 Yanjun Wang1
Yanjun Wang1 Jiahui Fang1
Jiahui Fang1 Bin Li1
Bin Li1 Chongyang Li2Shan Liu3Juan He1Lvyan Tao1Cuifen Li1Ya Yang1Li Li1*Shufang Xiao1*
Chongyang Li2Shan Liu3Juan He1Lvyan Tao1Cuifen Li1Ya Yang1Li Li1*Shufang Xiao1*- 1Kunming Children’s Hospital, Kunming, China
- 2Department of Oncology, The Affiliated Hospital of Yunnan University, Kunming, China
- 3Yunnan Cancer Hospital, Kunming, China
Background: Wilson’s disease (WD) is an autosomal recessive disease that is caused by mutations in the ATP7B (a copper-transporting P-type ATPase) gene. The disease has a low prevalence and is characterized by a copper metabolism disorder. However, various characteristics of the disease are determined by race and geographic region. We aimed to discover novel ATP7B mutations in pediatric patients with WD from Yunnan province, where there is a high proportion of ethnic minorities. We also performed a comprehensive analysis of ATP7B mutations in the different ethnic groups found in Southwest China.
Methods: We recruited 45 patients who had been clinically diagnosed with WD, from 44 unrelated families. Routine clinical examinations and laboratory evaluations were performed and details of age, gender, ethnic group and symptoms at onset were collected. Direct sequencing of the ATP7B gene was performed in 39 of the 45 patients and their families.
Results: In this study, participants came from seven different ethnic groups in China: Han, Bai, Dai, Zhuang, Yi, Hui and Jingpo. Three out of ten patients from ethnic minorities presented with elevated transaminases, when compared to the majority of the Han patients. Forty distinct mutations (28 missense, six splicing, three non-sense, two frameshift and one mutation of uncertain significance) were identified in the 39 patients with WD. Four of the mutations were novel and the most frequent mutation was c.2333G > T (p.R778L, allelic frequency: 15.38%). Using the phenotype-genotype correlation analysis, patients from ethnic minorities were shown to be more likely to have homozygous mutations (p = 0.035) than Han patients. The patients who carried the c.2310C > G mutation had lower serum ceruloplasmin levels (p = 0.012). In patients with heterozygous mutations, c.3809A > G was significantly associated with ethnic minorities (p = 0.042). The frequency of a protein-truncating variant (PTV) in Han patients was 34.38% (11/32), while we did not find PTV in patients from ethnic minorities.
Conclusion: This study revealed genetic defects in 39 pediatric patients with WD from Yunnan province. Four novel mutations were identified and have enriched the WD database. We characterized the genotypes and phenotypes in different minorities, which will enhance the current knowledge on the population genetics of WD in China.
Introduction
Wilson’s disease (WD; OMIM 277900) is an inherited, autosomal recessive copper metabolism disorder that is caused by mutations in the ATP7B gene (Cai et al., 2022). The age of onset ranges from eightmonths to 74 years (Ohura et al., 1999; Hahn et al., 2002; Ala et al., 2007; Abuduxikuer et al., 2015). The worldwide prevalence of this disease has been estimated to be approximately 1/100,000 to 3/100,000, whilst it is more prevalent in East Asia, where it is estimated to occur at a prevalence of 1/1,500 to 1/3,000 (Sandahl et al., 2020; Wallace and Dooley, 2020; Li et al., 2022). The carrier frequency of WD is 1/90 and heterozygous mutations are found in up to 2.5% of the general population (Czlonkowska et al., 2018; Sanchez-Monteagudo et al., 2021). Due to the accumulation of copper ions in the body (liver, brain and cornea), typical symptoms of WD include liver function injury, extrapyramidal symptoms, mental symptoms and Kayser-Fleischer rings (K-F ring) (Collins et al., 2021; Shribman et al., 2021). Patients with WD usually present with a variety of clinical subtypes that are characterized by hepatic and/or nervous system manifestations. Due to its various clinical manifestations and the fact that it is one of the few treatable neurogenetic disorders, genetic testing is an important diagnostic tool for this disease (Espinos and Ferenci, 2020). The development of irreversible sequelae can be prevented by timely diagnosis and early interventions in patients with WD (Harada, 2014; Espinos and Ferenci, 2020).
In 1993, the gene that encodes a copper-transporter P-type ATPase (ATP7B) was defined as the causative gene of WD. It is composed of 1,465 amino acids that contain a phosphatase domain, a phosphorylation domain, a nucleotide-binding domain, six N-terminal metal binding domains and eight transmembrane ion channels (Telianidis et al., 2013). It is located on chromosome 13q14.3 and contains 21 exons and 20 introns (Tanzi et al., 1993). The protein is used to expel copper ions from the cell and it plays an important role in regulating copper ion metabolism (Tanzi et al, 1993). Worldwide, more than 1,000 different mutations have been identified in the ATP7B gene, most of which are missense, non-sense or frameshift mutations (Human Gene Mutation Database, HGMD, http://www.hgmd.cf.ac.uk/ac/index.php). When ATP7B mutations lead to the abnormal function of the P-type copper transporter ATPase, the ability of ceruloplasmin to bind and transport copper ions is decreased or removed, which results in the various clinical symptoms of WD (Chen et al., 2015; Ferenci et al., 2019).
To date, several studies have reported the spectrum and frequency of mutations in the ATP7B gene in the Chinese WD population. However, genetic profiling in different ethnic groups is still lacking, particularly in pediatric patients with WD. Due to the higher prevalence and more complicated correlation between genotype and phenotype in the Chinese population, when compared with Caucasians (Sanchez-Monteagudo et al., 2020; Couchonnal et al., 2021), it is necessary to study the ATP7B profiles of different ethnic groups in China. In this study of WD patients from Yunnan province, we conducted a mutation analysis in 39 patients and their family members and identified four novel mutations in the ATP7B gene.
