 Ariska van der Nest1
Ariska van der Nest1 Michael J. Wingfield1
Michael J. Wingfield1 Dušan Sadiković2,3
Dušan Sadiković2,3 Martin S. Mullett4
Martin S. Mullett4 Benoit Marçais5Valentin Queloz6*Katarina Adamčíková7
Benoit Marçais5Valentin Queloz6*Katarina Adamčíková7 Kateryna Davydenko8,9
Kateryna Davydenko8,9 Irene Barnes1*
Irene Barnes1*- 1Department of Biochemistry, Genetics and Microbiology, Forestry and Agricultural Biotechnology Institute (FABI), University of Pretoria, Pretoria, South Africa
- 2Slovenian Forestry Institute, Ljubljana, Slovenia
- 3Southern Swedish Forest Research Centre, Swedish University of Agricultural Science, Alnarp, Sweden
- 4Phytophthora Research Centre, Mendel University in Brno, Brno, Czechia
- 5Université de Lorraine, INRAE-Grand-Est, UMR1136 Interactions Arbres, Microorganismes, Nancy, France
- 6Swiss Federal Research Institute WSL, Birmensdorf, Switzerland
- 7Institute of Forest Ecology Slovak Academy of Sciences, Department of Plant Pathology and Mycology, Nitra, Slovakia
- 8Department of Forest Mycology and Plant Pathology, Swedish University of Agricultural Science, Uppsala, Sweden
- 9Ukrainian Forest Research Institute, Forestry and Forest Melioration, Kharkiv, Ukraine, Slovakia
Dothistroma needle blight (DNB) is an important disease of Pinus species that can be caused by one of two distinct but closely related pathogens; Dothistroma septosporum and Dothistroma pini. Dothistroma septosporum has a wide geographic distribution and is relatively well-known. In contrast, D. pini is known only from the United States and Europe, and there is a distinct lack of knowledge regarding its population structure and genetic diversity. The recent development of 16 microsatellite markers for D. pini provided an opportunity to investigate the diversity, structure, and mode of reproduction for populations collected over a period of 12 years, on eight different hosts in Europe. In total, 345 isolates from Belgium, the Czech Republic, France, Hungary, Romania, Western Russia, Serbia, Slovakia, Slovenia, Spain, Switzerland, and Ukraine were screened using microsatellite and species-specific mating type markers. A total of 109 unique multilocus haplotypes were identified and structure analyses suggested that the populations are influenced by location rather than host species. Populations from France and Spain displayed the highest levels of genetic diversity followed by the population in Ukraine. Both mating types were detected in most countries, with the exception of Hungary, Russia and Slovenia. Evidence for sexual recombination was supported only in the population from Spain. The observed population structure and several shared haplotypes between non-bordering countries provides good evidence that the movement of D. pini in Europe has been strongly influenced by human activity in Europe.
1 Introduction
Dothistroma needle blight (DNB) is recognized as one of the most important diseases of Pinus spp., both in planted and native forests, worldwide. The disease has a long history of having damaged plantations in the Southern Hemisphere dating back to the 1960s (Gibson, 1972), but during the course of the last three decades, it has also increased in severity and incidence in the Northern Hemisphere (Drenkhan and Hanso, 2009; Welsh et al., 2009; Fabre et al., 2012; Boroń et al., 2016; Drenkhan et al., 2016; Ghelardini et al., 2020). Dothistroma needle blight has been reported on 113 taxa, of which 99 are in the genus Pinus (Drenkhan et al., 2016; Jánošíková-Hečková et al., 2018; Barnes et al., 2022) and reports of the disease on new hosts and in new geographical regions are increasing (Jánošíková-Hečková et al., 2018; Matsiakh et al., 2018; Mullett et al., 2018; Ondrušková et al., 2018; EPPO, 2019; Mesanza et al., 2021). The disease has been reported on Abies, Cedrus, Larix, Picea, and Pseudotsuga (Drenkhan et al., 2016), although in most cases, infection has occurred when high inoculum load of the pathogen was present on Pinus species in close proximity to these hosts (Barnes et al., 2022).
For many years, the identity of the causal agents of DNB was confused and strongly debated (Barnes et al., 2016). This was due to a single distinct symptom (red bands on infected needles) and taxonomy reliant on morphological characteristics of the associated pathogen. Almost 110 years after the first description of DNB in France (Vuillemin, 1896), it was conclusively shown that two distinct species can cause this disease. These include Dothistroma septosporum (Dorogin) M. Morelet and Dothistroma pini Hulbary that are most effectively distinguished based on molecular identification (Barnes et al., 2004; Barnes et al., 2016). In an attempt to consolidate existing knowledge, an extensive collaboration of pathologists participating in the DIAROD (Determining Invasiveness And Risk Of Dothistroma: DIAROD, COST Action FP1102) project documented, as far as possible, the geographic distribution, hosts and mating type distribution of these two Dothistroma species (Drenkhan et al., 2016).
Dothistroma septosporum has been the most extensively studied of the two DNB pathogens. This is at least in part due to its accidental introduction into various countries of the Southern Hemisphere where it became one of the most important constraints to plantation forestry based on non-native Pinus radiata (Gibson, 1972). Dothistroma septosporum has now been recorded in both the Southern and Northern Hemispheres in 48 countries (Drenkhan et al., 2016; Matsiakh et al., 2018; Mullett et al., 2018; Ghelardini et al., 2020) and its population structure and diversity in many of these areas is well understood (Drenkhan et al., 2013; Barnes et al., 2014b; Mullett et al., 2015; Adamson et al., 2018; Oskay et al., 2020; Capron et al., 2021; Mullett et al., 2021). Several genomes of the pathogen have been sequenced and population genomics studies (Ennos et al., 2020), as well as investigations considering factors affecting its pathogenicity have been conducted (Bradshaw et al., 2019; Guo et al., 2020). In contrast, very little is known regarding the biology or ecology of D. pini.
Dothistroma pini is known only in the Northern Hemisphere where it has been recorded in 17 countries on 19 different Pinus hosts as well as Picea abies (Drenkhan et al., 2016; Jánošíková-Hečková et al., 2018; Matsiakh et al., 2018; Mullett et al., 2018; Ondrušková et al., 2018). The pathogen was first described on non-native Pinus nigra J.F. Arnold collected in Michigan (1960s), Minnesota and Nebraska in the United States (Barnes et al., 2004). At that time, it was thought to be restricted to the North American continent. Since then, D. pini has been reported in four additional states of the United States (Barnes et al., 2014a; Mullett et al., 2018).
Dothistroma pini was first discovered in Europe when it was found in the Ukraine and Russia in 2008 on non-native P. nigra subsp. pallasiana (Lamb.) Holmboe (Barnes et al., 2008b). However, molecular analysis of herbarium samples collected in France have shown that the pathogen has been present on the European continent at least since 1907 (Fabre et al., 2012). Since the first molecular identification of D. pini in Europe in 2008, the pathogen has also been confirmed as present in Belgium (Schmitz et al., 2013), Czech Republic (Bergová and Kryštofová, 2014), France (Ioos et al., 2010), Georgia (Matsiakh et al., 2018), Germany (EPPO, 2019), Hungary (Barnes et al., 2011), Montenegro (Lazarević et al., 2017), Poland (Wartalska et al., 2021), Romania (Barnes et al., 2016), Serbia (Pap et al., 2015), Slovenia (Piškur et al., 2013), Slovakia (Ondrušková et al., 2017), Spain (Iturritxa et al., 2015) and Switzerland (Queloz et al., 2014).
Very little is known regarding the genetic diversity and population structure of D. pini. In a preliminary study testing 16 microsatellite markers developed for D. pini (Siziba et al., 2016), high levels of genetic diversity were found in populations of the pathogen in France, at least indicating the presence of the pathogen in that country for many years. In contrast, populations in other European countries such as Slovakia displayed low genetic diversity and strong signals of clonality, which suggests that D. pini was introduced into Slovakia (Adamčíková et al., 2021).
Collections of D. pini made over a 12-year period, and including those obtained while documenting the presence of both this species and D. septosporum in Europe by the DIAROD cost action, has resulted in a collection of 345 isolates. This collection provided an opportunity to expand on previous, relatively small-scale studies (Siziba et al., 2016; Adamčíková et al., 2021), and to more comprehensively consider the population structure and diversity of D. pini in Europe. The aims of this study were thus to 1) investigate the genetic diversity and population structure of the pathogen including countries or specific locations where the pathogen has been reported in Europe, and 2) determine its mode of reproduction and likely means of dispersal in Europe.
