 Wenqi Chen1†
Wenqi Chen1† Jing Zhang
Jing Zhang Kai Yang
Kai Yang- 1Prenatal Diagnosis Center, Shijiazhuang Obstetrics and Gynecology Hospital, Key Laboratory of Maternal and Fetal Medicine of Hebei Province, Shijiazhuang, Hebei, China
- 2Department of Pediatric Orthopaedic, The Third Hospital of Hebei Medical University, Shijiazhuang, Hebei, China
- 3Prenatal Diagnosis Center, Beijing Obstetrics and Gynecology Hospital, Beijing Maternal and Child Healthcare Hospital, Capital Medical University, Beijing, China
- 4Reproductive Medicine Center, Shijiazhuang Obstetrics and Gynecology Hospital, Shijiazhuang, Hebei, China
Background and aims: Short-rib thoracic dysplasia 3 with or without polydactyly (SRTD3) represents a type of severe fetal skeletal dysplasia (SD) characterized by shortened limbs, narrow thorax with or without polydactyly, which is caused by the homozygous or compound heterozygous mutations in the DYNC2H1 gene. SRTD3 is a recessive disorder, identification of the responsible genetic variation would be beneficial to an accurate prenatal diagnosis and well-grounded counseling for the affected families.
Material and methods: Two families having experienced recurrent fetal SDs were recruited and submitted to a multiplatform genetic investigation. Whole-exome sequencing (WES) was performed with samples collected from the probands. Sanger sequencing and fluorescent quantitative PCR (qPCR) were conducted as validation assays for suspected variations.
Results: WES identified two compound heterozygous variations in the DYNC2H1(NM_001080463.2) gene, namely c.2386C>T (p.Arg796Trp) and c.7289T>C (p.Ile2430Thr) for one; and exon (64–83)del and c.8190G>T (p.Leu2730Phe) for the other, respectively. One variant in them, exon (64–83)del, was novelly identified.
Conclusion: The study detected two compound heterozygous variation in DYNC2H1 including one novel deletion: exon (64–83) del. Our findings clarified the cause of fetal skeletal dysplasia in the subject families, provided guidance for their future pregnancies, and highlighted the value of WES in diagnosis of skeletal dysplasia with unclear prenatal indications.
Introduction
Short-rib thoracic dysplasia (SRTD) with or without polydactyly is an umbrella term of a group of genetically heterogeneous skeletal dysplasias consistent with the autosomal recessive (AR) inheritance pattern. SRTDs are characterized by short ribs, short limbs, constricted thoracic cage, a ‘trident’ appearance of the acetabular roof and anomalies in kidney, heart, liver, pancreas, genitalia and intestine (Chen and Tzen, 2001; Chen et al., 2002; Chen et al., 2003; Chen et al., 2005; Chen et al., 2012a; Chen et al., 2012b). Currently, SRTD are classified into the categories of short-rib thoracic dysplasia with or without polydactyly type 1–21 (SRTD1-21) (Table 1).
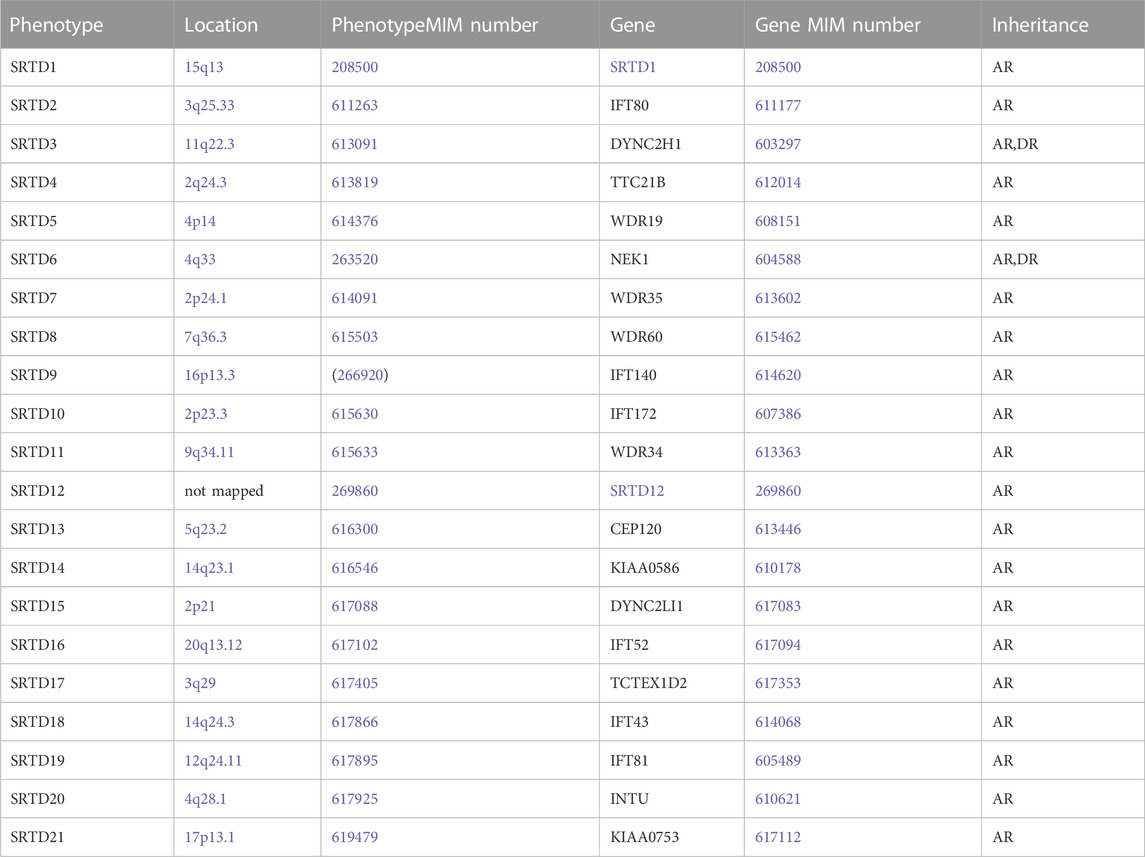
TABLE 1. SRTD are classified into the categories of SRTD1-21.
