
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 13 April 2023
Sec. Neurogenomics
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1126836
This article is part of the Research TopicInsights in Neurogenomics: 2022View all 5 articles
 Zheng Yongsheng1
Zheng Yongsheng1 Sun Chong1Liu Bingyou1
Sun Chong1Liu Bingyou1 Hu Jianian1
Hu Jianian1 Chen Haofeng1
Chen Haofeng1 Zhao Chongbo1
Zhao Chongbo1 Victor Wei Zhang2
Victor Wei Zhang2 Lin Jie1*
Lin Jie1*Introduction: Amyloid transthyretin (ATTR) is divided into either hereditary (ATTRv) or sporadic (ATTRwt) and ATTRv is a rare hereditary disease transmitted as an autosomal dominant manner. Its global prevalence is traditionally estimated as 5,000 to 10,000 persons. However, it may be underestimated and the exact prevalence of ATTRv in China mainland remains unknown.
Methods: The Genome Aggregation database (gnomAD) database (containing 125,748 exomes) and two genomic sequencing databases——China Metabolic Analytics Project (ChinaMAP) (containing 10588 individuals) and Amcarelab gene database (containing 45392 exomes), were integrated to estimate the prevalence of ATTRv in the world and mainland Chinese populations. Pathogenic variants allele frequency and the prevalence of ATTRv was calculated.
Results: Six variants, counting 470 alleles, were defined as pathogenic variants in gnomAD. The prevalence of ATTRv in the world population was 57.4/100,000. Two variants (2 allele counts) and 15 variants (34 individuals) were defined as pathogenic variants in the ChinaMAP database and the Amcarelab exome database, respectively. Thus, the estimated prevalence interval of ATTRv in mainland China was 18.9/100,000-74,9/100,000.
Conclusion: The present study demonstrated that the previous prevalence was greatly underestimated using traditional methods. Therefore, raising awareness of the disease is essential for recognizing ATTRv in its early stage.
Amyloid transthyretin (ATTR) amyloidosis is characterized by pathologic accumulation of extracellular protein arising from unstable transthyretin (TTR) tetramers, which dissociate into monomers that misfold, aggregate, and form insoluble fibrils that are resistant to proteolysis (Finsterer et al., 2019). TTR misfolding can lead to two distinct forms of amyloidosis: hereditary (ATTRv) and wild-type (ATTRwt) (Yamamoto and Yokochi, 2019). ATTRv is a rare hereditary disease transmitted as an autosomal dominant manner (Adams et al., 2016; Adams et al., 2021a). It is caused by mutations in the transthyretin gene located on chromosome 18 (18q12.1). There is considerable heterogeneity in the clinical presentation of ATTRv, ranging from primarily cardiac, primarily neuropathic, or other progressive multisystem disorders (Rapezzi et al., 2010). ATTRv usually proves fatal within 7–12 years from the onset of symptoms, causing a heavy economic burden for families and society (Takeshima et al., 2019; Galan et al., 2021).
ATTRv is an underdiagnosed disease and the true incidence and prevalence are currently unknown (Obi et al., 2022). Its global prevalence is traditionally and somewhat anecdotally estimated as 5,000 to 10,000 persons but only several countries in Europe and Japan have relatively exact prevalence (Sousa et al., 1995; Kato-Motozaki et al., 2008; Dardiotis et al., 2009; Ines et al., 2018; Lauppe et al., 2021). In epidemic regions such as Portugal, Sweden, and Japan, the prevalence varies from 10/100,000 to 100/100,000 people. Recently, the prevalence of transthyretin familial amyloid polyneuropathy (ATTR-FAP) in China was estimated to be approximately 2000 (range 435–10,134) [1,347.7 million persons in total] (Schmidt et al., 2018). However, the prevalence estimation and information supporting prevalence calculations were extracted from records yielded by reference-databases searches, conference proceedings and published cases or case series, which certainly had it underestimated.
