 Jian Tang1†
Jian Tang1† Xingyang Wan
Xingyang Wan JunXiao Zhang
JunXiao Zhang Na Diao
Na Diao Caibin Zhang
Caibin Zhang Xiang Gao
Xiang Gao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 30 May 2023
Sec. Genetics of Common and Rare Diseases
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1130529
This article is part of the Research Topic Genetics of Inflammatory and Immune Diseases View all 19 articles
Background: Crohn’s disease (CD), a chronic gastrointestinal inflammatory disease, is increasing in China. With a focus on Han Chinese families with CD, the aim of this study was to find genetic variations that increase CD susceptibility by genome sequencing, genetic association, expression, and functional research.
Materials and methods: We performed family-based genome sequencing (WGS) analysis on 24 patients with CD from 12 families and then filtered shared potential causal variants by incorporating association results from meta-analyses of CD GWAS and immunology genes and in silico variant effect prediction algorithms. Replication analyses were performed in an independent cohort including 381 patients with CD and 381 control subjects.
Results: There were 92 genetic variants significantly associated with CD in Chinese individuals. Among them, 61 candidate loci were validated in replication analyses. As a result, patients carrying a rare frameshift variant (c.1143_1144insG; p. Leu381_Leu382fs) in gene SIRPB1 had significantly higher risk to develop CD (p = 0.03, OR 4.59, 95% CI 0.98–21.36, 81.82% vs. 49.53%). The frameshift variation induced tyrosine phosphorylation of Syk, Akt, and Jak2, elevated the expression of SIRPB1 at the mRNA and protein levels, activated DAP12, and controlled the activation of NF-κB in macrophages. Additionally, it promoted the synthesis of the pro-inflammatory cytokines IL-1, TNF-, and IL-6.
Conclusion: Our results suggest that the rare gain-of-function frameshift variant in SIRPB1 is associated in Han Chinese patients with CD. The functional mechanism of SIRPB1 and its downstream inflammatory pathways was preliminarily explored in CD.
Inflammatory bowel disease (IBD), which includes Crohn’s disease (CD) and ulcerative colitis, is a complex polygenic disorder brought on by the improper activation of effector immunologic pathways in those with a genetic predisposition. Genome-wide association studies (GWAS) that look for genetic factors influencing disease start and progression have found 240 IBD-associated loci, which have greatly improved our understanding of the biology underlying these conditions (Anderson et al., 2011; de Lange et al., 2017; de Lange et al., 2017; Franke et al., 2010; Liu et al., 2015). The success of the GWAS approach is supported by the link between neighboring common variations in human populations, but it also makes it challenging to determine with accuracy which variant is causative, the molecular consequences of that variant, and frequently even which gene is perturbed. The association of uncommon protein-altering mutations that are expected to impart a higher risk of disease may help to explain some of the missing heritability in IBD. Because they are linked with fewer neighboring variants, rare variants with greater impact sizes may be easier to understand mechanistically. It is still unknown, nevertheless, how much of the heritability of complex disorders can be attributed to uncommon variations. The promise to better comprehend the molecular and genetic architecture of an exemplary complex disease is thus provided by well-powered investigations of uncommon variations in IBD.
IBD is assumed to have a significant genetic link because the most significant risk factor for the condition at any age is a family history of IBD (Childers et al., 2014; Kuwahara et al., 2012). Patients with IBD who have a family history of the condition frequently present it at a younger age, are more likely to have extra-intestinal manifestations, develop perforating disease, and need longer follow-up than patients without a family history, showing significant an increased genetic susceptibility to the condition (Kuwahara et al., 2012). Therefore, family-based IBD cohort genomic analyses are helpful for understanding the genetic architecture of IBD. IBD susceptibility is influenced by a variety of genetic variables. Whole genome sequencing (WGS), in conjunction with recent significant technological advancements, has made it possible to identify uncommon and novel harmful variants in IBD, providing a deeper understanding of genetic differences within the human genome. Due to this, GWAS have been created to objectively identify genetic risk factors for complex polygenic disorders. According to our hypothesis, rare or novel variations, such as those in genes linked to the innate immune system, are more likely to play a role in the development of CD in patients with a family history of the disease. We used WGS to investigate particular genes or pathways implicated in this disease state. Our capacity to research uncommon variations and ascertain the disease’s genetic origin has been transformed by WGS. To address this question, in this study, we identified and replicated a novel rare causative frameshift variant associated with CD and explored into how it affected functionality.
The genome sequence of 24 Han Chinese patients with CD from 12 families were obtained (Table 1). Written informed consent was provided by the attending parents or legal guardians of the pediatric participants. This study was approved by the Research Ethics Committee (REC) of the Sixth Affiliated Hospital of Sun Yet Sen University (SYSU) (N0.2020ZSLYEC-006).