2 Materials and methods
2.1 Sample collection, fungal isolations and identifications
Pine needles that displayed DNB symptoms were collected between 2008 and 2019 from 30 locations in 11 countries of Europe (Supplementary Table S2, Figure 1). Additionally, the data generated for the 10 locations in Slovakia by Adamčíková et al. (2021) were incorporated in this study. For most samples, isolations were made from the collected samples as described by Barnes et al. (2004). Single germinating conidia were selected and plated onto 2% Dothistroma Sporulating Media (DSM: 5 g yeast extract (Biolab, Merck, Modderfontein, South Africa), 20 g malt extract (Biolab) and 15 g agar (BD Difco™, Sparks, MD)) per liter of distilled water with 100 mg/l streptomycin (Sigma-Aldrich, St Louis, MO). The plates were incubated for 4–6 weeks at 23°C under natural day/night light cycles. All isolates are either maintained as cultures or freeze-dried material in the culture collection (CMW) of the Forestry and Agricultural Biotechnology Institute (FABI) in Pretoria, South Africa (Supplementary Table S2).
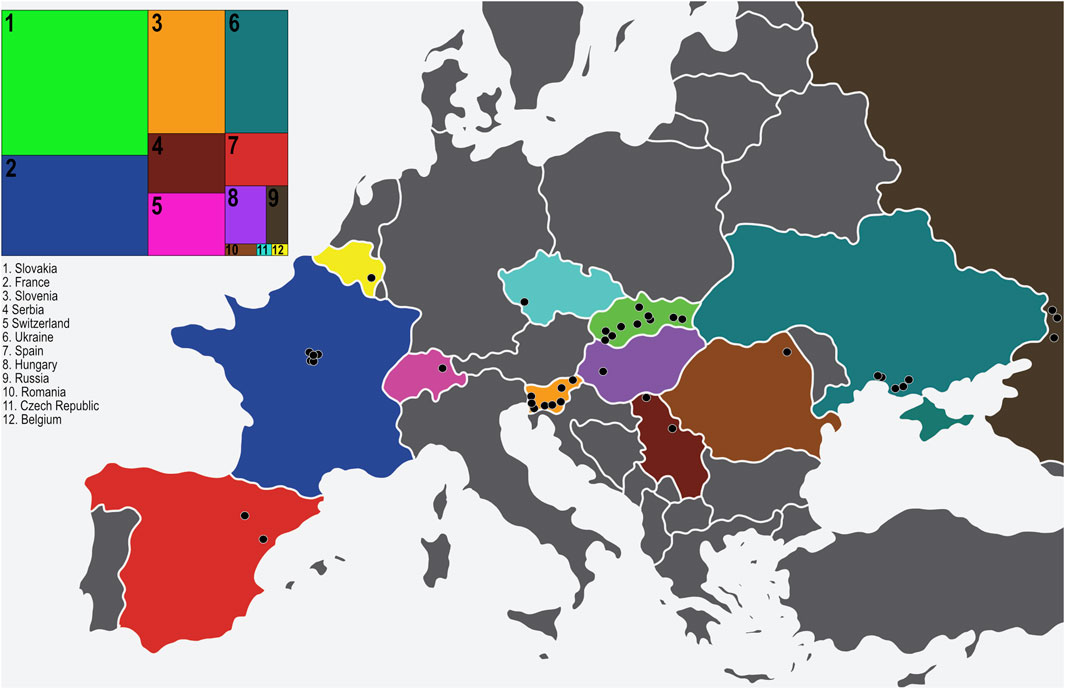
FIGURE 1. The 40 sampling locations of D. pini in Europe. The insert to the left indicates the proportion of isolates obtained per country in relation to other countries (numbered from 1–12). The countries are colour coded on the map to match the insert (original map obtained from https://www.vecteezy.com/free-vector/europe-map) and each sampling location is indicated with a black circle.
Fungal tissue was freeze dried and DNA extracted using a Zymo Research ZR fungal/Bacterial DNA MiniPrep™ kit (Irvine, CA) as described by van der Nest et al. (2019b). The identity of the isolates was determined by amplifying and sequencing the internal transcribed spacers (ITS) 1 and 2 and the 5.8 S rDNA region with the ITS1 and ITS4 primers (White et al., 1990) and using the protocols described in Barnes et al. (2004). The PCR amplicons were sequenced in both directions using the BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific) and the product was run on an ABI PRISM 3500xl capillary auto sequencer (Thermo Fisher Scientific).
CLC Main workbench version 8.0 (CLC Bio, https://www.qiagenbioinformatics.com/products/clc-main-workbench/) was used to create consensus sequences using the forward and reverse sequences of the ITS region for each isolate. All consensus sequences were compared in a BLAST analysis against the GenBank database (NCBI; http://www.ncbi.nlm.nih.gov/genbank/) to confirm the identity of each isolate. To determine the ITS haplotype for each confirmed isolate of D. pini, sequences were compared to those reported in Barnes et al. (2016); Mullett et al. (2018) using MEGA 7.0.14 (Kumar et al., 2016).
2.2 Microsatellite amplification and haplotype determination
Sixteen labelled microsatellite markers (Siziba et al., 2016) were used to amplify all isolates considered in this study. An additional marker (Doth_A; Barnes et al. (2008a)) was included as an internal diagnostic marker. PCR reactions were performed, and where needed, optimized as described by Adamčíková et al. (2021) (see also Supplementary Table S3) to produce single PCR products. PCR reactions were carried out on an Applied Biosystems® Veriti® 96 well Thermal cycler (Thermo Fisher Scientific, Waltham, MA). The fragments were amplified using the same cycling conditions described by Barnes et al. (2014b) with primer pair annealing temperatures as described by Adamčíková et al. (2021) (see also Supplementary Table S3). To determine amplification success, 5 µl PCR product was stained with 1 µl GelRed nucleic acid gel stain (Biotium), separated by gel electrophoresis on 2% SeaKem LE agarose gel (Lonza) for 15 min at 90 V and visualized under a UV light using a GelDoc EZ Imager (BioRad).
PCR products were pooled in two panels for fragment analysis as described by Siziba et al. (2016) and with adjusted dilutions as indicated in Supplementary Table S3. In preparation for analysis, 1 μl of the pooled product was added to 0.14 μl GENESCAN™ -500 LIZ® (Life Technologies, Applied Biosystems, Warrington, United Kingdom) size standard and 12 μl formamide. Fragment analyses of the prepared reactions was conducted at the University of Pretoria in South Africa with an ABI PRISM 3500xl capillary auto sequencer (Thermo Fisher Scientific). Allele sizes were scored using GENEMAPPER® Software version 5.0 (Applied Biosystems, Foster City, CA).
Alleles scored for each marker were combined to obtain a multilocus haplotype (MLH) for each isolate. Individual isolates were considered clones if they had the same combination of alleles for each marker analyzed. The R package poppr (Kamvar et al., 2014) was used to determine the number of MLHs in the dataset. Two datasets were generated for further analyses; the dataset that had not been clone-corrected included all individuals and the clone-corrected dataset contained single representatives of each unique MLH per population. Individuals from each particular country were grouped as populations.
2.3 Genetic diversity
The R package poppr (Kamvar et al., 2014) was used to calculate the number of MLHs, the expected number of MLHs based on rarefaction (Hurlbert, 1971), the Shannon-Wiener Index (Shannon, 2001), the Stoddart and Taylor’s Index (Stoddart and Taylor, 1988), the Simpson’s Index (Simpson, 1949) and genotypic evenness (Grünwald et al., 2003) for the populations using the non-clone-corrected dataset, as well as the genetic diversity (Nei, 1978) per population using the clone-corrected dataset. The clonal fraction was calculated as in Barnes et al. (2014b). Furthermore, allelic richness (AR) and private allelic richness (PAR) were determined using ADZE (Szpiech et al., 2008) that uses rarefaction to allow for comparisons between populations with varying sample sizes. Calculations were standardized corresponding to the country with the smallest population size (Russia, N = 6). A minimum spanning network using Bruvo’s genetic diversity (Bruvo et al., 2004) comparing the MLHs over 16 microsatellite loci was also drawn using the ismn function in the poppr package.
