 Hamdi Hameed Almaramhy1Firoz Abdul Samad2
Hamdi Hameed Almaramhy1Firoz Abdul Samad2 Ghadeer Al-Harbi3Dimah Zaytuni3Syed Nazar Imam1
Ghadeer Al-Harbi3Dimah Zaytuni3Syed Nazar Imam1 Tariq Masoodi4
Tariq Masoodi4 Monis Bilal Shamsi3,5*
Monis Bilal Shamsi3,5*- 1College of Medicine, Taibah University, Medina, Saudi Arabia
- 2College of Applied Medical Science, Taibah University, Medina, Saudi Arabia
- 3Centre for Genetics and Inherited Diseases, Taibah University, Medina, Saudi Arabia
- 4Translational Medicine Department, Research Branch, Sidra Medicine, Doha, Qatar
- 5Department of Biochemistry, College of Medicine, Taibah University, Medina, Saudi Arabia
Introduction: Hypospadias [MIM: 300633] is one of the most frequent congenital malformations of male external genitalia. The spectrum of genetic variants causing hypospadias is varied, with studies commonly implicating genes critical in the fetal steroidogenic pathway. This is the first genetic study on hypospadias from the Yemen ethnicity and the second to report HSD3B2 mutations in more than one affected individual from the same family.
Material and methods: Surgical hypospadias repair was performed on two hypospadias-affected siblings from a consanguineous family. Whole-exome sequencing (WES) was performed to identify the potential pathogenic variant for hypospadias, which was later confirmed by Sanger sequencing. The identified variant was further analyzed for its pathogenicity by using in silico tools such as SIFT, PolyPhen-2, MutationAssessor, MutationTaster, FATHMM, and ConSurf.
Results: We identified a novel missense mutation (Chr1:119964631T>A, c.507T>A, p. N169K) in 3β-hydroxysteroid 2-dehydrogenase (HSD3B2) gene by WES. Sanger sequencing confirmed that the variant segregated the disease in the family between the affected and non-affected individuals. Both patients are homozygous, while parents and two unaffected siblings are heterozygous carriers, indicating an autosomal recessive pattern of inheritance. The in silico analysis by all six in silico tools (SIFT, PolyPhen-2, MutationAssessor, MutationTaster, FATHMM, and ConSurf) predicted the variant to be pathogenic/deleterious.
Discussion: An abnormal fetal steroidogenic pathway due to genetic influences may affect the development of the male genital tract, including the urethral tract closure and morphogenesis of male genitalia. Furthermore, the pathogenicity of the observed variant in this study, confirmed by multiple in silico tools, characterizes the influence HSD3B2 gene variants may have in the etiology of hypospadias.
Conclusion: Understanding of pathogenic manifestation and inheritance of confounding genetic variants in hypospadias is a matter of great concern, especially in familial cases.
Introduction
Hypospadias is congenital hypoplasia of the penis, leading to a ventrally displaced urinary meatus, often associated with a dorsal-headed foreskin and chordee (Leung and Robson, 2007). It is the second most common male urogenital abnormality after cryptorchidism (Ságodi et al., 2014). The prevalence of hypospadias varies widely between different populations (0.26–47 per 10,000 births), with reports suggesting higher frequency in white people than in black people and Asians (KŠllŽn et al., 1986; Paulozzi, 1999; Kurahashi et al., 2004; Yang et al., 2004; Carmichael et al., 2007).
Hypospadias is characterized by incomplete fusion of the urethral folds during early embryonic development (Baskin and Ebbers, 2006). From the 8th week onwards, when gonads have differentiated into testes in XY embryonic males, the external genitalia development and urethral modifications are mainly dependent on the secretion and action of androgens (Baskin and Ebbers, 2006; Bouty et al., 2015), in particular, by testosterone produced from the fetal testes and transformed peripherally into dihydrotestosterone that binds to the androgen receptors. The cascades of hormonal activities influenced by numerous genetic factors induce fusion of the urethral folds along the ventral surface of the penis and produce a series of morphological changes in the developing external genitalia (Baskin and Ebbers, 2006). During this hypospadias-critical gestational window (8–14 weeks), the synthesis, concentration, timing, and metabolism of hormones/androgens are important for proper urethral closing and external genitalia development, and any abnormality may result in hypospadias (Bouty et al., 2015).
Numerous lines of evidence suggest a genetic component as an important etiology, with the presence of an affected family member being a high risk factor (van der Zanden et al., 2012; Thorup et al., 2014). First-, second-, or third-degree relatives are affected in 7% of patients with familial clustering of hypospadias (Fredell et al., 2002). In most familial cases, hypospadias shows equal transmission through both maternal and paternal lines with similar recurrence risks between the brothers and sons of patients (Schnack et al., 2008). The chances of hypospadias increase by 9%–17% for the brother of a male with hypospadias. Pedigree and twin studies have reported a higher genetic component of 57%–77% in the heritability of hypospadias (Schnack et al., 2008).
