 Yanzi Gao
Yanzi Gao Xiaohui Jiang3
Xiaohui Jiang3 Hu Long
Hu Long Wenli Lai
Wenli Lai- 1State Key Laboratory of Oral Diseases & National Clinical Research Center for Oral Diseases, West China Hospital of Stomatology, Sichuan University, Chengdu, China
- 2Department of Orthodontics, West China Hospital of Stomatology, Sichuan University, Chengdu, China
- 3Human Sperm Bank, Key Laboratory of Birth Defects and Related Diseases of Women and Children (Sichuan University), Ministry of Education, West China Second University Hospital, Sichuan University, Chengdu, China
Non-syndromic tooth agenesis (NSTA) is one of the most common dental developmental malformations affected by genetic factors predominantly. Among all 36 candidate genes reported in NSTA individuals, EDA, EDAR, and EDARADD play essential roles in ectodermal organ development. As members of the EDA/EDAR/NF-κB signaling pathway, mutations in these genes have been implicated in the pathogenesis of NSTA, as well as hypohidrotic ectodermal dysplasia (HED), a rare genetic disorder that affects multiple ectodermal structures, including teeth. This review provides an overview of the current knowledge on the genetic basis of NSTA, with a focus on the pathogenic effects of the EDA/EDAR/NF-κB signaling pathway and the role of EDA, EDAR, and EDARADD mutations in developmental tooth defects. We also discuss the phenotypic overlap and genetic differences between NSTA and HED. Ultimately, this review highlights the importance of genetic analysis in diagnosing and managing NSTA and related ectodermal disorders, and the need for ongoing research to improve our understanding of these conditions.
1 Introduction
Tooth agenesis (TA), one of the most common dental developmental malformations in humans, negatively impacts aesthetics, mastication, and enunciation (Wong et al., 2014). TA can be divided into syndromic tooth agenesis (STA), which involves concomitant symptoms other than the congenital absence of teeth, and non-syndromic tooth agenesis (NSTA), which affects dentition solely. It has been known that NSTA is a multi-etiological developmental anomaly. Compared with exogenous factors, compelling evidence has indicated that genetic factors play a predominant role in the etiology of NSTA (Nieminen, 2009; Yin and Bian, 2015; Ye and Attaie, 2016). NSTA can be inherited in an autosomal or gonosomal pattern and occur sporadically or in families (Sarkar et al., 2014; Wang et al., 2016; Al-Ani et al., 2021; Albu et al., 2021; Asano et al., 2021; Khan et al., 2022). It has been confirmed that more than 80 genes were associated with TA (Yin and Bian, 2015), and 36 candidate genes have been identified in individuals with NSTA (Letra, 2022). Of these reported genes, EDA, EDAR, and EDARADD are candidate genes of both NSTA and STA (Ruiz-Heiland et al., 2016; Zhang et al., 2021a). Ectodysplasin-A (EDA), ectodysplasin-A receptor (EDAR), and EDAR-associated death domain (EDARADD), encoded by EDA, EDAR, and EDARADD respectively, play an important role in odontogenesis, regulating tooth number, crown shape, and enamel formation (Arte et al., 2013; He et al., 2013; Fons Romero et al., 2017). As members of the EDA/EDAR/NF-κB signaling pathway, alterations occurring in any part of the signaling can affect the transduction, contributing to NSTA potentially. The fact that mutations in EDAR result in an identical phenotype to the loss of function of EDA further supports that this pathogenic signaling is one of the causes of NSTA (Monreal et al., 1998).
In addition to TA, changes in the EDA/EDAR/NF-κB signaling pathway are also involved in other ectodermal structure development disorders, leading to ectodermal dysplasia (ED) (Chassaing et al., 2006). Hypohidrotic ectodermal dysplasia (HED) is the most highly represented ED and is proven to be affected by mutations in EDA, EDAR, and EDARADD in most cases (Cluzeau et al., 2011; Martínez-Romero et al., 2019; Ahmed et al., 2021). HED is often characterized by TA, sparse hair and eyelashes (hypotrichosis), abnormal swear glands (hypohidrosis), and dry thin skin (Tariq et al., 2007; Mues et al., 2010; Bergendal et al., 2011) (Figure 1). Sharing a common genetic etiology, it was hypothesized that the distinction between STA and NSTA is not apparent (Mues et al., 2010; Zhang et al., 2011). Epigenetic regulation may explain this phenomenon, although further research is required to confirm this theory (Zhang et al., 2021a). The phenotypic overlap between NSTA and HED has led to confusion and controversy over the classification and diagnosis of these conditions. It underscores the need for a better understanding of their genetic and clinical features.
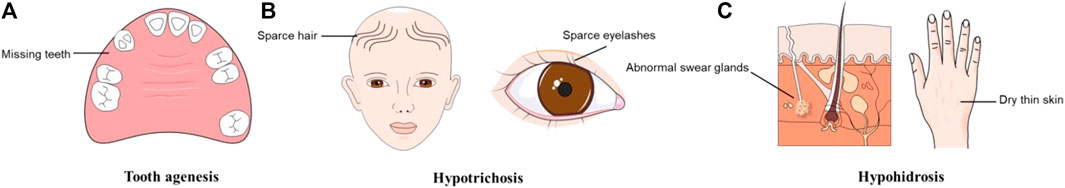
FIGURE 1. Typical symptoms of hypohidrotic ectodermal dysplasia (HED). (A)Tooth agenesis. (B) Hypotrichosis. (C) Hypohidrosis.
In this review, we will provide an overview of the current state of knowledge regarding the genetic basis of NSTA related to the EDA/EDAR/NF-κB pathway and its phenotypic overlap with HED, aiming at illustrating the importance of genetic analysis in the diagnosis of NSTA and related ectodermal disorders, and the need for ongoing research to improve our understanding of these conditions.
2 Genetic basis of the EDA/EDAR/NF-κB signaling pathway
2.1 The signaling mechanism
The EDA-encoded ligand EDA belongs to the tumor necrosis factor (TNF) ligand superfamily. During tooth development, EDA is continuously expressed in the dental epithelium from the epithelial thickening to the bud stage and then secreted into the mesenchyme, where it continues to be expressed until the end of the cap stage (Tucker et al., 2000). It has been established that EDA controls the formation of enamel knots, which operate as dental signaling centers, and facilitates communication between different epithelial compartments (Laurikkala et al., 2001; Häärä et al., 2012). EDA has 8 isoforms due to alternative splicing (Fan et al., 2008). However, only EDA-A1 (a 391-amino-acid protein) and EDA-A2 (a 389-amino-acid protein) have a receptor-binding domain among all isoforms (Kurban et al., 2010; Park et al., 2019; Liu et al., 2022). The EDA trimers in both isoforms are formed by three jelly-roll β-sandwich monomers (Hymowitz et al., 2003). EDA-A1 is a type II transmembrane protein with 4 functional domains: an N-terminal intracellular domain, a furin protease recognition sequence, a collagen-like repeat domain, and a C-terminal TNF homology domain (Ezer et al., 1999; Schneider et al., 2001; Mikkola and Thesleff, 2003; Andreoni et al., 2020). The C-terminal portion comprising the collagen and TNF homology domains cleaves off at the furin consensus site, releasing homotrimers that can bind to its particular receptor EDAR (Yan et al., 2000; Bodmer et al., 2002; Mikkola and Thesleff, 2003). EDA-A2, however, binding with its receptor X-linked ectodysplasin A (XEDAR), which is also encoded by EDAR, is not involved in TA (Newton et al., 2004; Mues et al., 2009). Although XEDAR can also recruit TRAF3 and TRAF6 to activate the NF-κB pathway (Verhelst et al., 2015), an intact EDA-A2/XEDAR signaling pathway cannot replace the blocked EDA/EDAR/NF-κB signaling pathway.