Previous prevalence estimation was given using traditional methods, namely, case reports, epidemiological surveys and patient registries (Ikeda et al., 2002; Ines et al., 2018). These traditional methods are prone to underestimations due to diagnostic bias and small sample size. In recent years, population-based genomic sequencing projects provided a new genetic perspective on the estimation of disease-related prevalences (Lahuerta Pueyo et al., 2019). To date, several studies have employed publicly available large-population databases to infer pathogenicity, penetrance or prevalence of rare diseases (Rutten et al., 2016; Lahuerta Pueyo et al., 2019; Liu et al., 2019). Herein, we utilized an integrated world population and Chinese population genomic sequencing databases to reevaluate the ATTRv prevalence in the mainland Chinese population.
GnomAD contained 125,748 exomes from unrelated individuals and from different populations, sequenced as part of various disease-specific and population genetic studies. All TTR (canonical transcript ENST00000237014) variants were selected from the gnomAD version 2.1.1 (http://gnomad.broadinstitute.org/). The analysis of the gnomAD data was carried out from June 2022 to August 2022. In terms of populations, all variants analyzed were classified according to the diverse ethnicities defined in the gnomAD database. The population classifications were as follows: non-Finnish European, African/African American, Latino, Finnish, Ashkenazi Jewish, East Asian, South Asian and other. Population in mainland China was not included in the gnomAD.
The ChinaMAP was based on cohort studies across diverse regions and ethnic groups in China. ChinaMAP (beta version, 2020-03) provided allele frequencies data of variants identified in the Chinese population (n = 10588). It randomly selected participants from 8 ethnic groups (Han, Hui, Manchu, Miao, Mongolian, Yi, Tibetan and Zhuang) across 27 provinces of China without biased selection or filtration (Cao et al., 2020). Therefore, the ChinaMAP database may be considered as a genetic database for the general Chinese population. All TTR (transcript NM_000371.3) variants were extracted from www.mBiobank.com on August 2022.
The Amcarelab gene database was provided by the Amcarelab genetic commercial company. It is composed of 45392 individuals who were suspected to have gene related disorders by medical practitioners from different clinical specialties. Therefore, this database included a subpopulation with negative results from genetic testing as well as patients with definite genetic disorders. All TTR (transcript NM_000371.4) variants were extracted on June 2022.
The present study is focused on missense mutations, genetic variations that result in the substitution of one amino acid with another. The disease-associated variants analyzed in this study specifically affect protein folding, which leads to the accumulation of misfolded proteins, rather than the truncation of protein function. For this reason, the study excluded splicing variants, stop codons, and frameshift changes, as these variants may lead to the production of truncated or non-functional proteins. Additionally, intronic variants, variants in non-coding regions, and synonymous variants were not considered in the study. Following the recommendations of American College of Medical Genetics and Genomics (ACMG), the variants were divided into the following categories: pathogenic, likely pathogenic, uncertain significance, likely benign, benign. In present study, pathogenic and likely pathogenic variants were further defined as disease-causing variant. The calculation of the prevalence of disease-causing variants is our main goal.
About 66 missense variants were found in the TTR gene, with an average of 125,780 exomes studied. According to our definition, six disease-causing variants (including 470 alleles count) were identified and their minor allelic frequency (MAF) was described in Table 1. There were 3 homozygotes identified. The disease-causing amyloidogenic variants MAF was 0.00186 and so the overall prevalence was 371.3/100,000. This is about 4-fold–40-fold higher than what would be expected based on the current epidemic regions prevalence estimation of 10/100,000 (actually from 10/100,000 to 100/100,000) (Ikeda et al., 2002; Kato-Motozaki et al., 2008). This huge prevalence gap between the estimation based on gene databases and traditional methods led us to conduct a further investigation. At first, we analyzed the relationship between population ethnicities and variants. Interestingly, we found a strong relation between the African population and the variant c.424G>A (p. (Val142Ile)) (Figure 1). Among the amyloidogenic TTR pathogenic variants, the point change c.424G>A is unusual for its overwhelmingly predominant occurrence in individuals of documented African descent. Previous studies have shown that the c.424G>A variant was reported in the European population with an allele frequency of 0.00004, while in the African population it was reported with an allele frequency of 0.016 (Lahuerta Pueyo et al., 2019). According to more recent data, the worldwide prevalence of this variant ranges from 0.3% to 1.6% in the general population. However, studies have reported higher prevalence rates of the c.424G>A variant among people of African descent, ranging from 1.1% to 9.8%. It is important to note that the prevalence of the c.424G>A variant in a specific region is highly dependent on the percentage of people with African ancestry residing in that region (Chandrashekar et al., 2021). For its predominance in African/African-American, we further classified pathogenic missense variants found in gnomAD into two groups: African/African-American population and non-African. Then, the allele counts of the African/African-American population group based on gnomAD was 402 in 12487 exomes, and the allele frequency was 0.016, which was consistent with the prevalence previously reported. At last, the allele counts of the non-African group was 65 in 113261 exomes and the allele frequency was 0.0000287 (prevalence: 57.4/100,000).