The risk of congenital abnormalities is higher in children born from consanguineous parents. Investigating such affected families provides a higher prospect of genetic findings. Therefore, we investigated a consanguineous family with two hypospadias-affected siblings using whole-exome sequencing. The bioinformatics tools for in silico predictions were applied to assess the structural and functional consequences of the variants observed in the study.
Subjects and methods
Patients
The patients (two male siblings) were brought to the Saudi German Hospital in Medina, Saudi Arabia, for surgical correction of hypospadias. The family also had two non-affected (normal) male siblings. The family was ethnically from Yemen, and the parents were consanguineous. The affected siblings responded well to the treatment, and no complications during or pre/post-surgery were observed. The Institute’s ethical committee approved the study, and written informed consent was taken from study participants.
Whole-exome sequencing (WES)
Whole-exome sequencing (WES) was performed on venous blood DNA from the parents and three siblings (two affected and one normal child) as per the manufacturer’s instructions (Exome RDY Kit Cat. No: A38262, Thermo Fisher Scientific, USA). Sequence reads were aligned to a reference genome (hg19) (http://genome.ucsc.edu/), and variants were identified using the Ion Torrent pipeline (Thermo Fisher Scientific, USA). Variants that show an effect on the protein sequence (missense, nonsense, frameshift, splicing, etc.) and variants with minor allele frequency ≤0.05 in the 1000 Genomes Project (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp) were considered in downstream analysis. We then identified the suspected pathogenic variants shared between the affected children by comparing each gene variant among the family members. The identified variant was further validated by Sanger sequencing among all the family members (both parents, two affected, and two non-affected siblings) (Figure 1). The primers (Supplementary Table S1) were designed using Primer3 software (https://primer3.ut.ee/).
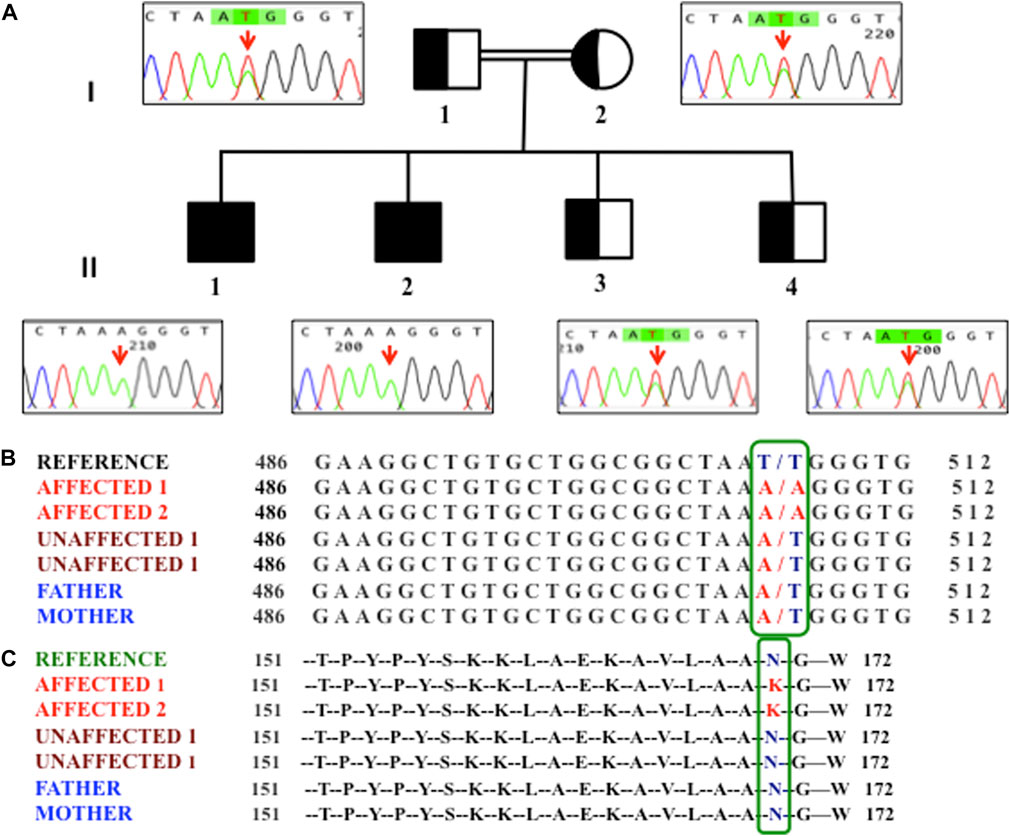
FIGURE 1. (A) Pedigree of the family showing homozygosity (II1 and II2) and hetrozygosity (I1, I2, II3, and II4). An arrow in the chromatogram highlights the T507A variant. (B) Nucleotide variants observed at the c.507 position of the HSD3B2 gene in members of the family. (C) Amino acid variant observed at the 169th position in members of the family.