EDAR, a member of the TNF receptor superfamily, is a type I transmembrane protein containing 448 amino acids (Wang et al., 2020). EDAR is expressed in the dental epithelium at early stages and then in the enamel knot during the cap stage (Yamashiro et al., 2007). It has three cysteine-rich domains in the extracellular region (LBD), a transmembrane region, a death domain in its intracellular region, and a signal peptide (Kowalczyk-Quintas and Schneider, 2014; Zhang et al., 2021a; Yang et al., 2022). The homotrimers formed from EDA-A1 bind specifically to the LBD on the surface of dental epithelial cells and interact with each other through their death domains (Sadier et al., 2014). This interaction activates the recruitment of the EDARADD adaptor protein (Kumar et al., 2001). Subsequently, this process can recruit a member of the TNF receptor-associated factor (Traf) family into the signaling complex, which can recruit other molecules into the signaling complex (Courtney et al., 2005). The Traf family of scaffold proteins has six known members (Wajant et al., 2001), among them, Traf1, Traf2, Traf3, Traf4, and Traf6 are all expressed in the developing tooth germ, exhibiting dynamic spatiotemporal patterns (Courtney et al., 2005).
EDARADD is a 208-amino-acid protein that consists of a C-terminal death domain and an N-terminal Traf-binding consensus sequence (Kuramoto et al., 2011). The death domain participates in its self-association and the interaction with EDAR, and the Traf-binding consensus sequence combines with the TRAF6/TAK1/TAB2 complex to mediate the activation of a multimeric complex (IκB kinase (IKK)) and ultimately activates NF-κB (Cui and Schlessinger, 2006). As a scaffold molecule, the Traf is able to recruit the IKK complex, which begins the process of NF-kB activation (Courtney et al., 2005). The IKK complex is composed of two kinase subunits (IKK1/α and IKK2/β) and a structural component (NEMO/IKKγ) (Israël, 2000). Research has demonstrated that both the IKKα and β subunits exhibit catalytic activity, while NEMO/IKKγ is involved in the recruitment of IκBs (Barczewski et al., 2019). Moreover, IKKβ has been identified as the predominant kinase responsible for IκB phosphorylation (Yamada et al., 2020; Jimi and Katagiri, 2022). The phosphorylation of IκBs triggers their polyubiquitination, proteasomal degradation by the 26s proteasome, and eventually the release of NF-κB transcription factor into the nucleus (Chen and Greene, 2004). NF-κB is a homo or heterodimer that can be formed from different combinations of RelA (p65), c-Rel, RelB, p50/p105 (NF-κB1), and p52/p100 (NF-κB2) (Courtney et al., 2005; Yamada et al., 2020; Jimi and Katagiri, 2022). After the nuclear translocation of NF-κB, it induces the expression of genes essential for initiating and differentiating skin appendages such as hair, teeth, and sweat glands (Figure 2). Studies have suggested that NF-κB plays an essential role in regulating the expression of proteins related to amelogenesis and can impair the enamel formation (Liang et al., 2019; Yamada et al., 2020).
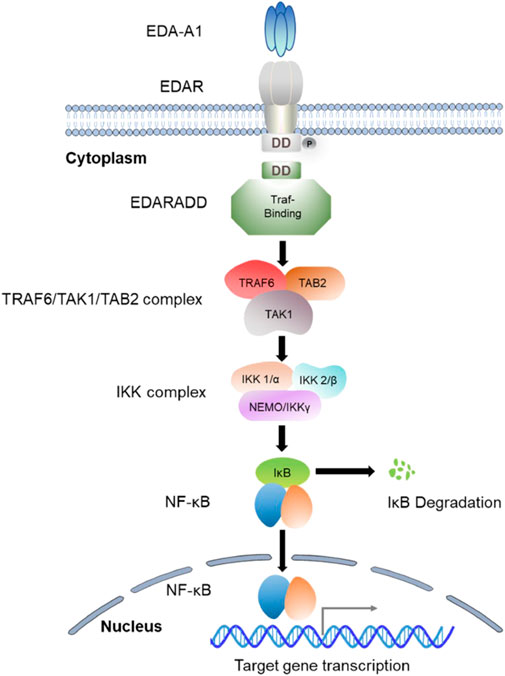
FIGURE 2. The signaling mechanism of the EDA/EDAR/NF-κB Pathway.
2.2 EDA mutations underlying NSTA
The EDA gene is located as a 425-kb segment on the long arm of the X-chromosome (Xq12-13.1) (Zonana et al., 1988). It can be inherited as XLR (OMIM 305100) or XLD (OMIM 313500). It was first discovered as a candidate gene in an NSTA individual in 2001 (Schneider et al., 2001) and was first identified in an X-linked inherited NSTA family in 2006 (Tao et al., 2006). To date, 45 mutations in EDA have been linked to NSTA (Schneider et al., 2001; Tao et al., 2006; Tarpey et al., 2007; Fan et al., 2008; Han et al., 2008; Li et al., 2008; Rasool et al., 2008; Mues et al., 2009; Song et al., 2009; Ayub et al., 2010; Mues et al., 2010; Arte et al., 2013; He et al., 2013; Nikopensius et al., 2013; Yang et al., 2013; Lee et al., 2014; Sarkar et al., 2014; Zhang et al., 2015; Gaczkowska et al., 2016; He et al., 2016; Shen et al., 2016; Bock et al., 2017; Martins et al., 2017; Yamaguchi et al., 2017; Zeng et al., 2017; Martínez-Romero et al., 2019; Parveen et al., 2019; Zhang et al., 2021a; Biedziak et al., 2022; Yu et al., 2022), with missense mutations accounting for the majority (39/45). In addition, deletion mutations (3/45), altered splicing (2/45), and non-sense mutation (1/45) were also found in NSTA cases.
