Study Group (2013). Biliary atresia in preterm infants in taiwan: A nationwide survey. J. Pediatr. 163 (1), 100–103.e1. doi:10.1016/j.jpeds.2012.12.085
Chu, A. S., Russo, P. A., and Wells, R. G. (2012). Cholangiocyte cilia are abnormal in syndromic and non-syndromic biliary atresia. Mod. Pathol. 25 (5), 751–757. doi:10.1038/modpathol.2011.212
Cui, S., Leyva-Vega, M., Tsai, E. A., EauClaire, S. F., Glessner, J. T., Hakonarson, H., et al. (2013). Evidence from human and zebrafish that GPC1 is a biliary atresia susceptibility gene. Gastroenterology 144 (5), 1107–1115. doi:10.1053/j.gastro.2013.01.022
Feldman, A. G., and Sokol, R. J. (2019). Neonatal cholestasis: Emerging molecular diagnostics and potential novel therapeutics. Nat. Rev. Gastroenterol. Hepatol. 16 (6), 346–360. doi:10.1038/s41575-019-0132-z
Fortini, M. E. (2009). Notch signaling: The core pathway and its posttranslational regulation. Dev. Cell. 16 (5), 633–647. doi:10.1016/j.devcel.2009.03.010
Garcia-Barceló, M. M., Yeung, M. Y., Miao, X. P., Tang, C. S. M., Cheng, G., Chen, G., et al. (2010). Genome-wide association study identifies a susceptibility locus for biliary atresia on 10q24.2. Hum. Mol. Genet. 19 (14), 2917–2925. doi:10.1093/hmg/ddq196
Han, Y., Zhu, K., Wu, H., Chen, B., Hu, S., Lai, D., et al. (2022). Case Report: Novel JAG1 gene mutations in two infants with alagille syndrome characterized by cholestasis. Front. Pediatr. 10, 1017647. doi:10.3389/fped.2022.1017647
Hartley, J. L., Davenport, M., and Kelly, D. A. (2009). Biliary atresia. Lancet (London, Engl. 374 (9702), 1704–1713. doi:10.1016/S0140-6736(09)60946-6
Jin, Y., Li, C., Xu, D., Zhu, J., Wei, S., Zhong, A., et al. (2020). Jagged1-mediated myeloid Notch1 signaling activates HSF1/Snail and controls NLRP3 inflammasome activation in liver inflammatory injury. Cell. Mol. Immunol. 17 (12), 1245–1256. doi:10.1038/s41423-019-0318-x
Kinugasa, Y., Nakashima, Y., Matsuo, S., Shono, K., Suita, S., and Sueishi, K. (1999). Bile ductular proliferation as a prognostic factor in biliary atresia: An immunohistochemical assessment. J. Pediatr. Surg. 34 (11), 1715–1720. doi:10.1016/s0022-3468(99)90652-8
Kohsaka, T., Yuan, Z. R., Guo, S. X., Tagawa, M., Nakamura, A., Nakano, M., et al. (2002). The significance of human jagged 1 mutations detected in severe cases of extrahepatic biliary atresia. Hepatology 36 (4), 904–912. doi:10.1053/jhep.2002.35820
Lakshminarayanan, B., and Davenport, M. (2016). Biliary atresia: A comprehensive review. J. Autoimmun. 73, 1–9. doi:10.1016/j.jaut.2016.06.005
Lam, W. Y., Tang, C. S., So, M. T., Yue, H., Hsu, J. S., Chung, P. H. Y., et al. (2021). Identification of a wide spectrum of ciliary gene mutations in nonsyndromic biliary atresia patients implicates ciliary dysfunction as a novel disease mechanism. EBioMedicine 71, 103530. doi:10.1016/j.ebiom.2021.103530
Leyva-Vega, M., Gerfen, J., Thiel, B. D., Jurkiewicz, D., Rand, E. B., Pawlowska, J., et al. (2010). Genomic alterations in biliary atresia suggest region of potential disease susceptibility in 2q37.3. Am. J. Med. Genet. Part A 152a (4), 886–895. doi:10.1002/ajmg.a.33332
Li, S. X., Zhang, Y., Sun, M., Shi, B., Xu, Z. Y., Huang, Y., et al. (2008). Ultrasonic diagnosis of biliary atresia: A retrospective analysis of 20 patients. World J. Gastroenterol. 14 (22), 3579–3582. doi:10.3748/wjg.14.3579
Loomes, K. M., Taichman, D. B., Glover, C. L., Williams, P. T., Markowitz, J. E., Piccoli, D. A., et al. (2002). Characterization of Notch receptor expression in the developing mammalian heart and liver. Am. J. Med. Genet. 112 (2), 181–189. doi:10.1002/ajmg.10592