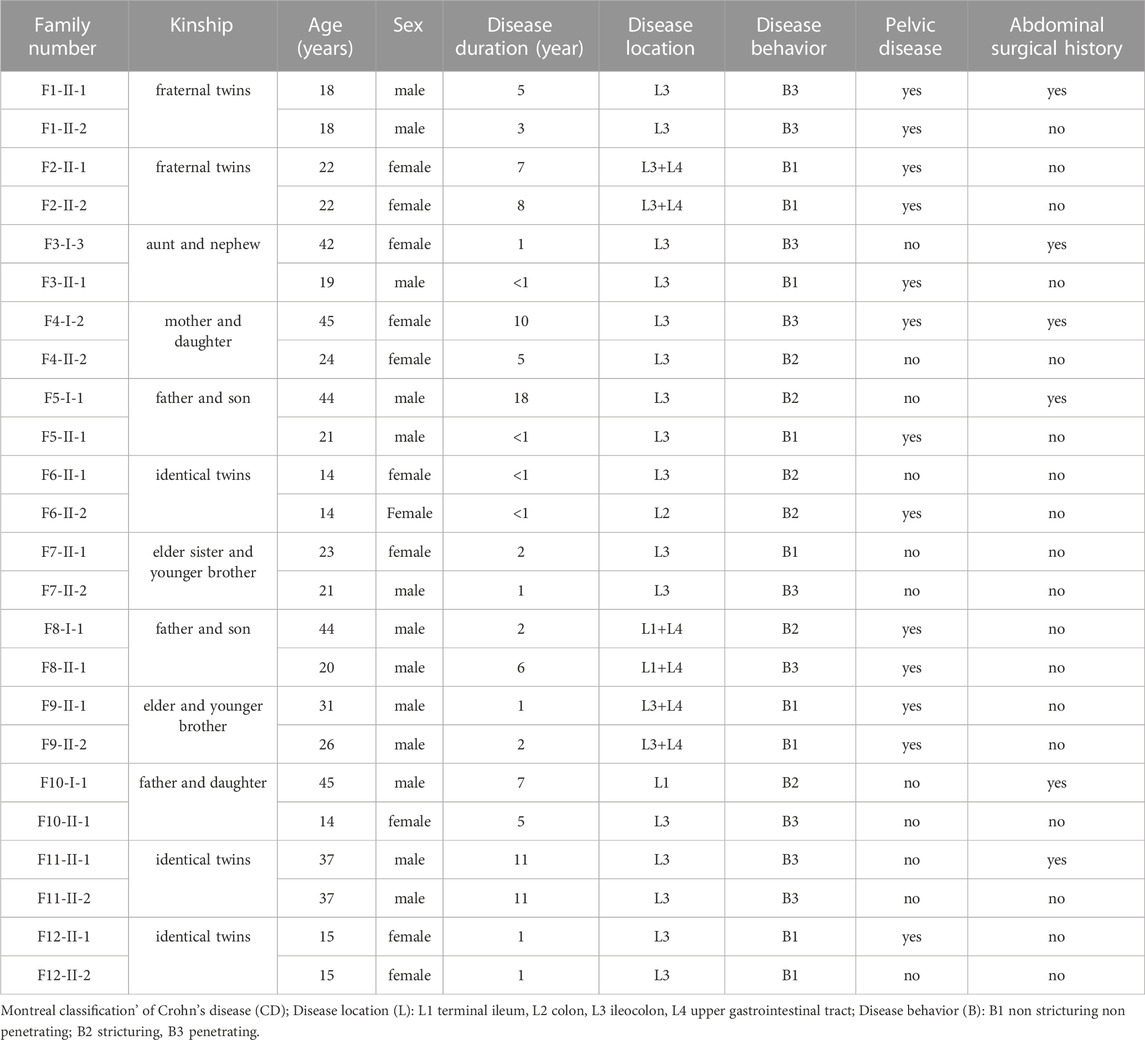
TABLE 1. The clinical characteristics of enrolled families of Crohn’s disease.
Using a QIAamp DNA Kit (QIAGEN, Hilden, Germany), genomic DNA was extracted from peripheral blood cells in accordance with the manufacturer’s protocol. DNA samples were quantified using a Qubit (Thermo Fisher Scientific). A total of 2 μg of each DNA sample was sent to the Beijing Genome Institute (BGI, Shenzhen, China) for WGS using the BGISEQ-500, according to the manufacturer’s guidelines. According to the manufacturer’s recommendations, the genomic DNA was briefly split by ultrasound on a Covaris E220 (Covaris) to DNA segments between 50 bp—800 bp. The fragmented DNA was then exposed to end-repair, phosphorylation, and A-tailing procedures after being further chosen to 100bp-300bp using AMpure XP Beads (Beckman Coulter, Indiana, United States). The A-tailed segments were ligated to the BGISEQ-500 platform-specific adaptors, and the ligated fragments were then purified and amplified using PCR. Finally, single-stranded DNA circles were produced by the circularization process. The libraries were sequenced using 50 bp paired-end reads on the BGISEQ-500 platform following quantification and qualifying.
SOAPnuke was used to filter the raw sequencing reads (Li et al., 2009b) (N rating >10%, low quality rating >50%, and quality rating<5) and Burrows-Wheeler Aligner (BWA v0.7.17) to align to the UCSC human reference genome (hg19) (Li and Durbin, 2009; Li and Durbin, 2010). The coordinates were sorted using Samtools (version 1.3.1) and duplicates were identified using Picard (version 1.129, http://picard.sourceforge.net) (Li et al., 2009a). Using GATK HaplotypeCaller (McKenna et al., 2010), single nucleotide substitution variants (SNV) and brief insertions and deletions (indels) were identified (McKenna et al., 2010). We used the GATK Variant Quality Score Recalibration (VQSR) that uses machine learning algorithm to filter the raw variant callset. The GATK VQSR used high-quality known variant sets as training and truth resources and built a predictive model to filter spurious variants. The SNPs and InDels marked PASS in the output VCF file were high-confident variation set. For SNPs recalibration strategy, we used the following datasets and features to train the model. (a) Training sets: HapMap V3.3, Omni2.5 M genotyping array data and high-confidence SNP sites produced by the 1000 Genomes Project. (b) Features: Coverage (DP), Quality/depth (QD), Fisher test on strand bias (FS), Odds ratio for strand bias (SOR), Mapping quality rank sum test (MQRankSum), Read position rank sum test (ReadPosRankSum), RMS mapping quality (MQ). For InDels recalibration strategy, we used the following datasets and features to train the model. (a) Training sets: Mills 1000G gold standard InDel set. (b) Features: Coverage (DP), Quality/depth (QD), Fisher test on strand bias (FS), Odds ratio for strand bias (SOR), Mapping quality rank sum test (MQRankSum), Read position rank sum test (ReadPosRankSum).
All germline SNV and indels were annotated using an in-house annotation pipeline, as described previously (Neveling et al., 2013; de Voer et al., 2013; Vissers et al., 2010). High-confidence calls (i.e., ≥10 reads, ≥5 variant reads, and ≥20% variant reads) were subsequently prioritized for variants that were non-synonymous and were absent in our in-house variant database (2,037 in-house analyzed exomes, mostly from European ancestry). Next, we removed all variants present with a MAF of >0.001 in dbSNPv138, the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project database (ESP, 6503 exomes, http://evs.gs.washington.edu/EVS/), the Exome Aggregation Consortium (Lek et al., 2016) and Thousand Genome Project. Subsequently, family index patients shared non-synonymous variants that result in alterations in protein function, including protein truncation, splice site defects and missense mutations at highly conserved (phyloP ≥ 3.0) nucleotide positions, were included in our analyses. Alamut v.2.0 software (Interactive Biosoftware) and integrated mutation prediction software (align GVDV, SIFT and PolyPhen-2) (Adzhubei et al., 2010; Kumar et al., 2009; Vissers et al., 2010)packages were used for analyses of the identified variants. The prediction of splicing effects was evaluated based on five different algorithms (SpliceSiteFinder, MaxEntScan, NNSPLICE, GeneSplicer, Human Splicing Finder) through the bioinformatics tools of the Alamut v.2.0 software.