Interestingly, we noted that nearly 80% of the mutations related to NSTA happened in the TNF homology domain of EDA, indicating that this region is crucial for EDA structure and functions. Specifically, the majority of the residues associated with NSTA were found close to the surface of the TNF homology domain (Yang et al., 2013). Previous studies have demonstrated that the position of amino acids in the TNF homology domain can affect the multimerization of EDA, leading to unnatural signaling (Song et al., 2009; Yang et al., 2013). The TNF homology domain has two distinct regions; one contains seven trimer-forming residues (H252, F302, Y304, Y347, C352, F379, and I383), and the other has five receptor binding residues (S275, I277, S319, 326E, and 328P). Specific amino acid changes in this region can cause the hydrogen bonds, hydrophilic or hydrophobic interaction, or electrostatic interactions between the monomers to be eliminated or damaged and the residues to be altered or reduced (Song et al., 2009; Zhang et al., 2015). According to 3D structural analysis, the stability of EDA homotrimers was partially disrupted by variations in the TNF domain, resulting in the inability of EDA to bind with EDAR (Elomaa et al., 2001; Tarpey et al., 2007; Li et al., 2008; Ayub et al., 2010; Lee et al., 2014). Another hotspot for EDA mutations is the collagen domain, which is also important for the function of EDA. It participates in the multimerization of EDA trimers, which is necessary for efficient signaling via high-valency receptor clustering (Mikkola and Thesleff, 2003). Thus, it was suggested that alterations in this region could inhibit multimerization of the TNF homology domain (Nikopensius et al., 2013). Two deletion mutations (c.612-629del; c.663-680del) were discovered in this region (Arte et al., 2013; Gaczkowska et al., 2016), and the deletions resulting in the shortening of the collagen domain ultimately affected its function. Moreover, mutations in the junction of transmembrane and extracellular domains changed the polarity of amino acids of EDA and affected its interactions with EDAR (Fan et al., 2008; Mues et al., 2009). The last mutant spot of EDA is the furin consensus site. The release of the TNF homology domain may be affected, and mutations in this region may impair proteolytic processing.
In conclusion, mutations in EDA can affect EDA/EDAR/NF-κB signaling pathway in the following ways: (Wong et al., 2014): aberrant multimerization altering the overall structure of EDA; (Ye and Attaie, 2016); Impairment of the EDA-EDAR binding activity; (Nieminen, 2009); abnormal transmembrane trafficking and proteolytic cleavage (Tao et al., 2006).
2.3 EDAR mutations implicated in the development of NSTA
The EDAR gene is a 425-kb segment located on chromosome 2q11-13. In total, 24 EDAR variants have been identified in NSTA since 2013 (Arte et al., 2013; Chen et al., 2017; Yamaguchi et al., 2017; Zeng et al., 2017; Jonsson et al., 2018; Mumtaz et al., 2020; Zhang et al., 2020; Zhang et al., 2021a; Zhang et al., 2021b; Biedziak et al., 2022; Yu et al., 2022; Zhao et al., 2022). As an integral component of the EDA/EDAR/NF-κB signaling pathway, EDAR binds to EDA through the extracellular LBD upstream and interacts with EDARADD through the intracellular death domain downstream. Consistent with this, the majority of the observed variation sites occurred in these two regions precisely, affecting either the upstream or the downstream. It was proposed that the mutant LBD can present conformational changes as a result of the substitution of amino acids, thus reducing its affinity with EDA subsequently (Zhang et al., 2020; Zhang et al., 2021a). Similarly, the mutant death domain could also cause the unstable structure of EDAR, reducing its ability to bind with EDARADD. As a result, the TRAF6/TAK1/TAB2 complex recruitment was decreased, which further affected the hydrolysis of NF-κB inhibitors and eventually reduced the activation of NF-κB (Sharp et al., 2015).
Similar to mutations in EDA, missense mutations accounted for the vast majority (21/24) of mutant types, while non-sense mutations made up a comparatively small proportion (3/24). However, no other types of mutations in EDAR have yet been described as NSTA-associated. By structural modeling analysis and molecular dynamics simulations, the structural dysfunction of missense mutant EDAR could be attributed to the following possible reasons: (Wong et al., 2014): alterations of the charge distribution of protein surface and declination of the strength and numbers of hydrogen bonds and hydrogen bonds; (Ye and Attaie, 2016); limited protein backbone motions trigger certain conformation changes of EDAR, making the protein unstable consequently; (Nieminen, 2009); loss of the attenuation of affinity with EDARADD (Zhao et al., 2022).
Regarding the non-sense mutations, it has been suggested that they can result in an early termination codon that causes the deletion of the wild-type EDAR’s residues. The first investigation into an NSTA-related non-sense EDAR mutation showed that the mutation (c.73C>T) truncated 424 amino acids of EDAR and eliminated the entire transmembrane domain and death domain (Zeng et al., 2017). The altered protein is, therefore, not anticipated to have any function. Likewise, the other two non-sense mutations (c.1302G>A; c.1072C>T), resulting in the deletion of certain residues, would prevent EDAR from binding to EDARADD (Mumtaz et al., 2020; Zhang et al., 2020).
2.4 EDARADD mutations as a contributing factor to NSTA
The EDARADD gene is a 423-kb segment located on chromosome 1q42.3-43. Despite the fact that only four NSTA-linked missense mutations in EDARADD have been reported (Bergendal et al., 2011; Arte et al., 2013; Gabrikova et al., 2016; Salvi et al., 2016; Martínez-Romero et al., 2019), the significance of EDARADD in tooth development is non-negligible. Mutant EDARADD can affect the signaling and lead to NSTA by losing its affinity for EDAR or reducing its capacity to bind to TRAF6. Compared with NSTA, however, more mutations of EDARADD were found in HED patients and usually with a more severe phenotype of the congenital tooth absence than non-syndromic ones. Nevertheless, it remains unknown why EDARADD mutations in NSTA individuals did not generate any ectodermal symptoms other than TA (Bergendal et al., 2011). It was proposed that the EDARADD inheritance pattern had an impact on the degree of EDARADD function loss (Asano et al., 2021). On the one hand, dominantly inherited EDARADD significantly diminished the ability to activate the downstream NF-κB due to the inability to bind with TRAF6. On the other hand, the recessively inherited mutant EDARADD only mildly diminished it on account of the mild reduction in the binding affinity with TRAF6 (Asano et al., 2021). However, a dominant inheritance pattern has also been found in the NSTA family with mutant EDARADD (Arte et al., 2013), thus the presence of the NSTA phenotype instead of the STA phenotype could not be explained by inheritance modes simply, and further investigations are needed.