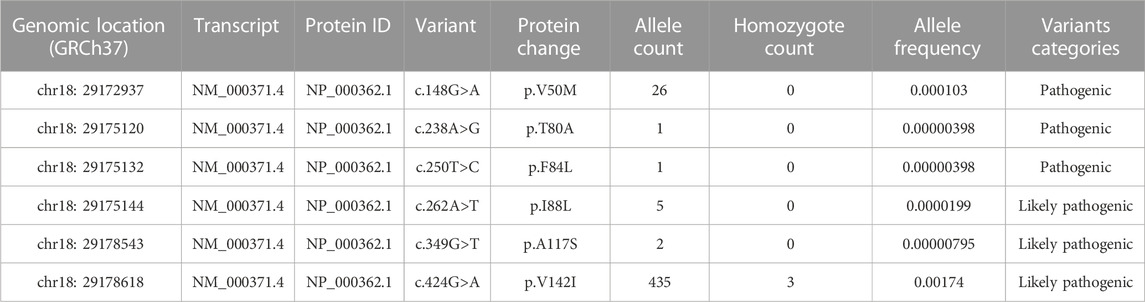
TABLE 1. Variants in the TTR gene classified as disease-causing variants from the gnomAD database global population.
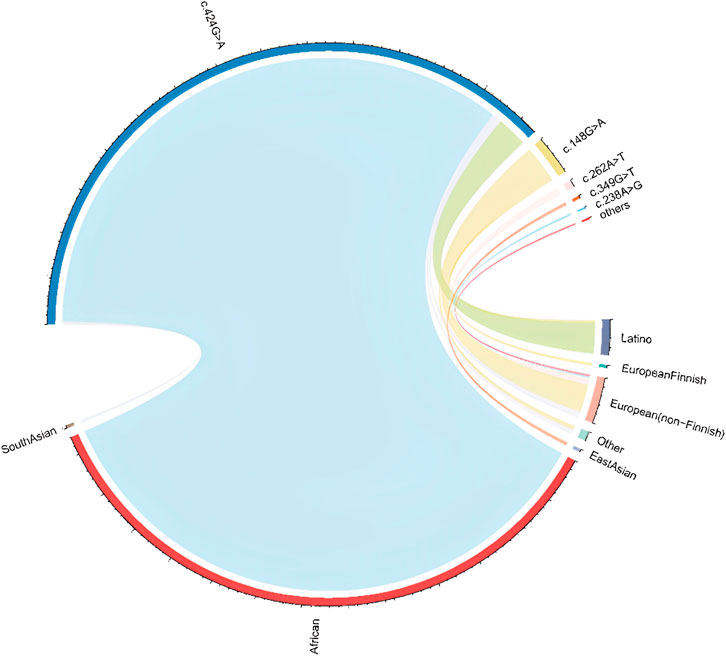
FIGURE 1. Population and pathogenic variant correlation in gnomAD Abbreviations: The ethical information was achieved from gnomAD database. The line connecting represent the pathogenic variant present in the ethnic population. The thicker the line, the higher the proportion of the corresponding variant in that ethnic population.
Analysis of the 10588 Chinese individuals identified 32 TTR missense mutation variants (Supplementary Material S1). Importantly, 2 variants (allele counts: 2), c.148 > A (p.Val50Met) and c.424G>A (p.Val142Ile) were classified as disease-causing variants according to this study. The allele frequency of disease-causing variants in the TTR within the Chinese population was estimated to be 0.0000944 (prevalence: 18.9/100,000).