By using GWAS, we first targeted germline variations in genes at susceptibility loci known to be linked to IBD (Anderson et al., 2011; de Lange et al., 2017; Ellinghaus et al., 2016; Huang et al., 2017; Julia et al., 2014; Kenny et al., 2012; Liu et al., 2015; Parkes et al., 2007; Yamazaki et al., 2013; Yang et al., 2014). Genetic defects in innate immunity that impair intestinal bacterial sensing are linked to the development of IBD (Cananzi et al., 2021). Recent developments in molecular biology have uncovered crucial details about the genetic basis of numerous inflammatory diseases. Next to the identification of variants in known IBD GWAS genes, we searched for potential pathogenic variants in novel candidate genes using the remaining genome data of our CD family cohort. We concentrated on genes that fulfilled the following criteria while choosing these variants: (Supplementary Table S2): 1) genes with variations that caused protein truncation (such as putative frameshifts, nonsense variations, and variations at canonical splice sites), as well as non-synonymous variations with a PhyloP score of more than 3.0, were chosen; 2) the International Union of Immunological Societies Expert Committee on Primary Immunodeficiency’s collection of primary immune deficiency (PID) genes (Picard et al., 2015); and 3) genes involved in pathways implicated in IBD pathogenesis, including innate immune system, immune system, and neutrophil degranulation pathway (Belinky et al., 2015; Kanegane, 2018).
After PCR amplification, WGS-identified candidate variant of SIRPB1 was verified using Sanger sequencing. The Primer3 software program was used to build PCR primers in silico. Standard PCR procedures were used on an Applied Biosystems Dual 96-Well GeneAmp PCR System 9,700 (primer sequences available upon request). Using the software package Vector NTI, variant analyses were carried out (Invitrogen, Paisley, United Kingdom).
Candidate variant validation analysis was performed on 381 probands with IBD and 381 unrelated individuals recruited by the Sixth Affiliated Hospital of SYSU using MassARRAY (Derkach et al., 2013). All individuals recruited were peotected by the REC of the Sixth Affiliated Hospital of SYSU (N0.2020ZSLYEC-006). The phenotypes of the controls were assessed, and no known gastrointestinal or immunological findings were reported. Fisher’s exact testing was conducted, and statistical significance was set at p < 0.05 (Derkach et al., 2013).
Both historical standard hematoxylin and eosin histological sections from the patient who was found to have the SIRPB1 p. Leu381 Leu382fs variation and IBD controls were assessed.
On 4 μm segments of formalin-fixed paraffin-embedded (FFPE) tissue samples containing terminal ileum tissue from the patient identified as harboring the SIRPB1 p. Leu381 _Leu382fs variation and IBD controls, SIRPB1 expression was analyzed by immunohistochemistry (IHC). Using a BenchMark XT automated tissue staining machine (Ventana Medical Systems, Tucson, AZ, United States), IHC staining was carried out in accordance with the manufacturer’s verified protocols.
All the microarray samples used in this study were systematically searched and downloaded from NCBI-GEO (Barrett et al., 2013) after the manual curation of the sample details. We obtained gene expression data of mucosal biopsies in CD patients and normal controls from following array data series: GSE75214, GSE36807 and GSE59071. The bioinformatics online tool GEO2R was used to analyze the mRNA expression of SIRPB1.
To study the variant type in vitro to understand its functional implications in more detail, we established THP-1 cell lines that stably expressed wild-type SIRPB1 (SIRPB1wt) and mutant SIRPB1 (SIRPB111143iG, c.1143_1144insG; p. Leu381_Leu382fs). Human SIRPB1wt and SIRPB111143iG expression vectors (pEZ-M02/SIRPB1wt and pEZ-M02/SIRPB111143iG, respectively) were established. According to the manufacturer’s instructions, Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Waltham, MA, United States) was used to transfect THP-1 cells with 0.5 µg of either pEZ-M02/SIRPB1wt and pEZ-M02/SIRPB111143iG. Continuous neomycin treatment at 450 μg/mL was used to select transfectants that could consistently express the inserted vector plasmid (Thermo Fisher Scientific). The limited dilution approach was used to clone neomycin-resistant cells, which were then kept alive in medium containing neomycin.
By administering THP-1 monocytes 100 ng/mL phorbol 12-myristate 13-acetate (PMA; Sigma) for 48 h, the macrophage-like state was generated. Then, THP-1 macrophages were transfected with 0.5 µg pEZ-M02/SIRPB1wt and pEZ-M02/SIRPB111143iG plasmid for 4 h. After transfection, THP-1 macrophages were incubated in complete medium for 48 h and stimulated with LPS (100 ng/mL). THP-1 cells were lysed in ice-cold lysis buffer for western blotting, and proteins were separated on 10% SDS page. The main antibodies against DAP12, p-Syk/Syk, p-Akt/Akt, p-Jak2/Jak2, and anti-GAPDH were then used to probe the membranes. According to the manufacturer’s instructions, the culture medium was suspended for ELISA assays and TNF-α, IL-1, and IL-6 ELISA kits were used to detect the substances.
Prism version 8.0 software (GraphPad) was used to perform the statistical analysis. Both the Student’s t-test and the Dunnett’s test were used to determine the significance of differences. When more than two groups were compared, a one-way ANOVA was performed. The data are shown as mean ± SE, and a significance level of 0.05 was used.