Yq microdeletion analysis
Y-chromosome microdeletions were analyzed as described by Shamsi et al. (2012). Briefly, multiplex analysis using STS primer sets, which spanned the AZFa, AZFb, and AZFc regions of the Y chromosome for sY84, sY86 (AZFa); sY127, sY134 (AZFb); and sY254, sY255 (AZFc) was performed on genomic blood DNA of hypospadias-affected and non-affected siblings. sY14 (SRY) was used as an internal control, while a male DNA and a female DNA sample were used as positive and negative controls, respectively.
Computational analysis for pathogenicity prediction
We used six different in silico tools to predict the pathogenicity potential of the variant. Each in silico tool predicted the pathogenicity potential based on distinct criteria and parameters. The tools used for pathogenicity prediction are SIFT (https://sift.bii.a-star.edu.sg/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), MutationTaster (http://www.mutationtaster.org/), MutationAssessor (http://mutationassessor.org/r3/), FATHMM (http://fathmm.biocompute.org.uk/index.html), and ConSurf Analysis (https://www.bio-sof.com/consurf) (https://consurf.tau.ac.il/).
Results
Clinical presentation and family history
The eldest two sons, aged 11 and 9 years, had hypospadias, while the younger two sons (6 and 4 years) were normal on clinical examination. At the time of sample collection, the father was 38 years old and the mother was 27 years old. The meatal position was proximal (penoscrotal), and the penile chordee was present in both the affected siblings. The testes were descended and normal. We did not observe Y-chromosome microdeletions in the hypospadias-affected or non-affected siblings. No medical history of hypospadias or any other genetic disorder or congenital malformation is reported in the family.
Genomic analysis
We performed WES in two hypospadias-affected siblings, one unaffected sibling, and both parents. A total of 6,072 exonic variants were observed; the majority were found to be missense (5,541 variants). We also observed 172 variants as homozygous, where both alleles were altered (Supplementary Table S2). After annotation and filtering, we observed a novel variant T507A in the 3β-hydroxysteroid 2-dehydrogenase (HSD3B2) gene, which segregated the disease in the family between the affected and non-affected individuals. Furthermore, we validated the T507A variant by Sanger sequencing among all members of the family, including one unaffected sibling who was not investigated by WES. We observed that the other unaffected sibling was also heterozygous for the same variant (Figure 1). We checked this T507A variant in an additional age- and gender-matched control cohort containing whole-exome sequencing of 127 males, and, interestingly, this variant was absent (0/127) in all control individuals. We also screened for this variant in the Greater Middle East (GME) Variome database (http://igm.ucsd.edu/gme/index.php), which has WES data for 2,497 healthy individuals (controls) as a genomic reference of the Middle Eastern population, and could not find any prior occurrence of our variant in their cohort. Additionally, we also searched in the Genome Aggregation Database (gnomAD), which has whole-exome and whole-genome data on 125,748 and 15,708 individuals, respectively, and our variant did not match with any of the early reported variants. No Yq microdeletion was observed in the affected or non-affected male siblings.
HSD3B2 is related to the etiology of hypospadias and was reported as causative or contributory to hypospadias in earlier studies (Codner et al., 2004a; Wang et al., 2007; Rabbani et al., 2012) due to its role in the synthesis of endocrinal factors that regulate fetal urogenital system development (van der Zanden et al., 2012). However, the T507A variant found in our study has not yet been reported. We confirmed the novelty of our T507A variant by a search in previous publications, various databases (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp), (https://www.ncbi.nlm.nih.gov/snp/), and particularly in the Middle-East-specific database (http://igm.ucsd.edu/gme/data-browser.php) due to the ethnically Yemen origin of our study subjects.
The HSD3B2 gene has four exons and three introns, and the candidate variant T507A is located in exon 4 of the HSD3B2 gene. The non-synonymous T507A variant caused an amino acid change at the 169th position from asparagine (N) to lysine (K). The T507A variant in the HSD3B2 gene is located in the evolutionarily conserved region of the genome among the closely related primates, such as chimpanzees, gorillas, and macaques, which represents its functional significance (Table 1).

TABLE 1. Comparison of evolutionarily conserved regions corresponding to the N169K change of HSD3B2 observed in our patients with closely related primates, such as chimpanzees, gorillas, and macaques.
Pathogenicity prediction
The in silico pathogenicity predictions used in this study were based on a combination of nucleotide/amino acid characteristics as primary DNA sequence features (PolyPhen-2), biochemical properties of substituted/variant amino acid (SIFT), protein structural features (PolyPhen-2), homology evolutionary conservation (SIFT, PolyPhen-2, MutationAssessor, FATHMM, and ConSurf Analysis), association/susceptibility to disease (MutationTaster and FATHMM), and the presence/proximity of the variant to the substrate binding/catalytic site (ConSurf Analysis) (Adzhubei et al., 2010; Schwarz et al., 2010; Sim et al., 2012; Carter et al., 2013; Shihab et al., 2013; Kircher et al., 2014; Quang et al., 2015; Rogers et al., 2018) (https://sift.bii.a-star.edu.sg/; https://ionreporter.thermofisher.com/ionreporter/help/GUID-57A60D00-0654-4F80-A8F9-F6B6A48D0278.html; http://www.mutationtaster.org/info/documentation.html; https://omictools.com/mutationassessor-tool; http://fathmm.biocompute.org.uk/inherited.html, http://fathmm.biocompute.org.uk/index.html; http://consurf.tau.ac.il/gallery.php). The in silico analysis of the six applied tools concordantly predicted the T507A variant as potentially deleterious considering the structural, functional, disease susceptibility, phylogenetic, and evolutionary aspects of the substitution (Table 2).
