 Babajan Banaganapalli1,2
Babajan Banaganapalli1,2 Ibrahim Fallatah1Fai Alsubhi1
Ibrahim Fallatah1Fai Alsubhi1 Preetha Jayasheela Shetty3
Preetha Jayasheela Shetty3 Zuhier Awan4
Zuhier Awan4 Ramu Elango1,2*
Ramu Elango1,2* Noor Ahmad Shaik1,2*
Noor Ahmad Shaik1,2*- 1Department of Genetic Medicine, Faculty of Medicine, King Abdulaziz University, Jeddah, Saudi Arabia
- 2Princess Al-Jawhara Al-Brahim Center of Excellence in Research of Hereditary Disorders, King Abdulaziz University, Jeddah, Saudi Arabia
- 3Department of Biomedical Sciences, College of Medicine, Gulf Medical University, Ajman, United Arab Emirates
- 4Department of Clinical Biochemistry, Faculty of Medicine, King Abdulaziz University, Jeddah, Saudi Arabia
Paget’s disease of bone (PDB) is the second most prevalent metabolic bone disorder worldwide, with a prevalence rate of 1.5%–8.3%. It is characterized by localized areas of accelerated, disorganized, and excessive bone production and turnover. Typically, PDB develops in the later stages of life, particularly in the late 50s, and affects men more frequently than women. PDB is a complex disease influenced by both genetic and environmental factors. PDB has a complex genetic basis involving multiple genes, with SQSTM1 being the gene most frequently associated with its development. Mutations affecting the UBA domain of SQSTM1 have been detected in both familial and sporadic PDB cases, and these mutations are often associated with severe clinical expression. Germline mutations in other genes such as TNFRSF11A, ZNF687 and PFN1, have also been associated with the development of the disease. Genetic association studies have also uncovered several PDB predisposing risk genes contributing to the disease pathology and severity. Epigenetic modifications of genes involved in bone remodelling and regulation, including RANKL, OPG, HDAC2, DNMT1, and SQSTM1, have been implicated in the development and progression of Paget’s disease of bone, providing insight into the molecular basis of the disease and potential targets for therapeutic intervention. Although PDB has a tendency to cluster within families, the variable severity of the disease across family members, coupled with decreasing incidence rates, indicates that environmental factors may also play a role in the pathophysiology of PDB. The precise nature of these environmental triggers and how they interact with genetic determinants remain poorly understood. Fortunately, majority of PDB patients can achieve long-term remission with an intravenous infusion of aminobisphosphonates, such as zoledronic acid. In this review, we discuss aspects like clinical characteristics, genetic foundation, and latest updates in PDB research.
Introduction
Paget’s disease of bone (PDB) is a chronic and progressive bone disease that is characterized by bone pain, deformities, and fractures. The term “osteitis deformans” was first coined by Sir James Paget, an English physician, in 1877 (Paget, 1877). However, evidence of the disease dates back to 3,000 years, as suggested by lesions resembling PDB found in dinosaur vertebrae from the late Paleozoic to the middle Mesozoic periods (Haridy et al., 2019; Gennari et al., 2022). PDB is characterized by abnormal activation of osteoclasts, which leads to improper bone resorption and compensatory osteogenic sclerosis (Tuck, 2020). The disease is associated with increased bone remodeling and mass, with abnormal osteoclast activity leading to increased metabolic osteolytic activity while osteoblasts produce bone normally (Alonso et al., 2017).
Epidemiology of PDB
PDB is the second most prevalent disorder of bone remodeling, after osteoporosis, despite it being asymptomatic in many with variable late age of onset. The incidence of PDB varies depending on the population studied and the diagnostic criteria used, but it is generally considered to be a rare disease. The global prevalence of PDB ranges from 1.5% to 8.3%, highest in Europeans living in the United Kingdom, followed by Australia, New Zealand, North America, France, Germany, Spain and Italy. Conversely, it appears rare among Scandinavians, Africans, Asians, and non-European immigrants living in Europe (Vallet and Ralston, 2016; Nebot Valenzuela and Pietschmann, 2017; Abdulla et al., 2018). Among Middle East Arabians in southern Israel revealed a 1% prevalence of PDB, comparable to southern Europe. In Saudi Arabia, Iran, Iraq, and Turkey, few isolated case reports of PDB have been recorded (Alshaikh et al., 2011; Merashli and Jawad, 2015; Michou and Orcel, 2019). The above epidemiological data confirms that there is marked geographical variation in the occurrence of PDB. But whether this is linked to genetic susceptibility of specific ethnic or racial population groups, and/or potential environmental influences like diet (mineral and vitamin deficiencies), lifestyle (tobacco smoking) exposure to the pollutants (lead), and an infection (paramyxovirus) is unclear.
Clinical manifestations
The classical PDB usually appears at the age of forty and is rarely diagnosed before the age of fifty, with a slight male predominance (Vallet and Ralston, 2016). In around 70% of patients, PDB is asymptomatic and is typically discovered incidentally through elevated alkaline phosphatase (ALP) values (Kravets, 2018). When symptoms are present, the most frequent symptom is bone pain (73.8%), followed by morphological conditions (18.1%), hearing loss (7.9%), and pathological fractures (5.7%) (Ralston and Albagha, 2018; Michou and Orcel, 2019). Bone deformities, such as bowing of the legs, skull enlargement, and kyphosis, may also occur. Hearing loss, vision problems, and headaches can result from cranial nerve involvement. Increased blood flow to the bones can cause warmth and redness in affected areas. In rare cases, pathological fractures may occur. Leontiasis ossea, a condition in which the facial bones become deformed, can also occur. The most severe consequences of PDB include osteosarcomas and other sarcomas (chondrosarcoma, fibrosarcoma), though their incidence is less than 1% (Makaram and Ralston, 2021). In addition, PDB primarily affects the skeletal system, with certain regions being more prone to its impact than others (Makaram et al., 2020), as seen in Figure 1, the axial skeleton is the most commonly affected area (Figure 2).
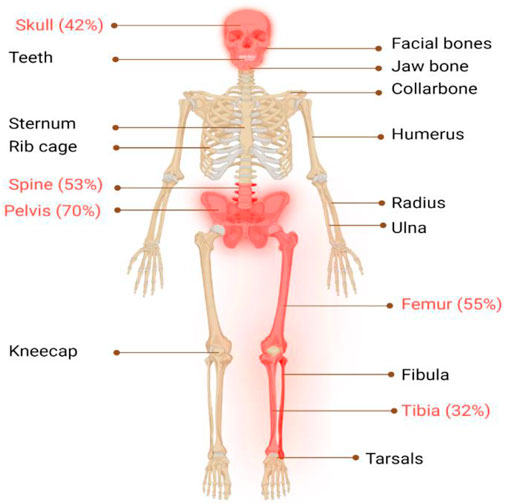
FIGURE 1. Illustrates the major skeletal locations affected by PDB: “The disorder predominantly affects the axial skeleton, with the highest incidence observed in the pelvis (70%), femur (55%), lumbar spine (53%), cranium (42%), and tibia (32%). To provide a visual representation of the most commonly affected areas in PDB, these locations are highlighted in red."