Studies of twins and familial clustering of disease clearly indicate that IBD, especially CD, is a hereditary disorder (Halfvarson et al., 2003). Therefore, we hypothesized that family-based CD is more probable to have occurred as a result of uncommon or unique variations and may play a role in the disease’s development. Thus, 24 patients with CD from 12 families in the case database of our IBD center were enrolled for WGS analysis. Table 1 provides a summary of the clinical traits of the individuals that were enrolled. Among them, there were two pairs of fraternal twins, three pairs of identical twins, four pairs of father or mother and daughter or son, two pairs of elder sister or brother and younger brother, and one pair of aunt and niece (Figure 1). All the patients were of Han origin, including 13 men (54.17%) and 11 women (45.83%). The median age of participants in this study was 22 years (range 14–45 years). Regarding disease location, 20 patients were diagnosed with ileocolonic inflammation (L3, 83.33%), one patient with colonic inflammation (L2, 4.17%), 3 patients with ileal inflammation (L1, 12.50%), and six patients had concomitant upper gastrointestinal disease (25.00%). Regarding disease behavior, nine patients were accompanied with non-stricturing non-penetrating lesions (B1, 37.50%), six patients had stricturing lesions (B2, 25.00%), and nine patients had penetrating lesions (B3, 37.50%). A total of 13 patients (54.17%) had pelvic disease and six patients (25.00%) had a history of abdominal surgery.
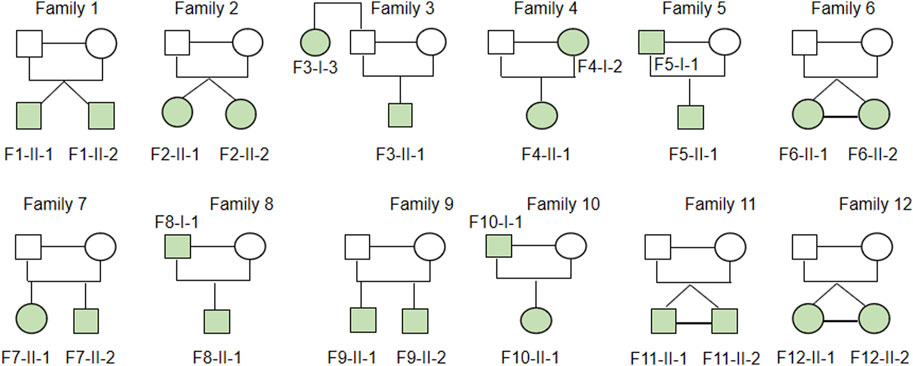
FIGURE 1. Heritability of enrolled CD family. Two pairs of fraternal twins, three pairs of identical twins, four pairs father or mother and daughter or son, two pairs of elder sister or brother and younger brother, one pair of aunt and niece were included in family based WGS analysis.
To further understand the genetic basis of IBD, we used WGS to look for uncommon, possibly disease-causing coding variations in familiar CD patients (Table1). A substantial percentage of coding variants discovered across individuals in a family were prioritized for further exploration using a series of progressive variant filters. We identified on average 3,430,329 variants (range: 3,393,972–3,466,204) per genome. A prioritization scheme was applied to identify candidate variants as shown in table 2. After rigorous bioinformatics analysis steps (see Methods), there were 92 genetic variants significantly associated with CD in Chinese individuals including several known IBD related gene including NOD2, ZPBP2, TNFSF15, LSP1, SDCCAG3, MUC19. Then, we adopted the MassArray Analyzer system (Sequenom, Inc., SanDiego, CA, United States) for genotyping those genetic variants. We designed primers for 89 variants in Sequenom official websites, while, there is suitable primer for the remaining 3 variants. However, those 89 primers were distributed in 5 panels, due to the limited budget, only the top 2 largest panels (a total of 61 variants were included, about 30 variants per panel) used for the subsequently assay. We finally generated a candidate variants list including 61 variants for replication analyses with an independent cohort including 381 patients with CD and 381 control subjects (Table 3).

TABLE 2. Prioritization scheme for genome data analysis of 24 CD patients.
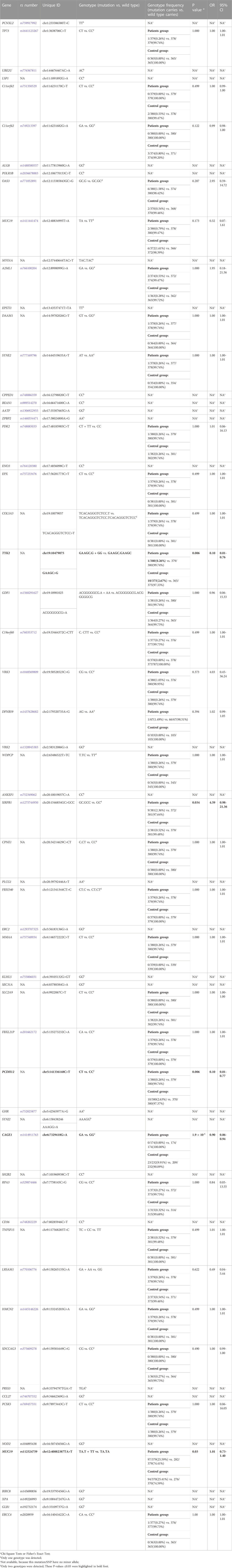
TABLE 3. IBD susceptibility analysis of identified gene variants.
Notably, we identified the frameshift variant SIRPB1 p. Leu381_Leu382fs in two index patients, F7-II-1 and F7-II-2, from Family 7 (Figure 2). Patient F7-II-1 was the elder sister of patient F7-II-2, whose age was 23 years with a disease duration of about 2 years, and patient F7-II-2 was the younger brother whose age was 21 years with a disease duration of approximately 1 year. Both manifested as ileocolonic inflammation (L3). The disease behavior of F7-II-1 was non-stricturing non-penetrating (B1), whereas the disease behavior of F7-II-2 was penetrating (B3). For replication analysis, the blood sample of 384 CD patients including 104 females and 280 males in our database were used for microarray analysis to verify the susceptibility gene mutation. The mean age of these patients was 27.0 ± 10.4 years-old. The 384 blood samples of control group were collected from healthy testing population including 170 female and 214 female and the mean age of these patients was 34.7 ± 9.1 years-old. The unique uncommon frameshift variant in SIRPB1 was considerably enriched in CD patients compared to controls, according to genotyping analysis (p = 0.03, OR 4.59, 95% CI 0.98–21.36, 81.82% vs. 49.53%). (Table 3). The clinical characteristics of patients with CD identified to harbor the SIRPB1 p. Leu381_Leu382fs variant are listed in Table 4.