The short-rib thoracic dysplasia 3 with or without polydactyly (SRTD3; OMIM #613091) refers to a rare subtype of SRTDs, characterized by the constricted thoracic cage, shortened limbs, and associated visceral abnormalities with or without polydactyly. SRTD3 belongs to the “ciliopathies with major skeletal involvement” conditions according to the revised consensus workshop (Bonafe et al., 2015), and is caused by homozygous or compound heterozygous mutations in the DYNC2H1 gene (dynein, cytoplasmic 2, heavy chain 1; OMIM #603297), which encodes a component of the cytoplasmic dynein complex (Dagoneau et al., 2009; McInerney-Leo et al., 2015). This complex is associated with the ciliary intraflagellar transport (IFT), an evolutionarily conserved process that is necessary for ciliogenesis and plays an important role in Hedgehog (Hh), Wnt, Platelet-derived growth factor (PDGF), Notch, G-Protein coupled receptor (GPCR), Mammalian target of rapamycin (mTOR), Transforming growth factor beta (TGF-β) and Calcium signaling pathways (Schmidts et al., 2013a; Schmidt et al., 2015).
In general, SRTD3 is lethal in the neonatal period due to respiratory insufficiency secondary to the severely restricted thoracic cage, whereas other SRTD subtypes are compatible with life (Huber and Cormier-Daire, 2012; Schmidts et al., 2013b). Although prenatal ultrasonography could detect the skeletal abnormalities of SRTD3, this condition was often difficult to be precisely diagnosed before birth. Since SRTD3 is a recessive disorder, identification of the responsible genetic variation would be beneficial to an accurate prenatal diagnosis and well-grounded counseling for the affected families.
In the present study, two families with experiences of multiple adverse gestations including recurrent fetal skeletal dysplasias were recruited. Prenatal ultrasonography examination and genetic detection were conducted to identify the causes of these manifestations in affected fetuses.
Materials and methods
Subjects
Two unrelated cases were recruited between March 2020 and April 2021 at the Prenatal Diagnosis Center, Shijiazhuang Obstetrics and Gynecology Hospital. A comprehensive prenatal ultrasonic examination was conducted on the patients. We carried out a thorough clinical survey. Subsequently, the peripheral blood samples of trio family members in the two pedigrees were collected for the following genetic detection. The studies involving human participants were reviewed and approved by The Ethics Committee of Shijiazhuang Obstetrics and Gynecology Hospital.
Genomic DNA extraction
Amniocentesis was performed to obtain the fetal cell samples, along with 3 ml of peripheral blood collected from the parents using BD Vacutainer™ tubes (BD Biosciences, New Jersey, United States). Genomic DNA was extracted with the QIAamp DNA Blood Mini-Kit (Qiagen Sciences, New York, United States), and the DNA quality was validated by 1% agarose gels and the Qubit® 2.0 Flurometer (Life Technologies, CA, United States).
Whole-exome sequencing
WES was performed by MyGenomics, Inc. (Changping, Beijing, China) as described in our previous study (Zhang et al., 2021). Briefly, the enrichment of the exonic region sequences was conducted by the Sure Select Human Exon Sequence Capture Kit (Agilent, United States). The sequencing library was quantified using the Illumina DNA Standards and Primer Premix Kit (Kapa Biosystems, United States), and was massively parallel-sequenced using the Illumina Novaseq6000 platform. After sequencing and filtering out the low-quality reads, the high-quality data (with general quality level Q30 reads >89%) was aligned to the human genome reference sequence [hg19] using Burrows-Wheeler Aligner tool. The third-party software GATK (https://softw are.broad institute.org/gatk/) and the Verita Trekker® Variants Detection system (Berry Genomics, China) were employed for variant calling. Variants with lower quality (read depth<10x, allele fraction<30%) were eliminated. The variations were identified by sequence alignment with the NCBI Reference Sequence (NG_016423.2, NP_001073932.1, NM_001080463.2) using Chromas v2.33. The pathogenicity of the identified variants was then assessed according to the common guidelines issued by the American Association of Medical Genetics and Genomics (ACMG) (Richards et al., 2015) referring to multiple databases (1000g2015aug_eas, https://www.internationalgenome.org/; ExAC_EAS, http://exac.broadinstitute.org; gnomAD_exome_EAS, http://gnomad.broadinstitute.org/; HGMD®: Human Gene Mutation Database Professional v.2021.10) with the Enliven® Variants Annotation Interpretation (Berry Genomics, China) system.
Validation experiments
The suspected diagnostic variants were validated by Sanger sequencing using ABI 3730 Automated Sequencer (Applied Biosystems, United States) according to the manufacturer’s protocol. Fluorescent quantitative PCR (qPCR) was also carried out to verify the suspected deletion variant identified in Case 2.
Analysis of missense variants
The evolutionary conservatism of amino acid (AA) residues affected by specific missense variants was analyzed using UGENE (http://ugene.net/) with default parameters.
Structural analysis
Referring to the crystal structure of 6rla. 1. A (Toropova et al., 2019) protein, the DYNC2H1 protein structures at 2241–2520 and 2611–2880 regions were constructed by Swiss-model program. The Swiss-Pdb Viewer program was referred to modeling the wild-type (WT) and DYNC2H1: p. Ile2430Thr and p. Leu2730Phe mutant models of DYNC2H1 protein segments. The molecular dynamics (MD) prediction analysis was generated by GROMACS (version 2020.6) (Rakhshani et al., 2019). We carried out 60 ns MD simulations on the DYNC2H1-WT, DYNC2H1-Ile2430Thr, and DYNC2H1-Leu2730Phe models. The CHARMM36 force field was applied to add hydrogen atoms and N-terminal and C-terminal patches to the models (Soteras Gutiérrez et al., 2016). The wild type or the mutant structure of the protein was immersed in cubic boxes which contains water and placed at least 1.0 nm from the box edge. Na+ and Cl− ions were used for neutralization. The MD simulations were performed at a temperature of 300K for 60 ns after energy minimization, equilibration. The following GROMACS distribution programs were used in MD trajectories: gmxrms, gmxrmsf, gmx gyrate, gmxsasa, and gmxhbond. These MD analyses generated parameters values for root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), radius of gyration, solvent accessible surface area (SASA), and number of h-bonds.
Results
Clinical manifestations
Case 1. A 32-year-old woman was referred to our center in March 2021 for previous multiple adverse gestations, when she was at the 17th+gestational week. Her husband was 31 years old, with no consanguineous relation to her. Based on the medical record and her personal dictation, we combed through the couple’s complete medical history and illustrated the pedigree in a diagram (Figure 1A). They had 3 previous pregnancies: in October 2016 and July 2017, they went through one spontaneous abortion and one stillbirth, respectively; in November 2018, the prenatal sonography revealed a 22-week fetus with short limbs and a narrow chest, which was diagnosed as thanatophoric dwarfism. That pregnancy was subsequently terminated.