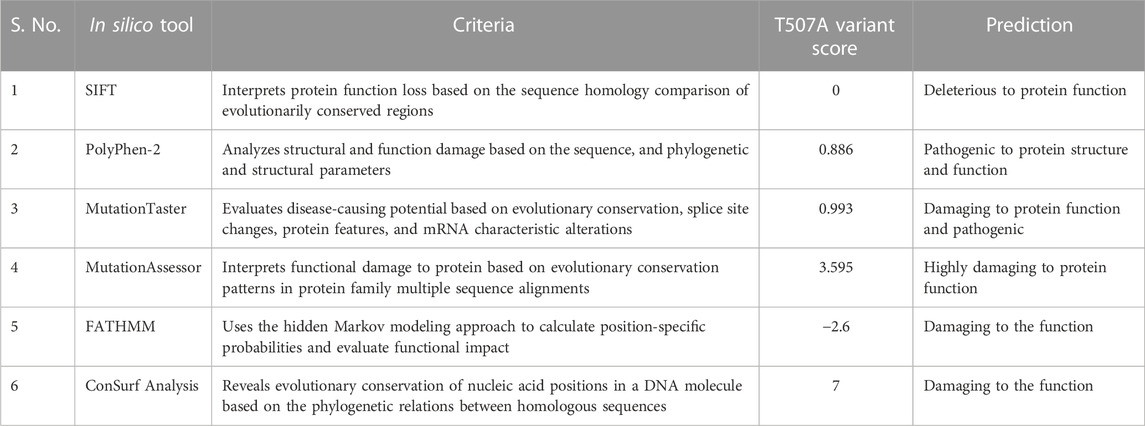
TABLE 2. Computational analysis for pathogenicity prediction of the T507A variant by in silico tools (SIFT, PolyPhen-2, MutationTaster, MutationAssessor, FATHMM, and ConSurf Analysis).
Discussion
The identified novel homozygous variant (T507A) in HSD3B2 genetically segregates the affected and non-affected individuals of the family. Both the parents and the two non-affected male siblings were heterozygous carriers (T/A), suggesting an autosomal recessive pattern of inheritance.
3β-Hydroxysteroid 2-dehydrogenase (3β-HSD2), a product of the HSD3B2 gene and an isozyme of 3β-hydroxysteroid dehydrogenase (3βHSD), is important for the biosynthesis of steroids, in particular, testosterone (Morrison et al., 1991; Bain et al., 1993). It is expressed predominantly in the adrenal gland, the ovary, and the testis (Lachance et al., 1992). Its role and function are implicated in several studies on hypospadias (Codner et al., 2004a; Carmichael et al., 2014; Kalfa et al., 2019). In embryonic males, the enzymatic steps that contribute to steroidogenesis mainly occur in the fetal Leydig cells via the steroidogenic pathway, which converts cholesterol into sex hormones, especially testosterone and its derivative dihydrotestosterone. The product of HSD3B2 is involved in enzymatic conversion of numerous precursors, such as pregnenolone to progesterone, 17αOH pregnenolone to 17αOH progesterone, dehydroepiandrosterone (DHEA) to androstenedione, and androstenedione to testosterone (Carmichael et al., 2014; Bouty et al., 2015). These are critical for the hormone-dependent development of the male genital tract, including the urethral tract closure and morphogenesis of male genitalia. Abnormalities in this development cause hypospadias (Kotula-Balak et al., 2012). Physiologically, the T507A variant observed in our probands may have affected the normal fetal steroidogenic pathway, leading to hypospadias in the affected siblings.
Numerous loci variants for HSD3B2 are reported in earlier studies (Codner et al., 2004a; Wang et al., 2007; Rabbani et al., 2012; Kon et al., 2015). However, the T507A variant (N169K amino acid change) observed in our study is novel and not reported earlier. A ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) search for the HSD3B2 variant suggests that most genetic changes in HSD3B2 are associated with abnormalities in the urogenital system (Supplementary Table S3). In a systematic mutation screening of 25 known causative/candidate/susceptible genes in Japanese and Vietnamese patients, Kon M et al. suggested that HSD3B2 mutations may potentially lead to non-syndromic hypospadias as a sole clinical manifestation (Kon et al., 2015). In another study, HSD3B2 was genotyped in 90 hypospadias patients and 101 healthy controls. Two missense heterozygous mutations (S213T and S284R) and three heterozygous variants (A238, T259, and T320) with no amino acid change were observed in patients but not in controls. These findings suggested that subtle molecular abnormalities in the HSD3B2 gene may be observed in idiopathic hypospadias patients (Codner et al., 2004a). A 27-bp deletion (687del27) in exon IV, deleting the terminal base pair of codon 229 and the entire codons 230–237, in addition to the first two base pairs of codon 238, is also reported in a normozoospermic man with hypospadias (Donadille et al., 2018). A homozygous missense mutation of A82P in exon 3 of the HSD3B2 gene is also reported in a hypospadias patient (Rabbani et al., 2012). Therefore, mutations of HSD3B2 underlie various types of hypospadias, as shown by the evidence of these earlier studies.