Excluding synonymous, non-sense, splice variants, intron variants and variants in non-coding regions, analysis of the 45392 exomes identified 107 missense TTR variants (Supplementary Material S2) and 15 variants were pathogenic and likely pathogenic (34 individuals) (Table 2). Thus, the prevalence of disease-causing variants in Chinese population was estimated to be 34 in 45392, namely 74.9/100,000. Herein, the estimated prevalence interval of ATTRv in mainland China was 18.9∼74.9/100,000, based on the public exome, ChinaMAP, and the commercial exome databases.
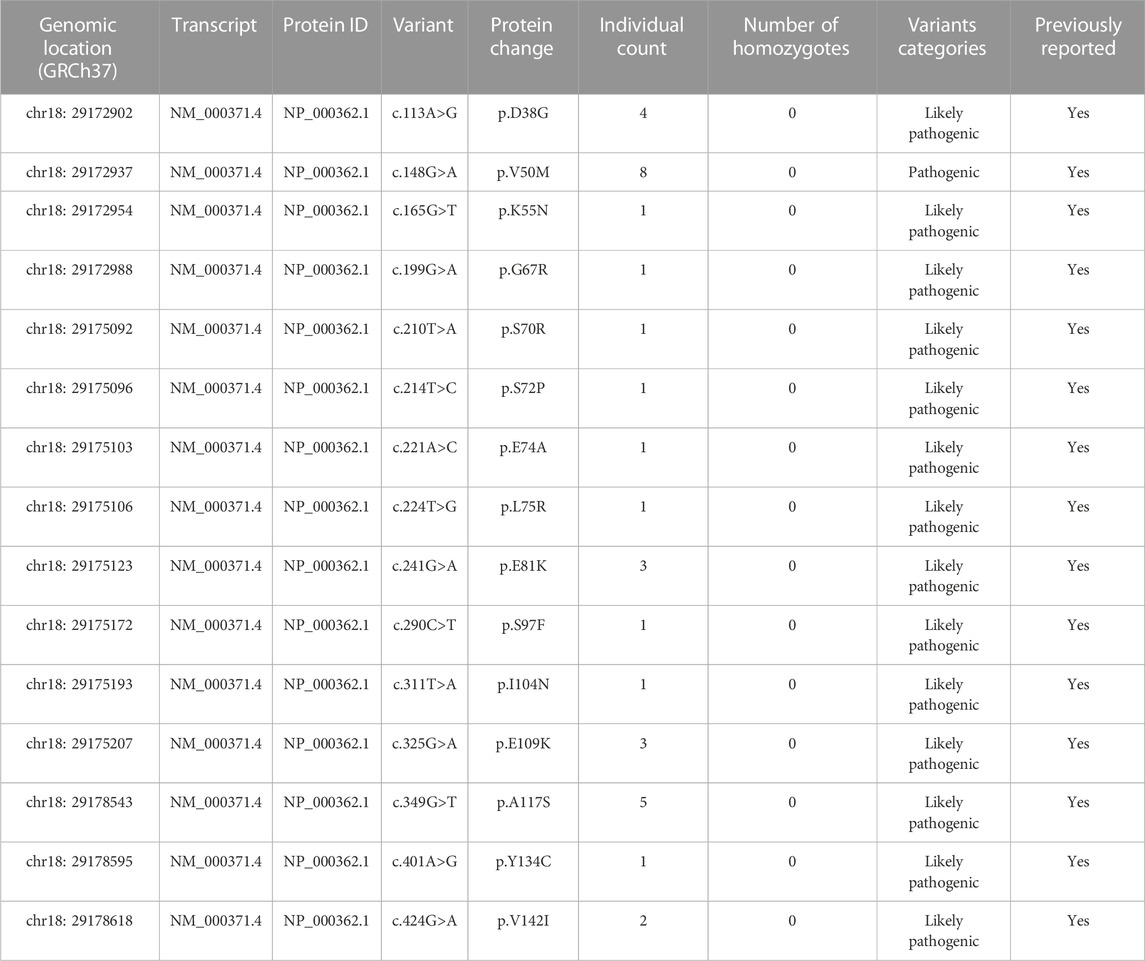
TABLE 2. Variants in the TTR gene classified as pathogenic variants from the Amcarelab database.