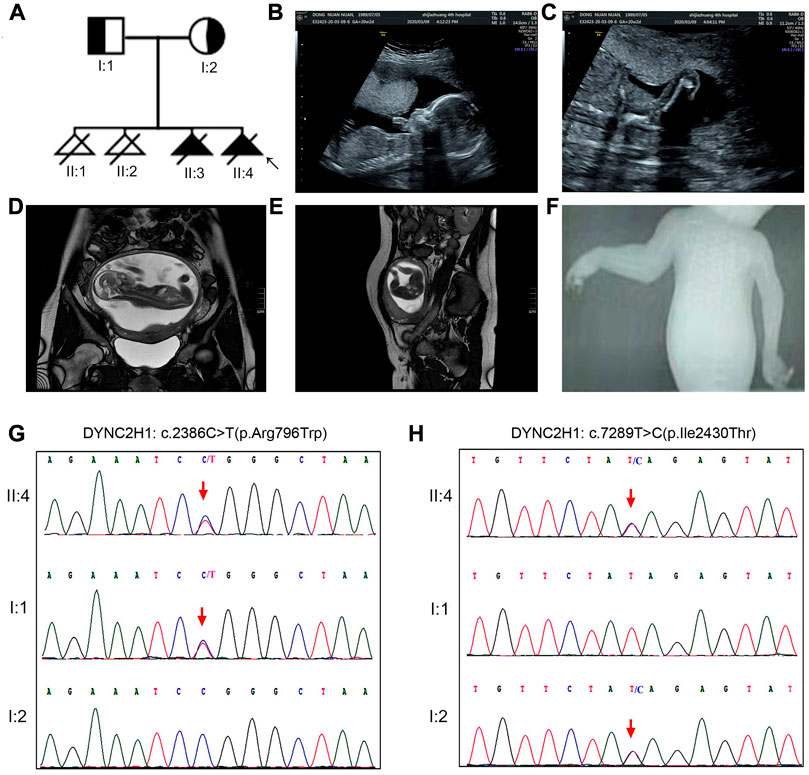
FIGURE 1. The clinical findings and genetic variation in case 1. (A) Pedigree diagram of the family with STRD3. (B–E) Ultrasonographic and MRI indications of the fetus in case 1: the fetus had extremely short limbs and a small, narrow thorax. (F) X-ray indications of the fetus in case 1: the fetus had a narrow thorax and short limbs, but no polydactyly. (G, H) The genetic variation identified in this case: proband 1 (II-4 in Case 1) carried two missense variants, namely c.2386C>T (p.Arg796Trp) and c.7289T>C (p.Ile2430Thr). Validation with Sanger sequencing demonstrated that the variants these probands carried were all inherited from their asymptomatic heterozygous carrier parents.
In 2020, they had the 4th gestation, and the serological screening and NIPT (non-invasive prenatal testing) results were normal. At the 17th week, ultrasonic examination and MRI revealed that the fetus had extremely short limbs and small, bell-shaped chest, short ribs, rhizomelic shortening in all extremities (Figures 1B–E). Prenatal ultrasound did not identify abnormalities in the brain, heart, kidneys, or liver. No exposure history to tobacco smoke, alcohol, ir-radiation, or infectious diseases during the pregnancy were admitted. Pregnancy termination was performed at 22 gestational weeks with informed consent. Afterwards, X-ray imaging result demonstrated that the fetus had a narrow thorax and short limbs, but no polydactyly (Figure 1F). And whole-exome sequencing (WES) was introduced afterwards.
Case 2. A 31-year-old pregnant woman was referred to our center in April 2021. Her husband and her were non-consanguineous. The pedigree diagram was depicted in Figure 2A: in November 2018, a fetus was diagnosed with constricted thoracic cage, extremely shortened tubular bones and bowing of long bones, and then aborted at 12th week; in April 2021, ultrasonography revealed that their second fetus had extremely short limbs at the 13th week (Figures 2B, C). No brain, heart, kidney, or liver abnormality was found by prenatal ultrasonography. The woman denied to be exposed to tobacco smoke, alcohol, radiation or infectious diseases during pregnancy. After informed consent by the couple, genetic analysis including WES was also performed after induction.
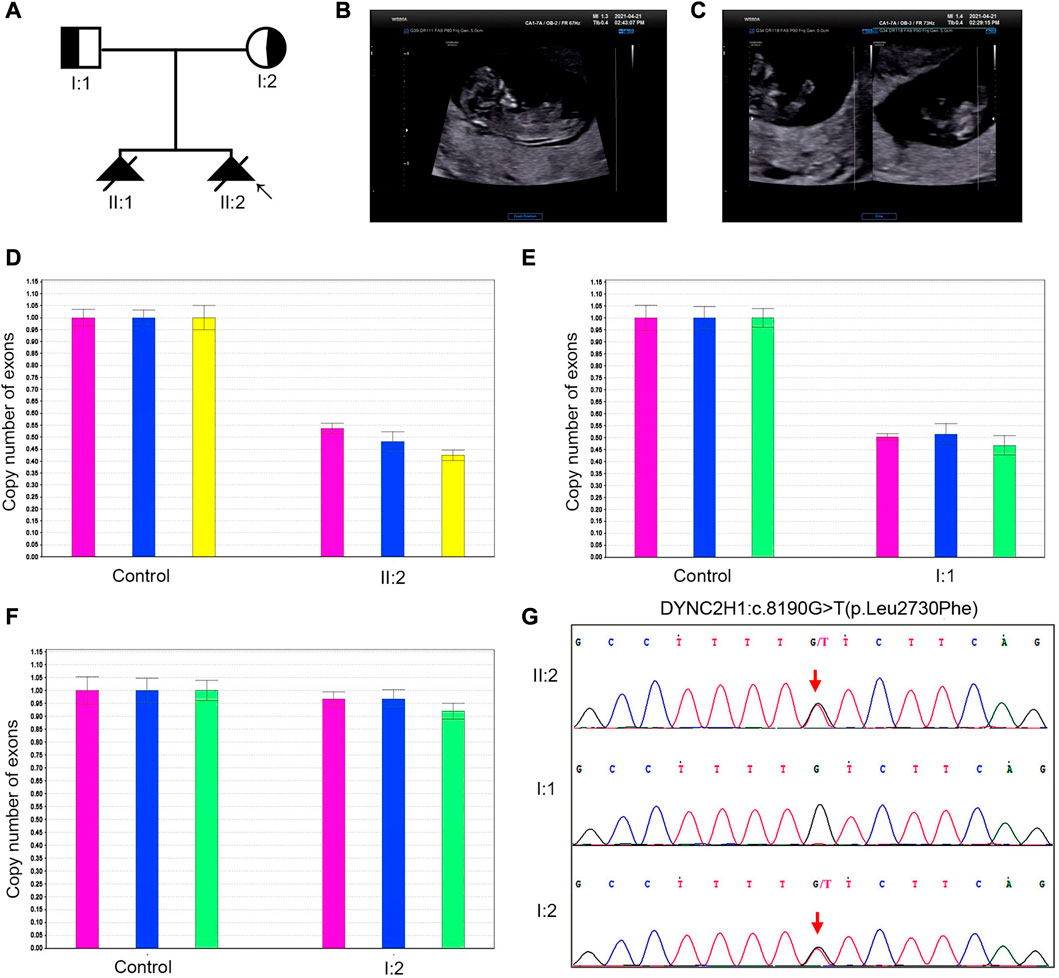
FIGURE 2. The clinical findings and genetic variation in case 2. (A) Pedigree diagram of the family with STRD3. (B, C) Ultrasonographic indications of the fetus in case 2: the fetus had extremely short limbs and bowing of long bones. (D–G) The genetic variation identified in case 2: proband 2 (II-2 in Case 2) carried an exonic deletion and a missense variant: exon (64–83)del and c.8190G>T (p.Leu2730Phe). qPCR validation confirmed one copy loss of the 64-83exons in DYNC2H1of proband 2 (D). Validation with Sanger sequencing and qPCR demonstrated that the variants these probands carried were all inherited from their asymptomatic heterozygous carrier parents.