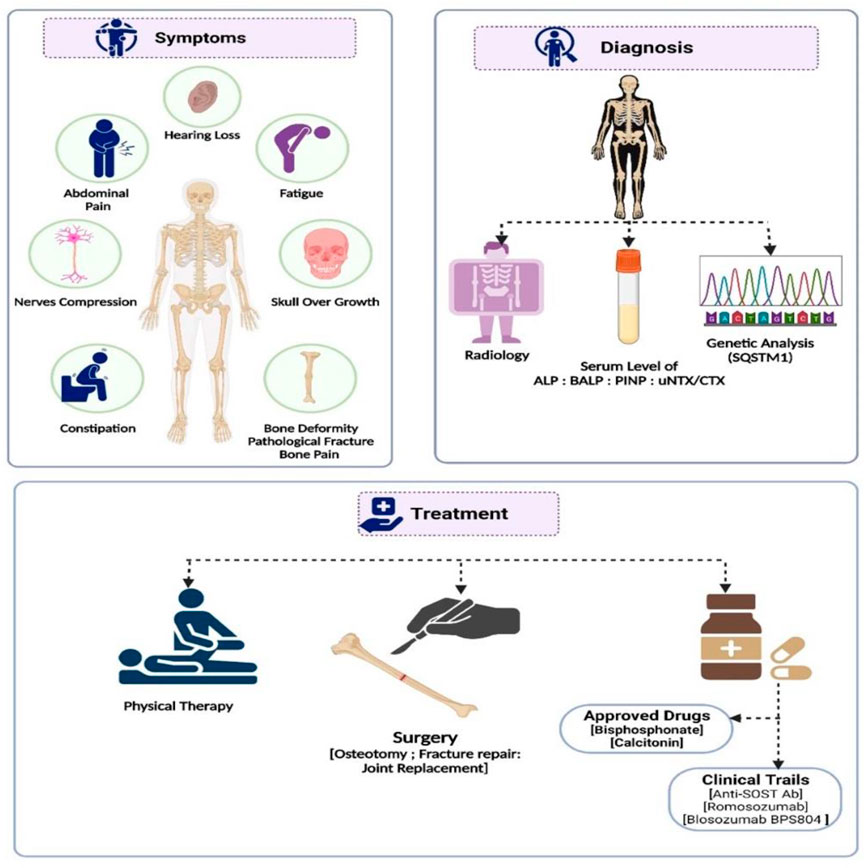
FIGURE 2. A summary of the diagnosis, management, and treatment approaches to Paget’s disease of bone.
Biochemical assessment of PDB
Paget’s disease of bone (PDB) can be diagnosed through clinical, radiographic, and biochemical assessments. Biochemical assessment involves measuring serum alkaline phosphatase (ALP) and urinary N-telopeptide (NTx) levels. ALP is an enzyme produced by osteoblasts, responsible for bone formation. In PDB, bone formation increases, leading to elevated ALP levels (Gennari et al., 2019). However, ALP levels can also be elevated in other conditions, such as liver disease or during pregnancy, making additional tests necessary for diagnosis confirmation (Seton, 2013; Gennari et al., 2019). In contrast, NTx is a specific marker of bone resorption, where osteoclasts break down bone tissue. In PDB, there is an increase in both bone formation and resorption, leading to elevated NTx levels. However, NTx levels can be affected by other factors, such as age, gender, and menopausal status, necessitating interpretation in the context of clinical findings (Delmas, 1999). Additional tests, including bone scans and imaging studies, may also be useful in PDB diagnosis to assess the extent of the disease and risk of fractures. Serum procollagen type 1 amino-terminal peptide (P1NP), osteocalcin, and bone-specific ALP (BALP) are more sensitive biochemical markers for bone production, while peptides of the collagen type I cross-linking domains, including NTx or CTX, are more specific for bone resorption. Despite the characteristic elevations in ALP and NTx levels in individuals with PDB, these markers are not specific to the condition (Cook and Wall, 2021). Thus, diagnostic confirmation and treatment response monitoring require interpretation in the context of clinical findings and other diagnostic tests (Alonso et al., 2017).
Radiological presentations in PDB
Radiographic changes can help diagnose Paget’s disease of bone (PDB). Indicators of increased bone resorption include a decrease in bone density, wedge-shaped bone resorption in long bones, and significant osteolytic regions in the skull. Early-stage PDB may show primarily lytic lesions, while older lesions tend to have a mixed sclerotic and lytic appearance. Late-stage PDB is characterized by sclerotic lesions, enlarged and distorted bones, and distinct radiographic patterns (Gennari et al., 2019; Ralston et al., 2019). The damaged bone enlargement in diameter is a distinguishing feature of PDB. A set of plain X-rays and bone scintigraphy are used to evaluate PDB, but bone scintigraphy may be negative in some cases. The presence of aberrant trabeculae, irregular cementation lines, increased vascularity, and an increase in the number and size of osteoclasts are the most distinctive findings (Alonso et al., 2017). Since genetic or bone biomarkers alone may lack sensitivity, combining many diagnostic markers is preferable to detect PDB at early stages and in asymptomatic cases. The PDB phenotype may be detected more accurately by integrating a screen for SQSTM1 gene mutations, followed by either a gene panel or a combination of genetic and biochemical tests (Guay-Bélanger et al., 2016). Approximately 70% of PDB patients have no symptoms, making early diagnosis challenging (Merchant et al., 2009). However, 15%–40% of patients have a positive family history, and first-degree relatives have a higher risk of developing PDB. Screening with a serum alkaline phosphatase test every two to 3 years is recommended for at-risk family members (Seton, 2013). Paget’s disease can lead to complications such as bone deformities, fractures, osteoarthritis, and an increased risk of developing bone cancer (Sabharwal et al., 2014). For instance, patients with PDB have an elevated chance of developing osteosarcoma, and despite its rarity (0.3% of PDB individuals), vast majority of osteosarcomas (OS) diagnosed in adulthood occur in patients with PDB. Similarly, there have been observation of families in which PDB is accompanied by giant cell tumours (Makaram and Ralston, 2021). Early detection and treatment are crucial in preventing these complications and improving patient outcomes. Radiographs should be taken to confirm the diagnosis (Ralston et al., 2019), and targeted genetic testing can be offered for at-risk family members (Seton, 2013; Guay-Bélanger et al., 2016).
Etiopathogenesis
The primary cause of PDB is believed to be the abnormal activation of osteoclasts, leading to improper bone resorption and compensatory osteogenic sclerosis. This results in increased bone remodeling and mass, with the osteoclasts being larger in size, number, and with more nuclei compared to normal cells (Albagha, 2015; Gennari et al., 2019). This increased metabolic osteolytic activity leads to bone destruction, but the normal osteoblasts continue to produce new bone. The result is structurally abnormal and weakened bones that are prone to fractures and deformities (Gennari et al., 2019). It is worth noting that while the activation of osteoclasts is the primary cause of PDB, the etiology of this activation is not yet fully understood. However, PDB is largely considered a multifactorial disease due to the combination of genetic and environmental factors contribute to its disease pathology.
Molecular factors in PDB
Normal adult skeleton remodeling involves osteoclasts destroying bone and osteoblasts generating new bone tissue at locations of past bone resorption (Kenkre and Bassett, 2018). The remodeling cycle is highly controlled and stereotypical, with five overlapping phases of activation, resorption, reversal, formation, and termination in cortical and trabecular bone respectively, within a period of 120–200 days. Osteocytes orchestrate the bone remodeling process by controlling osteoclast and osteoblast differentiation, and hence bone resorption and synthesis (Bellido et al., 2014). Several genes have been linked to osteoclast differentiation and activation which in turn leads to bone resorption (Albagha, 2015; Chen et al., 2018; Ralston and Albagha, 2018).
At the cellular level, normal bone remodeling is regulated by the receptor activator of nuclear factor kappa B (NF-kB) ligand (RANKL)/receptor activator of NF-kB (RANK)/Osteoprotegerin (OPG) system, which also controls the production and activity of osteoclasts (Kenkre and Bassett, 2018; Bolamperti et al., 2022). OPG, a decoy receptor, binds to RANKL to prevent RANK binding. Thereby, OPG suppresses the differentiation of osteoclasts. RANKL is expressed in the marrow stroma and on osteoblasts, when RANK binds on osteoclast precursors, it increases osteoclast proliferation and differentiation. It leads to the activation of a variety of downstream signaling pathways, including the nuclear factor kB (NF-kB), protein kinase B, c-jun N-terminal kinase, p38 mitogen-activated protein kinase and ERK pathways (Figure 3) (Tekkesin et al., 2011; Bellido et al., 2014).
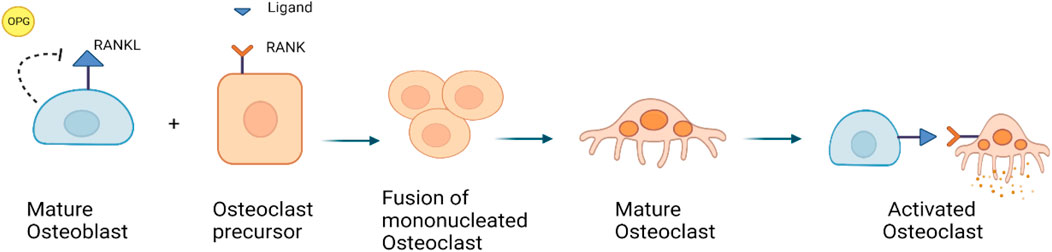
FIGURE 3. RANKL/RANK/OPG signaling pathway: RANKL is a receptor activator of nuclear factor kappa-B ligand, that is expressed by osteoblasts. OPG is also expressed by osteoblasts, it can bind to and inhibit RANKL and act as a protector against bone loss.