2.4 Population structure
The clone-corrected dataset was used to determine the most likely number of population clusters based on microsatellite allele sizes for all the individuals using STRUCTURE 2.3.4 (Falush et al., 2003). The program assigns individuals to clusters (K) using a Bayesian clustering algorithm. Thirty independent runs of K = 1–20 were performed, with a burn-in value of 100,000 and 500,000 iterations. An admixture model with correlated allele frequencies was selected with no additional priors such as information on the host or location.
The optimal number of clusters was estimated with StructureSelector (Li and Liu, 2018). StructureSelector implements the Evanno method that includes delta (K) and LnP (K) (Evanno et al., 2005) with the additional four Puechmaille methods (MAXMEAK, MAXMEDK, MEDMEDK and MEDMEAK) that provide a more accurate estimate of K in populations with uneven sizes (Puechmaille, 2016). In order to implement the Puechmaille methods, countries were assigned as populations in the dataset and the analysis was repeated twice. First a threshold of 0.5 was selected and second a threshold of 0.8 was selected to apply more stringent assignment of individuals into clusters. After the optimal K was determined, isolates were assigned into the optimal K clusters with a final STRUCTURE run with 30 independent runs, a burn-in value of 100,000 and 1,000 000 iterations. CLUMPAK (Kopelman et al., 2015) was used to converge all 30 runs of the optimal K and the output was visualized using the DISTRUCT program (Rosenberg, 2004). Both CLUMPAK and DISTRUCT were implemented using the StructureSelector website (https://lmme.qdio.ac.cn/StructureSelector/).
The adegenet package in R studio (Jombart and Ahmed, 2011) was used to perform discriminant analysis of principal components (DAPC) (Jombart et al., 2010) to additionally visualize the population genetic structure of the European samples. The find.clusters function was used to determine the optimal number of clusters by assessment of the Bayesian information criterion (BIC). The optimal number of principal components retained in the analysis was determined by cross-validation using the xvalDapc function.
An Analysis of Molecular Variance (AMOVA) test was implemented in GENALEX version 6.5 (Peakall and Smouse, 2012). The test was used to evaluate if there was genetic differentiation among and within groups according to host species, countries and locations. One thousand permutations of the dataset were used to test significance. The null hypothesis of no genetic difference was rejected at p < 0.05.
2.5 Mating type determination and random mating
The mating type of the D. pini isolates was determined by using the primers of Groenewald et al. (2007) or in some cases the primer set of Janoušek et al. (2014). Each reaction consisted of 2 μl template DNA (20 ng/μl concentration), 0.08 μl Faststart Taq DNA polymerase, 0.25 μl of each of the primers as specified by either Groenewald et al. (2007) or Janoušek et al. (2014), 0.6 μl of a mix of 200 mM dNTPs, 1.5 μl of 2.5 mM MgCl2, 1.25 μl 10x PCR reaction buffer and the volume was adjusted to 12.5 μl with sterile SABAX water.
PCR reactions were carried out on an Applied Biosystems® Veriti® 96 well Thermal cycler (Thermo Fisher Scientific, Waltham, MA). The cycling conditions for all microsatellite fragments included an initial denaturation step at 95°C for 4 min, 10 cycles consisting of 94°C for 20 s, a 45 s annealing step with the temperature set according to the protocols by Groenewald et al. (2007) or Janoušek et al. (2014), and an elongation step of 45 s at 72°C. This was followed by a further 25 cycles of 94°C for 20 s, 45 s with a 5 s extension step per cycle at the annealing temperature, a 72°C extension for 45 s and a final extension step of 72°C for 30 min. The amplified products were visualized by staining 10 µl of each product with GelRed™ nucleic acid gel stain. The fragments were separated on 2% SeaKem® LE agarose gel for 50 min at 90 V and viewed under a UV light using the GelDoc™ EZ Imager (BioRad, Hercules, CA). When using the Groenewald et al. (2007) primers, isolates that had an amplicon size of 820 bp were assigned as MAT1-1 and those with a size of 480 bp were assigned as MAT1-2. The Janoušek et al. (2014) primer sets produced amplicon sizes of approximately 560–634 bp for MAT1-1 and 288–323 bp for MAT1-2.
The possibility of sexual recombination was investigated using three methods. An exact binomial test, using two-tailed p-values (http://www.biostathandbook.com/exactgof.html) was used to test if the mating type ratios deviated from a 1:1 ratio (at p < 0.05) in the non-clone-corrected dataset, which provides evidence of random mating. The index of association (IA) (Brown et al., 1980; Smith et al., 1993) and rBarD (
3 Results
3.1 Sample collection, fungal isolations and identification
A total of 345 cultures included in this study were obtained from collections made in Europe. All of these isolates screened with the Doth_A marker (Siziba et al., 2016) produced an allele size of 111 bp and were thus confirmed as D. pini. These included representatives from 12 (Belgium, Czech Republic, France, Hungary, Romania, Western Russia, Serbia, Slovakia, Slovenia, Spain, Switzerland and Ukraine) of the 16 European countries where D. pini has been reported. The isolations were made from plant material obtained from 10 different Pinus species or sub-species with P. nigra being the most common of these (Supplementary Table S1).
Three of the six known D. pini ITS haplotypes (Barnes et al., 2016; Mullett et al., 2018) were identified in the collection of isolates (Supplementary Table S1). Individuals having the ITS Haplotype 1 were the most abundant and were present in eight of the twelve countries (Czech Republic, France, Hungary, Slovakia, Slovenia, Spain, Switzerland, Ukraine) including 25 different locations. ITS Haplotype 2 was the second most abundant and was present in eight of the twelve countries (France, Romania, Western Russia, Serbia, Slovakia, Spain, Switzerland, Ukraine) and at 20 different locations. ITS Haplotype 4 individuals were present at nine locations in five countries (Belgium, France, Serbia, Slovakia and Spain). All three haplotypes were present in France, Spain and Slovakia.
3.2 Microsatellite amplification and haplotype determination
A total of 109 alleles were detected across the 16 polymorphic microsatellite loci. The number of alleles at each locus ranged from 2 at DP-MS4 and DP-MS18 to 19 at DP-MS12 (Supplementary Table S3). Isolates from Spain, Ukraine and Russia had the highest percentage (87.5%) of polymorphic loci (Supplementary Table S2) and those from Hungary had the lowest percentage (31.2%) of polymorphic loci (excluding countries for which only single isolates were available).
A total of 109 unique multilocus haplotypes (MLHs) were identified in the 345 isolates analyzed (Table 1; Figure 2, Supplementary Table S2) of which eight MLHs occurred in multiple, often non-bordering countries (Supplementary Figure S1). Some individuals sharing the same microsatellite MLH in different populations were of opposite mating type or of different ITS haplotypes, which suggests that they were not true clones. For example, MLH 52 (Supplementary Figure S1) occurred in isolates from four countries (Hungary, Slovakia, Slovenia and Ukraine) and at seven different locations, covering a distance of approximately 1500 km. This MLH was represented by individuals with the MAT1-1 idiomorph in Ukraine and the MAT1-2 idiomorph in the other three countries. The fifth most commonly occuring MLH (MLH 83, Supplementary Figure S1) was shared by individuals from the Czech Republic, France (La Bouyale, La Ferté-Imbault, and Villefranche-sur-Cher), and Hola Prystan in Ukraine. All of these individuals were of ITS Haplotype 1, except for an individual from La Ferté-Imbault (ITS Haplotype 4) and the individuals from Hola Prystan in Ukraine (ITS Haplotype 2). Furthermore, all individuals were MAT1-1, except for two MAT1-2 individuals; one individual from La Bouyale in France and one individual from Hola Prystan in Ukraine. The population from Russia included an individual having ITS Haplotype 2 that shared MLH 47 (Supplementary Figure S1) with an ITS Haplotype 1 individual in Hungary (1150 km apart) also of opposite mating types.