Genetic variations
Karyotyping by G-bandeding showed that the results of the fetuses in both Case1 and 2 were normal, and array-CGH analysis did not reveal any genomic abnormality associated with known microdeletion or microduplication syndromes, either. On the other hand, according to the WES results, the two fetuses were recognized as positive with compound heterozygous variation in the DYNC2H1 gene. To be specific, Proband 1 (II-4 in Case 1) carried two heterozygous missense variants, namely c.2386C>T (p.Arg796Trp) and c.7289T>C (p.Ile2430Thr) (Figures 1G, H); while Proband 2 (II-2 in Case 2) carried an exonic deletion, exon (64–83)del, and a missense variant, c.8190G>T (p.Leu2730Phe) (Figure 2G). qPCR validation confirmed the one copy loss of 64–83 exons in DYNC2H1 of Proband 2 (Figure 2D). Validation with Sanger sequencing and qPCR demonstrated that the variants these probands carried were inherited from their asymptomatic heterozygous carrier parents, respectively (Figures 1G, H; Figures 2D–G). The location of each variant was illuminated in the gene and peptide diagrammatic sketches (Figure 3A).
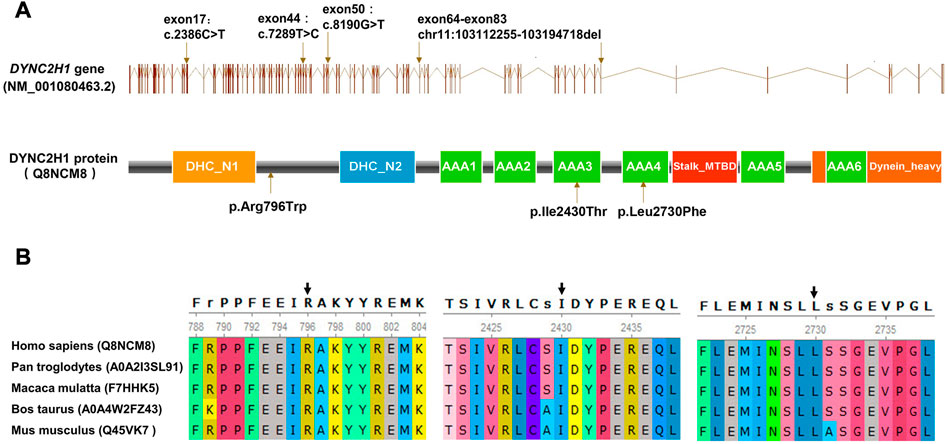
FIGURE 3. (A) Schematic diagram of the DYNC2H1 protein and the locations of the variants detected. (B) Conservation analysis of DYNC2H1 indicated that the protein at position 796, 2430 and 2730 are highly conserved in various species.
Conservatism analysis of missense variants
In this study, three missense variants were detected, which were DYNC2H1: c.2386C>T (p.Arg796Trp), c.7289T>C (p.Ile2430Thr) and c.8190G>T (p.Leu2730Phe). We analyzed the evolutionary conservatism of AA residues they affected. Results indicated that all three AAs maintained highly conserved among species (Figure 3B).
Structural analysis and molecular dynamics simulation
To investigate the intramolecular effect of these missense variants, we performed structural analysis and MD simulation. Due to the structural inaccuracy of the region where p. Arg796Trp was located, we only analyzed the DYNC2H1 protein structures at 2241–2520 region (violet part in Figure 4A) and 2611–2880 region (orange part in Figure 4A), where p. Ile2430Thr and p. Leu2730Phe were located.
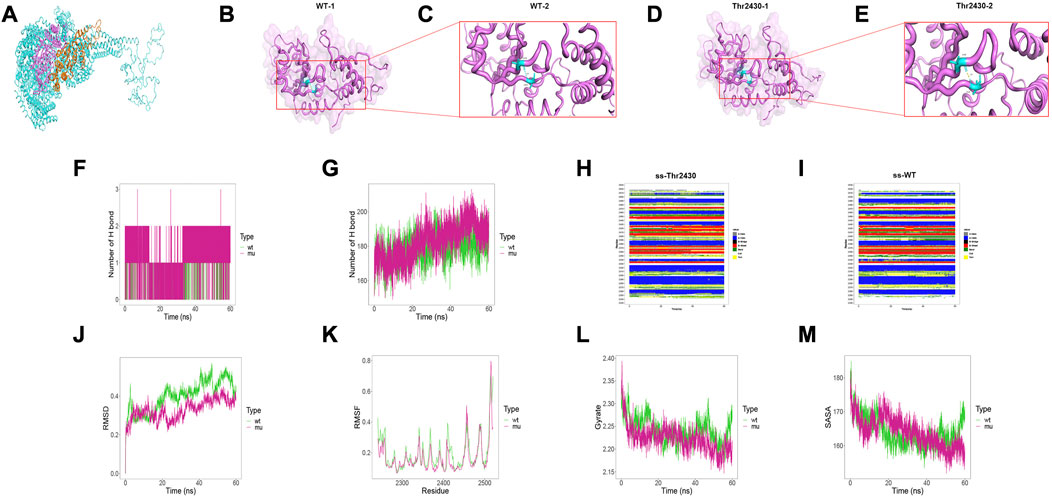
FIGURE 4. Results of structural analysis and molecular dynamics simulation of DYNC2H1: c.7289T>C (p.Ile2430Thr) variation. (A) Protein structure of DYNC2H1. (B, C) The wild-type structure of DYNC2H1: protein (DYNC2H1), and enlarged image of the segment containing Ile2430 residue. (D, E) The mutant structure of DYNC2H1, and segment containing the variant Thr2430 residue. (F) The number of hydrogen bonds formed between the target amino acid (meaning Ile2430 or Thr2430) and other residues. (G) Total number of hydrogen bonds in the wild-type model and the mutant model, respectively. (H, I) Comparison of local secondary structure data between Ile2430Thr mutant and wild-type. (J) RMSD: a numerical measurement indicating the difference between two structures. (K) RMSF: is a numerical measure similar to RMSD, but instead of indicating differences in position over time between entire structures; it calculates the flexibility of individual residues, or the extent to which a particular residue moves (fluctuates) during a simulation. (L) Gyrate: is a measure of the structural displacement of a protein atom with its common center of mass over the course of the simulation and provides comprehensive information about protein tightness over time. (M) SASA: measures the exposed surface in a protein structure accessible to solvent molecules.