Loomes, K. M., Russo, P., Ryan, M., Nelson, A., Underkoffler, L., Glover, C., et al. (2007). Bile duct proliferation in liver-specific Jag1 conditional knockout mice: Effects of gene dosage. Hepatol. Baltim. Md) 45 (2), 323–330. doi:10.1002/hep.21460
Louis, A. A., Van Eyken, P., Haber, B. A., Hicks, C., Weinmaster, G., Taub, R., et al. (1999). Hepatic jagged1 expression studies. Hepatol. Baltim. Md) 30 (5), 1269–1275. doi:10.1002/hep.510300512
Lu, Y. J., Yu, W. W., Cui, M. M., Yu, X. X., Song, H. L., Bai, M. R., et al. (2021). Association analysis of variants of DSCAM and BACE2 with hirschsprung disease susceptibility in han Chinese and functional evaluation in zebrafish. Front. Cell. Dev. Biol. 9, 641152. doi:10.3389/fcell.2021.641152
Malik, A., Thanekar, U., Mourya, R., and Shivakumar, P. (2020). Recent developments in etiology and disease modeling of biliary atresia: A narrative review. Dig. Med. Res. 3, 59. doi:10.21037/dmr-20-97
Matthews, R. P., Arias, I. M., Alter, H. J., Boyer, J. L., Cohen, D. E., Fausto, N., et al. (2009). “Zebra as a model system for the study of liver development and disease,” in The liver: Biology and pathobiology. 5th ed274 (Hoboken, NJ: Wiley), 245–259.
McCurley, A. T., and Callard, G. V. (2008). Characterization of housekeeping genes in zebrafish: Male-female differences and effects of tissue type, developmental stage and chemical treatment. BMC Mol. Biol. 9, 102. doi:10.1186/1471-2199-9-102
Mitchell, E., Gilbert, M., and Loomes, K. M. (2018). Alagille syndrome. Clin. liver Dis. 22 (4), 625–641. doi:10.1016/j.cld.2018.06.001
Ningappa, M., So, J., Glessner, J., Ashokkumar, C., Ranganathan, S., Min, J., et al. (2015). The role of ARF6 in biliary atresia. PLoS One 10 (9), e0138381. doi:10.1371/journal.pone.0138381
Oda, T., Elkahloun, A. G., Pike, B. L., Okajima, K., Krantz, I. D., Genin, A., et al. (1997). Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat. Genet. 16 (3), 235–242. doi:10.1038/ng0797-235
Ong, C. T., and Corces, V. G. (2011). Enhancer function: New insights into the regulation of tissue-specific gene expression. Nat. Rev. Genet. 12 (4), 283–293. doi:10.1038/nrg2957
Roos, F. J. M., van Tienderen, G. S., Wu, H., Bordeu, I., Vinke, D., Albarinos, L. M., et al. (2022). Human branching cholangiocyte organoids recapitulate functional bile duct formation. Cell. stem Cell. 29 (5), 776–794.e13. doi:10.1016/j.stem.2022.04.011
Sangkhathat, S., Laochareonsuk, W., Maneechay, W., Kayasut, K., and Chiengkriwate, P. (2018). Variants associated with infantile cholestatic syndromes detected in extrahepatic biliary atresia by whole exome studies: A 20-case series from Thailand. J. Pediatr. Genet. 7 (2), 67–73. doi:10.1055/s-0038-1632395
Schoenfelder, S., and Fraser, P. (2019). Long-range enhancer-promoter contacts in gene expression control. Nat. Rev. Genet. 20 (8), 437–455. doi:10.1038/s41576-019-0128-0
So, J., Khaliq, M., Evason, K., Ninov, N., Martin, B. L., Stainier, D. Y. R., et al. (2018). Wnt/β-catenin signaling controls intrahepatic biliary network formation in zebrafish by regulating notch activity. Hepatology 67 (6), 2352–2366. doi:10.1002/hep.29752
So, J., Ningappa, M., Glessner, J., Min, J., Ashokkumar, C., Ranganathan, S., et al. (2020). Biliary-atresia-associated mannosidase-1-alpha-2 gene regulates biliary and ciliary morphogenesis and laterality. Front. Physiol. 11, 538701. doi:10.3389/fphys.2020.538701