Genetic factors in PDB
Multiple lines of evidence suggest that inherited factors play an important role in PDB. The cumulative risk for developing PDB for a first-degree relative of an individual with PDB is estimated to be 9%, compared to 2% for those with unaffected relatives (Gennari et al., 2019). Recent evidence suggests that up to 40% of the PDB patients have a positive family history of PDB related symptoms or diseases. (Morales-Piga et al., 1995; Gennari et al., 2019; Gennari et al., 2022). At least one-third of patients have an autosomal dominant inheritance pattern with higher penetrance with age, risk in first-degree relatives will be as high as 50% (Ralston and Albagha, 2018; Gennari et al., 2019; Gennari et al., 2022). Though ∼40% of PDB patients had a positive family history for the illness, exclusion of undiagnosed, asymptomatic individuals makes it hard to determine the true incidence of familial disease (Morales-Piga et al., 1995). Intriguingly, the proportion of patients with a family history varies significantly between countries, ranging from approximately 5% in the Netherlands to 50% in the French-Canadian population. Between 12% and 15% of PDB patients in the United Kingdom and Italy have a family history (Merlotti et al., 2005; Langston et al., 2010). The marked ethnic differences in PDB prevalence support a strong genetic basis (Morales-Piga et al., 1995; Hocking et al., 2000; Morissette et al., 2006).
The genetic basis of PDB is complex and involves multiple genes. One of the most commonly implicated genes is SQSTM1, which encodes a multifunctional p62 protein (Shaik et al., 2021). Mutations in this gene have been found in up to 50% of people with familial PDB (Appelman-Dijkstra and Papapoulos, 2018). The p62 protein plays a crucial role in autophagy, a process involved in removing damaged cellular components. When this function is compromised, it can lead to the accumulation of damaged proteins in bone cells. The mutations in the SQSTM1 gene are responsible for both sporadic (10%–15%) and familial (25%–40%) forms of PDB. Studies have shown that the Pro392Leu mutation is present in about 46% of familial PDB cases of French-Canadian origin from Quebec, while two more mutations were found in a family of predominantly British background. In both studies, the P392L mutation was identified in 16% and 8.9% of sporadic PDB patients (Hocking et al., 2002; Laurin et al., 2002). Although SQSTM1 mutations are typically heterozygous, rare instances of homozygosity have also been reported (Rea et al., 2009). Over 30 distinct missense or truncating SQSTM1 mutations have been detected in up to 50% of familial and 20% of sporadic PDB cases in diverse populations (Gennari et al., 2019). In Hungary, about 21.95% of PDB patients carry the common p. Pro392Leu mutation in the SQSTM1 gene (Donáth et al., 2021).
Genotype-phenotype correlation studies have suggested that patients with nonsense mutations causing partial translation of UBA domain, have more severe and extensive disease than patients with missense mutations (Gennari et al., 2010; Visconti et al., 2010). Missense mutations are mostly restricted to exons 7 (29.41%) and 8 (70.59%) of the SQSTM1 gene (Shaik et al., 2021). However, even among patients with the same mutation and within the same family, clinical heterogeneity is reported (Laurin et al., 2002) (Gennari et al., 2010). Also somatic mutations in the SQSTM1 gene have been reported in sporadic papillary osteosarcoma (Merchant et al., 2009). In around 5% of PDB patients, somatic mutation in SQSTM1 (P392L) was observed in monocyte lineage only. Interestingly, PDB patients with this somatic mutation had a milder bone phenotype than those with the same mutation as a germline mutation (Guay-Bélanger et al., 2015). In germline mutations, the defect is present in every cell, while somatic mutations are seen in a subset of cells only, leading to variable expression of normal and abnormal proteins with reduced function. This might explain the milder phenotype in the somatic mutation carriers. Although SQSTM1 mutations are detected in about 50% of familial cases from various countries, their occurrence is relatively low in sporadic cases. Genome-wide association studies have revealed seven unique potential loci that account for ∼13% of the family risk of PDB in SQSTM1-negative individuals (Albagha et al., 2010; Albagha et al., 2011; Database, 2022).
Other PDB-associated genes
The genome wide scans have revealed several susceptibility loci for PDB and related syndromes (Makaram and Ralston, 2021; Gennari et al., 2022). Most of these gene loci are linked to osteoclast differentiation or function. These genetic loci are identified by few rare and common genetic variants, which collectively increase the PDB risk. Table 1 shows the list of different genetic loci and corresponding genes involved in the predisposition of individuals to PDB (Albagha et al., 2010; Albagha et al., 2011; Albagha, 2015). The TNFRSF11A gene mutations were first reported in isolated cases of PDB and other PDB-like illnesses, such as Familial expansile osteolysis (FEO) and Expansile skeletal hyperphosphatasia (ESH). Mutation of the TNFRSF11A gene revealed several insertions at the exon 1, resulting in the duplication of amino acid sequences in the RANK signal peptide (Hughes et al., 2000). Until now, many heterozygous in-frame tandem duplications of varied length, resulting in longer RANK signal peptide, have been reported, with the majority are related to uncommon PDB-like illnesses (Ralston and Taylor, 2019). This gene encodes a protein belonging to the TNF-receptor superfamily. This receptor can interact with many TRAF family members, thus activating NF-kappa B and MAPK8/JNK. This receptor is also a key osteoclast development mediator. The receptor activator of NFkB (RANK) is encoded by the TIFRSF11A gene.
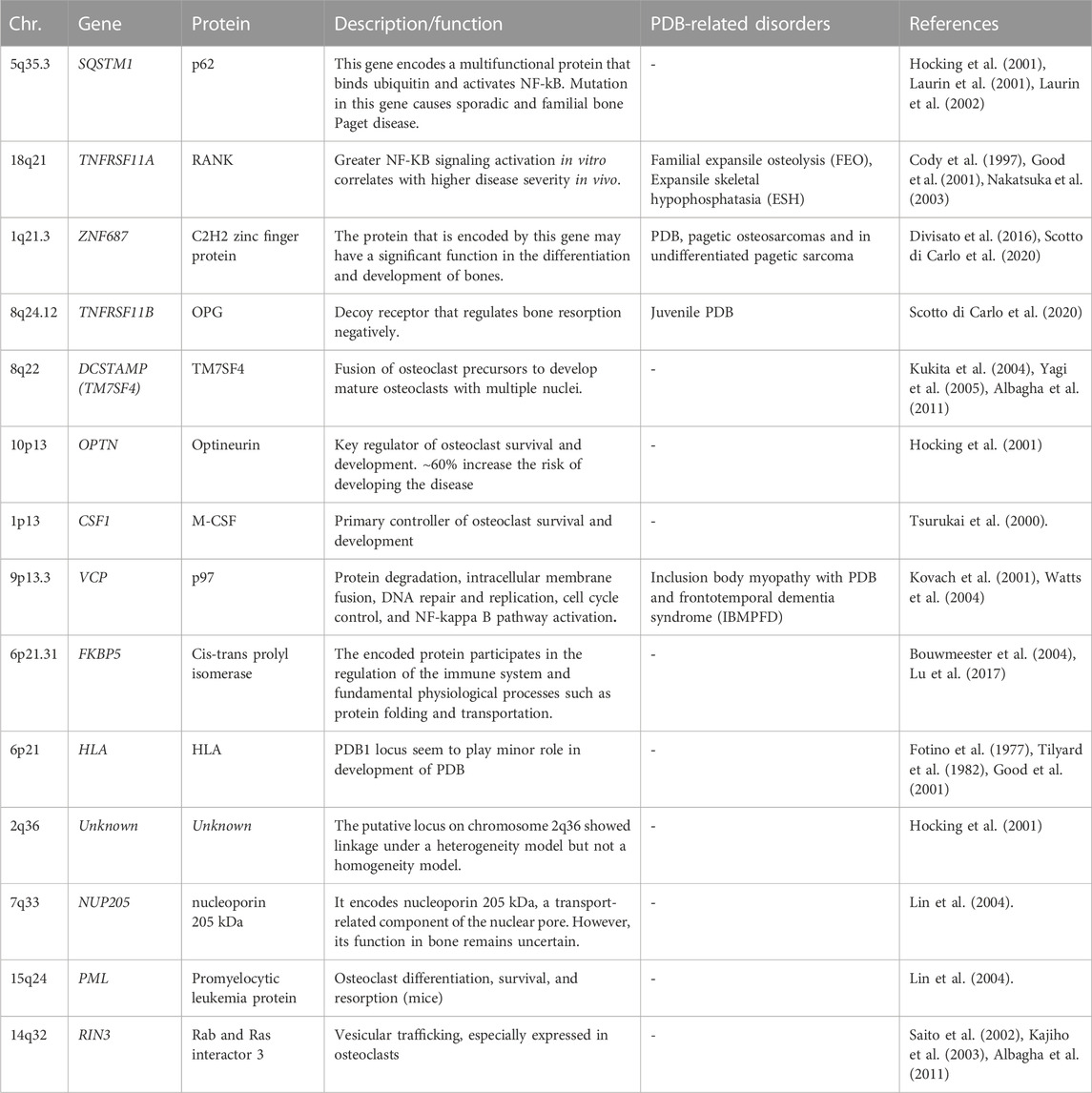
TABLE 1. PDB-predisposing risk genes with chromosomal location, encoded proteins, function, and diseases distinct from PDB (PDB-related disorders).