Materials and methods
Patients and data collection
Forty-five patients with WD, from 44 families, were recruited between January 2017 and October 2022 at the Children’s Hospital Affiliated to Kunming Medical University. The age range of pediatric patients was 0–18 years. The medical history, physical examination results, laboratory test data and imaging results were collected as clinical data. Physical examinations included tests for jaundice, hepatomegaly, K-F rings and neurological symptoms. Laboratory tests included routine blood tests, biochemical liver tests, renal function tests, coagulation function tests, virological tests (hepatitis virus, cytomegalovirus and Epstein-Barr virus), ceruloplasmin and 24-hour urinary copper level. Imaging included liver ultrasound and brain magnetic resonance imaging (MRI). The patients were diagnosed in accordance with their clinical symptoms, biochemical parameters and/or genetic analysis (Czlonkowska et al., 2018). All patients were assessed using the Leipzig score and the total score was ≥4 in every patient. There were two groups used for classifying the patients: Group 1 represented the Han ethnicity and ethnic minorities, while Group 2 represented the general group (patients from general departments) and the severely affected group (patients from the pediatric intensive care unit). Direct sequencing of the ATP7B gene was performed in 39 of the 45 patients and their family members. Written informed consent was signed by custodians of the child participants. This study adhered to the Declaration of Helsinki and was approved by the Ethics Committee of the Children’s Hospital Affiliated to Kunming Medical University, Kunming, Yunnan, China (No.202303034K01).
Light and electron microscopy analysis
Liver specimens from patient six were fixed in 10% formalin and embedded in paraffin. The thickness of the paraffin sections was 3 μm and they were stained with hematoxylin and eosin and Masson’s trichrome stain. The sections were observed under an Olympus BX53 microscope (Olympus, Tokyo, Japan). The sample for transmission electron microscopy was fixed in 2.5% glutaraldehyde for two hours, then fixed in 1% osmotic acid for one to two hours, dehydrated in acetone and embedded in epoxy resin. Ultrathin sections were sliced and stained with 3% uranyl acetate and lead citrate. The sections were observed under a transmission electron microscope (H7700, Hitachi, Tokyo, Japan).
DNA extraction and genetic analysis
Peripheral blood was collected from each patient (5 mL) and their parents (2 mL). The blood samples were sent to Beijing Kangso Medical Inspection for ATP7B genetic testing. The Qiagen FlexiGene DNA kit was used to extract genomic DNA from blood samples. The primers were designed using the Primer Z website (http://genepipe.ncgm.sinica.edu.tw/primerz/primerz4.do). The mutation sites were amplified by PCR and then sequenced by first-generation sequencing. The PCR amplification conditions were: 10 min at 95°C; 35 cycles of 30 s at 95°C, 30 s at 60°C and 45 s at 72°C; followed by 5 min at 72°C. The PCR products were analyzed by electrophoresis on a 1% agarose gel and then sequenced using an ABI PRISM 3730XL DNA automated sequencer (Applied Biosystems; Thermo Fisher Scientific, Inc.).
The mutations identified in this study were compared to the list of reported pathogenic mutations in the Human Gene Mutation Database (https://www.hgmd.cf.ac.uk/ac/index.php), the Wilson Disease Mutation Database (http://www.wilsons-disease.org.uk/) and the gnomAD database (http://gnomad-sg.org/). The pathogenicity of the mutations was evaluated in strict accordance with the American College of Medical Genetics and Genomics (ACMG) Standards and Guidelines (Richards et al., 2015), using Sorting Intolerant From Tolerant (SIFT, http://sift-dna.org), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/index.shtml) and Mutation Taster (https://www.mutationtaster.org/). Splice Ai (https://spliceailookup.broadinstitute.org/) was used to evaluate splice sites. The Alpha Fold 2 (Alpha Fold Protein Structure Database, https://alphafold.ebi.ac.uk/) and NetGene2-2.42 (https://mybiosoftware.com/netgene2-2-42-intron-splice-sites-human-c-elegans-a-thaliana-dna.html) sites were used for protein analysis.
Statistical analysis
Statistical analyses were performed using SPSS version 23. Quantitative data were expressed as the mean ± SD or median and interquartile range (IQR). Categorical variables were given as absolute (number) and relative frequencies (%). To compare quantitative data, the Student’s t-test or Mann-Whitney U test was used for 2-group comparisons. The X2 test or Fisher’s exact test was used to compare categorical variables, as appropriate. The criterion for a significant difference was a p-value of less than 0.05.
Results
Clinical features and laboratory data for the WD patients
Clinical data and laboratory findings for the WD patients are summarized in Table 1. In this study, we examined 45 pediatric patients with WD, of whom 17 (37.78%) were female and 28 (62.22%) were male. Their mean age of onset was 7.62 ± 3.46 years, with a range of 2.00–13.58 years. Simple elevated transaminases were found in 64.44% (29/45) of patients. The median ceruloplasmin level was 30 mg/L (range: 10–55 mg/L) and the median urine ketone in 24 h level was 171.2 ug (range: 99-445.65 ug). Ten patients, including one each from Bai, Dai, Zhuang, Yi, Hui and Jingpo, were from ethnic minorities, while the majority of patients were Han Chinese. Liver disease was found in 43 (95.56%) individuals and nine (20%) had brain disease. One patient (2.22%) presented with simple neurological symptoms. We found abnormalities in the basal ganglia, thalamus or brainstem in the craniocerebral MRI in seven out of the 45 (15.56%) patients. Eight (17.78%) patients presented with K-F rings. There was no significant difference in age of onset between Han patients and patients from ethnic minorities (p > 0.05). Male patients were more prevalent in our overall sample of pediatric patients, regardless of whether they were Han or members of an ethnic minority.