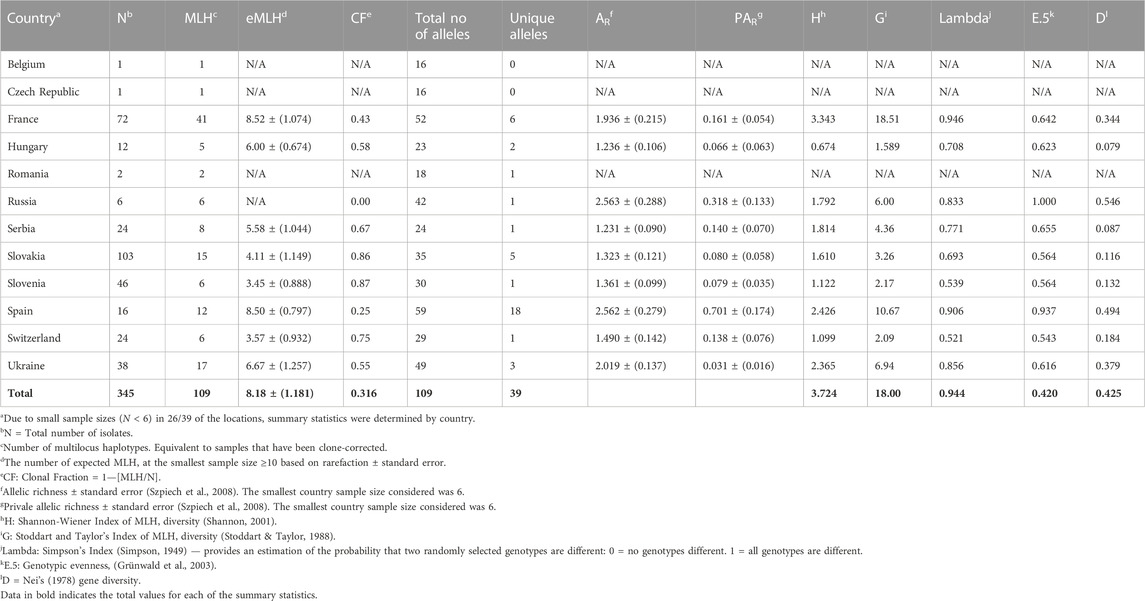
TABLE 1. Summary diversity statistics of Dothistroma pini isolates within populations by country in Europe.
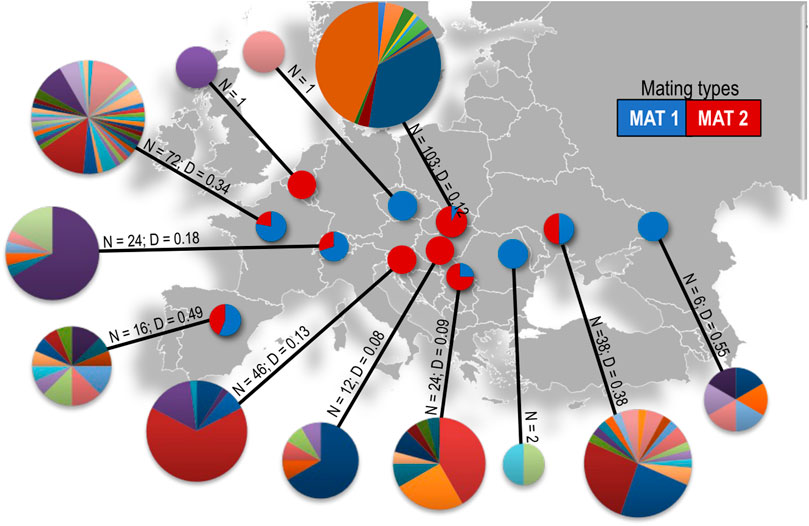
FIGURE 2. Microsatellite haplotype diversity and mating type ratios of D. pini in each of the sampled countries in Europe. Each colour in the pie charts represents a different multilocus haplotype. The size of each MLH pie chart is proportional to the number of isolates per country where Belgium N = 1 and Slovakia N = 103. N = number of isolates, D = Nei’s genetic diversity.
3.3 Genetic diversity
Collections from France had the greatest number of MLHs, followed by the isolates from the Ukraine. When considering populations with a sample size of six and higher, Hungary had the fewest MLHs (five) followed by Russia, Slovenia and Switzerland, which had six each (Table 1). When comparing the approximate number of haplotypes that would be expected for the largest shared sample size (N = 6) based on rarefaction (eMLG), the genotypic richness was the highest in the populations from France and Spain (8.52 and 8.50). The populations from Slovenia and Switzerland had the lowest genetic diversity (3.45 and 3.57 respectively) (Table 1). The Slovenian and Slovakian populations had the highest clonal fractions (0.87 and 0.86) followed by those from Switzerland 0.75 (Table 1). The lowest clonal fraction was found in populations from Russia (0) followed by those from Spain (0.25) and France (0.43). For populations collected within France, the clonal fraction ranged from 0 (Nueng-sur-Beuvron) to 0.61 (Villefranche-sur-Cher). In isolates from Slovenia, the clonal fraction also ranged from 0 (Ribnica) to 0.90 (Panovec). The clonal fraction of 0.55 in Ukraine was due to the high clonal fraction (0.67) in Tsjurupinsk (Supplementary Table S4). The genetic diversity of isolates from all locations is summarized in Supplementary Table S4.
Varying levels of genotypic diversity and genotypic richness were observed for the isolates considered in this study (Table 1). Populations from France followed by Spain displayed the highest level of genetic diversity and richness, based on the Simpson index (H), Stoddart Taylor’s index (G) and allelic richness (AR) and rarefaction of MLGs. The genotypic evenness (E.5) observed in the populations from Russia and Spain were the closest to having equal abundance. Using Nei’s unbiased gene diversity, the Russian population had the highest gene diversity (0.546) followed by those from Spain (0.494), Ukraine (0.379) and France (0.344). This could be due to the uneven sample sizes obtained at the different locations because the algorithm does not correct for small population sizes. Populations from Slovenia and Switzerland had the lowest genotypic diversity and genotypic richness. Countries for which only one or two isolates were available (i.e., Romania, Belgium and the Czech Republic) were not considered in the analyses.
The population from Spain had the highest number of private alleles (PAR) (16.51%) followed by those from France (5.50%) and Slovakia (4.59%). Populations from Russia, Serbia, Slovenia and Switzerland had the lowest number of private alleles (0.90%). Within Slovakia, private alleles were from Arboretum Mlyňany, Jahodná, Košice and Zvolen and in France the private alleles were only from Souesmes (Supplementary Table S4).
3.4 Population structure
There was no consensus between different methods of determining the optimal number of clusters in the STRUCTURE analysis. The Evanno ΔK supported nineteen (K = 19) clusters, which indicates that this method failed to detect population structure. LnP (K) suggested K = 10 as the optimal scenario. The four Puechmaille methods suggested that 5–8 clusters are most likely the optimal number of clusters depending on the threshold that was set (Supplementary Figure S2). The STRUCTURE barplots for K = 2 to K = 9 for the major modes are illustrated in Supplementary Figure S3. The barplots for K = 5–8, together with the geographical distribution of the clusters are represented in Figure 3. In order to conduct the DAPC analysis, the find. clusters function in the adegenet package in R was used and this showed that K resides between 8 and 12. After several runs, K = 10 was proposed as the optimal scenario.
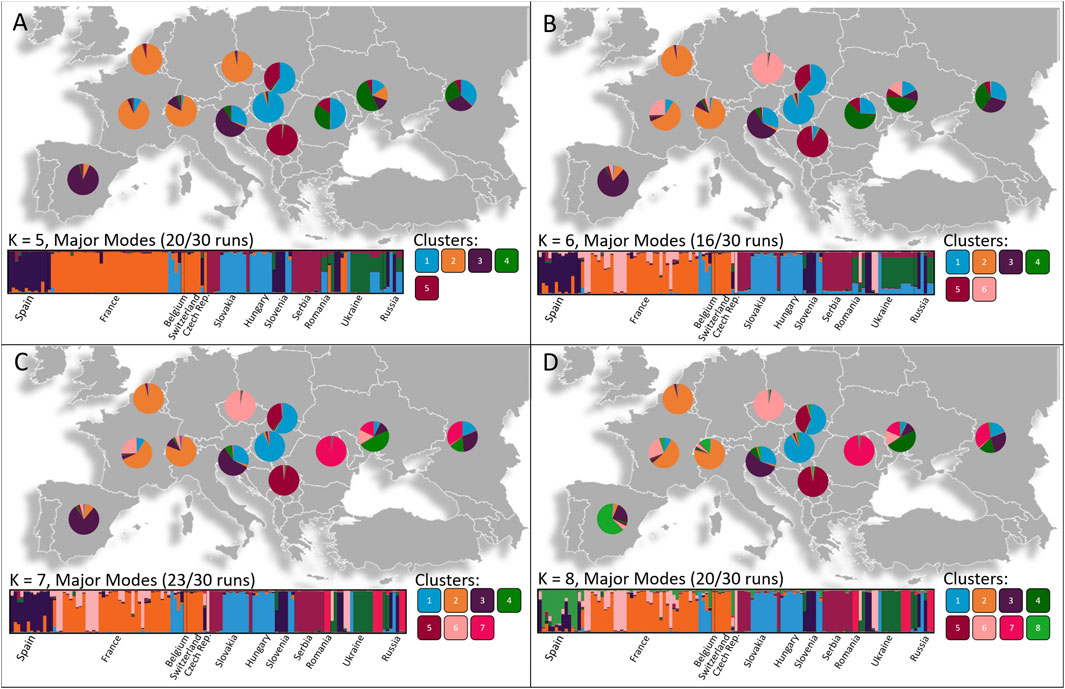
FIGURE 3. Geographical patterns of population divisions observed using STRUCTURE analyses based on the most likely K values determined by the Puecemaille methods with (A) K = 5, (B) K = 6, (C) K = 7, (D) K = 8. The number of individuals belonging to each Cluster is represented as pie charts in each respective country. Four main genetic groups are spread throughout Western, Central and Eastern Europe with several smaller scattered genetic groups residing among the populations.