The ZNF687 gene encodes for C2H2 zinc finger protein that may play a role in bone differentiation and development. Mutation in ZNF687 gene was identified for the first time in large Italian family many affected family members by whole exome analysis, followed by other studies confirms it as causal gene for PDB (Divisato et al., 2016; Scotto di Carlo et al., 2020). The ZNF687 gene mutation was also associated with polyostotic PDB, pagetic osteosarcomas and in undifferentiated pagetic sarcoma tissue as well (Scotto di Carlo et al., 2020). For instance, minority of PDB patients develop malignant giant cell tumors (GCTs) of the bone (PDB/GCTs) with an early onset (Wei et al., 2021). ZNF687 is the only gene currently proven to cause PDB/GCT; however, Profilin 1 gene have recently been identified as the cause of early-onset Paget’s disease of bone with GCT in Italian and Chinese patients (Wei et al., 2021). Meanwhile, individuals with mutations in the ZNF687 gene experience severe PDB at multiple sites. In vitro studies have revealed that ZNF687 mutant osteoclasts can have up to 150 nuclei, a finding unique to PDB patients with ZNF687 mutations (Divisato et al., 2016; Scotto di Carlo et al., 2020). Knock-in mouse model of ZNF687 mutation supports the crucial role it plays in the PDB development and its potential oncogenic property by having high incidence of hepatocellular carcinomas (Russo et al., 2023).
Complete TNFRSF11B gene deletion is associated with JPD (Juvenile Paget Disease) (Scotto di Carlo et al., 2020; Naot et al., 2014). Also, partial TNFRSF11B gene resulting in loss of a conserved aspartate residue at codon 192 was also identified (Middleton-Hardie et al., 2006). Nevertheless, TNFRSF11B mutations have not yet been discovered in classical PDB, and the 8q24 locus did not emerge as a susceptibility gene in GWAS. There is some indication that common polymorphisms at the TNFRSF11B gene may predispose women, but not men, to classical PDB; however, this must be validated by a large-scale investigation (Solomon, 1979; Beyens et al., 2007). This gene, encodes Osteoprotegerin (OPG) protein, belongs to the TNF-receptor superfamily; an osteoblast-secreted decoy receptor that regulates bone resorption negatively. Also, this protein controls osteoclast development and function by inhibiting the stimulatory effects of RANKL on osteoclast differentiation (Makaram and Ralston, 2021; Gennari et al., 2022). The mutant OPG is unable to block osteoclastic resorption in a bone culture system, demonstrating that it is a loss-of-function mutation (Chong et al., 2003).
The DCSTAMP gene (Dendrocyte Expressed Seven Transmembrane Protein, also known as TM7SF4), encodes for a seven-pass transmembrane protein and is expressed on cells that is involved in osteoclastogenesis, immunological activity, and myeloid differentiation. In addition, the fusing of osteoclast precursors into mature osteoclasts is facilitated by the DCSTAMP protein. During the osteoclast formation, expression of DCSTAMP is crucial. Variants in the genes that predispose to PDB may increase DCSTAMP expression, leading to the formation of massive, multinucleated, granulocytic osteoclasts (Kukita et al., 2004; Yagi et al., 2005; Albagha et al., 2011).
The OPTN gene encodes the protein optineurin, which has coiled-coil structures; highly expressed cytoplasmic protein with many physiological activities, including NFB signaling, autophagy, and innate immunity (Zhu et al., 2007; Wild et al., 2011). Previous investigations have found a negative regulatory function for OPTN in osteoclast formation by altering in vitro and in vivo NF-kB and interferon signaling in mouse models (Obaid et al., 2015) (Silva et al., 2018a; Silva et al., 2018b).
The chromosome 1p13 hosts a recombination hotspot in the vicinity of the CSF1 gene. Notably, CSF1 exclusively encodes M-CSF, a cytokine that plays a pivotal role in regulating osteoclastogenesis and modulating the activity and survival of macrophages (Tsurukai et al., 2000; Bouyer et al., 2007). It is a strong candidate for PDB susceptibility based on its function (Tsurukai et al., 2000). The function of CSF1 in the etiology of PDB is supported by the observation that individuals with PDB have elevated blood levels of M-CSF (Neale et al., 2002). Variations in this gene that predispose to PDB is still unknown, although it is hypothesized that they may cause PDB via enhancing osteoclast production through CSF1 activity. A predisposing variation for PDB in CSF1 was found by a GWAS analysis (Albagha et al., 2010) and confirmed by a follow-up study the same group (Albagha et al., 2011). In parallel, a missense mutation in the CSF1 gene was identified in JPD (Donáth et al., 2015). Moreover, the SNPs linked with PDB are situated upstream of the gene in a region rich in regulatory elements, suggesting that their influence on control of CSF1 expression; however, the precise mechanism by which these variations predispose to PDB is yet to be determined (Neale et al., 2002).
VCP gene, encodes valosin-containing protein, which is a member of the AAA (+) ATPase family of chaperone-like proteins. It is a multifunctional protein involved in several intracellular processes including NF-KB signaling, DNA repair, and autophagy. Mutations in the VCP gene were discovered as the cause of the autosomal dominant Inclusion Body Myopathy with PDB and Frontotemporal Dementia (IBMPFD), characterized by skeletal defects identical to the classical PDB (Kovach et al., 2001; Watts et al., 2004; Columbres et al., 2023)
The VCP mutational effects include a modulatory influence on the NF-KB signaling pathway due to VCP’s involvement in the degradation of phosphorylated IkB (Woodman, 2003; Tresse et al., 2010). In addition, there is evidence that protein-coding mutations of VCP may arise infrequently in people with classical PDB who lack other components of IBMPFD syndrome; however, there is no conclusive data that common mutations at the VCP gene make the individuals susceptible to PDB (Albagha et al., 2014). Furthermore, syndromic IBMPFD linked with VCP mutations has been categorized as one of the multisystem proteinopathies, in which neurological and muscular abnormalities sometimes accompany the PDB (Taylor, 2015). Moreover, Myopathy occurs in up to 90% of IBMPFD families, whereas PDB and dementia have been identified in 43% and 37% of cases, respectively (Ralston and Taylor, 2019).
Another gene reported to be associated with the PDB is FKBP5 gene. Mutation in a Chinese family with PDB and supported by mutant mouse model. The functional study of the mutant mouse shows hyperresponsive osteoclast precursor cells to RANK with significantly high bone resorbing function (Liu and Zhang, 2015). The FKBP5 gene is a member of the immunophilin protein family, which is involved in immunoregulation and in fundamental physiological processes including protein folding and transport (Bouwmeester et al., 2004; Lu et al., 2017) (Zheng et al., 2021).