In addition, in silico analysis (using SIFT, PolyPhen-2, MutationTaster, MutationAssessor, FATHMM, and ConSurf) consistently predicted the severely deleterious nature of the observed T507A variant in our patients. We employed six in silico tools to increase the stringency of our analysis because individual tools often disagree, in part because they use different predictive features. Together, these tools encompass a broad range of non-synonymous variant/amino acid classification criteria and methods, collectively making the final predicted outcome more accurate. Three important features that concern the mutant missense variant (N169K) observed in our study are as follows: 1) our variant is in close proximity to one of the two catalytic sites (amino acid 154–158 and 269–273) reported in earlier mutagenesis and structural studies (Morel et al., 1997; Thomas et al., 2002; Simard et al., 2005). The amino acid substitutions located at or near the binding interface or active site could hinder the active site, change the recognition, alter the specificity, or affect the binding affinity (Parker, 2001; Mata et al., 2006). Therefore, the deleterious influence of amino acid substitution on HSD3B2 enzyme activity in our patients cannot be overruled (Figure 2). 2) The N169K variant is located in a locus that is evolutionarily conserved across closely related species (Table 1; Figure 2). The degree of evolutionary conservation of an amino acid reflects the structural and functional importance of the domain. The higher the evolutionary conservation, the more critical is the function of the domain/protein and the need to retain the domain/protein’s structural integrity (Ashkenazy et al., 2016). 3) The T507A mutation substituted the neutral amino acid asparagine with positively charged (basic) lysine.

FIGURE 2. ConSurf Analysis predicting the pathogenicity score for variations in individual amino acids in HSD3B2.
Although endocrinal disorder has been reported in hypospadias, the percentage of patients with identified endocrinal etiology remains low (Kalfa et al., 2009). Cassorla et al., 2004 reported that isolated patients with hypospadias are more likely to have normal endocrine testicular and androgen end-organ functions (Codner et al., 2004b). In a study by Rey et al. (2005), of 61 patients, only 13 (21.31%) were found to have an androgen synthesis defect (Rey et al., 2005). As development of the urethra occurs during 8–16 weeks gestation, the postnatal hormonal milieu may not reflect early gestation events (Baskin, 2000; Baskin et al., 2001; Holmes et al., 2004). Unfortunately, we could not assess the hormone levels in our patients. However, any relatable clinical feature or symptom associated with any enzyme or hormone imbalance, in particular, adrenal insufficiency, was not observed in our patients.
Inconsistent findings have been reported for the presence of Y-chromosome microdeletions in hypospadias patients (Tateno et al., 2000; Juniarto et al., 2018). We did not observe Y chromosome microdeletions in any of the hypospadias-affected or non-affected siblings in our study.
To the best of our knowledge, this is the second reported familial hypospadias case with HSD3B2 mutations in more than one affected individual from the same family (siblings). The previously published HSD3B2 mutation in hypospadias siblings was published by Ladjouze et al. (2022). In addition, this is the first genetic study on hypospadias from the Yemen ethnicity. Interestingly, an ethnicity-specific difference in gene function is also reported for HSD3B2 variants between Caucasian-American, African-American, Han Chinese-American, and Mexican-American populations (Wang et al., 2007).
In summation, we described a novel variant in the HSD3B2 gene with an autosomal recessive inheritance that probably affected fetal androgen synthesis and caused hypospadias in two siblings. This clinical case re-emphasizes the importance of structurally and functionally intact HSD3B2 in proper urethral folding and external genitalia morphogenesis during fetal life. In silico analysis predicted the variant to be potentially pathogenic to the structure and function of the HSD3B2 gene product. While we have unraveled a single locus for hypospadias, future genetic and functional studies, particularly in patients from the same parents, will provide a better understanding of disease etiology and inheritance. Additionally, it will aid in developing strategies for improved management of future pregnancies in high-risk familial cases.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA982072 and https://www.ncbi.nlm.nih.gov/clinvar/docs/submit/, SUB12530918.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethical Committee for Scientific Research of Taibah University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
Conceptualization: MS and FA. Experimental work: GA-H and DZ. Data analysis: MS, TM, and FA. Clinical support: HHA and SI. Manuscript writing: MS, FA, HHA, and SI.
Funding
The authors are thankful to Deputyship for Research & Innovation, Ministry of Education, Kingdom of Saudi Arabia for providing funding for this research work (Project No. 442-158).