Table 3 showed the prevalence of ATTRv in previous reports. In epidemic regions such as Portugal, Japan, and Sweden, the prevalence ranges from 1.1/100,000 to 151/100,000 according to different estimation methods. In non-epidemic regions, the prevalence various from 0.433/100,000 to 3.72/100,000. Specially, ATTRv prevalence in Saudi was estimated to 21.5/100,000 using genome sequencing database.
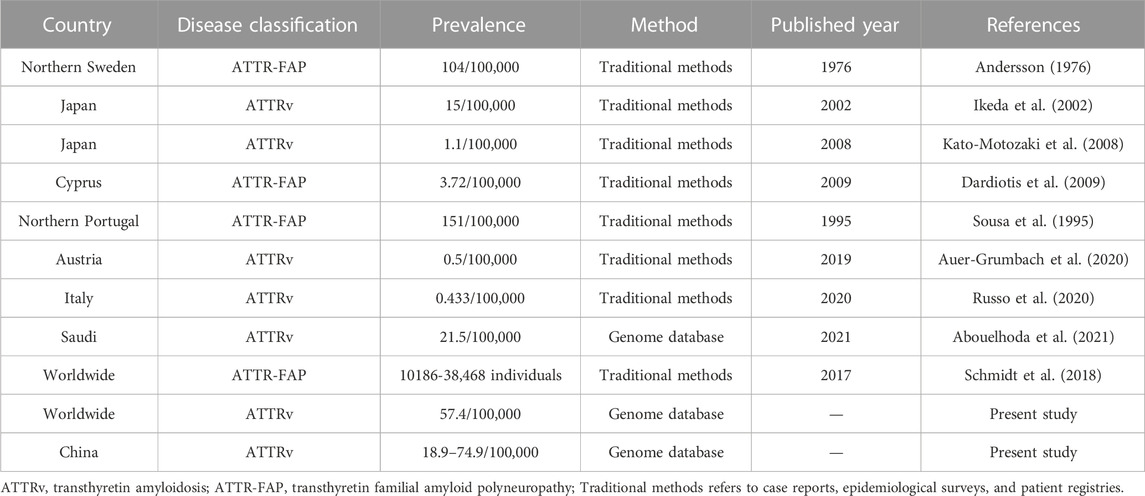
TABLE 3. Previous ATTRv prevalence estimations.
This is the first study to identify and investigate the prevalence of disease-causing TTR variants in the Chinese mainland population. The estimated prevalence interval of ATTRv was 18.9–74.9/100,000 based on the public exome ChinaMAP and commercial exome databases. The lower limit of the prevalence interval was as high as or near 20-fold higher than that in Japan (prevalence: 1/100,000) concluded by traditional epidemiological methods (Kato-Motozaki et al., 2008). Our results suggested that ATTRv in the Chinese was observed much more prevalent than previously recognized.
To determine whether the prevalence in China is high, we estimated the prevalence of different populations and various cohorts using gnomAD data. The gnomAD database consisted of eight ethnic groups but not all populations were equally represented, with the European population being the most represented and the Asian and Latino the least represented (Collins et al., 2020). Noteworthy, these databases did not contain the Chinese population. Comparisons to other population may aid in determine the prevalence gap of a number of disorders that exist between the Chinese and world populations. Not surprisingly, the ATTRv prevalence was 57.4 per 100,000 persons estimated by the genomic sequencing projects approach. It was equal to or even higher than that in epidemic regions such as Portugal, Sweden, and Japan, whose prevalence were calculated by case reports, epidemiological surveys and patient registries. A recent study using a national population-based exome sequence database reported the prevalence of 21.5 per 100,000 persons in Saudi (Abouelhoda et al., 2021), an Asian country that was not reported as an ATTRv epidemic region. This further supported that the true prevalence of ATTRv was much higher than that reported in previous studies.
There are four plausible explanations for the unexpected high prevalence: 1) the pathogenic variants in the ChinaMAP and Amcarelab databases differ from those found in ATTRv in reality and variants were not pathogenic but were polymorphisms, 2) all of the individuals with a TTR pathogenic variant in the ChinaMAP and Amcarelab databases have an undiagnosed or unreported classical TTR variant; 3) a late onset, mild TTR phenotype which was less readily diagnosed, may be much more prevalent than recognized to date. However, it is highly unlikely that any of the 15 disease-causing amyloidogenic TTR variants found in ChinaMAP and Amcarelab databases were polymorphisms as most the 15 pathogenic variants have been previously reported in TTR pedigrees. Taking the results from the gnomAD and Saudi databases into consideration, low prevalence estimation from previous traditional methods may be from penetrance TTR variability, misdiagnosis or missed diagnosis, delayed diagnosis and/or for other reasons (Rapezzi et al., 2010).