Epigenetics in PDB
In the last two to three decades, researchers have revealed that epigenetic modifications can influence the development of Paget’s disease (Leach et al., 2001; Galson and Roodman, 2014). Epigenetic alterations are chemical changes that occur in DNA and the proteins that bind to it without altering the actual DNA sequence. Environmental factors such as food, stress, pollutants, and infections can all influence these alterations, which can affect gene expression. Recent research has discovered various epigenetic changes that contribute to the development of PDB. For example, low DNA methylation of the RANKL gene promoter region was discovered in Paget’s disease patients compared to healthy controls, resulting in higher RANKL expression and associated bone resorption (Gennari et al., 2022). Similarly, increasing DNA methylation of the OPG gene promoter was linked to lower OPG expression, which accelerates bone resorption. Another important epigenetic component in PDB is histone modification. Histones are proteins that wrap DNA into nucleosomes, which are structural units. Histone acetylation and deacetylation are important gene expression regulators (Xu et al., 2020). Hypomethylation of histone H3 lysine 4 (H3K4me3) and hypermethylation of histone H3 lysine 9 (H3K9me3) have been linked to enhanced bone resorption. Factors like aging and viral infections are also known to play a role in PDB epigenetic alterations. Aging is a well-known risk factor for PDB, and studies have indicated that epigenetic changes associated with aging, such as increased DNA methylation, influence to the disease development (Saul and Kosinsky, 2021) and (Maatallah et al., 2021). PDB has also been related to viral infections, such as the measles virus, and studies have shown that the virus can modify the epigenetic landscape of bone cells, contributing to the disease development. DNA methylation and histone modification play important roles in bone resorption and creation, while environmental variables including aging and viral infections also contribute to epigenetic alterations (Diboun et al., 2021; Diboun et al., 2022). Therefore, epigenetic changes play a significant role in the development of Paget’s disease. With a greater understanding of these changes, researchers may be able to develop novel treatments that target the epigenetic abnormalities caused by Paget’s disease and aid in preventing the disease’s progression.
Role of environmental factors
Environmental factors are frequently proposed as an explanation for the variability in Paget’s disease epidemiology. For instance, river water contamination by arsenic in a pesticide used to treat cotton bales may have contributed to the high frequency of PDB in Lancashire during the 1970s. Because the nuclear inclusion bodies in osteoclasts appear to reflect viral nucleocapsids of one of the paramyxoviruses, it has been hypothesized that viral infections may be one of the causes of PDB (Visconti et al., 2017). Other environmental factors, such as cigarette smoke and wood stove smoke during childhood, have also been linked to Paget’s disease. These exposures have decreased due to changes in lifestyle over the time (Audet et al., 2017). Lifestyle factors such as diet and physical activity also play a role in the development of PDB. A diet that is high in saturated fats and low in calcium and vitamin D can contribute to the development of the disease, as can a sedentary lifestyle. Exercise and a diet that is rich in calcium and vitamin D can help to prevent the development of PDB and slow the progression of the disease (Numan et al., 2019). Furthermore, experiments revealed a relationship between the SQSTM1 gene and exposure to cadmium and tobacco smoke condensates, in certain patients with non-familial PDB (Numan et al., 2019). Addressing these environmental factors could prevent PDB development and improve outcomes. However, more research is needed to establish the exact mechanisms by which environmental factors contribute to PDB development.
Treatment of PDB
PDB does not have a specific treatment. However, some drugs can significantly improve the clinical symptoms and disease management. Bisphosphonates are the most used and FDA-approved drugs for treating PDB, and nitrogen-containing bisphosphonates have been found to be more effective than non-nitrogen-containing ones (Ralston et al., 2019). Patients with moderate-to-severe Paget’s disease of bone (PDB) have achieved successful outcomes with either a 6-month daily dose of alendronate (40 mg) or a 2-month daily dose of risedronate (30 mg). In approximately 60%–70% of cases, treatment normalized the ALP levels, and some patients were able to maintain biochemical remission for over a year (Gennari et al., 2009). Zoledronic acid is the most potent nitrogen-containing bisphosphonate and is the preferred choice for treating PDB due to its extended bone retention and intermittent dosing (Reid, 2016). However, its potential renal toxicity must be considered (Reid, 2016; Gennari et al., 2019). Oral alendronate or risedronate can also normalize alkaline phosphatase (ALP) levels in patients with moderate-to-severe PDB, but they may cause esophageal irritation and upper gastrointestinal tract (GI) discomfort. If patients cannot tolerate bisphosphonates, calcitonin is a safe, well-studied drug that can help lower the metabolic activity of pagetic bone and relieve GI discomfort (Reid, 2016). Surgery may be necessary in severe osteoarthritis cases but requires careful planning and preventative bisphosphonate medication. It is also crucial to tailor exercise programs and consume daily doses of vitamin D (400 IU) to maintain skeletal health (Ralston et al., 2019).
Conclusion
PDB has long been regarded as the second most frequent disease of bone metabolism, after osteoporosis. However, a remarkable and ongoing drop in its frequency and severity in several nations, making PDB a rare form of bone disease. The cause of this phenomena is unknown, however the rapid change in countries with high PDB, such as the United Kingdom, suggests an environmental impact. Genetic studies have changed our understanding of the pathophysiology of PDB and uncovered novel genes and signaling networks that govern the development and activity of osteoclasts. Despite these advances, the environmental triggers of PDB remain poorly understood. It is unknown why PDB attacks some bones but not others. In addition to gaining a better biological understanding of PDB, it will be useful for elucidating disease processes in other illnesses that affect the skeleton in a targeted manner. Now, gene expression and epigenetics studies highlight that PDB shares some targetable biomarkers, however, the development of targeted molecular drugs for PDB patients is lacking. Therefore, molecular novel drug targets must be identified using a complete molecular diagnostic profile so personalized medicine and clinical management can be achieved in the coming decades.
Author contributions
BB and NS, conceptualization; BB, IF, and FA data curation; funding acquisition; BB, RE NS, supervision; BB, IF, and FA: validation; BB, visualization; BB, IF, FA, PS, RE, and NS: writing original draft; BB, RE, and NS, writing review and editing.
Funding
This research work was funded by institutional Fund Projects under grant no. (IFPRP: 388-140-1442).
Acknowledgments
The authors extend their appreciation to the Deputyship for Research and Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project number IFPRP: 388-140-1442 and King Abdulaziz University, DSR, Jeddah, Saudi Arabia.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdulla, O., Naqvi, M. J., Shamshuddin, S., Bukhari, M., and Proctor, R. (2018). Prevalence of paget's disease of bone in Lancaster: Time for an update. Rheumatol. Oxf. 57, 931–932. doi:10.1093/rheumatology/kex505
Albagha, O., Lakshminarayan, R., and Ralston, S. (2014). A novel VCP mutation in a patient with paget's disease of bone without myopathy and neurological involvement, in Annual Meeting of the American-Society-for-Bone-and-Mineral-Research.
Albagha, O. M. (2015). Genetics of Paget's disease of bone. Bonekey Rep. 4, 756. doi:10.1038/bonekey.2015.125
Albagha, O. M., Visconti, M. R., Alonso, N., Langston, A. L., Cundy, T., Dargie, R., et al. (2010). Genome-wide association study identifies variants at CSF1, OPTN and TNFRSF11A as genetic risk factors for Paget's disease of bone. Nat. Genet. 42, 520–524. doi:10.1038/ng.562
Albagha, O. M., Wani, S. E., Visconti, M. R., Alonso, N., Goodman, K., Brandi, M. L., et al. (2011), Genome-wide association identifies three new susceptibility loci for Paget's disease of bone. Nat. Genet. 43, 685–689. doi:10.1038/ng.845
Alonso, N., Calero-Paniagua, I., and Del Pino-Montes, J. (2017). Clinical and genetic advances in paget's disease of bone: A review. Clin. Rev. Bone Min. Metab. 15, 37–48. doi:10.1007/s12018-016-9226-0
Alshaikh, O. M., Almanea, H., and Alzahrani, A. S. (2011). Paget disease of the bone: Does it exist in Saudi Arabia? Ann. Saudi Med. 31, 305–310. doi:10.4103/0256-4947.75588
Appelman-Dijkstra, N. M., and Papapoulos, S. E. (2018). Paget's disease of bone. Best. Pract. Res. Clin. Endocrinol. Metab. 32, 657–668. doi:10.1016/j.beem.2018.05.005
Audet, M. C., Jean, S., Beaudoin, C., Guay-Bélanger, S., Dumont, J., Brown, J. P., et al. (2017). Environmental factors associated with familial or non-familial forms of Paget's disease of bone. Jt. Bone Spine 84, 719–723. doi:10.1016/j.jbspin.2016.11.010
Bellido, T., Plotkin, L. I., and Bruzzaniti, A. (2014). “Chapter 2 - bone cells,” in Basic and applied bone biology. Editors D. B. Burr, and M. R. Allen (San Diego: Academic Press), 27–45.