FIGURE 2. Identification of a rare frameshift mutation of SIRPB1 gene A. rare frameshift mutation (c.1143_1144insG; p. Leu381_Leu382fs) was identified in an innate immune gene SIRPB1 in both probands of Family CD-7.
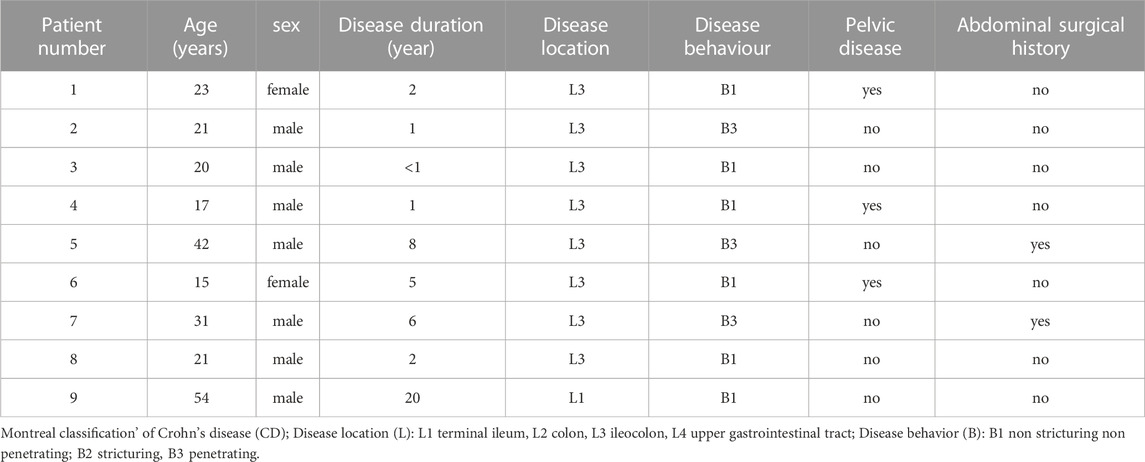
TABLE 4. The clinical characteristics of CD patients with SIRPB1 gene mutation.
A member of the family of signal-regulating proteins (SIRP) and of the immunoglobulin superfamily, SIRPβ (also known as CD172b) is encoded by SIRPB1 (van den Berg et al., 2005). Previous research on SIRPB1 mostly focused on its biochemical properties and functions and discovered that it stimulates DAP12 and Syk tyrosine phosphorylation, which then activates the mitogen-activated protein kinase (MAPK) pathway to increase phagocytosis in macrophages (Hayashi et al., 2004). However, the role of SIRPB1 in IBD pathogenesis has not yet been reported. Therefore, we first assessed the expression levels of SIRPB1 by analyzing publicly available data from patients with IBD and healthy individuals. According to the findings, patients with active CD (A-CD) had considerably higher levels of SIRPB1 expression in their ileocolonic tissue than either healthy individuals or patients with CD that was in remission (R-CD) (Figures 3G–I), indicating that SIRPB1 is possibly involved in the progression of CD. Next, we assessed the expression levels of SIRPβ in the ileocolonic tissue of variant and wild-type patients with CD using IHC (Supplementary Table S3). Notably, patients with CD and the SIRPB1 variant exhibited significantly higher levels of SIRPβ than those with CD and wild-type SIRPB1 (Figures 3A,D). Consistently, SIRPβ-mediated transduction signal molecules, such as DAP12 and p-Syk, were also significantly upregulated in patients with CD and the variant compared with WT controls (Figures 3B,C,E,F). Our data suggest that the higher expression of SIRPβ was caused by the frameshift variation of SIRPB1.
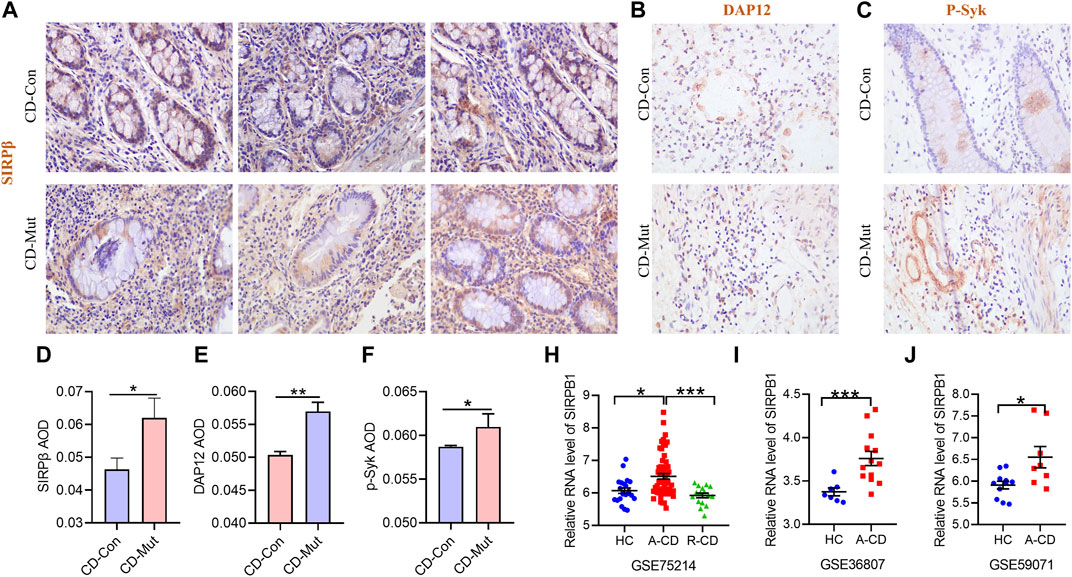
FIGURE 3. Higher expression of SIRPB1 in mutant CD patients. A/B/C Immunohistochemistry (IHC) staining of SIRPβ (A), DAP12 (B), and p-Syk (C) in ileocolonic tissue from wild type CD patients and control patients; (D,E,F) Statistical analyses analysis of IHC staining; G/H/I SIRPB1 expression was assessed using a publicly accessible data. The following array data series were analyzed to generate the human patient expression data: GSE75214; GSE36807; GSE59071. GSE75214 (G): mucosal biopsies of ileal mucosa of 11 health controls (HC), 15 inactive CD patients (R-CD) and 50 active CD patients (A-CD) were used to analyze mRNA expression of SIRPB1. GSE36807 (H): intestinal biopsies from 7 health controls (HC), and 15 CD patients were used to analyze mRNA expression of SIRPB1. GSE59071 (I) mucosal biopsies were obtained at endoscopy from 8 health controls (HC) and Crohn’s disease (CD) patients.