A high prevalence highlights the importance of diagnosing ATTRv clinically at an early stage. Recent years have seen the description of a series of novel and effective therapies for ATTRv. Tafamidis, patisiran, inotersen as well as the most promising NTLA-2001 have shown great benefits for patients with ATTRv. Specifically, these benefits are better received when ATTRv is diagnosed at an early stage of disease (Benson et al., 2018; Lamb and Deeks, 2019; Adams et al., 2021b; Gillmore et al., 2021). It challenges and encourage clinical practitioners to raise awareness and have a deeper and broader understanding of ATTRv because early diagnosis of amyloidosis remains an elusive goal that requires education of both physicians and patients (Wechalekar et al., 2016). Further examinations or even gene sequencing must be performed when there is any suspicion of hereditary amyloidosis with initial signs and symptoms, to enter the screening process for amyloidosis and improve early diagnosis (Conceicao et al., 2019).
Our study has several limitations. First, in the present study, only “pathogenic” and “likely pathogenic” variants were defined as disease-causing variants. In fact, some variants in the “uncertain significance” group may also be disease-causing. Thus, the prevalence of ATTRv may be even higher. Secondly, penetrance analysis of the TTR gene could not be determined. Lastly, the characteristics of the Chinese variants and the relationship between variants and clinical characteristics were not analyzed.
In conclusion, we confirmed the high prevalence of ATTRv in the Chinese mainland population, demonstrating that the previous prevalence was greatly underestimated obtained using traditional methods. Therefore, raising awareness of the disease is essential for recognizing ATTRv in its early stage.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
ZY wrote, checked and revised draft manuscripts; ZY, SC, LB, HJ, CH searched data from databases and collected and analyzed data together. LJ and VZ conceptualized the manuscript. LJ and ZC reviewed the draft and revised it.
We want to express our gratitude to the curators who help maintain the online genetic databases this study included.
Author VZ was employed by the company Amcarelab Genetic Commercial Company.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1126836/full#supplementary-material
ACMG, American College of Medical Genetics and Genomics; ATTR, amyloid transthyretin; ATTR-FAP, transthyretin familial amyloid polyneuropathy; ATTRv, hereditary amyloid transthyretin; ATTRwt, wild-type amyloid transthyretin; ChinaMAP, China Metabolic Analytics Project; GnomAD, Genome Aggregation database; HGVS, Human Genome Variation Society; TTR, transthyretin.
Abouelhoda, M., Mohty, D., Alayary, I., Meyer, B. F., Arold, S. T., Fadel, B. M., et al. (2021). Established and candidate transthyretin amyloidosis variants identified in the Saudi population by data mining. Hum. Genomics 15, 52. doi:10.1186/s40246-021-00351-2
Adams, D., Ando, Y., Beirao, J. M., Coelho, T., Gertz, M. A., Gillmore, J. D., et al. (2021). Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J. Neurol. 268, 2109–2122. doi:10.1007/s00415-019-09688-0
Adams, D., Polydefkis, M., Gonzalez-Duarte, A., Wixner, J., Kristen, A. V., Schmidt, H. H., et al. (2021). Long-term safety and efficacy of patisiran for hereditary transthyretin-mediated amyloidosis with polyneuropathy: 12-month results of an open-label extension study. Lancet Neurol. 20, 49–59. doi:10.1016/S1474-4422(20)30368-9
Adams, D., Suhr, O. B., Hund, E., Obici, L., Tournev, I., Campistol, J. M., et al. (2016). First European consensus for diagnosis, management, and treatment of transthyretin familial amyloid polyneuropathy. Curr. Opin. Neurol. 29 (1), S14–S26. doi:10.1097/WCO.0000000000000289
Andersson, R. (1976). Familial amyloidosis with polyneuropathy. A clinical study based on patients living in northern Sweden. Acta Med. Scand. Suppl. 590, 1–64.