Beyens, G., Daroszewska, A., De Freitas, F., Fransen, E., Vanhoenacker, F., Verbruggen, L., et al. (2007). Identification of sex-specific associations between polymorphisms of the osteoprotegerin gene, TNFRSF11B, and Paget's disease of bone. J. Bone Min. Res. 22, 1062–1071. doi:10.1359/jbmr.070333
Bolamperti, S., Villa, I., and Rubinacci, A. (2022). Bone remodeling: An operational process ensuring survival and bone mechanical competence. Bone Res. 10, 48. doi:10.1038/s41413-022-00219-8
Bouwmeester, T., Bauch, A., Ruffner, H., Angrand, P.-O., Bergamini, G., Croughton, K., et al. (2004). A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat. Cell Biol. 6, 97–105. doi:10.1038/ncb1086
Bouyer, P., Sakai, H., Itokawa, T., Kawano, T., Fulton, C. M., Boron, W. F., et al. (2007). Colony-Stimulating factor-1 increases osteoclast intracellular pH and promotes survival via the electroneutral Na/HCO3 cotransporter NBCn1. Endocrinology 148, 831–840. doi:10.1210/en.2006-0547
Chen, X., Wang, Z., Duan, N., Zhu, G., Schwarz, E. M., and Xie, C. (2018). Osteoblast–osteoclast interactions. Connect. Tissue Res. 59, 99–107. doi:10.1080/03008207.2017.1290085
Chong, B., Hegde, M., Fawkner, M., Simonet, S., Cassinelli, H., Coker, M., et al. (2003). Idiopathic hyperphosphatasia and TNFRSF11B mutations: Relationships between phenotype and genotype. J. Bone Min. Res. 18, 2095–2104. doi:10.1359/jbmr.2003.18.12.2095
Cody, J. D., Singer, F. R., Roodman, G. D., Otterund, B., Lewis, T. B., Leppert, M., et al. (1997). Genetic linkage of Paget disease of the bone to chromosome 18q. Am. J. Hum. Genet. 61, 1117–1122. doi:10.1086/301601
Columbres, R. C. A., Chin, Y., Pratti, S., Quinn, C., Gonzalez-Cuyar, L. F., Weiss, M., et al. (2023). Novel Variants in the VCP Gene Causing Multisystem Proteinopathy 1, 14.Genes (Basel)
Cook, S. J., and Wall, C. (2021). Paget's disease of bone: A clinical update. Aust. J. Gen. Pract. 50, 23–29. doi:10.31128/AJGP-10-20-5690
Delmas, P. D. (1999). Biochemical markers of bone turnover in paget's disease of bone. J. Bone Mineral Res. 14, 66–69. doi:10.1002/jbmr.5650140213
Diboun, I., Wani, S., Ralston, S. H., and Albagha, O. M. E. (2022). Epigenetic DNA methylation signatures associated with the severity of paget's disease of bone. Front. Cell Dev. Biol. 10, 903612. doi:10.3389/fcell.2022.903612
Diboun, I., Wani, S., Ralston, S. H., and Albagha, O. M. (2021). Epigenetic analysis of Paget's disease of bone identifies differentially methylated loci that predict disease status. Elife 10, e65715. doi:10.7554/eLife.65715
Divisato, G., Formicola, D., Esposito, T., Merlotti, D., Pazzaglia, L., Del Fattore, A., et al. (2016). ZNF687 mutations in severe paget disease of bone associated with giant cell tumor. Am. J. Hum. Genet. 98, 275–286. doi:10.1016/j.ajhg.2015.12.016
Donáth, J., Balla, B., Pálinkás, M., Rásonyi, R., Vastag, G., Alonso, N., et al. (2021). Pattern of SQSTM1 gene variants in a Hungarian cohort of paget's disease of bone. Calcif. Tissue Int. 108, 159–164. doi:10.1007/s00223-020-00758-4
Donáth, J., Speer, G., Kósa, J. P., Árvai, K., Balla, B., Juhász, P., et al. (2015). Polymorphisms of CSF1 and TM7SF4 genes in a case of mild juvenile Paget's disease found using next-generation sequencing. Croat. Med. J. 56, 145–151. doi:10.3325/cmj.2015.56.145
Fotino, M., Haymovits, A., and Falk, C. T. (1977). Evidence for linkage between HLA and Paget's disease. Transpl. Proc. 9, 1867–1868.
Galson, D. L., and Roodman, G. D. (2014). Pathobiology of paget's disease of bone. J. Bone Metab. 21, 85–98. doi:10.11005/jbm.2014.21.2.85
Gennari, L., Gianfrancesco, F., Di Stefano, M., Rendina, D., Merlotti, D., Esposito, T., et al. (2010). SQSTM1 gene analysis and gene-environment interaction in Paget's disease of bone. J. Bone Min. Res. 25, 1375–1384. doi:10.1002/jbmr.31
Gennari, L., Merlotti, D., Mossetti, G., Rendina, D., De Paola, V., Martini, G., et al. (2009). The use of intravenous aminobisphosphonates for the treatment of Paget's disease of bone. Mini Rev. Med. Chem. 9, 1052–1063. doi:10.2174/138955709788922683
Gennari, L., Rendina, D., Falchetti, A., and Merlotti, D. (2019). Paget's disease of bone. Calcif. Tissue Int. 104, 483–500. doi:10.1007/s00223-019-00522-3
Gennari, L., Rendina, D., Merlotti, D., Cavati, G., Mingiano, C., Cosso, R., et al. (2022). Update on the pathogenesis and genetics of Paget's disease of bone. Front. Cell Dev. Biol. 10, 932065. doi:10.3389/fcell.2022.932065
Good, D., Busfield, F., Duffy, D., Lovelock, P. K., Kesting, J. B., Cameron, D. P., et al. (2001). Familial paget's disease of bone: Nonlinkage to the PDB1 and PDB2 loci on chromosomes 6p and 18q in a large pedigree. J. Bone Min. Res. 16, 33–38. doi:10.1359/jbmr.2001.16.1.33
Guay-Bélanger, S., Picard, S., Gagnon, E., Morissette, J., Siris, E. S., Orcel, P., et al. (2015). Detection of SQSTM1/P392L post-zygotic mutations in Paget's disease of bone. Hum. Genet. 134, 53–65. doi:10.1007/s00439-014-1488-3
Guay-Bélanger, S., Simonyan, D., Bureau, A., Gagnon, E., Albert, C., Morissette, J., et al. (2016). Development of a molecular test of Paget's disease of bone. Bone 84, 213–221. doi:10.1016/j.bone.2016.01.007
Haridy, Y., Witzmann, F., Asbach, P., and Reisz, R. R. (2019). Permian metabolic bone disease revealed by microCT: Paget's disease-like pathology in vertebrae of an early amniote. PLoS One 14, e0219662. doi:10.1371/journal.pone.0219662
Hocking, L. J., Herbert, C. A., Nicholls, R. K., Williams, F., Bennett, S. T., Cundy, T., et al. (2001). Genomewide search in familial Paget disease of bone shows evidence of genetic heterogeneity with candidate loci on chromosomes 2q36, 10p13, and 5q35. Am. J. Hum. Genet. 69, 1055–1061. doi:10.1086/323798
Hocking, L. J., Lucas, G. J., Daroszewska, A., Mangion, J., Olavesen, M., Cundy, T., et al. (2002). Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget's disease. Hum. Mol. Genet. 11, 2735–2739. doi:10.1093/hmg/11.22.2735
Hocking, L., Slee, F., Haslam, S. I., Cundy, T., Nicholson, G., Van Hul, W., et al. (2000). Familial paget’s disease of bone: Patterns of inheritance and frequency of linkage to chromosome 18q. Bone 26, 577–580. doi:10.1016/s8756-3282(00)00278-7
Hughes, A. E., Ralston, S. H., Marken, J., Bell, C., Macpherson, H., Wallace, R. G. H., et al. (2000). Mutations in TNFRSF11A, affecting the signal peptide of RANK, cause familial expansile osteolysis. Nat. Genet. 24, 45–48. doi:10.1038/71667