3 Differences between HED and EDA/EDAR/NF-κB signaling pathway-related NSTA: A genetic insight
3.1 The phenotypic overlaps between HED and EDA/EDAR/NF-κB signaling pathway-related NSTA
HED is an uncommon congenital disorder characterized by aberrant ectodermal-derived structure development, which leads to abnormalities in skin appendages (Chassaing et al., 2006). As mentioned above, the EDA/EDAR/NF-κB signaling pathway is essential for ectodermal structure development. A report has revealed that four genes, including EDA1, EDAR, and EDARADD, are responsible for 90% of ED cases (Cluzeau et al., 2011). Additionally, mutations in EDAR account for 25% of non-EDA-related HED cases (Chassaing et al., 2006). There is a phenotypic overlap between HED and EDA/EDAR/NF-κB signaling pathway-related NSTA, as both conditions can present with missing teeth. Due to ongoing discoveries and advancements in genetic research, more and more scientists propose that HED and EDA-related NSTA are the same diseases with different degrees of severity (Mues et al., 2010; Zhang et al., 2011; Zeng et al., 2015; Gaczkowska et al., 2016). An attenuated phenotype has been regarded as a non-syndromic trait when the patient is affected by only one defective ectoderm-derived structure (Martínez-Romero et al., 2019; Wright et al., 2019). In other words, NSTA is presumably a variable expression of HED (Zhang et al., 2011). However, when concomitant ectodermal symptoms are too moderate to be recognized in clinical practice, patients can be misdiagnosed as NSTA (Chassaing et al., 2006; Tarpey et al., 2007; Bergendal et al., 2011; Plaisancié et al., 2013). Differentiating between HED and NSTA has implications for treatment strategies and long-term management. HED patients may require multidisciplinary care to address the various systemic manifestations, whereas NSTA patients may primarily require dental interventions. This has brought us to wonder where the boundary between HED and EDA/EDAR/NF-κB signaling pathway-related NSTA lies due to the same potential pathogenic signaling pathway. There is a growing body of evidence suggesting that the genetic underpinnings of HED and NSTA are distinct (Cluzeau et al., 2011; Arte et al., 2013). Understanding the genetic differences between HED and NSTA is crucial for genetic counseling and family planning.
3.2 Genetic differences between HED and EDA/EDAR/NF-κB signaling pathway-related NSTA
One possible explanation for mutations in EDA, EDAR or EDARADD genes resulting in the NSTA rather than the full-spectrum HED phenotype is that these genes may be expressed at higher levels during tooth development compared to other ectodermal appendages (Fournier et al., 2018). From the expression patterns, the observed distinct phenotype of hypodontia rather than the full HED phenotype is caused when mutations in EDA only partially disrupt the interaction of the EDA homotrimers and their target receptors, significantly affecting their function only in the dental tissues (Tarpey et al., 2007; Li et al., 2008; Song et al., 2009; Mues et al., 2010). Moreover, studies have shown that STA-related EDA mutations completely eliminate the capacity of mutant EDA proteins to bind to their receptors, whereas NSTA-related EDA mutations only decrease this ability (Mues et al., 2010). From the mouse rescue experiments, a small amount of recombinant EDA is sufficient for rescuing hair and salivary gland defects. In contrast, a relatively large amount of recombinant EDA is needed to save the number of teeth (Casal et al., 2007). This indicates that EDA signaling is required to develop ectodermal organs in a tissue-specific and dose-dependent manner (Mustonen et al., 2004; Casal et al., 2007). In all, mutations that entirely disrupt the EDA-EDAR interaction lead to severe HED phenotypes, whereas those causing only a partial weakening of the interaction give rise to mild non-syndromic manifestations (Yu et al., 2023).
It has been found that different mutation sites can have different impacts on the structure of proteins. From a systematic 3D conformation analysis, most of the residues associated with EDA-related NSTA are found near the surface of the TNF homology domain, while a large portion of residues associated with HED is buried in the interior of the domain (Yang et al., 2013). Although non-surface-exposed mutations were also found in NSTA (Rasool et al., 2008), it seems reasonable to postulate that mutations at sites buried in the interior could drastically alter EDA’s structure and might have more significant impact than mutations at regions close to the surface (Yang et al., 2013).
However, these hypotheses are still being investigated and may be subject to further refinement or revision as more research is conducted. The two conditions may represent a spectrum of severity rather than distinct diseases. Correct recognition of their overlapping symptoms and distinguishment from the genetic level is conducive to correct diagnosis and treatment.
4 Summary
The EDA/EDAR/NF-kB signaling pathway is involved in the development of ectodermal structures, including teeth, hair, and sweat glands. Therefore, defects in the EDA/EDAR/NF-kB pathway can disrupt this process and prevent the proper development of the teeth and other ectodermal organs. This review concludes the signaling mechanism of the pathway and provides a detailed summary for the first time of the molecular mechanisms of EDA, EDAR, and EDARADD, elucidating how gene mutations at different domains lead to protein alterations and ultimately result in the development of NSTA. Moreover, this review highlights the phenotypic overlap between EDA/EDAR/NF-κB signaling pathway-related NSTA and HED and provides novel insight to differentiate these conditions at the genetic level. By focusing on the genetic analysis of these disorders, the review emphasizes the importance of accurate diagnosis and management of NSTA and related ectodermal disorders. However, more research is needed to fully understand the mechanisms by which these mutations lead to the development of NSTA. By better understanding how this pathway works, researchers hope to develop new treatments and targeted therapies to address these conditions.
Author contributions
YG and XJ contributed to the writing of the manuscript. YG, ZW, and HL contributed to the conceptualization, design, editing of the manuscript. WL supervised the project. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahmed, H. A., El-Kamah, G. Y., Rabie, E., Mostafa, M. I., Abouzaid, M. R., Hassib, N. F., et al. (2021). Gene mutations of the three ectodysplasin pathway key players (EDA, EDAR, and EDARADD) account for more than 60% of Egyptian ectodermal dysplasia: A report of seven novel mutations. Genes 12 (9), 1389. doi:10.3390/genes12091389
Al-Ani, A. H., Antoun, J. S., Thomson, W. M., Topless, R., Merriman, T. R., and Farella, M. (2021). Common variants of EDA are associated with non-syndromic hypodontia. Orthod. craniofacial Res. 24 (1), 155–163. doi:10.1111/ocr.12419