For both the K = 8 and K = 10 scenario, the DAPC (Figure 4) and STRUCTURE analysis (Figure 3) indicated that three or four major genetic groups reside between bordering countries in Western, Central, and Eastern Europe. Within these clusters, several smaller genetic groups were observed. The STRUCTURE analysis showed that populations in Western Europe (Belgium, Czech Republic, France and Switzerland) share a major cluster. In Central Europe, one cluster was shared between Hungary, Slovakia and Slovenia and a second genetic cluster was shared between Slovakia and Serbia. In Eastern Europe, isolates from Romania, Russia and Ukraine shared a cluster. Several smaller scattered genetic groups also resided among the populations and the Slovenian population, as well as the Spanish population, included unique genetic clusters.
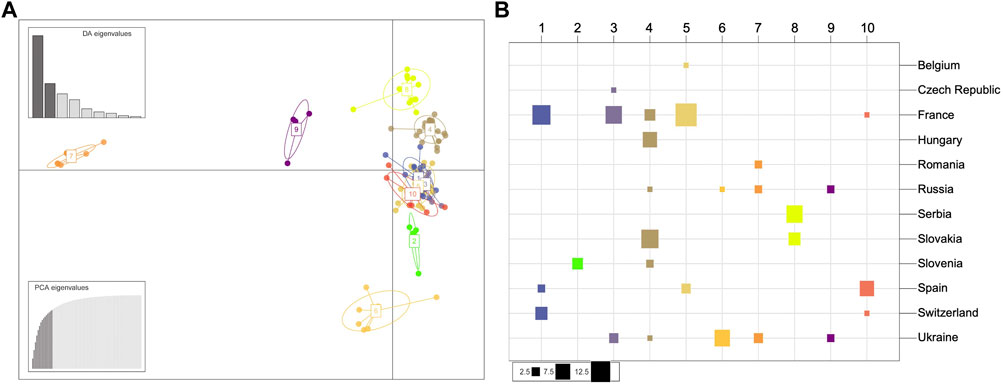
FIGURE 4. Population structure of the European Dothistroma pini collection of isolates. (A) Scatterplot of the discriminant analysis of principal components (DAPC) on European Dothistroma pini multilocus haplotypes. The number and colours represent the 10 groups delineated by the K-means method. Individual multilocus haplotypes are represented by dots and clusters as ellipses. At the top left the eigenvalues of the first nine axes are represented. (B) The composition of the DAPC clusters. The columns and colours correspond to the inferred clusters and the rows correspond to the countries where the populations were sampled. The size of the squares is proportional to the number of individuals comprising each cluster. Cluster one for instance is comprised of individuals isolated from France, Spain and Switzerland with the majority of the individuals in this cluster isolated from France.
The DAPC clusters (Figure 4) were mostly correlated with the geographic groups indicated by the STRUCTURE analysis with a Western group containing Cluster 1 (France, Spain, Switzerland), Cluster 3 (Czech Republic, France, Ukraine), Cluster 5 (Belgium, France and Spain) and Cluster 10 (France, Spain and Switzerland). A Central European group accommodated Cluster 4 (France, Hungary, Russia, Slovakia and Slovenia), Cluster 8 (Serbia and Slovakia) as well as a unique cluster (Cluster 2) having only individuals from Slovenia. The DAPC also indicated an Eastern European group with Cluster 6 (Russia, Ukraine), Cluster 7 (Romania, Russia and Ukraine) as well as Cluster 9 (Russia and Ukraine). The four distinct geographic groups suggested by both the STRUCTURE analysis and DAPC were also evident in a haplotype network drawn using Bruvo’s genetic distance (Supplementary Figure S4).
The AMOVA results (Table 2) indicated significant population differentiation according to country (variance among individuals 47%, variance among countries 53%) and even more so by location within countries (variance among individuals 41%, variance among countries 59%). Although this explained less of the variance found among populations, AMOVA also strongly supported the grouping by host species (27% between species and 73% among individuals).
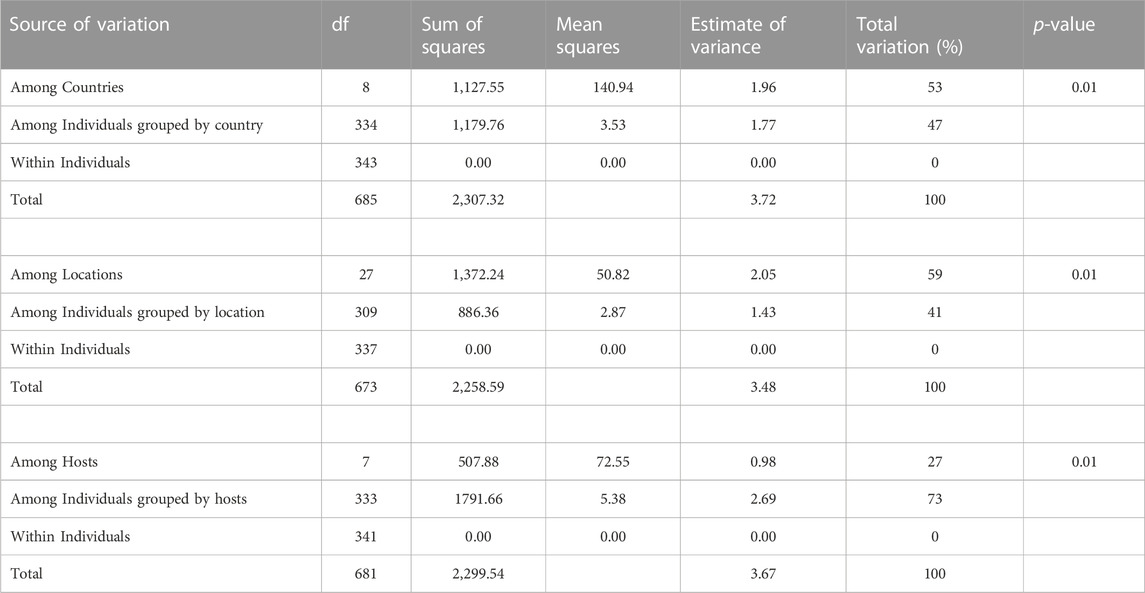
TABLE 2. Hierarchical analysis of molecular variance (AMOVA) of Dothistroma pini populations, grouped by countries, by locations and by host species.
3.5 Mating type determination and random mating
The mating types were successfully amplified for all but two isolates, both from Slovakia (Table 3). Both mating type idiomorphs were detected in isolates from France, Serbia, Slovakia, Spain, Switzerland and Ukraine (Table 3). However, in Nueng-sur-Beuvron in France only MAT1-1 individuals were detected and in Serbia only MAT1-2 individuals were present in isolates from Subotica Sands. Similarly, although both mating types were present in the Slovakian collections, either MAT1-1 or MAT1-2 individuals were detected at each of the 10 locations sampled in this country. In Ukraine, the population from Nova Zburivka included only one individual that was MAT1-2 and in Mykolaiv Kinburn, only MAT1-1 individuals were detected. Although both mating types were found in these countries, random mating was statistically supported only in the populations from Spain, Switzerland and Ukraine as well as in the sub-populations from Souesmes and La Bouyale in France, Deliblato Sands in Serbia, and Hola Prystan, Tsjurupinsk and Mykolaiv Kinburn in Ukraine. In isolates from the Czech Republic, Romania and Russia only MAT1-1 individuals were present and in those from Belgium, Hungary, and Slovenia only MAT1-2 individuals were present (Table 3, Supplementary Table S2).