Kajiho, H., Saito, K., Tsujita, K., Kontani, K., Araki, Y., Kurosu, H., et al. (2003). RIN3: A novel Rab5 GEF interacting with amphiphysin II involved in the early endocytic pathway. J. Cell Sci. 116, 4159–4168. doi:10.1242/jcs.00718
Kenkre, J. S., and Bassett, J. (2018). The bone remodelling cycle. Ann. Clin. Biochem. 55, 308–327. doi:10.1177/0004563218759371
Kovach, M. J., Waggoner, B., Leal, S. M., Gelber, D., Khardori, R., Levenstien, M. A., et al. (2001). Clinical delineation and localization to chromosome 9p13.3–p12 of a unique dominant disorder in four families: Hereditary inclusion body myopathy, paget disease of bone, and frontotemporal dementia. Mol. Genet. Metabolism 74, 458–475. doi:10.1006/mgme.2001.3256
Kravets, I. (2018). Paget's disease of bone: Diagnosis and treatment. Am. J. Med. 131, 1298–1303. doi:10.1016/j.amjmed.2018.04.028
Kukita, T., Wada, N., Kukita, A., Kakimoto, T., Sandra, F., Toh, K., et al. (2004). RANKL-induced DC-STAMP is essential for osteoclastogenesis. J. Exp. Med. 200, 941–946. doi:10.1084/jem.20040518
Langston, A. L., Campbell, M. K., Fraser, W. D., Maclennan, G. S., Selby, P. L., Ralston, S. H., et al. (2010). Randomized trial of intensive bisphosphonate treatment versus symptomatic management in Paget's disease of bone. J. Bone Min. Res. 25, 20–31. doi:10.1359/jbmr.090709
Laurin, N., Brown, J. P., Lemainque, A., Duchesne, A., Huot, D., Lacourcière, Y., et al. (2001). Paget disease of bone: Mapping of two loci at 5q35-qter and 5q31. Am. J. Hum. Genet. 69, 528–543. doi:10.1086/322975
Laurin, N., Brown, J. P., Morissette, J., and Raymond, V. (2002). Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am. J. Hum. Genet. 70, 1582–1588. doi:10.1086/340731
Leach, R. J., Singer, F. R., and Roodman, G. D. (2001). The genetics of Paget's disease of the bone. J. Clin. Endocrinol. Metab. 86, 24–28. doi:10.1210/jcem.86.1.7112
Lin, H.-K., Bergmann, S., and Pandolfi, P. P. (2004). Cytoplasmic PML function in TGF-β signalling. Nature 431, 205–211. doi:10.1038/nature02783
Liu, W., and Zhang, X. (2015). Receptor activator of nuclear factor-κB ligand (RANKL)/RANK/osteoprotegerin system in bone and other tissues (Review). Mol. Med. Rep. 11, 3212–3218. doi:10.3892/mmr.2015.3152
Lu, B., Jiao, Y., Wang, Y., Dong, J., Wei, M., Cui, B., et al. (2017). A FKBP5 mutation is associated with Paget’s disease of bone and enhances osteoclastogenesis. Exp. Mol. Med. 49, e336. doi:10.1038/emm.2017.64
Maatallah, K., Ben Nessib, D., Labbène, E., Ferjani, H., Bouaziz, M., Kaffel, D., et al. (2021). Paget's Disease of Bone in Patients under 40 Years: Two case reports and review of the literature. Sultan Qaboos Univ. Med. J. 21, e127–e131. doi:10.18295/squmj.2021.21.01.019
Makaram, N. S., and Ralston, S. H. (2021). Genetic determinants of paget's disease of bone. Curr. Osteoporos. Rep. 19, 327–337. doi:10.1007/s11914-021-00676-w
Makaram, N., Woods, L., Beattie, N., Roberts, S. B., and Macpherson, G. J. (2020). Long-term outcomes following total hip and total knee arthroplasty in patients with Paget's disease of bone (PDB) - a national study. Surgeon 18, 335–343. doi:10.1016/j.surge.2019.12.002
Merashli, M., and Jawad, A. (2015). Paget's disease of bone among various ethnic groups. Sultan Qaboos Univ. Med. J. 15, e22–e26.
Merchant, A., Smielewska, M., Patel, N., Akunowicz, J. D., Saria, E. A., Delaney, J. D., et al. (2009). Somatic mutations in SQSTM1 detected in affected tissues from patients with sporadic Paget's disease of bone. J. Bone Min. Res. 24, 484–494. doi:10.1359/jbmr.081105
Merlotti, D., Gennari, L., Galli, B., Martini, G., Calabrò, A., De Paola, V., et al. (2005). Characteristics and familial aggregation of Paget's disease of bone in Italy. J. Bone Min. Res. 20, 1356–1364. doi:10.1359/JBMR.050322
Michou, L., and Orcel, P. (2019). Has Paget's bone disease become rare? Jt. Bone Spine 86, 538–541. doi:10.1016/j.jbspin.2019.01.015
Middleton-Hardie, C., Zhu, Q., Cundy, H., Lin, J. M., Callon, K., Tong, P. C., et al. (2006). Deletion of aspartate 182 in OPG causes juvenile Paget's disease by impairing both protein secretion and binding to RANKL. J. Bone Min. Res. 21, 438–445. doi:10.1359/JBMR.051104
Morales-Piga, A. A., Rey-Rey, J. S., Corres-González, J., García-Sagredo, J. M., and López-Abente, G. (1995). Frequency and characteristics of familial aggregation of Paget's disease of bone. J. Bone Min. Res. 10, 663–670. doi:10.1002/jbmr.5650100421
Morissette, J., Laurin, N., and Brown, J. P. (2006). Sequestosome 1: Mutation frequencies, haplotypes, and phenotypes in familial paget's disease of bone. J. Bone Min. Res. 21 (2), P38–P44. doi:10.1359/jbmr.06s207
Nakatsuka, K., Nishizawa, Y., and Ralston, S. H. (2003). Phenotypic characterization of early onset Paget's disease of bone caused by a 27-bp duplication in the TNFRSF11A gene. J. Bone Min. Res. 18, 1381–1385. doi:10.1359/jbmr.2003.18.8.1381
Naot, D., Choi, A., Musson, D. S., Simsek Kiper, P. O., Utine, G. E., Boduroglu, K., et al. (2014). Novel homozygous mutations in the osteoprotegerin gene TNFRSF11B in two unrelated patients with juvenile Paget's disease. Bone 68, 6–10. doi:10.1016/j.bone.2014.07.034
Neale, S. D., Schulze, E., Smith, R., and Athanasou, N. A. (2002). The influence of serum cytokines and growth factors on osteoclast formation in Paget's disease. QJM Int. J. Med. 95, 233–240. doi:10.1093/qjmed/95.4.233
Nebot Valenzuela, E., and Pietschmann, P. (2017). Epidemiology and pathology of Paget's disease of bone - a review. Wien Med. Wochenschr 167, 2–8. doi:10.1007/s10354-016-0496-4
Numan, M. S., Jean, S., Dessay, M., Gagnon, E., Amiable, N., Brown, J. P., et al. (2019). Gene-environment interactions in Paget's disease of bone. Jt. Bone Spine 86, 373–380. doi:10.1016/j.jbspin.2018.12.007
Obaid, R., Wani, S. E., Azfer, A., Hurd, T., Jones, R., Cohen, P., et al. (2015). Optineurin negatively regulates osteoclast differentiation by modulating NF-κB and interferon signaling: Implications for paget's disease. Cell Rep. 13, 1096–1102. doi:10.1016/j.celrep.2015.09.071
Paget, J. (1877). On a form of chronic inflammation of bones (osteitis deformans). Med. Chir. Trans. 60, 37–64. doi:10.1177/095952877706000105
Ralston, S. H., and Albagha, O. M. (2018). “Genetics of paget’s disease of bone,” in Genetics of bone biology and skeletal disease (Elsevier), 439–452.