Albu, C. C., Pavlovici, R. C., Imre, M., Ţâncu, A. M. C., Stanciu, I. A., Vasilache, A., et al. (2021). Research algorithm for the detection of genetic patterns and phenotypic variety of non-syndromic dental agenesis. Romanian J. Morphol. embryology = Revue roumaine de Morphol. embryologie 62 (1), 53–62. doi:10.47162/RJME.62.1.05
Andreoni, F., Sgattoni, C., Bencardino, D., Simonetti, O., Forabosco, A., and Magnani, M. (2020). Missense mutations in EDA and EDAR genes cause dominant syndromic tooth agenesis. Mol. Genet. genomic Med. 9, e1555. doi:10.1002/mgg3.1555
Arte, S., Parmanen, S., Pirinen, S., Alaluusua, S., and Nieminen, P. (2013). Candidate gene analysis of tooth agenesis identifies novel mutations in six genes and suggests significant role for WNT and EDA signaling and allele combinations. PloS one 8 (8), e73705. doi:10.1371/journal.pone.0073705
Asano, N., Yasuno, S., Hayashi, R., and Shimomura, Y. (2021). Characterization of EDARADD gene mutations responsible for hypohidrotic ectodermal dysplasia. J. dermatology 48 (10), 1533–1541. doi:10.1111/1346-8138.16044
Ayub, M., Ur-Rehman, F., Yasinzai, M., and Ahmad, W. (2010). A novel missense mutation in the ectodysplasin-A (EDA) gene underlies X-linked recessive nonsyndromic hypodontia. Int. J. Dermatology 49 (12), 1399–1402. doi:10.1111/j.1365-4632.2010.04596.x
Barczewski, A. H., Ragusa, M. J., Mierke, D. F., and Pellegrini, M. (2019). The IKK-binding domain of NEMO is an irregular coiled coil with a dynamic binding interface. Sci. Rep. 9 (1), 2950. doi:10.1038/s41598-019-39588-2
Bergendal, B., Klar, J., Stecksen-Blicks, C., Norderyd, J., and Dahl, N. (2011). Isolated oligodontia associated with mutations in EDARADD, AXIN2, MSX1, and PAX9 genes. Am. J. Med. Genet. Part A 155A (7), 1616–1622. doi:10.1002/ajmg.a.34045
Biedziak, B., Firlej, E., Dabrowska, J., Bogdanowicz, A., Zadurska, M., and Mostowska, A. (2022). Novel candidate genes for non-syndromic tooth agenesis identified using targeted next-generation sequencing. J. Clin. Med. 11 (20), 6089. doi:10.3390/jcm11206089
Bock, N. C., Lenz, S., Ruiz-Heiland, G., and Ruf, S. (2017). Nonsyndromic oligodontia: Does the Tooth Agenesis Code (TAC) enable prediction of the causative mutation? J. Orofac. Orthop. = Fortschritte der Kieferorthopadie Organ/official J. Deutsche Gesellschaft fur Kieferorthopadie. 78 (2), 112–120. doi:10.1007/s00056-016-0056-y
Bodmer, J. L., Schneider, P., and Tschopp, J. (2002). The molecular architecture of the TNF superfamily. Trends Biochem. Sci. 27 (1), 19–26. doi:10.1016/s0968-0004(01)01995-8
Casal, M. L., Lewis, J. R., Mauldin, E. A., Tardivel, A., Ingold, K., Favre, M., et al. (2007). Significant correction of disease after postnatal administration of recombinant ectodysplasin A in canine X-linked ectodermal dysplasia. Am. J. Hum. Genet. 81 (5), 1050–1056. doi:10.1086/521988
Chassaing, N., Bourthoumieu, S., Cossee, M., Calvas, P., and Vincent, M. C. (2006). Mutations in EDAR account for one-quarter of non-ED1-related hypohidrotic ectodermal dysplasia. Hum. Mutat. 27 (3), 255–259. doi:10.1002/humu.20295
Chen, L. F., and Greene, W. C. (2004). Shaping the nuclear action of NF-kappaB. Nat. Rev. Mol. Cell Biol. 5 (5), 392–401. doi:10.1038/nrm1368
Chen, Y. T., Liu, H. C., Han, D., Liu, Y., and Feng, H. L. (2017). Association between EDAR polymorphisms and non-syndromic tooth agenesis in the Chinese han population. Chin. J. Dent. Res. official J. Sci. Sect. Chin. Stomatological Assoc. (CSA) 20 (3), 153–159. doi:10.3290/j.cjdr.a38770
Cluzeau, C., Hadj-Rabia, S., Jambou, M., Mansour, S., Guigue, P., Masmoudi, S., et al. (2011). Only four genes (EDA1, EDAR, EDARADD, and WNT10A) account for 90% of hypohidrotic/anhidrotic ectodermal dysplasia cases. Hum. Mutat. 32 (1), 70–72. doi:10.1002/humu.21384
Courtney, J. M., Blackburn, J., and Sharpe, P. T. (2005). The Ectodysplasin and NFkappaB signalling pathways in odontogenesis. Archives oral Biol. 50 (2), 159–163. doi:10.1016/j.archoralbio.2004.11.019
Cui, C. Y., and Schlessinger, D. (2006). EDA signaling and skin appendage development. Cell cycleGeorget. Tex) 5 (21), 2477–2483. doi:10.4161/cc.5.21.3403
Elomaa, O., Pulkkinen, K., Hannelius, U., Mikkola, M., Saarialho-Kere, U., and Kere, J. (2001). Ectodysplasin is released by proteolytic shedding and binds to the EDAR protein. Hum. Mol. Genet. 10 (9), 953–962. doi:10.1093/hmg/10.9.953
Ezer, S., Bayés, M., Elomaa, O., Schlessinger, D., and Kere, J. (1999). Ectodysplasin is a collagenous trimeric type II membrane protein with a tumor necrosis factor-like domain and co-localizes with cytoskeletal structures at lateral and apical surfaces of cells. Hum. Mol. Genet. 8 (11), 2079–2086. doi:10.1093/hmg/8.11.2079
Fan, H., Ye, X., Shi, L., Yin, W., Hua, B., Song, G., et al. (2008). Mutations in the EDA gene are responsible for X-linked hypohidrotic ectodermal dysplasia and hypodontia in Chinese kindreds. Eur. J. oral Sci. 116 (5), 412–417. doi:10.1111/j.1600-0722.2008.00555.x
Fons Romero, J. M., Star, H., Lav, R., Watkins, S., Harrison, M., Hovorakova, M., et al. (2017). The impact of the eda pathway on tooth root development. J. Dent. Res. 96 (11), 1290–1297. doi:10.1177/0022034517725692
Fournier, B. P., Bruneau, M. H., Toupenay, S., Kerner, S., Berdal, A., Cormier-Daire, V., et al. (2018). Patterns of dental agenesis highlight the nature of the causative mutated genes. J. Dent. Res. 97 (12), 1306–1316. doi:10.1177/0022034518777460
Gabrikova, D., Grejtakova, D., Bernasovska, J., Pavukova, A., Lewandowska, B., Bindasova, M., et al. (2016). Sequence analysis of candidate genes in two roma families with severe tooth agenesis. Belgr. 48 (3), 945–954. doi:10.2298/gensr1603945g
Gaczkowska, A., Abdalla, E. M., Dowidar, K. M. L., Elhady, G. M., Jagodzinski, P. P., and Mostowsk, A. (2016). De novo EDA mutations: Variable expression in two Egyptian families. Archives oral Biol. 68, 21–28. doi:10.1016/j.archoralbio.2016.03.015
Häärä, O., Harjunmaa, E., Lindfors, P. H., Huh, S. H., Fliniaux, I., Åberg, T., et al. (2012). Ectodysplasin regulates activator-inhibitor balance in murine tooth development through Fgf20 signaling. Dev. Camb. Engl. 139 (17), 3189–3199. doi:10.1242/dev.079558
Han, D., Gong, Y., Wu, H., Zhang, X., Yan, M., Wang, X., et al. (2008). Novel EDA mutation resulting in X-linked non-syndromic hypodontia and the pattern of EDA-associated isolated tooth agenesis. Eur. J. Med. Genet. 51 (6), 536–546. doi:10.1016/j.ejmg.2008.06.002
He, H., Han, D., Feng, H., Qu, H., Song, S., Bai, B., et al. (2013). Involvement of and interaction between WNT10A and EDA mutations in tooth agenesis cases in the Chinese population. PloS one 8 (11), e80393. doi:10.1371/journal.pone.0080393
He, H., Liu, Y., Han, D., Liu, H., Bai, B., and Feng, H. (2016). EDA mutation screening and phenotype analysis in patients with tooth agenesis. J. Peking Univ. Health Sci. 48 (4), 686–691.