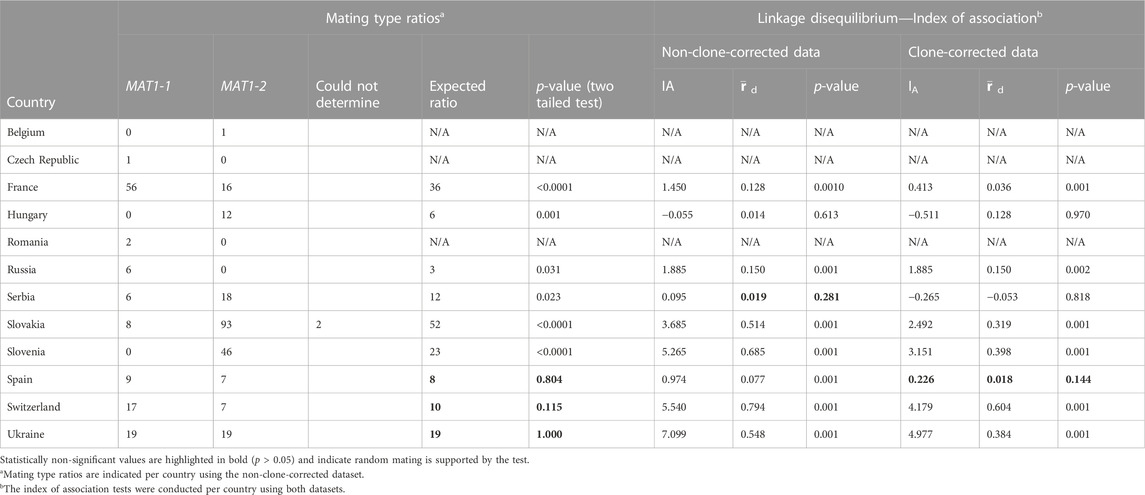
TABLE 3. Mating type ratios and index of association tests for the Dothistroma pini populations collected in Europe.
Testing linkage disequilibrium using the clone-corrected dataset, with the index of association and rbarD, provided evidence for sexual recombination only in the population from Spain (p-value of 0.144). Analysis of the non-clone-corrected dataset also supported evidence of sexual recombination in Serbia (p-values of 0.281). This result is however not plausible as the data for both Deliblato Sands and Subotica Sands in Serbia were pooled for this analysis and therefore do not reflect that single mating types were observed at each of these locations.
4 Discussion
This study provided the first insights into the population structure and genetic diversity of D. pini in Europe. Even though extensive sampling was conducted in the area over a 12-year period, due to the low incidence of D. pini, sampling was relatively unstructured and sample sizes were relatively small. This was also emphasized in reports in Switzerland (Dubach et al., 2018) as well as Spain (Ortíz De Urbina et al., 2017) where D. pini was less frequently detected than D. septosporum. Nonetheless, it was clear that D. pini is not new to the European continent and that movement of the pathogen was facilitated through anthropogenic activities.
Based on population structure analyses, the D. pini populations considered in this study grouped in four main geographic clusters including one in Western Europe, two in Central Europe, and one in Eastern Europe. Variable population diversity was observed between countries, with France, Spain and Ukraine having the highest levels of genetic diversity and the presence of both mating types. This suggests that D. pini has most likely been present in those countries for a long period of time and is in agreement with the identification of D. pini in France from herbarium specimens dating back to 1907 and 1965 (Fabre et al., 2012). In contrast, there were populations that were clonal and with a single mating type such as in Slovakia and Slovenia, suggesting more recent introductions. Additionally, the presence of the same MLHs over long distances suggests that human-mediated movement of D. pini is taking place in Europe, possibly through plant trade (Pautasso and Jeger, 2014).
Both mating types of D. pini were present in many populations considered in this study, but evidence for sexual recombination was supported only in the population from Spain. The fact that some isolates of the same MLHs were of different mating type suggests that sexual recombination could be occurring in other European populations of D. pini. This is not unusual and has been found in pathogens such as Teratosphaeria destructans (Havenga et al., 2021) as well as Verticillium dahliae, a clonally reproducing pathogen, having individuals of opposite mating types that were indicative of cryptic or ancestral sexual recombination events (Milgroom et al., 2014; Short et al., 2014).
Dothistroma pini has a limited host range and is currently confined to a particular latitudinal geographical range both in Europe as well as in North America. The majority of the isolates in the present study were from several sub-species of Pinus nigra with few collections from P. coulteri, P. jeffreyi, P. mugo, P. ponderosa, P. schwerinii and P. sylvestris. Many of the single isolates from hosts other than P. nigra were from urban areas or arboreta and not from the native ranges of the host trees. This suggests that D. pini is most likely not native to the areas where it was collected in Europe and could have been introduced to the continent. This is in contrast to the more commonly occurring D. septosporum that is hypothesized to be native to the P. sylvestris forests in Northern Europe (Adamson et al., 2018), Eastern Europe and Western Asia (Mullett et al., 2021).
The results of this study have provided no clues to the possible center of origin of the pathogen. The only other area of the world where D. pini is known to occur is North America (Barnes et al., 2004; Barnes et al., 2014a; Mullett et al., 2018). Dothistroma needle blight is widespread in the United States and has been reported in 35 states (Drenkhan et al., 2016; Mullett et al., 2018). However, most of the reports were from the time before D. septosporum and D. pini were conclusively separated based on phylogenetic inference in 2004 (Barnes et al., 2004). Thus, the presence of D. pini has been confirmed in only seven states in the Central regions of the United States (Barnes et al., 2004; Barnes et al., 2014a; Mullett et al., 2018) and D. septosporum in four states (Barnes et al., 2004; Barnes et al., 2016). The techniques available to discriminate between the two species with relative ease (Barnes et al., 2004; Groenewald et al., 2007; Barnes et al., 2008a; Ioos et al., 2010; Schneider et al., 2019; Aglietti et al., 2021; Myrholm et al., 2021) should simplify efforts to collect isolates known to be those of D. pini from the United States, and potentially other unsampled areas such as Asia. This would facilitate an opportunity to compare populations across continents, using either microsatellite markers or whole genome comparisons, in an effort to understand global pathways of spread and potential native areas. The extensive data assembled in the present study will provide a solid foundation for these comparisons.
An intriguing question pertaining to DNB is why D. septosporum has spread from the Northern Hemisphere to many Southern Hemisphere countries but that the closely related D. pini has not done so. This could be related to host range where D. septosporum has mainly been a problem on P. radiata in the Southern Hemisphere (Gibson, 1972; Barnes et al., 2014b; Drenkhan et al., 2016), although it has recently emerged as a serious constraint in plantations of P. tecunumanii in Colombia (Rodas et al., 2016). Both Dothistroma species have relatively wide host ranges and as greater numbers of Pinus spp. are being tested and propagated in Southern Hemisphere countries, it seems plausible to suggest that D. pini poses an important threat to these resources. Based on experience with D. septosporum as well as the increasingly important pine needle pathogen Lecanosticta acicola (van der Nest et al., 2019a), and apparently D. pini as was found in this study, there is good reason to emphasize the importance of quarantine when moving Pinus germplasm between countries and continents.
Data availability statement
The authors acknowledge that the data presented in this study must be deposited and made publicly available in an acceptable repository, prior to publication. Frontiers cannot accept a manuscript that does not adhere to our open data policies.