The results demonstrated that the Ile2430Thr variant affected the hydrogen bonding between amino acids inside the protein. Particularly, in the wild-type, Ile2430 formed hydrogen bonds with Val2290 residues, with the hydrogen bond length of 2.9Å; while the Ile2430Thr mutant formed a hydrogen bond with Val2290 with a hydrogen bond length of 1.9Å (Figures 4B–E; 4C and 4E are local amplifications of 4B and 4D, respectively). As for the MD results, first, the Thr2430 mutant formed more hydrogen bonds with other amino acids in the protein than the wild-type residue Ile2430 (Figure 4F); for the total number of hydrogen bonds inside the models within 60ns, the amount of Ile2430Thr mutant was more than that of the wild-type (Figure 4G). Besides, the Ile2430Thr variant resulted in a change in secondary structure around 2350th and 2400th residue (Figures 4H, I). Specifically, in wild-type, the secondary structure at 2350 position was dominated by BEND; yet in the mutant, the secondary structure alternated between BEND and TURN and was dominated by TURN. In wild-type, the secondary structure at 2400 position was dominated by BEND; yet in the mutant, the secondary structure alternated between BEND and TURN and was dominated by TURN. Finally, the Ile2430Thr variant resulted in decreased changes in protein structure (Figure 4J), decreased flexibility of amino acids in protein (Figure 4K), increased protein compactness (Figure 4L), and decreased surface area accessible to protein solvent (Figure 4M).
Structural result demonstrated that Leu2730Phe variant affected the hydrogen bonding between amino acids inside the protein. Particularly, Leu2730 and Phe2730 both formed hydrogen bonds with Ile2726 and Lys2802 residues, yet in the Leu2730Phe mutant the hydrogen bond length is shorter than that in the wild-type (Figures 5A–D; 5B and 5D are local amplifications of 5A and 5C, respectively). As for the MD results, there was no difference between Leu2730 and Phe2730 in the number of hydrogen bonds formed with other amino acids in the protein (Figure 5E); for the number of total hydrogen bonds inside the models within 60ns, the amount of Leu2730Phe mutant was more than that of the wild-type (Figure 5F). In wild-type, the secondary structure at 2690 position was dominated by H-helix; yet in the mutant, the secondary structure showed a high rate of BEND and TURN (Figures 5G, H). The Leu2730Phe mutation has little influence on RMSD, RMSF, Gyrate, and SASA (Figures 5I–L).
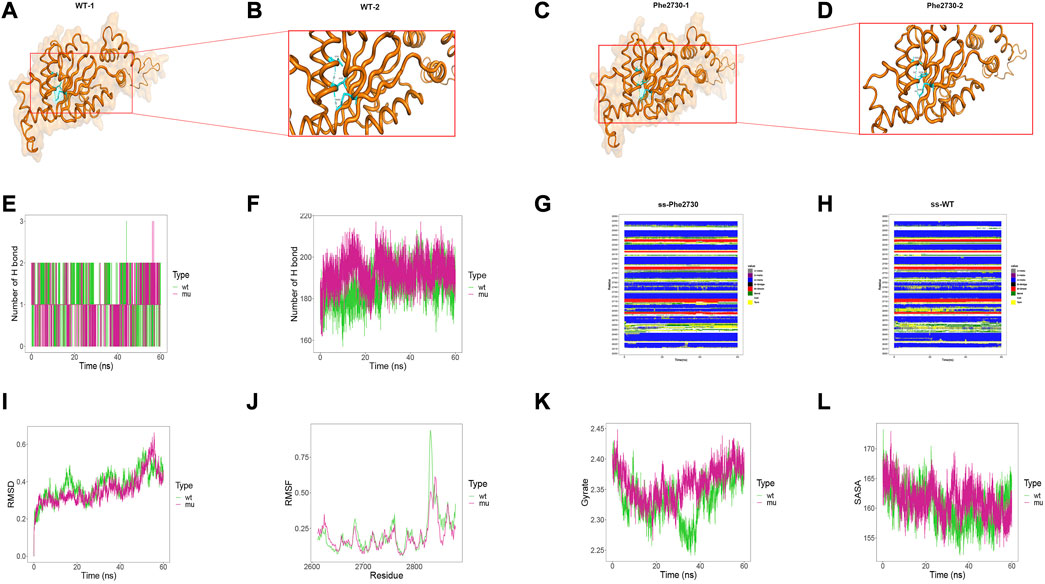
FIGURE 5. Results of structural analysis and molecular dynamics simulation of DYNC2H1: c.8190G>T (p.Leu2730Phe) variation. (A, B) The wild-type structure of DYNC2H1: protein (DYNC2H1), and enlarged image of the segment containing Leu2730 residue. (C, D) The mutant structure of DYNC2H1, and segment containing the variant Phe2730 residue. (E) The number of hydrogen bonds formed between the target amino acid (meaning Leu2730 or Phe2730) and other residues. (F) Total number of hydrogen bonds in the wild-type model and the mutant model, respectively. (G, H) Comparison of local secondary structure data between Leu2730Phe mutant and wild-type. (I) RMSD: a numerical measurement indicating the difference between two structures. (J) RMSF: is a numerical measure similar to RMSD, but instead of indicating differences in position over time between entire structures; it calculates the flexibility of individual residues, or the extent to which a particular residue moves (fluctuates) during a simulation. (K) Gyrate: is a measure of the structural displacement of a protein atom with its common center of mass over the course of the simulation and provides comprehensive information about protein tightness over time. (L) SASA: measures the exposed surface in a protein structure accessible to solvent molecules.