Hymowitz, S. G., Compaan, D. M., Yan, M., Wallweber, H. J., Dixit, V. M., Starovasnik, M. A., et al. The crystal structures of EDA-A1 and EDA-A2: Splice variants with distinct receptor specificity. Struct. Lond. Engl. 1993). 2003;11(12):1513–1520. doi:10.1016/j.str.2003.11.009
Israël, A. (2000). The IKK complex: An integrator of all signals that activate NF-kappaB? Trends Cell Biol. 10 (4), 129–133. doi:10.1016/s0962-8924(00)01729-3
Jimi, E., and Katagiri, T. (2022). Critical roles of NF-κB signaling molecules in bone metabolism revealed by genetic mutations in osteopetrosis. Int. J. Mol. Sci. 23 (14), 7995. doi:10.3390/ijms23147995
Jonsson, L., Magnusson, T. E., Thordarson, A., Jonsson, T., Geller, F., Feenstra, B., et al. (2018). Rare and common variants conferring risk of tooth agenesis. J. Dent. Res. 97 (5), 515–522. doi:10.1177/0022034517750109
Khan, M. I., Ahmed, N., Neela, P. K., and Unnisa, N. (2022). The human genetics of dental anomalies. Glob. Med. Genet. 09 (02), 76–81. doi:10.1055/s-0042-1743572
Kowalczyk-Quintas, C., and Schneider, P. (2014). Ectodysplasin A (EDA) - EDA receptor signalling and its pharmacological modulation. Cytokine & growth factor Rev. 25 (2), 195–203. doi:10.1016/j.cytogfr.2014.01.004
Kumar, A., Eby, M. T., Sinha, S., Jasmin, A., and Chaudhary, P. M. (2001). The ectodermal dysplasia receptor activates the nuclear factor-kappaB, JNK, and cell death pathways and binds to ectodysplasin A. J. Biol. Chem. 276 (4), 2668–2677. doi:10.1074/jbc.M008356200
Kuramoto, T., Yokoe, M., Hashimoto, R., Hiai, H., and Serikawa, T. (2011). A rat model of hypohidrotic ectodermal dysplasia carries a missense mutation in the Edaradd gene. BMC Genet. 12, 91. doi:10.1186/1471-2156-12-91
Kurban, M., Michailidis, E., Wajid, M., Shimomura, Y., and Christiano, A. M. (2010). A common founder mutation in the EDA-A1 gene in X-linked hypodontia. Dermatol. (Basel, Switz. 221 (3), 243–247. doi:10.1159/000314329
Laurikkala, J., Mikkola, M., Mustonen, T., Aberg, T., Koppinen, P., Pispa, J., et al. (2001). TNF signaling via the ligand-receptor pair ectodysplasin and edar controls the function of epithelial signaling centers and is regulated by Wnt and activin during tooth organogenesis. Dev. Biol. 229 (2), 443–455. doi:10.1006/dbio.2000.9955
Lee, K. E., Ko, J., Shin, T. J., Hyun, H. K., Lee, S. H., and Kim, J. W. (2014). Oligodontia and curly hair occur with ectodysplasin-a mutations. J. Dent. Res. 93 (4), 371–375. doi:10.1177/0022034514522059
Letra, A. (2022). Rethinking the genetic etiology of nonsyndromic tooth agenesis. Curr. Osteoporos. Rep. 20, 389–397. doi:10.1007/s11914-022-00761-8
Li, S., Li, J., Cheng, J., Zhou, B., Tong, X., Dong, X., et al. (2008). Non-syndromic tooth agenesis in two Chinese families associated with novel missense mutations in the TNF domain of EDA (ectodysplasin A). PloS one 3 (6), e2396. doi:10.1371/journal.pone.0002396
Liang, Y., Chen, G., Yang, Y., Li, Z., Chen, T., Sun, W., et al. (2019). Effect of canonical NF-κB signaling pathway on the differentiation of rat dental epithelial stem cells. Stem Cell Res. Ther. 10 (1), 139. doi:10.1186/s13287-019-1252-7
Liu, X., Zhao, Y., and Zhu, J. (2022). A novel mutation in the collagen domain of EDA results in hypohidrotic ectodermal dysplasia by impacting the receptor-binding capability. Mol. Genet. genomic Med., e2119. doi:10.1002/mgg3.2119
Martínez-Romero, M. C., Ballesta-Martínez, M. J., López-González, V., Sánchez-Soler, M. J., Serrano-Antón, A. T., Barreda-Sánchez, M., et al. (2019). EDA, EDAR, EDARADD and WNT10A allelic variants in patients with ectodermal derivative impairment in the Spanish population. Orphanet J. rare Dis. 14 (1), 281. doi:10.1186/s13023-019-1251-x
Martins, L., Machado, R. A., Araujo, D. S., Giovani, P. A., Rebouças, P. D., Rodrigues, L. P., et al. (2017). EDA mutation by exome sequencing in non-syndromic X-linked oligodontia. Clin. Genet. 92 (2), 227–229. doi:10.1111/cge.12961
Mikkola, M. L., and Thesleff, I. (2003). Ectodysplasin signaling in development. Cytokine & growth factor Rev. 14 (3-4), 211–224. doi:10.1016/s1359-6101(03)00020-0
Monreal, A. W., Zonana, J., and Ferguson, B. (1998). Identification of a new splice form of the EDA1 gene permits detection of nearly all X-linked hypohidrotic ectodermal dysplasia mutations. Am. J. Hum. Genet. 63 (2), 380–389. doi:10.1086/301984
Mues, G., Tardivel, A., Willen, L., Kapadia, H., Seaman, R., Frazier-Bowers, S., et al. (2010). Functional analysis of Ectodysplasin-A mutations causing selective tooth agenesis. Eur. J. Hum. Genet. EJHG 18 (1), 19–25. doi:10.1038/ejhg.2009.127
Mues, G. I., Griggs, R., Hartung, A. J., Whelan, G., Best, L. G., Srivastava, A. K., et al. (2009). From ectodermal dysplasia to selective tooth agenesis. Am. J. Med. Genet. Part A 149A (9), 2037–2041. doi:10.1002/ajmg.a.32801
Mumtaz, S., Nalbant, G., Yıldız Bölükbaşı, E., Huma, Z., Ahmad, N., Tolun, A., et al. (2020). Novel EDAR mutation in tooth agenesis and variable associated features. Eur. J. Med. Genet. 63 (9), 103926. doi:10.1016/j.ejmg.2020.103926
Mustonen, T., Ilmonen, M., Pummila, M., Kangas, A. T., Laurikkala, J., Jaatinen, R., et al. (2004). Ectodysplasin A1 promotes placodal cell fate during early morphogenesis of ectodermal appendages. Dev. Camb. Engl. 131 (20), 4907–4919. doi:10.1242/dev.01377