Ralston, S. H., Corral-Gudino, L., Cooper, C., Francis, R. M., Fraser, W. D., Gennari, L., et al. (2019). Diagnosis and management of paget's disease of bone in adults: A clinical guideline. J. Bone Min. Res. 34, 579–604. doi:10.1002/jbmr.3657
Ralston, S. H., and Taylor, J. P. (2019). Rare inherited forms of paget’s disease and related syndromes. Calcif. Tissue Int. 104, 501–516. doi:10.1007/s00223-019-00520-5
Rea, S. L., Walsh, J. P., Ward, L., Magno, A. L., Ward, B. K., Shaw, B., et al. (2009). Sequestosome 1 mutations in paget's disease of bone in Australia: Prevalence, genotype/phenotype correlation, and a novel non-UBA domain mutation (P364S) associated with increased NF-kappaB signaling without loss of ubiquitin binding. J. Bone Mineral Res. 24, 1216–1223. doi:10.1359/jbmr.090214
Reid, I. R. (2016). “Chapter 9 - treatment of paget’s disease of bone,” in Advances in pathobiology and management of Paget's disease of bone. Editor S. V. Reddy (Academic Press), 119–136.
Russo, S., Scotto Di Carlo, F., Maurizi, A., Fortunato, G., Teti, A., Licastro, D., et al. (2023). A mutation in the ZNF687 gene that is responsible for the severe form of Paget's disease of bone causes severely altered bone remodeling and promotes hepatocellular carcinoma onset in a knock-in mouse model. Bone Res. 11, 16. doi:10.1038/s41413-023-00250-3
Sabharwal, R., Gupta, S., Sepolia, S., Panigrahi, R., Mohanty, S., Subudhi, S. K., et al. (2014). An insight in to paget's disease of bone. Niger. J. Surg. 20, 9–15. doi:10.4103/1117-6806.127098
Saito, K., Murai, J., Kajiho, H., Kontani, K., Kurosu, H., and Katada, T. (2002). A novel binding protein composed of homophilic tetramer exhibits unique properties for the small GTPase Rab5. J. Biol. Chem. 277, 3412–3418. doi:10.1074/jbc.M106276200
Saul, D., and Kosinsky, R. L. (2021). Epigenetics of aging and aging-associated diseases. Int. J. Mol. Sci. 22, 401. doi:10.3390/ijms22010401
Scotto Di Carlo, F., Whyte, M. P., and Gianfrancesco, F. (2020). The two faces of giant cell tumor of bone. Cancer Lett. 489, 1–8. doi:10.1016/j.canlet.2020.05.031
Seton, M. (2013). Paget disease of bone: Diagnosis and drug therapy. Cleve Clin. J. Med. 80, 452–462. doi:10.3949/ccjm.80a.12142
Shaik, N. A., Nasser, K. K., Alruwaili, M. M., Alallasi, S. R., Elango, R., and Banaganapalli, B. (2021). Molecular modelling and dynamic simulations of sequestosome 1 (SQSTM1) missense mutations linked to Paget disease of bone. J. Biomol. Struct. Dyn. 39, 2873–2884. doi:10.1080/07391102.2020.1758212
Silva, I. a. L., Conceicao, N., Gagnon, E., Brown, J. P., Cancela, M. L., and Michou, L. (2018a). Molecular effect of an OPTN common variant associated to Paget's disease of bone. PLoS One 13, e0197543. doi:10.1371/journal.pone.0197543
Silva, I. a. L., Conceicao, N., Gagnon, E., Caiado, H., Brown, J. P., Gianfrancesco, F., et al. (2018b). Effect of genetic variants of OPTN in the pathophysiology of Paget's disease of bone. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 143–151. doi:10.1016/j.bbadis.2017.10.008
Solomon, L. R. (1979). Billiard-player's fingers: An unusual case of paget's disease of bone. Br. Med. J. 1, 931. doi:10.1136/bmj.1.6168.931
Taylor, J. P. (2015). Multisystem proteinopathy: Intersecting genetics in muscle, bone, and brain degeneration. Intersecting Genet. muscle, bone, Brain Degener. 85, 658–660. doi:10.1212/WNL.0000000000001862
Tekkesin, M. S., Mutlu, S., and Olgac, V. (2011). The role of RANK/RANKL/OPG signalling pathways in osteoclastogenesis in odontogenic keratocysts, radicular cysts, and ameloblastomas. Head Neck Pathology 5, 248–253. doi:10.1007/s12105-011-0271-1
Tilyard, M. W., Gardner, R. J., Milligan, L., Cleary, T. A., and Stewart, R. D. (1982). A probable linkage between familial Paget's disease and the HLA loci. Aust. N. Z. J. Med. 12, 498–500. doi:10.1111/j.1445-5994.1982.tb03830.x
Tresse, E., Salomons, F. A., Vesa, J., Bott, L. C., Kimonis, V., Yao, T.-P., et al. (2010). VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 6, 217–227. doi:10.4161/auto.6.2.11014
Tsurukai, T., Udagawa, N., Matsuzaki, K., Takahashi, N., and Suda, T. (2000). Roles of macrophage-colony stimulating factor and osteoclast differentiation factor in osteoclastogenesis. J. Bone Mineral Metabolism 18, 177–184. doi:10.1007/s007740070018
Tuck, S. P. (2020). Adult paget disease of bone: A tale of two guidelines. Rheumatology 59, 2197–2198. doi:10.1093/rheumatology/keaa345
Vallet, M., and Ralston, S. H. (2016). Biology and treatment of paget's disease of bone. J. Cell Biochem. 117, 289–299. doi:10.1002/jcb.25291
Visconti, M. R., Langston, A. L., Alonso, N., Goodman, K., Selby, P. L., Fraser, W. D., et al. (2010). Mutations of SQSTM1 are associated with severity and clinical outcome in paget disease of bone. J. Bone Min. Res. 25, 2368–2373. doi:10.1002/jbmr.132
Visconti, M. R., Usategui-Martín, R., and Ralston, S. H. (2017). Antibody response to paramyxoviruses in paget’s disease of bone. Calcif. Tissue Int. 101, 141–147. doi:10.1007/s00223-017-0265-4
Watts, G. D. J., Wymer, J., Kovach, M. J., Mehta, S. G., Mumm, S., Darvish, D., et al. (2004). Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36, 377–381. doi:10.1038/ng1332
Wei, Z., Li, S., Tao, X., Zhu, G., Sun, Z., Wei, Z., et al. (2021). Mutations in Profilin 1 cause early-onset paget's disease of bone with giant cell tumors. J. Bone Min. Res. 36, 1088–1103. doi:10.1002/jbmr.4275
Wild, P., Farhan, H., Mcewan, D. G., Wagner, S., Rogov, V. V., Brady, N. R., et al. (2011). Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333, 228–233. doi:10.1126/science.1205405
Woodman, P. G. (2003). p97, a protein coping with multiple identities. J. Cell Sci. 116, 4283–4290. doi:10.1242/jcs.00817
Xu, F., Li, W., Yang, X., Na, L., Chen, L., and Liu, G. (2020). The roles of epigenetics regulation in bone metabolism and osteoporosis. Front. Cell Dev. Biol. 8, 619301. doi:10.3389/fcell.2020.619301
Yagi, M., Miyamoto, T., Sawatani, Y., Iwamoto, K., Hosogane, N., Fujita, N., et al. (2005). DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J. Exp. Med. 202, 345–351. doi:10.1084/jem.20050645
Zheng, D. C., Shen, Y. T., Wei, Z. W., Wan, X., Xie, M. K., Yao, H. J., et al. (2021). Transcriptome sequencing reveals a lncRNA-mRNA interaction network in extramammary Paget's disease. BMC Med. Genomics 14, 291. doi:10.1186/s12920-021-01135-2
Keywords: Paget’s disease of bone, genetics factors, environmental factors, osteoclast (OCs), SQSTM1
Citation: Banaganapalli B, Fallatah I, Alsubhi F, Shetty PJ, Awan Z, Elango R and Shaik NA (2023) Paget’s disease: a review of the epidemiology, etiology, genetics, and treatment. Front. Genet. 14:1131182. doi: 10.3389/fgene.2023.1131182
Received: 24 December 2022; Accepted: 17 April 2023;
Published: 26 April 2023.
Edited by:
Luis Corral-Gudino, Universidad de Valladolid, SpainReviewed by:
Javier Del Pino, University of Salamanca, SpainSher Zaman Safi, Mahsa University, Malaysia
Copyright © 2023 Banaganapalli, Fallatah, Alsubhi, Shetty, Awan, Elango and Shaik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ramu Elango, cmVsYW5nb0BrYXUuZWR1LnNh; Noor Ahmad Shaik, bnNoYWlrQGthdS5lZHUuc2E=