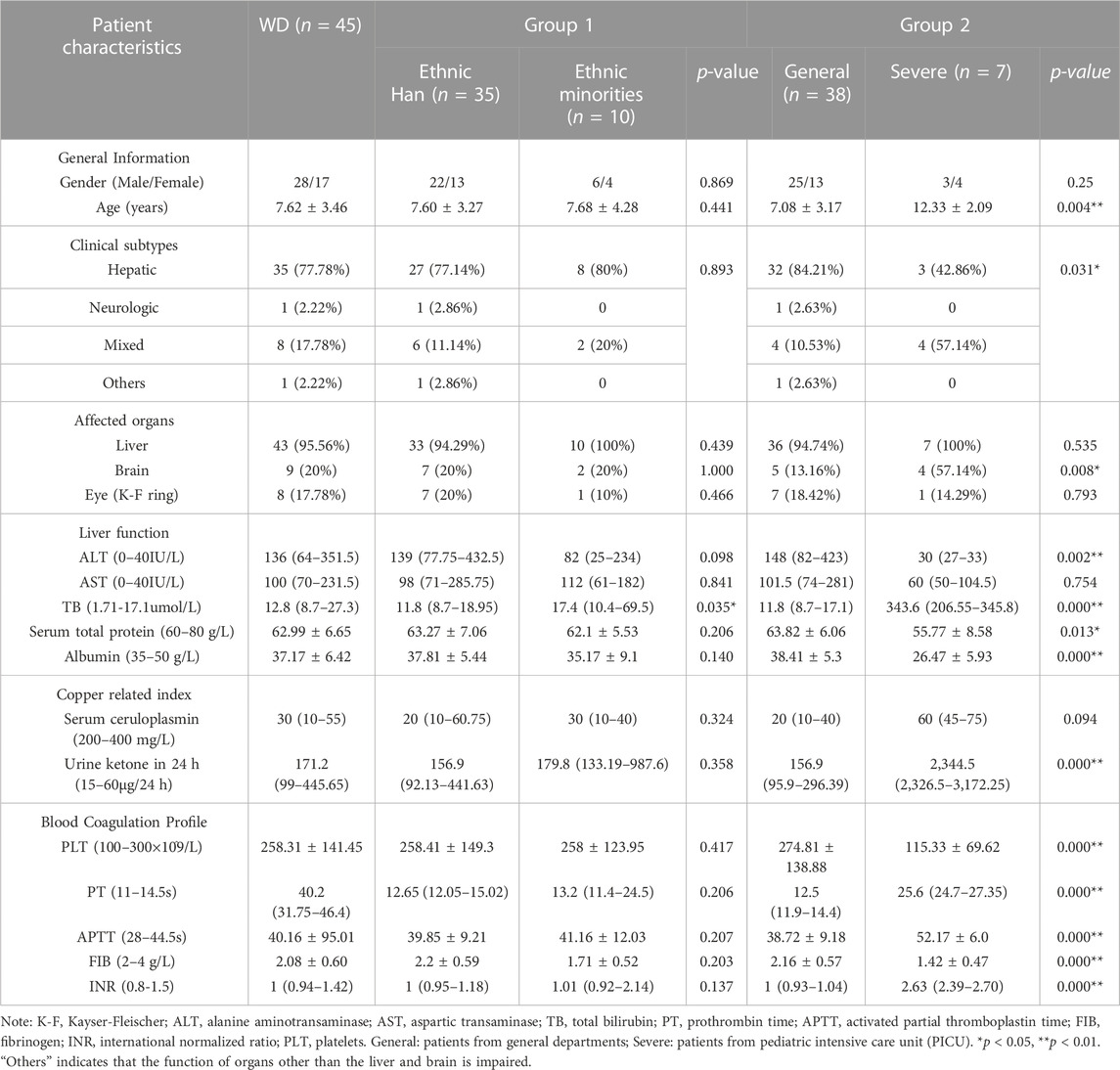
TABLE 1. Characteristics of 45 patients with Wilson’s disease clinical and laboratory data.
The laboratory data showed that, when compared to Han patients, ethnic minority patients had higher total bilirubin (TB) (p = 0.035). We also found that severely affected patients were significantly older at the onset of symptoms than general patients (p = 0.004). The severely affected patient group had a higher percentage of individuals with mixed subtype (p = 0.031) and symptoms that affected the brain (p = 0.008). General patients had increased alanine aminotransaminase (ALT), while severely affected patients had a normal ALT range (p = 0.002). Increased TB, prothrombin time (PT), activated partial thromboplastin (APTT), International Normalized Ratio (INR), urine ketone in 24 h and decreased platelets (PLT), serum total protein, albumin and fibrinogen (FIB) were observed in severely affected patients (p < 0.05).
Three pediatric patients underwent a liver biopsy during hospitalization. As shown inFigure 1, hepatocyte steatosis, cloudy swelling and ballooning were commonly observed. Glycogenated nuclei were present in hepatocytes and the Masson’s trichrome stain showed hepatic fibrotic lesions. Ultrastructural analysis showed pleomorphic dilated mitochondria.
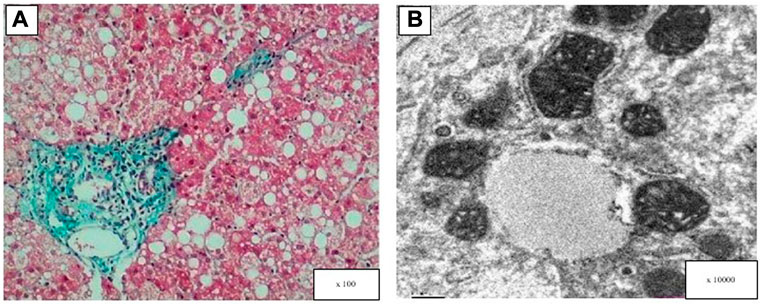
FIGURE 1. Light and electron microscopy analysis of the patient’s liver (patient 6). (A) Masson’s trichrome stain showed hepatic fibrotic lesions, diffuse hepatocellular steatosis (the size of the bubble fat gets mixed, 70%), and a few hepatocytes showed balloon-like. (B) Electron microscopy: hepatocyte swelling and variable lipid droplets are seen in diffuse hepatocytes.
Characterization of genetic mutations in the ATP7B gene
A total of 39 pediatric patients, from 38 unrelated families, were analyzed by direct sequencing. The results are shown in Table 2. Patient 20 and patient 21 were from the same family. Among the 39 patients, 25.64% (10/39) harbored homozygous mutations and 74.36% (29/39) harbored compound heterozygous mutations. Of the patients that harbored compound heterozygous mutations, 75.86% (22/29) had two mutations and 24.14% (7/29) had three mutations (Figure 2A). Interestingly, we observed that the c.2310C > G mutation always accompanied the c.2333G > T mutation (100%), while the c.2333G > T mutation was either a homozygous site or was found with other mutations. In the patients that had three mutations, it was noted that 71.43% (5/7) had the combination of c.2310C > G and c.2333G > T. Phenotype-genotype correlation analysis suggested that, when compared with Han patients, patients from ethnic minorities tended to have homozygous mutations (p = 0.035) (Figure 2B).

TABLE 2. Clinical and genetic data of the 39 patients with WD.

FIGURE 2. (A) Distribution of genotype in total patients with WD. (B) Distribution of genotype in patients from different ethnic groups (p = 0.035).