Newton, K., French, D. M., Yan, M., Frantz, G. D., and Dixit, V. M. (2004). Myodegeneration in EDA-A2 transgenic mice is prevented by XEDAR deficiency. Mol. Cell. Biol. 24 (4), 1608–1613. doi:10.1128/MCB.24.4.1608-1613.2004
Nieminen, P. (2009). Genetic basis of tooth agenesis. J. Exp. Zoology Part B-Molecular Dev. Evol. 312B (4), 320–342. doi:10.1002/jez.b.21277
Nikopensius, T., Annilo, T., Jagomägi, T., Gilissen, C., Kals, M., Krjutškov, K., et al. (2013). Non-syndromic tooth agenesis associated with a nonsense mutation in ectodysplasin-A (EDA). J. Dent. Res. 92 (6), 507–511. doi:10.1177/0022034513487210
Park, J. S., Ko, J. M., and Chae, J. H. (2019). Novel and private EDA mutations and clinical phenotypes of Korean patients with X-linked hypohidrotic ectodermal dysplasia. Cytogenet. genome Res. 158 (1), 1–9. doi:10.1159/000500214
Parveen, A., Khan, S. A., Mirza, M. U., Bashir, H., Arshad, F., Iqbal, M., et al. (2019). Deleterious variants in WNT10A, EDAR, and EDA causing isolated and syndromic tooth agenesis: A structural perspective from molecular dynamics simulations. Int. J. Mol. Sci. 20 (21), 5282. doi:10.3390/ijms20215282
Plaisancié, J., Bailleul-Forestier, I., Gaston, V., Vaysse, F., Lacombe, D., Holder-Espinasse, M., et al. (2013). Mutations in WNT10A are frequently involved in oligodontia associated with minor signs of ectodermal dysplasia. Am. J. Med. Genet. Part A 161a (4), 671–678. doi:10.1002/ajmg.a.35747
Rasool, M., Schuster, J., Aslam, M., Tariq, M., Ahmad, I., Ali, A., et al. (2008). A novel missense mutation in the EDA gene associated with X-linked recessive isolated hypodontia. J. Hum. Genet. 53 (10), 894–898. doi:10.1007/s10038-008-0323-x
Ruiz-Heiland, G., Jabir, S., Wende, W., Blecher, S., Bock, N., and Ruf, S. (2016). Novel missense mutation in the EDA gene in a family affected by oligodontia. J. Orofac. Orthopedics-Fortschritte Der Kieferorthopadie 77 (1), 31–38. doi:10.1007/s00056-015-0005-1
Sadier, A., Viriot, L., Pantalacci, S., and Laudet, V. (2014). The ectodysplasin pathway: From diseases to adaptations. Trends Genet. TIG 30 (1), 24–31. doi:10.1016/j.tig.2013.08.006
Salvi, A., Giacopuzzi, E., Bardellini, E., Amadori, F., Ferrari, L., De Petro, G., et al. (2016). Mutation analysis by direct and whole exome sequencing in familial and sporadic tooth agenesis. Int. J. Mol. Med. 38 (5), 1338–1348. doi:10.3892/ijmm.2016.2742
Sarkar, T., Bansal, R., and Das, P. (2014). Whole genome sequencing reveals novel non-synonymous mutation in ectodysplasin A (EDA) associated with non-syndromic X-linked dominant congenital tooth agenesis. PloS one 9 (9), e106811. doi:10.1371/journal.pone.0106811
Schneider, P., Street, S. L., Gaide, O., Hertig, S., Tardivel, A., Tschopp, J., et al. (2001). Mutations leading to X-linked hypohidrotic ectodermal dysplasia affect three major functional domains in the tumor necrosis factor family member ectodysplasin-A. J. Biol. Chem. 276 (22), 18819–18827. doi:10.1074/jbc.M101280200
Sharp, K. A., O'Brien, E., Kasinath, V., and Wand, A. J. (2015). On the relationship between NMR-derived amide order parameters and protein backbone entropy changes. Proteins 83 (5), 922–930. doi:10.1002/prot.24789
Shen, W., Wang, Y., Liu, Y., Liu, H., Zhao, H., Zhang, G., et al. (2016). Functional study of ectodysplasin-A mutations causing non-syndromic tooth agenesis. PloS one 11 (5), e0154884. doi:10.1371/journal.pone.0154884
Song, S., Han, D., Qu, H., Gong, Y., Wu, H., Zhang, X., et al. (2009). EDA gene mutations underlie non-syndromic oligodontia. J. Dent. Res. 88 (2), 126–131. doi:10.1177/0022034508328627
Tao, R., Jin, B., Guo, S. Z., Qing, W., Feng, G. Y., Brooks, D. G., et al. (2006). A novel missense mutation of the EDA gene in a Mongolian family with congenital hypodontia. J. Hum. Genet. 51 (5), 498–502. doi:10.1007/s10038-006-0389-2
Tariq, M., Wasif, N., Ayub, M., and Ahmad, W. (2007). A novel 4-bp insertion mutation in EDA1 gene in a Pakistani family with X-linked hypohidrotic ectodermal dysplasia. Eur. J. dermatology EJD 17 (3), 209–212. doi:10.1684/ejd.2007.0150
Tarpey, P., Pemberton, T. J., Stockton, D. W., Das, P., Ninis, V., Edkins, S., et al. (2007). A novel Gln358Glu mutation in ectodysplasin A associated with X-linked dominant incisor hypodontia. Am. J. Med. Genet. Part A 143 (4), 390–394. doi:10.1002/ajmg.a.31567
Tucker, A. S., Headon, D. J., Schneider, P., Ferguson, B. M., Overbeek, P., Tschopp, J., et al. (2000). Edar/Eda interactions regulate enamel knot formation in tooth morphogenesis. Dev. Camb. Engl. 127 (21), 4691–4700. doi:10.1242/dev.127.21.4691
Verhelst, K., Gardam, S., Borghi, A., Kreike, M., Carpentier, I., and Beyaert, R. (2015). XEDAR activates the non-canonical NF-κB pathway. Biochem. biophysical Res. Commun. 465 (2), 275–280. doi:10.1016/j.bbrc.2015.08.019
Wajant, H., Henkler, F., and Scheurich, P. (2001). The TNF-receptor-associated factor family: Scaffold molecules for cytokine receptors, kinases and their regulators. Cell. Signal. 13 (6), 389–400. doi:10.1016/s0898-6568(01)00160-7
Wang, B., Liang, Y., Chai, X., Chen, S., Ye, Z., Li, R., et al. (2020). Ectodysplasin A receptor (EDAR) promotes colorectal cancer cell proliferation via regulation of the Wnt/β-catenin signaling pathway. Exp. Cell Res. 395 (1), 112170. doi:10.1016/j.yexcr.2020.112170
Wang, J., Sun, K., Shen, Y., Xu, Y., Xie, J., Huang, R., et al. (2016). DNA methylation is critical for tooth agenesis: Implications for sporadic non-syndromic anodontia and hypodontia. Sci. Rep. 6, 19162. doi:10.1038/srep19162