Discussion
SRTD3 refers to a sort of autosomal recessive skeletal condition characterized by shortened limbs, narrow thorax, with or without polydactyly, non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia (Chen et al., 2018). The prenatal ultrasound features of SRTD3 are similar to other skeletal diseases, so it is difficult to establish a definite diagnosis, which proposes a challenge in prenatal diagnosis and management on fetuses with similar early manifestations.
In this study, two families with experiences of multiple adverse gestations including recurrent fetal skeletal dysplasia were enrolled. Ultrasonography detection of the two fetuses revealed skeletal abnormalities characteristics of SRTD3, including extreme shortness of the limbs, and narrow thorax. Signs of polydactyly and non-skeletal symptoms were not noted in the two cases. In Case 1, WES detected a compound heterozygous variation in the DYNC2H1 gene with two variants, c.2386C>T and c.7289T>C. The allele frequencies of c.2386C>T (p.Arg796Trp) and c.7289T>C (p.Ile2430Thr) in gnomAD database were 2.3822e-05 and 9.39391e-06 respectively. In agreement with autosomal-recessive segregation, the parents were heterozygous for these two variants, respectively: the father carried the heterozygous c.2386C>T variant while the mother carried the c.7289T>C variant. The c.2386C>T variant caused a replacement of Arg796 residue by a Trp amino acid, and c.7289T>C caused a substitution of Ile2430 residue by a Thr amino acid. According to the variant interpretation criteria by ACMG, c.2386C>T (p.Arg796Trp), and c.7289T>C (p.Ile2430Thr) variants were classified as VUS, with the evidence of PM2+PP3. The c.2386C>T (p.Arg796Trp) and c.7289T>C (p.Ile2430Thr) variants were predicted to be pathogenic by the SIFT algorithm, Mutation Taster, and PolyPhen-2.
In Case 2, a compound heterozygous variation in DYNC2H1 gene with 2 variants, exon (64–83) del and c.8190G>T, was identified and confirmed. The former one was a novel variant not indexed in the databases of 1000G (https://www.internationalgenome.org/), gnomAD (http://gnomad.broadinstitute.org/), ExAC_EAS (http://exac.broadinstitute.org) and Berry Genomics inhouse database, which expanded the variation spectrum of DYNC2H1 gene. The allele frequencies of c.8190G>T (p.Leu2730Phe) in the databases of EXAC and gnomAD were 2.7e-05 and 1.6 e-05 respectively. Consistent with autosomal-recessive segregation, the parents were heterozygous for the identified mutations: the father carried exon (64–83)del, while the mother carried c.8190G>T. According to the variant interpretation criteria by ACMG, the exon (64–83)del variant was classified as pathogenic, with the evidence of PVS1+PM2. According to the variant interpretation criteria by ACMG, the c.8190G>T (p.Leu2730Phe) variant was classified as VUS, with the evidence of PM2+PP3. The c.8190G>T variant caused a replacement of Leu2730 residue by a Phe amino acid, and this variant was predicted to be pathogenic by the SIFT algorithm, Mutation Taster, and PolyPhen-2. The three residues affected by c.2386C>T (p.Arg796Trp), c.7289T>C (p.Ile2430Thr) and c.8190G>T (p.Leu2730Phe) variants maintained conserved across species, which strongly supports the pathogenicity of these variants. The pathogenicity and ACMG classification of all the variants identified in DYNC2H1 were shown in Table 2.
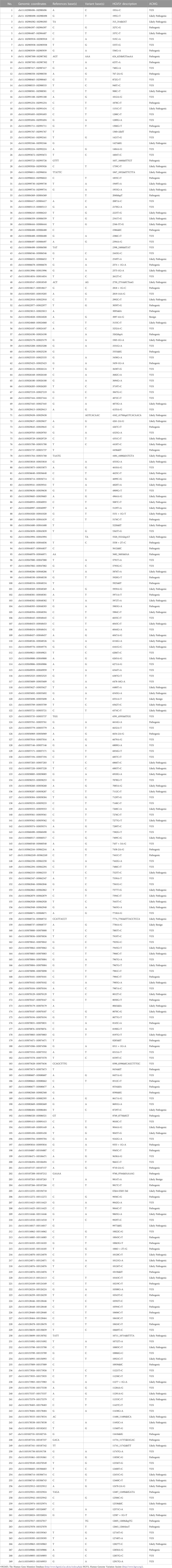
TABLE 2. DYNC2H1 variants (cited from HGMD† database, in order of mutation position).
The DYNC2H1 protein consists of an N-terminal tail (DHC_N1), a linker domain (DHC_N2), six identifiable AAA-ATPase domains, a stalk between AAA domains 4 and 5 in the microtubule binding domain (stalk MTBD), and a conserved C-terminal domain arranged on top of the ATPase ring (Carter et al., 2011). DYNC2H1 is essential for ciliogenesis and plays an important role in Hedgehog signaling events which are critical to human skeletal development (Krakow et al., 2000; Pazour et al., 2006). DYNC2H1 encodes a subunit of cytoplasmic dynein complex, a component of IFTA involved in the retrograde transport from the ciliary tip to the basal body of the ciliary axoneme and plays a role in the generation and maintenance of mammalian cilia (Baujat et al., 2013). Variants in DYNC2H1 have been associated with a heterogeneous spectrum of conditions related to altered primary cilium function that often involve polydactyly, abnormal skeletogenesis, and polycystic kidneys (Chen et al., 2016). Schmidts et al. (2013a) indicated that DYNC2H1 missense mutations altered protein function, yet the effects might be “mild” or submorphic. Merrill et al. (2009) hypothesized that homozygosity for two null alleles would lead to early embryonic lethality, but a series of phenotypes with various severity could result from a combination of multiple missense and null mutations. The genotype-phenotype correlation of DYNC2H1 is pending further elucidation along with larger genetic data.
The c.2386C>T variant affects the highly conserved arginine residues in the stem domain, while the c.7289T>C and the c.8190G>T missense variants are located within the ATP binding and hydrolysis domains (AAA3 and AAA4, respectively). These domains play an important regulatory role for ATP binding (Schmidts et al., 2013a). Blockage of ATP hydrolysis at AAA3 or AAA4 affects the catalytic and mechanical force production activities of dynein (Cho et al., 2008). Modeling the elimination of nucleotide binding at AAA2–4 domains cytoplasmic dynein indicated a severe slowed down in the microtubule sliding activity of the protein, implying dysfunctional motor activity (Zhang et al., 2018). The c.7289T>C and the c.8190G>T were missense changes affecting the ATP binding and hydrolysis domains of the protein (AAA3 and AAA4, respectively), which may disrupt the motor integrity, and interfering with proper retrograde IFT activity. According to the results of structural and MD analysis, the c.7289T>C (p.Ile2430Thr) variant was most likely to significantly affect both local and global hydrogen bond formation to alter protein stability, while also disrupting the desired secondary structure of the protein, thereby disrupting the binding of the protein to ATP. However, the c.8190G>T (p.Leu2730Phe) variant has less influence on the molecular dynamics. Further functional experiments are necessary, not only to clarify the effects of each variant on the protein itself, but also to understand the mechanistically contributes to the pathology seen in the skeletal ciliopathies.