The authors would also like to extend their gratitude to the Deanship of Scientific Research (DSR), Taibah University for its supervision and kind support.
Acknowledgments
The authors thank the Deanship of Scientific Research, Ministry of Education, Taibah University (Saudi Arabia), for funding this research work through Project Number 442-158. The authors also acknowledge the participating patients and their family for support and cooperation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1106933/full#supplementary-material
Abbreviations
DHEA, dehydroepiandrotone; HSD3B2, 3β-hydroxysteroid 2-dehydrogenase; WES, whole-exome sequencing.
References
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7 (4), 248–249. doi:10.1038/nmeth0410-248
Ashkenazy, H., Abadi, S., Martz, E., Chay, O., Mayrose, I., Pupko, T., et al. (2016). ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44 (W1), W344–W350. doi:10.1093/nar/gkw408
Bain, P. A., Meisler, M. H., Taylor, B. A., and Payne, A. H. (1993). The genes encoding gonadal and nongonadal forms of 3 beta-hydroxysteroid dehydrogenase/delta 5-delta 4 isomerase are closely linked on mouse chromosome 3. Genomics 16 (1), 219–223. doi:10.1006/geno.1993.1162
Baskin, L. S., and Ebbers, M. B. (2006). Hypospadias: Anatomy, etiology, and technique. J. Pediatr. Surg. 41 (3), 463–472. doi:10.1016/j.jpedsurg.2005.11.059
Baskin, L. S., Erol, A., Jegatheesan, P., Li, Y., Liu, W., and Cunha, G. R. (2001). Urethral seam formation and hypospadias. Cell Tissue Res. 305, 379–387. doi:10.1007/s004410000345
Baskin, L. S. (2000). Hypospadias and urethral development. J. Urol. 163, 951–956. doi:10.1097/00005392-200003000-00076
Bouty, A., Ayers, K. L., Pask, A., Heloury, Y., and Sinclair, A. H. (2015). The genetic and environmental factors underlying hypospadias. Sex. Dev. 9 (5), 239–259. doi:10.1159/000441988
Carmichael, S. L., Shaw, G. M., Laurent, C., Olney, R. S., Lammer, E. J., and National Birth Defects Prevention Study, (2007). Maternal reproductive and demographic characteristics as risk factors for hypospadias. Paediatr. Perinat. Epidemiol. 21 (3), 210–218. doi:10.1111/j.1365-3016.2007.00809.x
Carmichael, S. L., Witte, J. S., Ma, C., Lammer, E. J., and Shaw, G. M. (2014). Hypospadias and variants in genes related to sex hormone biosynthesis and metabolism. Andrology 2 (1), 130–137. doi:10.1111/j.2047-2927.2013.00165.x
Carter, H., Douville, C., Stenson, P. D., Cooper, D. N., and Karchin, R. (2013). Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genomics 14 (3), S3. doi:10.1186/1471-2164-14-S3-S3
Codner, E., Okuma, C., Iñiguez, G., Boric, M. A., Avila, A., Johnson, M. C., et al. (2004). Molecular study of the 3 beta-hydroxysteroid dehydrogenase gene type II in patients with hypospadias. J. Clin. Endocrinol. Metab. 89 (2), 957–964. doi:10.1210/jc.2002-020873
Codner, E., Okuma, C., Iñiguez, G., BoricM, A., Avila, A., Johnson, M. C., et al. (2004). Molecular study of the 3β-hydroxysteroid dehydrogenase gene type II in patients with hypospadias. J. Clin. Endocrinol. Metabolism 89 (2), 957–964. doi:10.1210/jc.2002-020873
Donadille, B., Houang, M., Netchine, I., Siffroi, J. P., and Christin-Maitre, S. (2018). Human 3beta-hydroxysteroid dehydrogenase deficiency associated with normal spermatic numeration despite a severe enzyme deficit. Endocr. Connect. 7 (3), 395–402. doi:10.1530/EC-17-0306
Fredell, L., Kockum, I., Hansson, E., Holmner, S., Lundquist, L., Lšckgren, G., et al. (2002). Heredity of hypospadias and the significance of low birth weight. J. Urol. 167 (3), 1423–1427. doi:10.1097/00005392-200203000-00069
Holmes, N. M., Miller, W. L., and Baskin, L. S. (2004). Lack of defects in androgen production in children with hypospadias. J. Clin. Endocrinol. Metabolism 89 (6), 2811–2816. doi:10.1210/jc.2003-032098
Juniarto, A., Yasari, N. A. L., Santosa, A., and Faradz, L. (2018). Sultana. Investigation of Azoospermia Factor (AZF) microdeletion of hypospadia patients in Indonesian population. Pak. J. Med. Health Sci. 12, 1350–1353.