Auer-Grumbach, M., Rettl, R., Ablasser, K., Agis, H., Beetz, C., Duca, F., et al. (2020). Hereditary ATTR amyloidosis in Austria: Prevalence and epidemiological hot spots. J. Clin. Med. 9, 2234. doi:10.3390/jcm9072234
Benson, M. D., Waddington-Cruz, M., Berk, J. L., Polydefkis, M., Dyck, P. J., Wang, A. K., et al. (2018). Inotersen treatment for patients with hereditary transthyretin amyloidosis. N. Engl. J. Med. 379, 22–31. doi:10.1056/NEJMoa1716793
Cao, Y., Li, L., Xu, M., Feng, Z., Sun, X., Lu, J., et al. (2020). The ChinaMAP analytics of deep whole genome sequences in 10,588 individuals. Cell Res. 30, 717–731. doi:10.1038/s41422-020-0322-9
Chandrashekar, P., Alhuneafat, L., Mannello, M., Al-Rashdan, L., Kim, M. M., Dungu, J., et al. (2021). Prevalence and outcomes of p.Val142Ile TTR amyloidosis cardiomyopathy: A systematic review. Circ. Genom Precis. Med. 14, e003356. doi:10.1161/CIRCGEN.121.003356
Collins, R. L., Brand, H., Karczewski, K. J., Zhao, X., Alföldi, J., Francioli, L. C., et al. (2020). A structural variation reference for medical and population genetics. Nature 581, 444–451. doi:10.1038/s41586-020-2287-8
Conceicao, I., Damy, T., Romero, M., Galán, L., Attarian, S., Luigetti, M., et al. (2019). Early diagnosis of ATTR amyloidosis through targeted follow-up of identified carriers of TTR gene mutations. Amyloid 26, 3–9. doi:10.1080/13506129.2018.1556156
Dardiotis, E., Koutsou, P., Papanicolaou, E. Z., Vonta, I., Kladi, A., Vassilopoulos, D., et al. (2009). Epidemiological, clinical and genetic study of familial amyloidotic polyneuropathy in Cyprus. Amyloid 16, 32–37. doi:10.1080/13506120802676948
Finsterer, J., Iglseder, S., Wanschitz, J., Topakian, R., Loscher, W. N., and Grisold, W. (2019). Hereditary transthyretin-related amyloidosis. Acta Neurol. Scand. 139, 92–105. doi:10.1111/ane.13035
Galan, L., Gonzalez-Moreno, J., Martinez-Sesmero, J. M., Muñoz-Beamud, F., Santos-Rubio, M. D., Tran, D., et al. (2021). Estimating the annual economic burden for the management of patients with transthyretin amyloid polyneuropathy in Spain. Expert Rev. Pharmacoecon Outcomes Res. 21, 967–973. doi:10.1080/14737167.2021.1900738
Gillmore, J. D., Gane, E., Taubel, J., Kao, J., Fontana, M., Maitland, M. L., et al. (2021). CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 385, 493–502. doi:10.1056/NEJMoa2107454
Ikeda, S., Nakazato, M., Ando, Y., and Sobue, G. (2002). Familial transthyretin-type amyloid polyneuropathy in Japan: Clinical and genetic heterogeneity. Neurology 58, 1001–1007. doi:10.1212/wnl.58.7.1001
Ines, M., Coelho, T., Conceicao, I., Duarte-Ramos, F., de Carvalho, M., and Costa, J. (2018). Epidemiology of transthyretin familial amyloid polyneuropathy in Portugal: A nationwide study. Neuroepidemiology 51, 177–182. doi:10.1159/000490553
Kato-Motozaki, Y., Ono, K., Shima, K., Morinaga, A., Machiya, T., Nozaki, I., et al. (2008). Epidemiology of familial amyloid polyneuropathy in Japan: Identification of a novel endemic focus. J. Neurol. Sci. 270, 133–140. doi:10.1016/j.jns.2008.02.019
Lahuerta Pueyo, C., Aibar Arregui, M. A., Gracia Gutierrez, A., Bueno Juana, E., and Menao Guillen, S. (2019). Estimating the prevalence of allelic variants in the transthyretin gene by analysing large-scale sequencing data. Eur. J. Hum. Genet. 27, 783–791. doi:10.1038/s41431-019-0337-1