For any future pregnancy of the couples in this study, the recurrent risk of SRTD3 condition would be 25%. Given such circumstances, the couples were informed of reproductive options such as prenatal testing and preimplantation genetic diagnosis (PGD). Proper genetic counseling for the affected family is essential in the case of rare genetic diseases. Furthermore, parenteral genetic screening/diagnosis is the best strategy for managing this disease, which currently has no therapy (Alyafee et al., 2021a; Alyafee et al., 2021b; Alyafee et al., 2022). Reporting additional cases associated with this gene would help identify genotype–phenotype correlations and lead to clinical trials in the future (Alfadhel et al., 2019).
In summary, this study detected two compound heterozygous variation in DYNC2H1 including one novel deletion: exon (64–83)del. Our findings clarified the cause of fetal skeletal dysplasias in the subject families, provided guidance for their future pregnancies, and highlighted the value of WES in diagnosis of skeletal dysplasia with unclear prenatal indications.
Data availability statement
The datasets presented in this study can be found in online repositories. The sequencing results have been deposited to the Figshare repository and can be accessed via the following links: https://doi.org/10.6084/m9.figshare.21738650.v3 and https://doi.org/10.6084/m9.figshare.21738068.v4.
Ethics statement
The studies involving human participants were reviewed and approved by Shijiazhuang Obstetrics and Gynecology Hospital. The patients/participants provided their written informed consent to participate in this study.
Author contributions
WC wrote the first draft of the manuscript. YzL and JZ organized the database. DS and JY performed the data curation and visualization. KY wrote sections of the manuscript. YiL and QG contributed to conception and design of the study. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
The present study was funded by Medical scientific research project of Health Commission of Hebei Province (No. 20231661) and Medical scientific research project of Health Commission of Hebei Province (No. 20231649).
Acknowledgments
We thank the patients and their families for their participation in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alfadhel, M., Umair, M., Almuzzaini, B., Alsaif, S., AlMohaimeed, S. A., Almashary, M. A., et al. (2019). Targeted SLC19A3 gene sequencing of 3000 Saudi newborn: A pilot study toward newborn screening. Ann. Clin. Transl. neurology 6 (10), 2097–2103. doi:10.1002/acn3.50898
Alyafee, Y., Al Tuwaijri, A., Alam, Q., Umair, M., Haddad, S., Alharbi, M., et al. (2021). Next generation sequencing based non-invasive prenatal testing (NIPT): First report from Saudi Arabia. Front. Genet. 12, 630787. doi:10.3389/fgene.2021.630787
Alyafee, Y., Al Tuwaijri, A., Umair, M., Alharbi, M., Haddad, S., Ballow, M., et al. (2022). Non-invasive prenatal testing for autosomal recessive disorders: A new promising approach. Front. Genet. 13, 1047474. doi:10.3389/fgene.2022.1047474
Alyafee, Y., Alam, Q., Tuwaijri, A. A., Umair, M., Haddad, S., Alharbi, M., et al. (2021). Next-generation sequencing-based pre-implantation genetic testing for aneuploidy (PGT-A): First report from Saudi Arabia. Genes 12 (4), 461. doi:10.3390/genes12040461
Baujat, G., Huber, C., El Hokayem, J., Caumes, R., Do Ngoc Thanh, C., David, A., et al. (2013). Asphyxiating thoracic dysplasia: Clinical and molecular review of 39 families. J. Med. Genet. 50 (2), 91–98. doi:10.1136/jmedgenet-2012-101282
Bonafe, L., Cormier-Daire, V., Hall, C., Lachman, R., Mortier, G., Mundlos, S., et al. (2015). Nosology and classification of genetic skeletal disorders: 2015 revision. Am. J. Med. Genet. Part A 167a (12), 2869–2892. doi:10.1002/ajmg.a.37365
Carter, A. P., Cho, C., Jin, L., and Vale, R. D. (2011). Crystal structure of the dynein motor domain. Sci. (New York, NY) 331 (6021), 1159–1165. doi:10.1126/science.1202393
Chen, C. P., Chang, T. Y., Chen, C. Y., Wang, T. Y., Tsai, F. J., Wu, P. C., et al. (2012). Short rib-polydactyly syndrome type II (majewski): Prenatal diagnosis, perinatal imaging findings and molecular analysis of the NEK1 gene. Taiwan. J. obstetrics Gynecol. 51 (1), 100–105. doi:10.1016/j.tjog.2012.01.020
Chen, C. P., Chang, T. Y., Tzen, C. Y., Lin, C. J., and Wang, W. (2002). Sonographic detection of situs inversus, ventricular septal defect, and short-rib polydactyly syndrome type III (Verma-Naumoff) in a second-trimester fetus not known to be at risk. Ultrasound obstetrics Gynecol. 19, 629. doi:10.1046/j.1469-0705.2002.00731_4.x
Chen, C. P., Chang, T. Y., Tzen, C. Y., and Wang, W. (2003). Second-trimester sonographic detection of short rib-polydactyly syndrome type II (Majewski) following an abnormal maternal serum biochemical screening result. Prenat. Diagn. 23, 353. doi:10.1002/pd.574
Chen, C. P., Chern, S. R., Chang, T. Y., Su, Y. N., Chen, Y. Y., Su, J. W., et al. (2012). Prenatal diagnosis and molecular genetic analysis of short rib-polydactyly syndrome type III (Verma-Naumoff) in a second-trimester fetus with a homozygous splice site mutation in intron 4 in the NEK1 gene. Taiwan. J. obstetrics Gynecol. 51 (2), 266–270. doi:10.1016/j.tjog.2012.04.018
Chen, C. P., Ko, T. M., Chang, T. Y., Chern, S. R., Chen, S. W., Lai, S. T., et al. (2018). Prenatal diagnosis of short-rib polydactyly syndrome type III or short-rib thoracic dysplasia 3 with or without polydactyly (SRTD3) associated with compound heterozygous mutations in DYNC2H1 in a fetus. Taiwan. J. obstetrics Gynecol. 57 (1), 123–127. doi:10.1016/j.tjog.2017.12.021