Author contributions
AN, MW, and IB contributed to conception and design of the study. IB, DS, MM, BM, VQ, KA, and KD contributed sample collections and KA contributed a dataset for inclusion. AN and IB conducted the lab work and organized the datasets. AN, DS, and IB did the data analysis and MW, MM, BM, VQ, KA, and KD made improvements to the analysis. AN wrote the first draft of the manuscript and MW and IB contributed significantly to editing the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
Financial support for this study was provided by the University of Pretoria, members of the Tree Protection Cooperative Program (TPCP), the National Research Foundation (Thuthuka Grant no 80670, and Grant no 95875), a Scarce Skills Doctoral Scholarship (Grant 89086) for A. van der Nest, and DIAROD: EU COST Action FP1102 DIAROD (Determining Invasiveness and Risk of Dothistroma, https://www.cost.eu/actions/FP1102/). The NRF acknowledge that opinions, findings, conclusions and/or recommendations expressed in any publication generated by the NRF supported research are that of the author (s), and that the NRF accepts no liability whatsoever in this regard. The NRF had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
We acknowledge that this manuscript was a research chapter of the PhD thesis of the leading author, A. van der Nest, and that the thesis will be electronically available in the University of Pretoria library repository as: van der Nest A. 2021. Research chapter 2: Population structure and diversity of the needle pathogen Dothistroma pini suggests human mediated movement in Europe. In: Species diversity of Lecanosticta and population genetics of Dothistroma species: important needle pathogens of Pinus. PhD(Genetics), Department of Biochemistry, Genetics and Microbiology, University of Pretoria, Pretoria. Pp 73–115. We thank Dr Zuzana Janošíková-Hečková and Dr Emília Ondrušková from the Institute of Forest Ecology Slovak Academy of Sciences in Slovakia for their assistance in obtaining the large collection of D. pini isolates from Slovakia.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1103331/full#supplementary-material
Supplementary Figure S1 | The 109 multilocus haplotypes (MLH) derived from microsatellite data in each country and shared between countries. Eight MLHs, indicated by coloured arrows are shared between multiple, often non-bordering countries. France contains the highest number of MLHs, followed by Ukraine. MLH 52 (navy blue) occurred in four countries (Hungary, Slovakia, Slovenia, and Ukraine) in 7 locations, covering a distance of approximately 1500 km. The single individual from the Czech Republic shared MLH 83 (dark green) with isolates from different locations in France and 1480 km away as well as two isolates from Hola Prystan in Ukraine. MLH 56 (purple) was the second most occurring MLH and was shared by individuals in Souesmes (France), Diszel (Hungary) and Arborétum Mlyňany, Gabčikovo, Jahodna and Trstice in Slovakia. MLH47 (red) occurred in the Russian population (ITS Haplotype 2 individual) and 1150 km apart in the Hungarian population (ITS Haplotype 1 individual). MLH 11 (light blue) occurred in the population in Russia as well as in Hola Prystan in Ukraine (620 km apart). MLH 34 (lime green) was present in both Arborétum Mlyňany and Zvolen (Slovakia) as well as Delibratski Pesak and Subotička Pescara (Serbia), while individuals of MLH 32 (yellow) were detected in Kováčová (Slovakia) and Subotička Pescara (Serbia). MLH 59 (orange) was found in both Selles-Saint-Denis and La Ferté-Imbault (France) as well as in the population from Switzerland.
Supplementary Figure S2 | The optimum number of clusters determined using STRUCTURESELECTOR. (A) The Evanno method (Delta K and LnP (K), suggested K= 19 and K = 10 respectively. When a threshold of 0.5 was set, the Puechmaille methods (B) determined the most optimal number of clusters as 7 or 8 and at a threshold of 0.8 (C), the most optimal number of clusters were determined to be 5 or 6.
Supplementary Figure S3 | STRUCTURE results of D. pini populations per country using the clone-corrected dataset. The structure bar plots show the results for the major clustering modes from K=2 to K=9. The bar plots are divided according to geographical location.
Supplementary Figure S4 | Haplotype network of D. pini collected in Europe drawn using Bruvo’s genetic distance. Each circle represents a multilocus haplotype. The larger the circle, the more individuals have the same haplotype. The circle size for 1, 22 and 47 individuals of a particular haplotype are indicated on the left hand side for scale. The same four major clusters are observed as with the STRUCTURE analysis with the Western Europe individuals (blue oval), Central European clusters (yellow and pink ovals) and Eastern European cluster (dark green) clustering together.
Supplementary Table S1 | Dothistroma pini collections from Europe used in this study.
Supplementary Table S2 | Allele sizes for 345 individuals of Dothistroma pini in Europe based on 17 microsatellite markers.
Supplementary Table S3 | Dothistroma pini microsatellite PCR annealing temperatures, MgCl2 adjustments, dilutions for fragment analysis for each microsatellite marker and number of alleles per microsatellite marker (Adamčíková et al., 2021).
Supplementary Table S4 | Summary diversity statistics of Dothistroma pini isolates within locations in Europe.
References
Adamčíková, K., Jánošíková, Z., Van Der Nest, A., Adamčík, S., Ondrušková, E., and Barnes, I. (2021). Population structure and genetic diversity suggest recent introductions of Dothistroma pini in Slovakia. Plant Pathol. 70, 1883–1896. doi:10.1111/ppa.13428
Adamson, K., Mullett, M. S., Solheim, H., Barnes, I., Müller, M. M., Hantula, J., et al. (2018). Looking for relationships between the populations of Dothistroma septosporum in northern Europe and Asia. Fungal Genet. Biol. 110, 15–25. doi:10.1016/j.fgb.2017.12.001
Agapow, P., and Burt, A. (2001). Indices of multilocus linkage disequilibrium. Mol. Ecol. Notes 1, 101–102. doi:10.1046/j.1471-8278.2000.00014.x
Aglietti, C., Meinecke, C. D., Ghelardini, L., Barnes, I., Van Der Nest, A., and Villari, C. (2021). Rapid detection of pine pathogens Lecanosticta acicola, Dothistroma pini and D. septosporum on needles by probe-based LAMP assays. Forests 12, 479. doi:10.3390/f12040479
Barnes, I., Cortinas, M. N., Wingfield, M. J., and Wingfield, B. D. (2008a). Microsatellite markers for the red band needle blight pathogen, Dothistroma septosporum. Mol. Ecol. Resour. 8, 1026–1029. doi:10.1111/j.1755-0998.2008.02142.x
Barnes, I., Crous, P. W., Wingfield, B. D., and Wingfield, M. J. (2004). Multigene phylogenies reveal that red band needle blight of Pinus is caused by two distinct species of Dothistroma, D. septosporum and D. pini. Stud. Mycol. 50, 551–565.
Barnes, I., Kirisits, T., Akulov, A., Chhetri, D. B., Wingfield, B. D., Bulgakov, T. S., et al. (2008b). New host and country records of the Dothistroma needle blight pathogens from Europe and Asia. For. Pathol. 38, 178–195. doi:10.1111/j.1439-0329.2007.00536.x
Barnes, I., Kirisits, T., Wingfield, M. J., and Wingfield, B. D. (2011). Needle blight of pine caused by two species of Dothistroma in Hungary. For. Pathol. 41, 361–369. doi:10.1111/j.1439-0329.2010.00689.x
Barnes, I., Van Der Nest, A., Granados, G. M., and Wingfield, M. J. (2022). “Chapter 11 - Dothistroma needle blight,” in Forest Microbiology. Editors F. O. ASIEGBU, and A. KOVALCHUK (Massachusetts, United States: Academic Press).
Barnes, I., Van Der Nest, A., Mullett, M. S., Crous, P. W., Drenkhan, R., Musolin, D. L., et al. (2016). Neotypification of Dothistroma septosporum and epitypification of D. pini, causal agents of Dothistroma needle blight of pine. For. Pathol. 46, 388–407. doi:10.1111/efp.12304
Barnes, I., Walla, J. A., Bergdahl, A., and Wingfield, M. J. (2014a). Four new host and three new state records of Dothistroma needle blight caused by Dothistroma pini in the United States. Plant Dis. 98, 1443. doi:10.1094/PDIS-06-14-0606-PDN
Barnes, I., Wingfield, M. J., Carbone, I., Kirisits, T., and Wingfield, B. D. (2014b). Population structure and diversity of an invasive pine needle pathogen reflects anthropogenic activity. Ecol. Evol. 4, 3642–3661. doi:10.1002/ece3.1200
Bergová, E., and Kryštofová, A. (2014). First occurrence of Dothistroma pini in the Czech republic. Prague: Conference Mikromyco, 17–18.