Sokol, R. J., Shepherd, R. W., Superina, R., Bezerra, J. A., Robuck, P., and Hoofnagle, J. H. (2007). Screening and outcomes in biliary atresia: Summary of a national institutes of health workshop. Hepatol. Baltim. Md) 46 (2), 566–581. doi:10.1002/hep.21790
Sparks, E. E., Huppert, K. A., Brown, M. A., and Washington, M. K. (2010). Notch signaling regulates formation of the three-dimensional architecture of intrahepatic bile ducts in mice. Hepatol. Baltim. Md) 51 (4), 1391–1400. doi:10.1002/hep.23431
Tang, V., Cofer, Z. C., Cui, S., Sapp, V., Loomes, K. M., and Matthews, R. P. (2016). Loss of a candidate biliary atresia susceptibility gene, add3a, causes biliary developmental defects in zebrafish. J. Pediatr. Gastroenterol. Nutr. 63 (5), 524–530. doi:10.1097/MPG.0000000000001375
Tharehalli, U., Svinarenko, M., Kraus, J. M., Kühlwein, S. D., Szekely, R., Kiesle, U., et al. (2018). YAP activation drives liver regeneration after cholestatic damage induced by rbpj deletion. Int. J. Mol. Sci. 19 (12), 3801. doi:10.3390/ijms19123801
Verkade, H. J., Bezerra, J. A., Davenport, M., Schreiber, R. A., Mieli-Vergani, G., Hulscher, J. B., et al. (2016). Biliary atresia and other cholestatic childhood diseases: Advances and future challenges. J. Hepatol. 65 (3), 631–642. doi:10.1016/j.jhep.2016.04.032
Wada, H., Muraji, T., Yokoi, A., Okamoto, T., Sato, S., Takamizawa, S., et al. (2007). Insignificant seasonal and geographical variation in incidence of biliary atresia in Japan: A regional survey of over 20 years. J. Pediatr. Surg. 42 (12), 2090–2092. doi:10.1016/j.jpedsurg.2007.08.035
Wang, J., Shelly, L., Miele, L., Boykins, R., Norcross, M. A., and Guan, E. (2001). Human Notch-1 inhibits NF-kappa B activity in the nucleus through a direct interaction involving a novel domain. J. Immunol. 167 (1), 289–295. doi:10.4049/jimmunol.167.1.289
Wang, J., Xu, Y., Chen, Z., Liang, J., Lin, Z., Liang, H., et al. (2020). Liver immune profiling reveals pathogenesis and therapeutics for biliary atresia. Cell. 183 (7), 1867–1883.e26. doi:10.1016/j.cell.2020.10.048
Yang, L., Mizuochi, T., Shivakumar, P., Mourya, R., Luo, Z., Gutta, S., et al. (2018). Regulation of epithelial injury and bile duct obstruction by NLRP3, IL-1R1 in experimental biliary atresia. J. hepatology 69 (5), 1136–1144. doi:10.1016/j.jhep.2018.05.038
Zani, A., Quaglia, A., Hadzić, N., Zuckerman, M., and Davenport, M. (2015). Cytomegalovirus-associated biliary atresia: An aetiological and prognostic subgroup. J. Pediatr. Surg. 50 (10), 1739–1745. doi:10.1016/j.jpedsurg.2015.03.001
Zhang, R. Z., Zeng, X. H., Lin, Z. F., Fu, M., Tong, Y. L., Lui, V. C., et al. (2018). Downregulation of Hes1 expression in experimental biliary atresia and its effects on bile duct structure. World J. Gastroenterol. 24 (29), 3260–3272. doi:10.3748/wjg.v24.i29.3260
Keywords: biliary atresia, JAG1, single nucleotide polymorphisms, zebrafish, bile ducts
Citation: Bai M-R, Pei H-Y, Zhou Y, Song H-L, Pan W-H, Gong Y-M, Wu W-J, Yu W-W, Cui M-M, Gu B-L, Chu X and Cai W (2023) Association analysis and functional follow-up identified common variants of JAG1 accounting for risk to biliary atresia. Front. Genet. 14:1186882. doi: 10.3389/fgene.2023.1186882
Received: 15 March 2023; Accepted: 05 May 2023;
Published: 15 May 2023.
Edited by:
Mohiuddin Mohammed Taher, Umm al-Qura University, Saudi ArabiaReviewed by:
Vincent Chi Hang Lui, The University of Hong Kong, Hong Kong SAR, ChinaCopyright © 2023 Bai, Pei, Zhou, Song, Pan, Gong, Wu, Yu, Cui, Gu, Chu and Cai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Cai, Y2FpdzE5NzhAMTYzLmNvbQ==; Xun Chu, Y2h1eHVuQHhpbmh1YW1lZC5jb20uY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.