In 39 patients with WD, 39 potential pathogenic ATP7B mutations, one known polymorphism (c.2310C > G) that does not disrupt gene function and one mutation of uncertain significance (c.1869 + 20A > G) were identified (Figure 3A). Four of the mutations were novel (c.1286-1delG, c.1869 + 20A > G, c.2570_2572del and c.3742A > G). The mutations consisted of 28 (70%) missense mutations, six (15%) splice-site mutations, three (7.5%) non-sense mutations, two (5%) frameshift mutations and one (2.5%) mutation of uncertain significance (Figure 3B). The mutations were distributed in all exons apart from exons 1, 2, 4, 9, 15, 17 and 21. We found that exon 8 (26.83%) had the highest frequency of mutations, followed by exon 18 (9.76%), exon 3 (7.32%), exon 11 (7.32%), exon 12 (7.32%) and exon 13 (7.32%). This suggested that these exons may be more susceptible to mutations that cause WD in Yunnan province (Figure 3C). The most frequent mutation was c.2333G > T (p.R778L, allelic frequency: 15.38%), followed by c.2975C > T (p.P992L, 5.26%), c.2621C > T (p.A874V, 5.13%), c.3809A > G (p.N1270S, 5.13%) and c.2804C > T (p.T935M, 3.85%) (Figure 3D). The allelic frequency of c.2333G > T and c.2975C > T showed the same trend, in terms of the Minor allele frequency in genomAD from East Asian (Minor allele frequency in genomAD from East Asian). The allelic frequency of c.2621C > T and c.3809A > G was 5.13% (4/78), while the MAF was 0.0000. We also found that the allelic frequency c.2785A > G was 1.28% (1/78), which was not consistent with the MAF (0.0166) (Table 3).
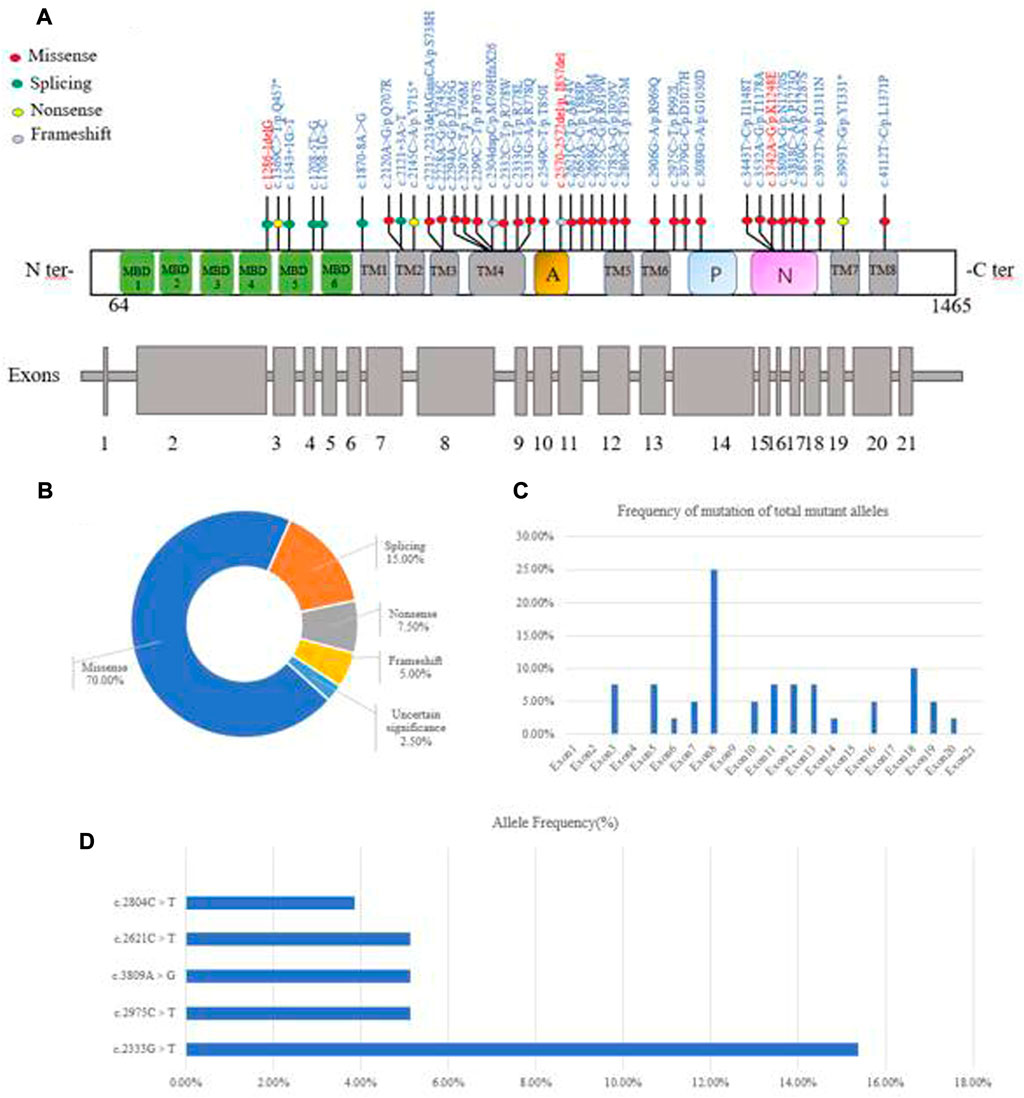
FIGURE 3. Characterization of genetic mutations in the ATP7B gene. (A) Novel mutations were observed. (B) Various mutants identified in this study and their percentage. (C) The frequency of mutations found in the 39 WD patients is given per exon as a percentage of the total mutant alleles. (D) Allele frequency of common ATP7B mutations in Yunnan province. MBDs, metal binding domains; Transmembrane segments are numbered TM1 to TM8; A: actuator domain; p: phosphorylation domain; N: nucleotide-binding domain.

TABLE 3. The potential pathogenic ATP7B mutations.