Chen, C. P., Shih, J. C., Tzen, C. Y., Lin, Y. H., and Wang, W. (2005). Recurrent short-rib polydactyly syndrome: Prenatal three-dimensional ultrasound findings and associations with congenital high airway obstruction and pyelectasia. Prenat. Diagn. 25 (5), 417–418. doi:10.1002/pd.976
Chen, C. P., and Tzen, C. Y. (2001). Short-rib polydactyly syndrome type III (Verma-Naumoff) in a third-trimester fetus with unusual associations of epiglottic hypoplasia, renal cystic dysplasia, pyelectasia and oligohydramnios. Prenat. Diagn. 21, 1101. doi:10.1002/pd.182
Chen, L. S., Shi, S. J., Zou, P. S., Ma, M., Chen, X. H., and Cao, D. H. (2016). Identification of novel DYNC2H1 mutations associated with short rib-polydactyly syndrome type III using next-generation panel sequencing. Genet. Mol. Res. GMR 15 (2). doi:10.4238/gmr.15028134
Cho, C., Reck-Peterson, S. L., and Vale, R. D. (2008). Regulatory ATPase sites of cytoplasmic dynein affect processivity and force generation. J. Biol. Chem. 283 (38), 25839–25845. doi:10.1074/jbc.M802951200
Dagoneau, N., Goulet, M., Geneviève, D., Sznajer, Y., Martinovic, J., Smithson, S., et al. (2009). DYNC2H1 mutations cause asphyxiating thoracic dystrophy and short rib-polydactyly syndrome, type III. Am. J. Hum. Genet. 84 (5), 706–711. doi:10.1016/j.ajhg.2009.04.016
Huber, C., and Cormier-Daire, V. (2012). Ciliary disorder of the skeleton. Am. J. Med. Genet. Part C, Seminars Med. Genet. 160c (3), 165–174. doi:10.1002/ajmg.c.31336
Krakow, D., Salazar, D., Wilcox, W. R., Rimoin, D. L., and Cohn, D. H. (2000). Exclusion of the Ellis-van Creveld region on chromosome 4p16 in some families with asphyxiating thoracic dystrophy and short-rib polydactyly syndromes. Eur. J. Hum. Genet. EJHG 8 (8), 645–648. doi:10.1038/sj.ejhg.5200507
McInerney-Leo, A. M., Harris, J. E., Leo, P. J., Marshall, M. S., Gardiner, B., Kinning, E., et al. (2015). Whole exome sequencing is an efficient, sensitive and specific method for determining the genetic cause of short-rib thoracic dystrophies. Clin. Genet. 88 (6), 550–557. doi:10.1111/cge.12550
Merrill, A. E., Merriman, B., Farrington-Rock, C., Camacho, N., Sebald, E. T., Funari, V. A., et al. (2009). Ciliary abnormalities due to defects in the retrograde transport protein DYNC2H1 in short-rib polydactyly syndrome. Am. J. Hum. Genet. 84 (4), 542–549. doi:10.1016/j.ajhg.2009.03.015
Pazour, G. J., Agrin, N., Walker, B. L., and Witman, G. B. (2006). Identification of predicted human outer dynein arm genes: Candidates for primary ciliary dyskinesia genes. J. Med. Genet. 43 (1), 62–73. doi:10.1136/jmg.2005.033001
Rakhshani, H., Dehghanian, E., and Rahati, A. (2019). Enhanced GROMACS: Toward a better numerical simulation framework. J. Mol. Model. 25 (12), 355. doi:10.1007/s00894-019-4232-z
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Schmidt, H., Zalyte, R., Urnavicius, L., and Carter, A. P. (2015). Structure of human cytoplasmic dynein-2 primed for its power stroke. Nature 518 (7539), 435–438. doi:10.1038/nature14023
Schmidts, M., Arts, H. H., Bongers, E. M., Yap, Z., Oud, M. M., Antony, D., et al. (2013). Exome sequencing identifies DYNC2H1 mutations as a common cause of asphyxiating thoracic dystrophy (Jeune syndrome) without major polydactyly, renal or retinal involvement. J. Med. Genet. 50 (5), 309–323. doi:10.1136/jmedgenet-2012-101284
Schmidts, M., Vodopiutz, J., Christou-Savina, S., Cortés, C. R., McInerney-Leo, A. M., Emes, R. D., et al. (2013). Mutations in the gene encoding IFT dynein complex component WDR34 cause Jeune asphyxiating thoracic dystrophy. Am. J. Hum. Genet. 93 (5), 932–944. doi:10.1016/j.ajhg.2013.10.003
Soteras Gutiérrez, I., Lin, F. Y., Vanommeslaeghe, K., Lemkul, J. A., Armacost, K. A., Brooks, C. L., et al. (2016). Parametrization of halogen bonds in the CHARMM general force field: Improved treatment of ligand-protein interactions. Bioorg. Med. Chem. 24 (20), 4812–4825. doi:10.1016/j.bmc.2016.06.034
Toropova, K., Zalyte, R., Mukhopadhyay, A. G., Mladenov, M., Carter, A. P., and Roberts, A. J. (2019). Structure of the dynein-2 complex and its assembly with intraflagellar transport trains. Nat. Struct. Mol. Biol. 26 (9), 823–829. doi:10.1038/s41594-019-0286-y
Zhang, J., Hu, H., Mu, W., Yu, M., Chen, W., Mi, D., et al. (2021). Case report: Exome sequencing identified a novel compound heterozygous variation in PLOD2 causing bruck syndrome type 2. Front. Genet. 12, 619948. doi:10.3389/fgene.2021.619948
Keywords: DYNC2H1 gene, short-rib thoracic dysplasia 3 (SRTD3), skeletal dysplasia, whole-exome sequencing, prenatal diagnosis
Citation: Chen W, Li Y, Zhang J, Yuan Y, Sun D, Yuan J, Yang K, Liang Y and Guo Q (2023) Genetic variations in the DYNC2H1 gene causing SRTD3 (short-rib thoracic dysplasia 3 with or without polydactyly). Front. Genet. 14:1125473. doi: 10.3389/fgene.2023.1125473
Received: 16 December 2022; Accepted: 27 March 2023;
Published: 06 April 2023.
Edited by:
Hu Hao, The Sixth Affiliated Hospital of Sun Yat-sen University, ChinaReviewed by:
Nadia Akawi, United Arab Emirates University, United Arab EmiratesMuhammad Umair, King Abdullah International Medical Research Center (KAIMRC), Saudi Arabia
Copyright © 2023 Chen, Li, Zhang, Yuan, Sun, Yuan, Yang, Liang and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing Guo, eWZndW9xaW5nQDE2My5jb20=; Ying Liang, eWluZ19saWFuZ2NuQGhvdG1haWwuY29t
†These authors have contributed equally to this work and share first authorship