As documented above, the frameshift variant in SIRPB1 led to higher expression of SIRPβ; however, the function of the rare SIRPB1 frameshift variant still needs to be elucidated. The interaction of SIRPβ with the activating adaptor protein DAP12, which carries an immunoreceptor tyrosine-based activation motif (ITAM) and transmits activating signals, depends on the presence of a basic amino acid side chain in the transmembrane domain of SIRPβ (Lanier and Bakker, 2000; Tomasello and Vivier, 2005). An earlier study showed that phosphorylating Syk and MAPK and crosslinking mouse SIRPβ with monoclonal antibodies increases neutrophil migration and macrophage phagocytosis (Hayashi et al., 2004; Liu et al., 2005). As shown in Figure 4, the frameshift variant of SIRPB1 gene led to an alteration of the amino acid sequence located both on the transmembrane and the cytoplasm. Therefore, we hypothesized that the frameshift variant of SIRPB1 could make functional contributions to the inflammation process.

FIGURE 4. Predicted amino-acid sequence from wild-type control and patients with frameshift SIRPB1 mutation.
To investigate the potential effects of the identified novel frameshift variant of SIRPB1, Plasmids carrying either SIRPB1wt and SIRPB111143iG sequence were transfected into THP-1 cells that had been stimulated with PMA for 48 h. In line with a previous report, compared with THP-1 cells transfected with the empty vector (NC), THP-1 cells with SIRPB1 overexpression (SIRPB1wt) exhibited increased activation of DAP12 and NF-κB and elicited tyrosine phosphorylation of Syk, Akt, and Jak2, causing increased IL-1β, TNF-α, and IL-6 release (Figures 5A,B,C,D,E). Furthermore, compared with SIRPB1wt cells, THP-1 cells expressing variant SIRPB1 (SIRPB111143iG) displayed higher SIRPβ expression, enhanced activation of DAP12 and NF-κB, increased tyrosine phosphorylation of Syk, AKT, and Jak2, and higher secretion of IL-1β, TNF-α, and IL-6 (Figures 5A,B,C,D,E). Taken together, our results show that the frameshift variant of SIRPB1 amplified the function of the wild-type SIRPB1 gene.

FIGURE 5. SIRPB1 mutation promote inflammatory response of macrophages in vitro. (A) NF-kB transcriptional activity of THP-1 cells from different groups was detected by a luciferase reporter assay; (B,C,D) IL-1β, IL-6 and TNFα levels in culture medium of THP-1 cells from different groups; (E) Western Blotting of signal transduction molecules in THP-1 cells from different groups.
SIRPs are a class of cell surface signaling receptors that are produced differently in myeloid and neural cells. They each include three extracellular Ig-like domains (Adams et al., 1998; Kharitonenkov et al., 1997). Despite having very identical extracellular domains, SIRPs can be distinguished as activating (α) or inhibitory (β) isoforms using conventional patterns in their cytoplasmic or transmembrane regions (Kharitonenkov et al., 1997). SIRPα, which binds to its ligand CD47, was found to inhibit signaling pathways that are mediated by receptor tyrosine kinase, as its cytoplasmic domains contain immunoreceptor tyrosine-based inhibitory motifs (ITIMs), which recruit the phosphatase SH2-domain-containing proteins SHP-1 and SHP-2 in vivo (Barclay and Brown, 2006). Contrary to SIRPα, SIRPβ lacks the sequence patterns necessary to attach SHP-1 and SHP-2 in its short six-amino acid cytoplasmic domain. The transmembrane domain of SIRPβ, however, has a charged amino acid residue that can bind to DAP12, which possesses a single cytoplasmic ITAM, and activate cell-mediated cytotoxicity and cytokine release (Dietrich et al., 2000; Liu et al., 2005; McVicar et al., 1998).
In the present study, a rare frameshift variant (c.1143_1144insG; p. Leu381_Leu382fs) in the innate immunity gene SIRPB1 was identified. Genotyping analysis revealed that the novel frameshift variant in SIRPB1 was significantly enriched in patients compared to that in healthy controls. Based on publicly available gene expression data, our research showed that patients with CD in the active stage had considerably higher relative expression of SIRPβ, indicating that SIRPβ is involved in the progression of intestinal inflammation. In addition, the frameshift variant of SIRPB1 leads to an alteration of the amino acid sequence located both in the transmembrane and cytoplasmic regions. Therefore, we tested the functional contribution of the frameshift variant of SIRPB1. Our data show that, compared with WT controls, THP-1 cells transfected with the variant SIRPB1 sequence displayed higher expression of SIRPβ and its adaptor protein DAP12, enhanced activation of subsequent signal transduction, and increased pro-inflammatory cytokines production and NF-κB expression. Accordingly, IHC data of ileocolonic tissue implied that patients with CD and variant SIRPB1 expressed higher levels of SIRPβ and its adaptor protein DAP12. Collectively, we found a rare frameshift variant (c.1143_1144insG; p. Leu381_Leu382fs) in SIRPB1 that leads to alterations in the amino acid sequence located both in the transmembrane and cytoplasmic domains, could contribute to inflammation exacerbation by promoting the expression of SIRPβ and its adaptor protein, DAP12. We hypothesized that the frameshift variant in SIRPB1 may result in conformational changes of SIRPβ or form a functional cytoplasmic domain owing to a longer amino acid sequence, which needs to be elucidated in future studies.