Characterization of novel mutations
The four novel mutations found in the pediatric patients with WD included one splice-site mutation, one missense mutation, one frameshift mutation and one mutation of uncertain significance. As shown in Figure 4A, the novel c.1286-1delG mutation was identified in the ATP7B gene of a patient and her mother. Using Splice Ai, we predicted the impact of the mutation on the protein. The NetGene2-2.42 site was used to predict that c.1286-1delG would cause a shift in the splice-site. We also identified the novel c.1869 + 20A > G mutation in a patient and his mother, as shown in Figure 4B. Splice Ai predicted that this mutation would not impact the splice-site. As shown in Figure 4C, the novel c.2570_2572del mutation was detected in a patient and his mother. Figure 4D shows the c.3742A > G mutation that was detected in another patient and his mother. The SIFT, PolyPhen-2 and Mutation Taster programs were used to predict the functional damage to the protein, caused by the mutations. The mutations shown in Figures 4B–D were classed as “Deleterious,” “Probably damaging” and “disease causing,” respectively. The structural analysis showed that the c.3742A > G (p.K1248E) mutation leads to the loss of the hydrogen bond between amino acids 1,027 and 1,271, which may affect the protein configuration (Figure 4D)
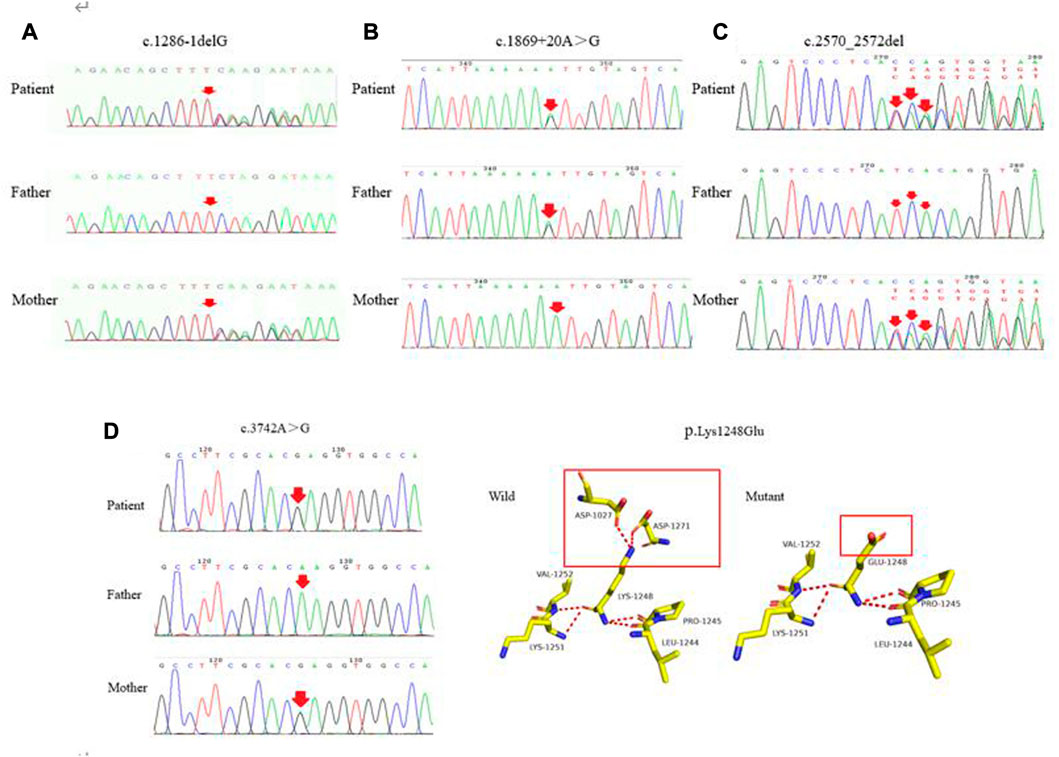
FIGURE 4. (A–C) represent the novel mutations (c.1286-1delG, c.1869 + 20A > G, c.2570_2572del), respectively. (D) c.3742A > G was detected in the patient and his mother. As shown in the structure of ATP7B protein, each color represents different atoms, yellow - C atom, gray - H atoms, blue - N atoms, red - O atoms, orange - S atom, the red dotted line for the hydrogen bond. The red rectangles represent areas of change.
In conclusion, in accordance with the ACMG Standards and Guidelines, c.1286-1delG was considered to be a “pathogenic variant,” c.2570_2572del and c.3742A > G were considered to be “likely pathogenic variants,” while c.1869 + 20A > G was classified as a “variant with uncertain significance.” Further studies are needed to confirm the changes predicted by the in silico tools.
Correlation between genotype and phenotype
To describe the correlation between the genotype and phenotype, we first studied the association between the exons and clinical subtypes in Yunnan province. The exons that were hotspots were examined in the 39 WD patients. The results showed that exons 8 and 18 harbored the highest percentage of mutations, which was not consistent with previous results that exons 8, 13 and 16 were the hotpot exons in the Chinese population (Li et al., 2021). This result may be due to the differences between Han individuals and ethnic minorities.
We then focused on the correlation between specific mutations and phenotypes. The most prevalent mutations were examined in the 39 WD patients. There was no significant difference, in terms of age of onset, between different clinical subtypes and several common mutations (c.2333G > T, c.2975C > T, c.2621C > T and c.3809A > G), in the different ethnic groups (p > 0.05). The results showed that the two most frequent variants were c.2333G > T (p.R778L) and c.2975C > T (p.P992L). We observed that the patients who carried the c.2310C > G mutation had lower serum ceruloplasmin levels than patients with other mutations (p = 0.012, Figure 5A). In patients who harbored compound heterozygous mutations, those from ethnic minorities tended to carry c.3809A > G (p = 0.042, Figure 5B). We also analyzed the relationship between the phenotypes and PTV (protein-truncated variants, e.g., frameshift, non-sense and splice-sites). The frequency of PTV in patients from Yunnan province was 28.2% (11/39). Interestingly, we found that the frequency of PTV in Han patients was 34.38% (11/32), while we did not find any PTV in patients from ethnic minorities (Figure 5C). Among the patients with PTV, 90.91% (10/11) presented with elevated transaminases and the p.T935M/PTV genotype had a frequency of 27.27% (3/11).
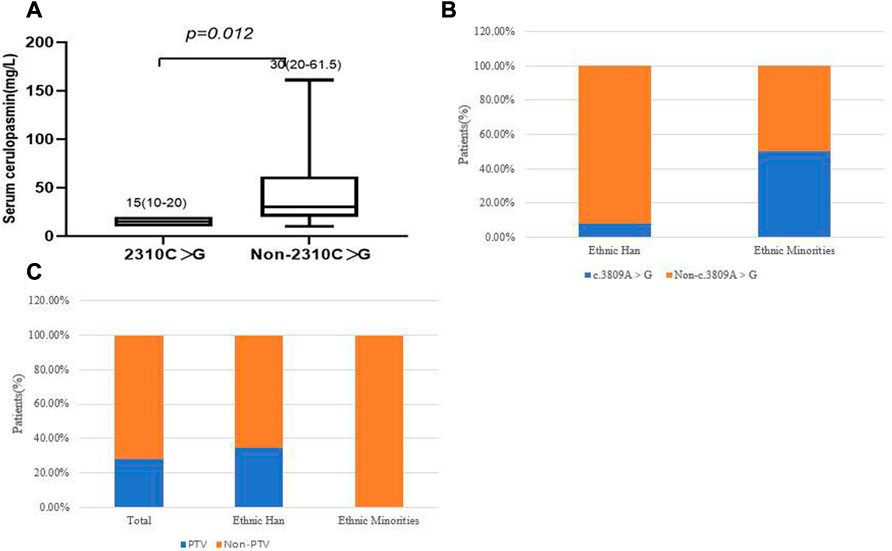
FIGURE 5. (A) Correlation of c.2310C > G and serum ceruloplasmin level. (B) correlation of c.3809A > G and ethnic groups. (C) The frequency of PTV in different ethnic groups.