Lamb, Y. N., and Deeks, E. D. (2019). Tafamidis: A review in transthyretin amyloidosis with polyneuropathy. Drugs 79, 863–874. doi:10.1007/s40265-019-01129-6
Lauppe, R. E., Liseth Hansen, J., Gerdeskold, C., Rozenbaum, M. H., Strand, A. M., Vakevainen, M., et al. (2021). Nationwide prevalence and characteristics of transthyretin amyloid cardiomyopathy in Sweden. Open Heart 8, e001755. doi:10.1136/openhrt-2021-001755
Liu, W., Pajusalu, S., Lake, N. J., Zhou, G., Ioannidis, N., Mittal, P., et al. (2019). Estimating prevalence for limb-girdle muscular dystrophy based on public sequencing databases. Genet. Med. 21, 2512–2520. doi:10.1038/s41436-019-0544-8
Obi, C. A., Mostertz, W. C., Griffin, J. M., and Judge, D. P. (2022). ATTR epidemiology, genetics, and prognostic factors. Methodist Debakey Cardiovasc J. 18, 17–26. doi:10.14797/mdcvj.1066
Rapezzi, C., Quarta, C. C., Riva, L., Longhi, S., Gallelli, I., Lorenzini, M., et al. (2010). Transthyretin-related amyloidoses and the heart: A clinical overview. Nat. Rev. Cardiol. 7, 398–408. doi:10.1038/nrcardio.2010.67
Russo, M., Obici, L., Bartolomei, I., Cappelli, F., Luigetti, M., Fenu, S., et al. (2020). ATTRv amyloidosis Italian registry: Clinical and epidemiological data. Amyloid 27, 259–265. doi:10.1080/13506129.2020.1794807
Rutten, J. W., Dauwerse, H. G., Gravesteijn, G., van Belzen, M. J., van der Grond, J., Polke, J. M., et al. (2016). Archetypal NOTCH3 mutations frequent in public exome: Implications for CADASIL. Ann. Clin. Transl. Neurol. 3, 844–853. doi:10.1002/acn3.344
Schmidt, H. H., Waddington-Cruz, M., Botteman, M. F., Carter, J. A., Chopra, A. S., Hopps, M., et al. (2018). Estimating the global prevalence of transthyretin familial amyloid polyneuropathy. Muscle Nerve 57, 829–837. doi:10.1002/mus.26034
Sousa, A., Coelho, T., Barros, J., and Sequeiros, J. (1995). Genetic epidemiology of familial amyloidotic polyneuropathy (FAP)-type I in Povoa do Varzim and Vila do Conde (north of Portugal). Am. J. Med. Genet. 60, 512–521. doi:10.1002/ajmg.1320600606
Takeshima, T., Wan, Q., Zhang, Y., Komori, M., Stretton, S., Rajan, N., et al. (2019). Prevalence, burden, and clinical management of migraine in China, Japan, and South Korea: A comprehensive review of the literature. J. Headache Pain 20, 111. doi:10.1186/s10194-019-1062-4
Wechalekar, A. D., Gillmore, J. D., and Hawkins, P. N. (2016). Systemic amyloidosis. Lancet 387, 2641–2654. doi:10.1016/S0140-6736(15)01274-X
Keywords: amyloid transthyretin (ATTR), ChinaMap, GnomAD, prevalence, genetic database
Citation: Yongsheng Z, Chong S, Bingyou L, Jianian H, Haofeng C, Chongbo Z, Zhang VW and Jie L (2023) Prevalence estimation of ATTRv in China based on genetic databases. Front. Genet. 14:1126836. doi: 10.3389/fgene.2023.1126836
Received: 18 December 2022; Accepted: 04 April 2023;
Published: 13 April 2023.
Edited by:
Sarah H. Elsea, Baylor College of Medicine, United StatesReviewed by:
Dario Ronchi, University of Milan, ItalyCopyright © 2023 Yongsheng, Chong, Bingyou, Jianian, Haofeng, Chongbo, Zhang and Jie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lin Jie, bGluamllMTVAZnVkYW4uZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.