Kalfa, N., Gaspari, L., Ollivier, M., Philibert, P., Bergougnoux, A., Paris, F., et al. (2019). Molecular genetics of hypospadias and cryptorchidism recent developments. Clin. Genet. 95 (1), 122–131. doi:10.1111/cge.13432
Kalfa, N., Philibert, P., and Sultan, C. (2009). Is hypospadias a genetic, endocrine or environmental disease, or still an unexplained malformation? Int. J. Androl. 32 (3), 187–197. doi:10.1111/j.1365-2605.2008.00899.x
Kircher, M., Witten, D. M., Jain, P., O'Roak, B. J., Cooper, G. M., and Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46 (3), 310–315. doi:10.1038/ng.2892
Kon, M., Suzuki, E., Dung, V. C., Hasegawa, Y., Mitsui, T., Muroya, K., et al. (2015). Molecular basis of non-syndromic hypospadias: Systematic mutation screening and genome-wide copy-number analysis of 62 patients. Hum. Reprod. 30 (3), 499–506. doi:10.1093/humrep/deu364
Kotula-Balak, M., Hejmej, A., and Barbara, B. (2012). Hydroxysteroid dehydrogenases - localization, function and regulation in the testis, dehydrogenases. London, United Kingdom: Rosa Angela Canuto, IntechOpen. doi:10.5772/47740
KšllŽn, B., Bertollini, R., Castilla, E., Czeizel, A., Knudsen, L. B., Martinez-Frias, M. L., et al. (1986). A joint international study on the epidemiology of hypospadias. Acta Paediatr. Scand. Suppl. 324, 1–52. doi:10.1111/j.1651-2227.1986.tb14935.x
Kurahashi, N., Murakumo, M., Kakizaki, H., Nonomura, K., Koyanagi, T., Kasai, S., et al. (2004). The estimated prevalence of hypospadias in Hokkaido, Japan. J. Epidemiol. 14 (3), 73–77. doi:10.2188/jea.14.73
Lachance, Y., Luu-The, V., Labrie, C., Simard, J., Dumont, M., de Launoit, Y., et al. (1992). Characterization of human 3 beta-hydroxysteroid dehydrogenase/delta 5-delta 4-isomerase gene and its expression in mammalian cells. J. Biol. Chem. 267 (5), 3551. doi:10.1016/s0021-9258(19)50764-5
Ladjouze, A., Donaldson, M., Plotton, I., Djenane, N., Mohammedi, K., Tardy-Guidollet, V., et al. (2022). Genotype, mortality, morbidity, and outcomes of 3β-hydroxysteroid dehydrogenase deficiency in Algeria. Front. Endocrinol. 13, 867073. doi:10.3389/fendo.2022.867073
Leung, A. K., and Robson, W. L. (2007). Hypospadias: An update. Asian J. Androl. 9 (1), 16–22. doi:10.1111/j.1745-7262.2007.00243.x
Mata, I. F., Wedemeyer, W. J., Farrer, M. J., Taylor, J. P., and Gallo, K. A. (2006). LRRK2 in Parkinson's disease: Protein domains and functional insights. Trends Neurosci. 29 (5), 286–293. doi:10.1016/j.tins.2006.03.006
Morel, Y., Mžbarki, F., RhŽaume, E., Sanchez, R., Forest, M. G., and Simard, J. (1997). Structure-function relationships of 3 beta-hydroxysteroid dehydrogenase: Contribution made by the molecular genetics of 3 beta-hydroxysteroid dehydrogenase deficiency. Steroids 62 (1), 176–184. doi:10.1016/s0039-128x(96)00178-x
Morrison, N., Nickson, D. A., McBride, M. W., Mueller, U. W., Boyd, E., and Sutcliffe, R. G. (1991). Regional chromosomal assignment of human 3-beta-hydroxy-5-ene steroid dehydrogenase to 1p13.1 by non-isotopic in situ hybridisation. Hum. Genet. 87 (2), 223–225. doi:10.1007/BF00204189
Parker, J. (2001) Amino Acid Subsitutions. In brenner's online encyclopedia of genetics (eBook ISBN: 9780080547909) 57–58. Available from: https://www.elsevier.com/books/T/A/9780122270802.