Discussion
To define the mutation spectrum, clinical characteristics and genotype-phenotype correlations in WD in various ethnic groups, we examined ATP7B mutations in 39 pediatric patients from Yunnan province. Patients from seven ethnic groups (Han, Bai, Dai, Zhuang, Yi, Hui and Jingpo) from China were involved in this study. Patients with neurologic symptoms were not predominant in our cohort, in contrast to previous studies in China (Dong et al., 2021; Zhang et al., 2022). This may be because patients with neurologic symptoms mainly present during adolescence. However, male pediatric patients were predominant in our study, which is consistent with some studies in China and Korea (Cheng et al., 2017; Seo et al., 2018; Zhang et al., 2022). Most Han patients presented with elevated transaminases, while only three out of 10 patients from ethnic minorities presented with elevated transaminases. Three out of the six severely affected patients were from ethnic minorities. Several studies have shown that clinically asymptomatic patients have an earlier age of onset than patients with typical clinical manifestations (Li et al., 2021; Zhang et al., 2022). In our study, most pediatric patients presented with abnormal liver enzymes, without symptoms. We also found that pediatric patients from ethnic minorities had more severe disease.
This study identified homozygous mutations in 10 pedigrees and compound heterozygous mutations in 29 pedigrees. The frequency of homozygous mutations was 25.64% (10/39), which is not consistent with the frequency of 15.67% (204/1,302) that was observed in a previous study (Zhang et al., 2022). Moreover, phenotype-genotype correlation analysis suggested that, when compared with Han patients, patients from ethnic minorities tend to have homozygous mutations. This may indicate the significant genetic characteristics of patients from ethnic minorities from China. Of the patients who had compound heterozygous mutations, seven had three mutations. We observed that the c.2310C > G mutation always accompanied the c.2333G > T mutation, which is consistent with a previous study (aL). The c.2333G > T mutation was found as a homozygous site and also accompanied other mutations. It is possible that the coexistence of c.2333G > T and c.2310C > G may affect the function of the protein in a specific way. Hence, it is necessary to further explore the functional implications of both mutations.
Forty potential pathogenic mutations were identified in this study. Thirty-six of these have already been reported as disease-causing mutations in the Wilson Disease Mutation Database. The other four mutations were novel and evidence indicated that c.1286-1delG is considered to be a “pathogenic variant,” c.2570_2572del and c.3742A > G are considered to be “likely pathogenic variants,” while c.1869 + 20A > G needs further functional analysis and is classified as a “variant with uncertain significance.” In the patients who carried three mutations, it was noted that 71.43% had the c.2310C > G mutation, whilst the accompanying two pathogenic mutations had previously been reported. The novel c.1869 + 20A > G mutation co-occurred with two other reported pathogenic mutations (c.3532A > G and c.2121+3A > G) and its pathogenicity should be explored further.
The top five most common mutations in our study were c.2333G > T (p.R778L), c.2975C > T (p.P992L), c.2621C > T (p.A874V), c.3809A > G (p.N1270S) and c.2804C > T (p.T935M), which is not consistent with other reported studies (p.R778L, p.P992L, p.A874V, p.R919G and p.V1216M) (Li et al., 2021). Mutation hotspots in ATP7B vary by geographic region, with a higher prevalence of specific mutations reported in certain populations (Wallace and Dooley, 2020). The predominant mutations in the Chinese population include c.2333G > T (p.R778L), c.2975C > T (p.P992L), c.3443T > C (p.I1148T) and c.2804C > T (p.T935M). In contrast, the most common mutation in European populations is p.H1069Q (Wang et al., 2011; Wei et al., 2014). Chinese populations from different regions also have different mutation types. Although p.R778L was always detected as the most common pathogenic mutation, a study of populations in Northern China found that p.A874V was the second most common pathogenic mutation, whilst p.I1148T was the second most common mutation found in Guangdong province, Southern China (Li et al., 2013; Wei et al., 2014). Another study found that in pediatric patients from Southern China, p.R778L and p.I1148T had the highest frequencies, at approximately 23.0% and 10.7%, respectively. This is not consistent with our study (Zhou et al., 2022). A previous study found that p.T935M was significantly associated with Fujian province, which hints at the possibility of a founder effect (Wei et al., 2014). In our study, p.T935M was found in Yunnan province. The gnomAD database also shows a low frequency of p.T935M in the East Asian population, which does not support the previous hypothesis of a founder effect. Due to the vast diversity of mutations in the ATP7B gene, analysis of the regional distribution of mutations can help to develop time-saving approaches and speed up the genetic diagnosis of WD in specific regions (Roy et al., 2020).
The overall genetic diagnosis rate in this study was 97.5% (39/40). Previous studies gave 78.4% in Caucasians, by exon-by-exon sequencing, and 87.9% in Poland, by whole-exome sequencing (Ferenci et al., 2019; Kluska et al., 2019). In China, one study showed that the genetic diagnosis rate was 90.0%, by sequencing of the 5’ untranslated region (UTR), 21 exons and their flanking regions (Dong et al., 2016). Another study estimated that the genetic diagnosis rate was 97.1%, by mutational analysis of 68 WD patients from China (aL). The reason that not all patients are genetically diagnosed may be related to a large hemizygous deletion, regulatory region variants and genetic alterations outside of the ATP7B gene (Dong et al., 2016; Ferenci et al., 2019).
We did not find a significant difference in terms of age of onset between different clinical subtypes and several common mutations (c.2333G > T, c.2975C > T, c.2621C > T and c.3809A > G) in different ethnic groups (p > 0.05). This may have been due to the small sample size. Therefore, the sample size in Yunnan province should be expanded further to study these correlations.
We also analyzed the relationship between phenotypes and PTV. The frequency of PTV in patients from Yunnan province was 28.2% (11/39), which is consistent with the previous finding that the frequency of PTV was 26.5% (345/1,302) in a large cohort of patients with WD (Zhang et al., 2022). Interestingly, we found that the frequency of PTV in Han patients was 34.38% (11/32), while we did not find PTV in patients from ethnic minorities. Among the patients with PTV, 90.91% (10/11) presented with elevated transaminases and a specific genotype (p.T935M/PTV) had a frequency of 27.27% (3/11). Previous studies have found that truncating variants are associated with an early onset of WD (Gromadzka et al., 2005; Merle et al., 2010). Recently, one study found that PTV was more common in patients with a younger age of onset in both the hepatic and neurological groups (Zhang et al., 2022). There has been no research into the role of PTV in different ethnic groups. However, our findings revealed that PTV are not common in ethnic minority patients.