Wong, S. W., Liu, H. C., Han, D., Chang, H. G., Zhao, H. S., Wang, Y. X., et al. (2014). A novel non-stop mutation in MSX1 causing autosomal dominant non-syndromic oligodontia. Mutagenesis 29 (5), 319–323. doi:10.1093/mutage/geu019
Wright, J. T., Fete, M., Schneider, H., Zinser, M., Koster, M. I., Clarke, A. J., et al. (2019). Ectodermal dysplasias: Classification and organization by phenotype, genotype and molecular pathway. Am. J. Med. Genet. Part A 179 (3), 442–447. doi:10.1002/ajmg.a.61045
Yamada, A., Kawasaki, M., Miake, Y., Yamada, Y., Blackburn, J., Kawasaki, K., et al. (2020). Overactivation of the NF-κB pathway impairs molar enamel formation. Oral Dis. 26 (7), 1513–1522. doi:10.1111/odi.13384
Yamaguchi, T., Hosomichi, K., Yano, K., Kim, Y-I., Nakaoka, H., Kimura, R., et al. (2017). Comprehensive genetic exploration of selective tooth agenesis of mandibular incisors by exome sequencing. Hum. Genome Var. 4, 17005. doi:10.1038/hgv.2017.5
Yamashiro, T., Zheng, L., Shitaku, Y., Saito, M., Tsubakimoto, T., Takada, K., et al. (2007). Wnt10a regulates dentin sialophosphoprotein mRNA expression and possibly links odontoblast differentiation and tooth morphogenesis. Differentiation 75 (5), 452–462. doi:10.1111/j.1432-0436.2006.00150.x
Yan, M., Wang, L. C., Hymowitz, S. G., Schilbach, S., Lee, J., Goddard, A., et al. (2000). Two-amino acid molecular switch in an epithelial morphogen that regulates binding to two distinct receptors. Sci. (New York, NY) 290 (5491), 523–527. doi:10.1126/science.290.5491.523
Yang, R., Mei, Y., Jiang, Y., Li, H., Zhao, R., Sima, J., et al. (2022). Ectodysplasin A (EDA) signaling: From skin appendage to multiple diseases. Int. J. Mol. Sci. 23 (16), 8911. doi:10.3390/ijms23168911
Yang, Y., Luo, L., Xu, J., Zhu, P., Xue, W., Wang, J., et al. (2013). Novel EDA p.Ile260Ser mutation linked to non-syndromic hypodontia. J. Dent. Res. 92 (6), 500–506. doi:10.1177/0022034513487557
Ye, X., and Attaie, A. B. (2016). Genetic basis of nonsyndromic and syndromic tooth agenesis. J. Pediatr. Genet. 5 (4), 198–208. doi:10.1055/s-0036-1592421
Yin, W., and Bian, Z. (2015). The gene network underlying hypodontia. J. Dent. Res. 94 (7), 878–885. doi:10.1177/0022034515583999
Yu, K., Dou, J., Huang, W., Wang, F., and Wu, Y. (2022). Expanding the genetic spectrum of tooth agenesis using whole-exome sequencing. Clin. Genet. 102, 503–516. doi:10.1111/cge.14225
Yu, K., Huang, C., Wan, F., Jiang, C., Chen, J., Li, X., et al. (2023). Structural insights into pathogenic mechanism of hypohidrotic ectodermal dysplasia caused by ectodysplasin A variants. Nat. Commun. 14 (1), 767. doi:10.1038/s41467-023-36367-6
Zeng, B., Lu, H., Xiao, X., Zhou, L., Lu, J., Zhu, L., et al. (2015). Novel EDA mutation in X-linked hypohidrotic ectodermal dysplasia and genotype-phenotype correlation. Oral Dis. 21 (8), 994–1000. doi:10.1111/odi.12376
Zeng, B., Zhao, Q., Li, S., Lu, H., Lu, J., Ma, L., et al. (2017). Novel EDA or EDAR mutations identified in patients with X-linked hypohidrotic ectodermal dysplasia or non-syndromic tooth agenesis. Genes 8 (10), 259. doi:10.3390/genes8100259
Zhang, H., Kong, X., Ren, J., Yuan, S., Liu, C., Hou, Y., et al. (2021). A novel EDAR missense mutation identified by whole-exome sequencing with non-syndromic tooth agenesis in a Chinese family. Mol. Genet. genomic Med. 9 (6), e1684. doi:10.1002/mgg3.1684
Zhang, H., Yang, L., Meng, L., Zheng, S., Ma, W., Yang, D., et al. (2021). A novel EDAR mutation identified in nonsyndromic oligodontia patients by whole exome sequencing. J. Pract. Stomatology 37 (6), 783–789.
Zhang, J., Han, D., Song, S., Wang, Y., Zhao, H., Pan, S., et al. (2011). Correlation between the phenotypes and genotypes of X-linked hypohidrotic ectodermal dysplasia and non-syndromic hypodontia caused by ectodysplasin-A mutations. Eur. J. Med. Genet. 54 (4), e377–e382. doi:10.1016/j.ejmg.2011.03.005
Zhang, L., Yu, M., Wong, S. W., Qu, H., Cai, T., Liu, Y., et al. (2020). Comparative analysis of rare EDAR mutations and tooth agenesis pattern in EDAR- and EDA-associated nonsyndromic oligodontia. Hum. Mutat. 41 (11), 1957–1966. doi:10.1002/humu.24104
Zhang, X. X., Wong, S. W., Han, D., and Feng, H. L. (2015). Simultaneous occurence of an autosomal dominant inherited MSX1 mutation and an X-linked recessive inherited EDA mutation in one Chinese family with non-syndromic oligodontia. Chin. J. Dent. Res. official J. Sci. Sect. Chin. Stomatological Assoc. (CSA) 18 (4), 229–234. doi:10.3290/j.cjdr.a35147
Zhao, Z., Zhang, T., Li, T., Ye, Y., Feng, C., Wang, H., et al. (2022). A novel EDAR variant identified in non-syndromic tooth agenesis: Insights from molecular dynamics. Archives oral Biol. 146, 105600. doi:10.1016/j.archoralbio.2022.105600
Keywords: non-syndromic tooth agenesis (NSTA), hypohidrotic ectodermal dysplasia (HED), genetic basis, signaling pathway, EDA, EDAR, EDARADD
Citation: Gao Y, Jiang X, Wei Z, Long H and Lai W (2023) The EDA/EDAR/NF-κB pathway in non-syndromic tooth agenesis: A genetic perspective. Front. Genet. 14:1168538. doi: 10.3389/fgene.2023.1168538
Received: 01 March 2023; Accepted: 23 March 2023;
Published: 03 April 2023.
Edited by:
Peter J. Koch, East Carolina University, United StatesReviewed by:
Maiko Kawasaki, Niigata University, JapanCopyright © 2023 Gao, Jiang, Wei, Long and Lai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenli Lai, d2VubGlsYWlAc2N1LmVkdS5jbg==