Paulozzi, L. J. (1999). International trends in rates of hypospadias and cryptorchidism. Environ. Health Perspect. 107 (4), 297–302. doi:10.1289/ehp.99107297
Quang, D., Chen, Y., and Xie, X. (2015). Dann: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 31 (5), 761–763. doi:10.1093/bioinformatics/btu703
Rabbani, B., Mahdieh, N., Haghi Ashtiani, M. T., Setoodeh, A., and Rabbani, A. (2012). In silico structural, functional and pathogenicity evaluation of a novel mutation: An overview of HSD3B2 gene mutations. Gene 503 (2), 215–221. doi:10.1016/j.gene.2012.04.080
Rey, R. A., Codner, E., Iñíguez, G., Bedecarrás, P., Trigo, R., Okuma, C., et al. (2005). Low risk of impaired testicular sertoli and Leydig cell functions in boys with isolated hypospadias. J. Clin. Endocrinol. Metabolism 90 (11), 6035–6040. doi:10.1210/jc.2005-1306
Rogers, M. F., Shihab, H. A., Mort, M., Cooper, D. N., Gaunt, T. R., and Campbell, C. (2018). FATHMM-XF: Accurate prediction of pathogenic point mutations via extended features. Bioinformatics 34 (3), 511–513. doi:10.1093/bioinformatics/btx536
Ságodi, L., Kiss, A., Kiss-Tóth, E., and Barkai, L. (2014). Prevalence and possible causes of hypospadias. Orvosi Hetil. 155 (25), 978–985. doi:10.1556/OH.2014.29858
Schnack, T. H., Zdravkovic, S., Myrup, C., Westergaard, T., Christensen, K., Wohlfahrt, J., et al. (2008). Familial aggregation of hypospadias: A cohort study. Am. J. Epidemiol. 167 (3), 251–256. doi:10.1093/aje/kwm317
Schwarz, J. M., Ršdelsperger, C., Schuelke, M., and Seelow, D. (2010). MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7 (8), 575–576. doi:10.1038/nmeth0810-575
Shamsi, M. B., Kumar, R., Malhotra, N., Singh, N., Mittal, S., Upadhyay, A. D., et al. (2012). Chromosomal aberrations, Yq microdeletion, and sperm DNA fragmentation in infertile men opting for assisted reproduction. Mol. Reprod. Dev. 79 (9), 637–650. doi:10.1002/mrd.22072
Shihab, H. A., Gough, J., Cooper, D. N., Stenson, P. D., Barker, G. L., Edwards, K. J., et al. (2013). Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 34 (1), 57–65. doi:10.1002/humu.22225
Sim, N. L., Kumar, P., Hu, J., Henikoff, S., Schneider, G., and Ng, P. C. (2012). SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40, W452–W457. doi:10.1093/nar/gks539
Simard, J., Ricketts, M. L., Gingras, S., Soucy, P., Feltus, F. A., and Melner, M. H. (2005). Molecular biology of the 3beta-hydroxysteroid dehydrogenase/delta5-delta4 isomerase gene family. Endocr. Rev. 26 (4), 525–582. doi:10.1210/er.2002-0050
Tateno, T., Sasagawa, I., Ashida, J., Nakada, T., and Ogata, T. (2000). Absence of Y-chromosome microdeletions in patients with isolated hypospadias. Fertil. Steril. 74 (2), 399–400. doi:10.1016/s0015-0282(00)00629-4
Thomas, J. L., Mason, J. I., Brandt, S., Spencer, B. R., and Norris, W. (2002). Structure/function relationships responsible for the kinetic differences between human type 1 and type 2 3beta-hydroxysteroid dehydrogenase and for the catalysis of the type 1 activity. J. Biol. Chem. 277 (45), 42795–42801. doi:10.1074/jbc.M208537200
Thorup, J., Nordenskjšld, A., and Hutson, J. M. (2014). Genetic and environmental origins of hypospadias. Curr. Opin. Endocrinol. Diabetes Obes. 21 (3), 227–232. doi:10.1097/MED.0000000000000063
van der Zanden, L. F., van Rooij, I. A., Feitz, W. F., Franke, B., Knoers, N. V., and Roeleveld, N. (2012). Aetiology of hypospadias: A systematic review of genes and environment. Hum. Reprod. Update 18 (3), 260–283. doi:10.1093/humupd/dms002
Wang, L., Salavaggione, E., Pelleymounter, L., Eckloff, B., Wieben, E., and Weinshilboum, R. (2007). Human 3beta-hydroxysteroid dehydrogenase types 1 and 2: Gene sequence variation and functional genomics. J. Steroid Biochem. Mol. Biol. 107 (1-2), 88–99. doi:10.1016/j.jsbmb.2007.03.037
Keywords: hypospadias, 3β-hydroxysteroid dehydrogenase 2, genetic risk factors, whole-exome sequencing, Y-chromosome microdeletion
Citation: Almaramhy HH, Abdul Samad F, Al-Harbi G, Zaytuni D, Imam SN, Masoodi T and Shamsi MB (2023) Identification of a novel candidate HSD3B2 gene variant for familial hypospadias by whole-exome sequencing. Front. Genet. 14:1106933. doi: 10.3389/fgene.2023.1106933
Received: 24 November 2022; Accepted: 27 March 2023;
Published: 13 June 2023.
Edited by:
Jared C. Roach, Institute for Systems Biology (ISB), United StatesReviewed by:
Marek Switonski, Poznan University of Life Sciences, PolandMeraj Alam Khan, University of Toronto, Canada
Mohammad Tanweer Alam, Technological University Dublin, Ireland
Dimitrios T. Papadimitriou, National and Kapodistrian University of Athens, Greece
Copyright © 2023 Almaramhy, Abdul Samad, Al-Harbi, Zaytuni, Imam, Masoodi and Shamsi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Monis Bilal Shamsi, bW9uaXNiaWxhbHNoYW1zaUBnbWFpbC5jb20=