In our study, the data analysis shows that the rare gain-of-function frameshift variant (c.1143_1144insG; p. Leu381_Leu382fs) in SIRPB1 is associated with Han Chinese patients with CD and provides insights that the variant in SIRPB1 upregulated the activation of NF-κB and increased the production of IL-1β, TNF-α, and IL-6 by inducing DAP12, Syk, Akt, and Jak2 to become tyrosine phosphorylated in macrophages, CD pathogenesis is facilitated. This study has several limitations. The familiar CD patients and the subsequent cohort for replication analyses are all Han Chinese from a single IBD center, so, further validation in other population is necessary. As well, due to limited budget and lack of suitable primers, only 61 variants were validated in replication analyses. Although the function of frameshift variant in SIRPB1 were explored in vitro using THP-1 cells, further investigation using other cell lines and also in vivo are required. And also, for replication analysis, a p-value <0.05 without multiple testing correction was used in validation, so, further function study was adopted to verify the susceptive possibility of the SIRPB1 variant in CD patients.
The data presented in the study are deposited in the SRA repository and can be accessed at: https://dataview.ncbi.nlm.nih.gov/object/PRJNA962047.
The studies involving human participants were reviewed and approved by Research Ethics Committee (REC) of the Sixth Affiliated Hospital of Sun Yet Sen University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
XG and DR as correspondent author: conception and design, revising the article, final approval of the version to be published. JT, XW, and JZ as co-first author: conception and design, drafting the article, final approval of the version to be published. ND: analysis and interpretation of data, drafting the article, final approval of the version to be published. CZ: analysis and interpretation of data, final approval of the version to be published.
This project was supported by the National Nature Science Foundation of China (81870382), the Major Scientific and Technological Project of Guangdong Province (A2020123), the Science and Technology Program of Guangzhou (201903010081), and the Youth Project for National Nature Science Foundation of China (82100547).
We thank the patients and their families contributing to this study.
Author JZ was empolyed by SequMed Biotech Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1130529/full#supplementary-material
Adams, S., van der Laan, L. J., Vernon-Wilson, E., Renardel, D. L. C., Dopp, E. A., Dijkstra, C. D., et al. (1998). Signal-regulatory protein is selectively expressed by myeloid and neuronal cells. J. Immunol. 161 (4), 1853–1859. doi:10.4049/jimmunol.161.4.1853
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods. 7 (4), 248–249. doi:10.1038/nmeth0410-248
Anderson, C. A., Boucher, G., Lees, C. W., Franke, A., D'Amato, M., Taylor, K. D., et al. (2011). Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 43 (3), 246–252. doi:10.1038/ng.764
Barclay, A. N., and Brown, M. H. (2006). The SIRP family of receptors and immune regulation. Nat. Rev. Immunol. 6 (6), 457–464. doi:10.1038/nri1859
Belinky, F., Nativ, N., Stelzer, G., Zimmerman, S., Iny, S. T., Safran, M., et al. (2015). PathCards: Multi-source consolidation of human biological pathways. Database (Oxford) 2015, 2015. doi:10.1093/database/bav006
Cananzi, M., Wohler, E., Marzollo, A., Colavito, D., You, J., Jing, H., et al. (2021). IFIH1 loss-of-function variants contribute to very early-onset inflammatory bowel disease. Hum. Genet. 140 (9), 1299–1312. doi:10.1007/s00439-021-02300-4
Childers, R. E., Eluri, S., Vazquez, C., Weise, R. M., Bayless, T. M., and Hutfless, S. (2014). Family history of inflammatory bowel disease among patients with ulcerative colitis: A systematic review and meta-analysis. J. Crohns Colitis. 8 (11), 1480–1497. doi:10.1016/j.crohns.2014.05.008
de Lange, K. M., Moutsianas, L., Lee, J. C., Lamb, C. A., Luo, Y., Kennedy, N. A., et al. (2017). Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 49 (2), 256–261. doi:10.1038/ng.3760
de Voer, R. M., Geurts, V. K. A., Weren, R. D., Ligtenberg, M. J., Smeets, D., Fu, L., et al. (2013). Germline mutations in the spindle assembly checkpoint genes BUB1 and BUB3 are risk factors for colorectal cancer. Gastroenterology 145 (3), 544–547. doi:10.1053/j.gastro.2013.06.001
Derkach, A., Lawless, J. F., and Sun, L. (2013). Robust and powerful tests for rare variants using Fisher's method to combine evidence of association from two or more complementary tests. Genet. Epidemiol. 37 (1), 110–121. doi:10.1002/gepi.21689
Dietrich, J., Cella, M., Seiffert, M., Buhring, H. J., and Colonna, M. (2000). Cutting edge: Signal-regulatory protein beta 1 is a DAP12-associated activating receptor expressed in myeloid cells. J. Immunol. 164 (1), 9–12. doi:10.4049/jimmunol.164.1.9
Ellinghaus, D., Jostins, L., Spain, S. L., Cortes, A., Bethune, J., Han, B., et al. (2016). Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat. Genet. 48 (5), 510–518. doi:10.1038/ng.3528
Franke, A., McGovern, D. P., Barrett, J. C., Wang, K., Radford-Smith, G. L., Ahmad, T., et al. (2010). Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat. Genet. 42 (12), 1118–1125. doi:10.1038/ng.717
Halfvarson, J., Bodin, L., Tysk, C., Lindberg, E., and Jarnerot, G. (2003). Inflammatory bowel disease in a Swedish twin cohort: A long-term follow-up of concordance and clinical characteristics. Gastroenterology 124, 1767–1773. doi:10.1016/s0016-5085(03)00385-8
Hayashi, A., Ohnishi, H., Okazawa, H., Nakazawa, S., Ikeda, H., Motegi, S., et al. (2004). Positive regulation of phagocytosis by SIRPbeta and its signaling mechanism in macrophages. J. Biol. Chem. 279, 29450–29460. doi:10.1074/jbc.M400950200