Conclusion
In this study, we performed mutation analysis of the ATP7B gene in 39 WD patients from Yunnan province. In summary, our study identified four novel mutations that expand the spectrum of pathogenic ATP7B mutations. In addition, phenotype-genotype correlation analysis suggested that, when compared with Han patients, patients from ethnic minorities tend to carry homozygous mutations. We observed that the patients who carried the c.2310C > G mutation had lower serum ceruloplasmin levels than patients with other mutations. In patients with heterozygous mutations, the c.3809A > G mutation was significantly associated with ethnic minorities. The frequency of PTV in patients from ethnic minorities was lower than in Han patients. Our research highlights the differences in genotypes of WD pediatric patients from different ethnic groups. This may provide valuable insights into the diagnosis, counseling and treatment of WD pediatric patients in China.
Data availability statement
The original contributions presented in the study are included in the article/supplementary materials, further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the Children’s Hospital Affiliated to Kunming Medical University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
YW designed the research study and drafted the manuscript. JF, BL, and CL performed the research and analysis. JH, CL, and YY performed routine examinations and assessments. SL and LT performed the validation. LL, and SX supervised the study and edited the manuscript.
Funding
The work was supported by the National Natural Science Foundation of China (No. 82160367), the Joint Special Fund for Basic Research from Yunnan Provincial Science and Technology Department and Kunming Medical University (No. 202001AY070001-269), Yunnan Provincial Key specialty (Critical Care Medicine Department) construction project, the Sixth Cycle Key Discipline of Kunming (Critical Care Medicine Department) and Kunming high-level talents Training Special Project- Spring City famous doctor Special project.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abuduxikuer, K., Li, L. T., Qiu, Y. L., Wang, N. L., and Wang, J. S. (2015). Wilson disease with hepatic presentation in an eight-month-old boy. World J. Gastroenterol. 21, 8981–8984. doi:10.3748/wjg.v21.i29.8981
Ala, A., Walker, A. P., Ashkan, K., Dooley, J. S., and Schilsky, M. L. (2007). Wilson's disease. Lancet 369, 397–408. doi:10.1016/S0140-6736(07)60196-2
Cai, H., Cheng, X., and Wang, X. P. (2022). ATP7B gene therapy of autologous reprogrammed hepatocytes alleviates copper accumulation in a mouse model of Wilson's disease. Hepatology 76, 1046–1057. doi:10.1002/hep.32484
Cater, M. A., La Fontaine, S., and Mercer, J. F. B. (2007). Copper binding to the N-terminal metal-binding sites or the CPC motif is not essential for copper-induced trafficking of the human Wilson protein (ATP7B). Biochem. J. 401, 143–153. doi:10.1042/BJ20061055
Chen, C., Shen, B., Xiao, J.-J., Wu, R., Duff Canning, S. J., and Wang, X. P. (2015). Currently clinical views on genetics of Wilson's disease. Chin. Med. J. 128, 1826–1830. doi:10.4103/0366-6999.159361
Cheng, N., Wang, H., Wu, W., Yang, R., Liu, L., Han, Y., et al. (2017). Spectrum of ATP7B mutations and genotype-phenotype correlation in large-scale Chinese patients with Wilson Disease. Clin. Genet. 92, 69–79. doi:10.1111/cge.12951
Collins, C. J., Yi, F., Dayuha, R., Duong, P., Horslen, S., Camarata, M., et al. (2021). Direct measurement of ATP7B peptides is highly effective in the diagnosis of Wilson disease. Gastroenterology 160, 2367–2382.e1. doi:10.1053/j.gastro.2021.02.052
Couchonnal, E., Lion-Francois, L., Guillaud, O., Habes, D., Debray, D., Lamireau, T., et al. (2021). Pediatric Wilson's disease: Phenotypic, genetic characterization and outcome of 182 children in France. J. Pediatr. Gastroenterol. Nutr. 73, e80–e86. doi:10.1097/MPG.0000000000003196
Czlonkowska, A., Litwin, T., Dusek, P., Ferenci, P., Lutsenko, S., Medici, V., et al. (2018). Wilson disease. Nat. Rev. Dis. Prim. 4, 21. doi:10.1038/s41572-018-0018-3
Dong, Y., Ni, W., Chen, W. J., Wan, B., Zhao, G. X., Shi, Z. Q., et al. (2016). Spectrum and classification of ATP7B variants in a large cohort of Chinese patients with Wilson's disease guides genetic diagnosis. Theranostics 6, 638–649. doi:10.7150/thno.14596
Dong, Y., Wang, R.-M., Yang, G.-M., Yu, H., Xu, W.-Q., Xie, J. J., et al. (2021). Role for biochemical assays and kayser-fleischer rings in diagnosis of Wilson’s disease. Clin. Gastroenterology Hepatology 19, 590–596. doi:10.1016/j.cgh.2020.05.044
Espinos, C., and Ferenci, P. (2020). Are the new genetic tools for diagnosis of Wilson disease helpful in clinical practice? JHEP Rep. 2, 100114. doi:10.1016/j.jhepr.2020.100114
Ferenci, P., Stremmel, W., Czlonkowska, A., Szalay, F., Viveiros, A., Stattermayer, A. F., et al. (2019). Age and sex but not ATP7B genotype effectively influence the clinical phenotype of Wilson disease. Hepatology 69, 1464–1476. doi:10.1002/hep.30280
Gromadzka, G., Schmidt, H. H., Genschel, J., Bochow, B., Rodo, M., Tarnacka, B., et al. (2005). Frameshift and nonsense mutations in the gene for ATPase7B are associated with severe impairment of copper metabolism and with an early clinical manifestation of Wilson's disease. Clin. Genet. 68, 524–532. doi:10.1111/j.1399-0004.2005.00528.x
Hahn, S. H., Lee, S. Y., Jang, Y.-J., Kim, S. N., Shin, H. C., Park, S. Y., et al. (2002). Pilot study of mass screening for Wilson's disease in Korea. Mol. Genet. metabolism 76, 133–136. doi:10.1016/s1096-7192(02)00026-4