Huang, H., Fang, M., Jostins, L., Umicevic, M. M., Boucher, G., Anderson, C. A., et al. (2017). Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 547 (7662), 173–178. doi:10.1038/nature22969
Julia, A., Domenech, E., Chaparro, M., Garcia-Sanchez, V., Gomollon, F., Panes, J., et al. (2014). A genome-wide association study identifies a novel locus at 6q22.1 associated with ulcerative colitis. Hum. Mol. Genet. 23 (25), 6927–6934. doi:10.1093/hmg/ddu398
Kanegane, H. (2018). Inflammatory bowel diseases and primary immunodeficiency diseases. Immunol. Med. 41 (4), 154–161. doi:10.1080/25785826.2018.1556025
Kenny, E. E., Pe'Er, I., Karban, A., Ozelius, L., Mitchell, A. A., Ng, S. M., et al. (2012). A genome-wide scan of Ashkenazi Jewish Crohn's disease suggests novel susceptibility loci. PLoS Genet. 8 (3), e1002559. doi:10.1371/journal.pgen.1002559
Kharitonenkov, A., Chen, Z., Sures, I., Wang, H., Schilling, J., and Ullrich, A. (1997). A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature 386 (6621), 181–186. doi:10.1038/386181a0
Kumar, P., Henikoff, S., and Ng, P. C. (2009). Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4 (7), 1073–1081. doi:10.1038/nprot.2009.86
Kuwahara, E., Asakura, K., Nishiwaki, Y., Inoue, N., Watanabe, M., Hibi, T., et al. (2012). Effects of family history on inflammatory bowel disease characteristics in Japanese patients. J. Gastroenterol. 47 (9), 961–968. doi:10.1007/s00535-012-0558-3
Lanier, L. L., and Bakker, A. B. (2000). The ITAM-bearing transmembrane adaptor DAP12 in lymphoid and myeloid cell function. Immunol. Today 21 (12), 611–614. doi:10.1016/s0167-5699(00)01745-x
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536 (7616), 285–291. doi:10.1038/nature19057
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26 (5), 589–595. doi:10.1093/bioinformatics/btp698
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009a). The sequence alignment/map format and SAMtools. Bioinformatics 25 (16), 2078–2079. doi:10.1093/bioinformatics/btp352
Li, R., Yu, C., Li, Y., Lam, T. W., Yiu, S. M., Kristiansen, K., et al. (2009b). SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 25 (15), 1966–1967. doi:10.1093/bioinformatics/btp336
Liu, Y., Soto, I., Tong, Q., Chin, A., Buhring, H. J., Wu, T., et al. (2005). SIRPbeta1 is expressed as a disulfide-linked homodimer in leukocytes and positively regulates neutrophil transepithelial migration. J. Biol. Chem. 280 (43), 36132–36140. doi:10.1074/jbc.M506419200
Liu, J. Z., van Sommeren, S., Huang, H., Ng, S. C., Alberts, R., Takahashi, A., et al. (2015). Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47 (9), 979–986. doi:10.1038/ng.3359
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20 (9), 1297–1303. doi:10.1101/gr.107524.110
McVicar, D. W., Taylor, L. S., Gosselin, P., Willette-Brown, J., Mikhael, A. I., Geahlen, R. L., et al. (1998). DAP12-mediated signal transduction in natural killer cells. A dominant role for the Syk protein-tyrosine kinase. J. Biol. Chem. 273 (49), 32934–32942. doi:10.1074/jbc.273.49.32934
Neveling, K., Feenstra, I., Gilissen, C., Hoefsloot, L. H., Kamsteeg, E. J., Mensenkamp, A. R., et al. (2013). A post-hoc comparison of the utility of sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum. Mutat. 34 (12), 1721–1726. doi:10.1002/humu.22450
Parkes, M., Barrett, J. C., Prescott, N. J., Tremelling, M., Anderson, C. A., Fisher, S. A., et al. (2007). Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat. Genet. 39 (7), 830–832. doi:10.1038/ng2061
Picard, C., Al-Herz, W., Bousfiha, A., Casanova, J. L., Chatila, T., Conley, M. E., et al. (2015). Primary immunodeficiency diseases: An update on the classification from the international union of immunological Societies Expert committee for primary immunodeficiency 2015. J. Clin. Immunol. 35 (8), 696–726. doi:10.1007/s10875-015-0201-1
Tomasello, E., and Vivier, E. (2005). KARAP/DAP12/TYROBP: Three names and a multiplicity of biological functions. Eur. J. Immunol. 35 (6), 1670–1677. doi:10.1002/eji.200425932
van den Berg, T. K., van Beek, E. M., Buhring, H. J., Colonna, M., Hamaguchi, M., Howard, C. J., et al. (2005). A nomenclature for signal regulatory protein family members. J. Immunol. 175 (12), 7788–7789. doi:10.4049/jimmunol.175.12.7788
Vissers, L. E., de Ligt, J., Gilissen, C., Janssen, I., Steehouwer, M., de Vries, P., et al. (2010). A de novo paradigm for mental retardation. Nat. Genet. 42 (12), 1109–1112. doi:10.1038/ng.712
Yamazaki, K., Umeno, J., Takahashi, A., Hirano, A., Johnson, T. A., Kumasaka, N., et al. (2013). A genome-wide association study identifies 2 susceptibility Loci for Crohn's disease in a Japanese population. Gastroenterology 144 (4), 781–788. doi:10.1053/j.gastro.2012.12.021
Yang, S. K., Hong, M., Zhao, W., Jung, Y., Baek, J., Tayebi, N., et al. (2014). Genome-wide association study of Crohn's disease in Koreans revealed three new susceptibility loci and common attributes of genetic susceptibility across ethnic populations. Gut. 63(1), 80–87. doi:10.1136/gutjnl-2013-305193
Keywords: Crohn ‘s disease, han Chinese patients, gene susceptibility, whole gene sequencing, SIRPB1
Citation: Tang J, Wan X, Zhang J, Diao N, Zhang C, Gao X and Ren D (2023) A frameshift variant in the SIRPB1 gene confers susceptibility to Crohn’s disease in a Chinese population. Front. Genet. 14:1130529. doi: 10.3389/fgene.2023.1130529
Received: 23 December 2022; Accepted: 17 April 2023;
Published: 30 May 2023.
Edited by:
Yonghu Sun, Shandong Provincial Hospital of Dermatology, ChinaReviewed by:
Koldo Garcia-Etxebarria, Biodonostia Health Research Institute (IIS Biodonostia), SpainCopyright © 2023 Tang, Wan, Zhang, Diao, Zhang, Gao and Ren. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiang Gao, Z3hpYW5nQG1haWwuc3lzdS5lZHUuY24=; Donglin Ren, cmVuZGxAbWFpbC5zeXN1LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.