Harada, M. (2014). Pathogenesis and management of Wilson disease. Hepatology Res. official J. Jpn. Soc. Hepatology 44, 395–402. doi:10.1111/hepr.12301
Kluska, A., Kulecka, M., Litwin, T., Dziezyc, K., Balabas, A., Piatkowska, M., et al. (2019). Whole-exome sequencing identifies novel pathogenic variants across the ATP7B gene and some modifiers of Wilson's disease phenotype. Liver Int. 39, 177–186. doi:10.1111/liv.13967
Li, K., Zhang, W. M., Lin, S., Wen, L., Wang, Z. F., Xie, D., et al. (2013). Mutational analysis of ATP7B in north Chinese patients with Wilson disease. J. Hum. Genet. 58, 67–72. doi:10.1038/jhg.2012.134
Li, M., Ma, J., Wang, W., Yang, X., and Luo, K. (2021). Mutation analysis of the ATP7B gene and genotype-phenotype correlation in Chinese patients with Wilson disease. BMC Gastroenterol. 21, 339. doi:10.1186/s12876-021-01911-5
Li, W. J., Chen, H. L., Wang, B., Yao, L., and Wang, X. P. (2022). Wilson's disease: Food therapy out of trace elements. Front. Cell Dev. Biol. 10, 1091580. doi:10.3389/fcell.2022.1091580
Merle, U., Weiss, K. H., Eisenbach, C., Tuma, S., Ferenci, P., and Stremmel, W. (2010). Truncating mutations in the Wilson disease gene ATP7B are associated with very low serum ceruloplasmin oxidase activity and an early onset of Wilson disease. BMC Gastroenterol. 10, 8. doi:10.1186/1471-230X-10-8
Ohura, T., Abukawa, D., Shiraishi, H., Yamaguchi, A., Arashima, S., Hiyamuta, S., et al. (1999). Pilot study of screening for Wilson disease using dried blood spots obtained from children seen at outpatient clinics. J. Inherit. metabolic Dis. 22, 74–80. doi:10.1023/a:1005455401076
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Roy, S., McCann, C. J., Ralle, M., Ray, K., Ray, J., Lutsenko, S., et al. (2020). Analysis of Wilson disease mutations revealed that interactions between different ATP7B mutants modify their properties. Sci. Rep. 10, 13487. doi:10.1038/s41598-020-70366-7
Rui, H., Fang, H., Yonggeng, J., Yu, P., Xu, Y., Shanshan, P., et al. (2016). Mutational analysis of ATP7B in Chinese Wilson disease patients. Am. J. Transl. Res. 8 (6), 2851–2861.
Sanchez-Monteagudo, A., Alvarez-Sauco, M., Sastre, I., Martinez-Torres, I., Lupo, V., Berenguer, M., et al. (2020). Genetics of Wilson disease and Wilson-like phenotype in a clinical series from eastern Spain. Clin. Genet. 97, 758–763. doi:10.1111/cge.13719
Sanchez-Monteagudo, A., Ripolles, E., Berenguer, M., and Espinos, C. (2021). Wilson's disease: Facing the challenge of diagnosing a rare disease. Biomedicines 9, 1100. doi:10.3390/biomedicines9091100
Sandahl, T. D., Laursen, T. L., Munk, D. E., Vilstrup, H., Weiss, K. H., and Ott, P. (2020). The prevalence of Wilson’s disease: An update. Hepatology 71, 722–732. doi:10.1002/hep.30911
Seo, G. H., Kim, Y.-M., Oh, S. H., Chung, S. J., Choi, I. H., Kim, G. H., et al. (2018). Biochemical and molecular characterisation of neurological Wilson disease. J. Med. Genet. 55, 587–593. doi:10.1136/jmedgenet-2017-105214
Shribman, S., Poujois, A., Bandmann, O., Czlonkowska, A., and Warner, T. T. (2021). Wilson's disease: Update on pathogenesis, biomarkers and treatments. J. Neurol. Neurosurg. Psychiatry 92, 1053–1061. doi:10.1136/jnnp-2021-326123
Tanzi, R. E., Petrukhin, K., Chernov, I., Pellequer, J. L., Wasco, W., Ross, B., et al. (1993). The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat. Genet. 5, 344–350. doi:10.1038/ng1293-344
Telianidis, J., Hung, Y. H., Materia, S., and Fontaine, S. L. (2013). Role of the P-Type ATPases, ATP7A and ATP7B in brain copper homeostasis. Front. Aging Neurosci. 5, 44. doi:10.3389/fnagi.2013.00044
Wallace, D. F., and Dooley, J. S. (2020). ATP7B variant penetrance explains differences between genetic and clinical prevalence estimates for Wilson disease. Hum. Genet. 139, 1065–1075. doi:10.1007/s00439-020-02161-3
Wang, L. H., Huang, Y. Q., Shang, X., Su, Q. X., Xiong, F., Yu, Q. Y., et al. (2011). Mutation analysis of 73 southern Chinese Wilson's disease patients: Identification of 10 novel mutations and its clinical correlation. J. Hum. Genet. 56, 660–665. doi:10.1038/jhg.2011.76
Wei, Z., Huang, Y., Liu, A., Diao, S., Yu, Q., Peng, Z., et al. (2014). Mutational characterization of ATP7B gene in 103 Wilson's disease patients from southern China: Identification of three novel mutations. Neuroreport 25, 1075–1080. doi:10.1097/WNR.0000000000000216
Zhang, S., Yang, W., Li, X., Pei, P., Dong, T., Yang, Y., et al. (2022). Clinical and genetic characterization of a large cohort of patients with Wilson’s disease in China. Transl. Neurodegener. 11, 13. doi:10.1186/s40035-022-00287-0
Keywords: Wilson’s disease, pediatric patients, ATP7B, genetic characterization, ethnic minorities
Citation: Wang Y, Fang J, Li B, Li C, Liu S, He J, Tao L, Li C, Yang Y, Li L and Xiao S (2023) Clinical and genetic characterization of pediatric patients with Wilson’s disease from Yunnan province where ethnic minorities gather. Front. Genet. 14:1142968. doi: 10.3389/fgene.2023.1142968
Received: 12 January 2023; Accepted: 08 March 2023;
Published: 20 March 2023.
Edited by:
Xiu-An Yang, Chengde Medical College, ChinaReviewed by:
Xiao-Ping Wang, Shanghai Jiao Tong University School of Medicine, ChinaAtchariya Chanpong, University College London, United Kingdom
Carmen Espinós, Principe Felipe Research Center (CIPF), Spain
Copyright © 2023 Wang, Fang, Li, Li, Liu, He, Tao, Li, Yang, Li and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Li, bGlsaUBldHl5LmNu; Shufang Xiao, eGlhb3NodWZhbmdAZXR5eS5jbg==