Boroń, P., Lenart-Boroń, A., and Mullett, M. (2016). The distribution of Dothistroma septosporum and its mating types in Poland. For. Pathol. 46, 489–496. doi:10.1111/efp.12262
Bradshaw, R. E., Sim, A. D., Chettri, P., Dupont, P.-Y., Guo, Y., Hunziker, L., et al. (2019). Global population genomics of the forest pathogen Dothistroma septosporum reveal chromosome duplications in high dothistromin-producing strains. Mol. Plant Pathol. 20, 784–799. doi:10.1111/mpp.12791
Brown, A. H., Feldman, M. W., and Nevo, E. (1980). Multilocus structure of natural populations of Hordeum Spontaneum. Genet. 96, 523–536. doi:10.1093/genetics/96.2.523
Bruvo, R., Michiels, N. K., D’souza, T. G., and Schulenburg, H. (2004). A simple method for the calculation of microsatellite genotype distances irrespective of ploidy level. Mol. Ecol. 13, 2101–2106. doi:10.1111/j.1365-294X.2004.02209.x
Capron, A., Feau, N., Heinzelmann, R., Barnes, I., Benowicz, A., Bradshaw, R. E., et al. (2021). Signatures of post-glacial genetic isolation and human-driven migration in the Dothistroma needle blight pathogen in Western Canada. Phytopathology® 111, 116–127. doi:10.1094/PHYTO-08-20-0350-FI
Drenkhan, R., and Hanso, M. (2009). Recent invasion of foliage fungi of pines (Pinus spp.) to the Northern Baltics. For. Stud. 51, 49–64. doi:10.2478/v10132-011-0077-7
Drenkhan, R., Hantula, J., Vuorinen, M., Jankovský, L., and Müller, M. M. (2013). Genetic diversity of Dothistroma septosporum in Estonia, Finland and Czech republic. Eur. J. Plant Pathology 136, 71–85. doi:10.1007/s10658-012-0139-6
Drenkhan, R., Tomešová-Haataja, V., Fraser, S., Bradshaw, R. E., Vahalík, P., Mullett, M. S., et al. (2016). Global geographic distribution and host range of Dothistroma species: A comprehensive review. For. Pathol. 46, 408–442. doi:10.1111/efp.12290
Dubach, V., Meyer, J. B., Schneider, S., Ruffner, B., and Queloz, V. (2018). Nationales Monitoring von zwei besonders gefährlichen Föhrenkrankheiten 2016. Suivi national de deux maladies du pin particulièrement dangereuses 2016. Monitoraggio nazionale di due malattie particolarmente pericolose del pino 2016. Phytopathologie, WSL and Im Auftrag des Bundesamtes für Umwelt. Birmensdorf: BAFU.
Ennos, R. A., Sjökvist, E. I., Piotrowska, M. J., Riddell, C., and Hoebe, P. N. (2020). Using genome resequencing to investigate racial structure, genetic diversity, sexual reproduction and hybridisation in the pine pathogen Dothistroma septosporum. Fungal Ecol. 45, e100921. doi:10.1016/j.funeco.2020.100921
EPPO (2019). First report of Dothistroma pini in Germany. EPPO reporting Service no. 2 Num. article: 2019/042. Available at: https://gd.eppo.int/reporting/article-6472.
Evanno, G., Regnaut, S., and Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. Resour. 14, 2611–2620. doi:10.1111/j.1365-294X.2005.02553.x
Fabre, B., Ioos, R., Piou, D., and Marçais, B. (2012). Is the emergence of Dothistroma needle blight of pine in France caused by the cryptic species Dothistroma pini? Phytopathology 102, 47–54. doi:10.1094/PHYTO-02-11-0036
Falush, D., Stephens, M., and Pritchard, J. K. (2003). Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 164, 1567–1587. doi:10.1093/genetics/164.4.1567
Ghelardini, L., Aglietti, C., Loria, F., Cerboneschi, M., Gionni, A., Goti, E., et al. (2020). Dothistroma Needle Blight in protected pine forests in Italy. Manag. Biol. Invasions 11, 689–702. doi:10.3391/mbi.2020.11.4.05
Gibson, I. a. S. (1972). Dothistroma blight of Pinus radiata. Annu. Rev. Phytopathology 10, 51–72. doi:10.1146/annurev.py.10.090172.000411
Groenewald, M., Barnes, I., Bradshaw, R. E., Anna, V., B., Dale, A., Groenewald, J. Z., et al. (2007). Characterization and distribution of mating type genes in the Dothistroma needle blight pathogens. Phytopathology 97, 825–834. doi:10.1094/PHYTO-97-7-0825
Grünwald, N. J., Goodwin, S. B., Milgroom, M. G., and Fry, W. E. (2003). Analysis of genotypic diversity data for populations of microorganisms. Phytopathology® 93, 738–746. doi:10.1094/PHYTO.2003.93.6.738
Guo, Y., Hunziker, L., Mesarich, C. H., Chettri, P., Dupont, P.-Y., Ganley, R. J., et al. (2020). DsEcp2-1 is a polymorphic effector that restricts growth of Dothistroma septosporum in pine. Fungal Genet. Biol. 135, 103300. doi:10.1016/j.fgb.2019.103300
Havenga, M., Wingfield, B. D., Wingfield, M. J., Marincowitz, S., Dreyer, L. L., Roets, F., et al. (2021). Genetic recombination in Teratosphaeria destructans causing a new disease outbreak in Malaysia. For. Pathol. 51, e12683. doi:10.1111/efp.12683
Hurlbert, S. H. (1971). The nonconcept of species diversity: A critique and alternative parameters. Ecology 52, 577–586. doi:10.2307/1934145
Ioos, R., Fabre, B., Saurat, C., Fourrier, C., Frey, P., and Marcais, B. (2010). Development, comparison, and validation of real-time and conventional PCR tools for the detection of the fungal pathogens causing Brown spot and red band needle blights of pine. Am. Phytopathological Soc. 100, 105–114. doi:10.1094/PHYTO-100-1-0105
Iturritxa, E., Mesanza, N., and Brenning, A. (2015). Spatial analysis of the risk of major forest diseases in Monterey pine plantations. Plant Pathol. 64, 880–889. doi:10.1111/ppa.12328
Jánošíková-Hečková, Z., Ondrušková, E., Barta, M., Ostrovský, R., Kádasi-Horáková, M., Pastirčáková, K., et al. (2018). The hosts and geographic range of Dothistroma needle blight in Slovakia. For. Pathol. 48, e12421. doi:10.1111/efp.12421
Janoušek, J., Krumbock, S., Kirisits, T., Bradshaw, R. E., Barnes, I., Jankovský, L., et al. (2014). Development of microsatellite and mating type markers for the pine needle pathogen Lecanosticta acicola. Australas. Plant Pathol. 43, 161–165. doi:10.1007/s13313-013-0256-5
Jombart, T., and Ahmed, I. (2011). Adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27, 3070–3071. doi:10.1093/bioinformatics/btr521
Jombart, T., Devillard, S., and Balloux, F. (2010). Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 11, 94. doi:10.1186/1471-2156-11-94
Kamvar, Z. N., Tabima, J. F., and Grünwald, N. J. (2014). Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2, e281. doi:10.7717/peerj.281
Kopelman, N. M., Mayzel, J., Jakobsson, M., Rosenberg, N. A., and Mayrose, I. (2015). Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 15, 1179–1191. doi:10.1111/1755-0998.12387
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi:10.1093/molbev/msw054
Lazarević, J., Davydenko, K., and Millberg, H. (2017). Dothistroma needle blight on high altitude pine forests in Montenegro. Balt. For. 23, 294–302.
Li, Y., and Liu, J. (2018). StructureSelector: A web-based software to select and visualize the optimal number of clusters using multiple methods. Mol. Ecol. Resour. 18, 176–177. doi:10.1111/1755-0998.12719
Matsiakh, I., Doğmuş-Lehtijärvi, H. T., Kramarets, V., Aday Kaya, A. G., Oskay, F., Drenkhan, R., et al. (2018). Dothistroma spp. in western Ukraine and Georgia. For. Pathol. 48, e12409. doi:10.1111/efp.12409
Mesanza, N., Raposo, R., Elvira-Recuenco, M., Barnes, I., Van Der Nest, A., Hernández, M., et al. (2021). New hosts for Lecanosticta acicola and Dothistroma septosporum in newly established arboreta in Spain. For. Pathol. 51, e12650. doi:10.1111/efp.12650
Milgroom, M. G., Jiménez-Gasco, M. D. M., Olivares García, C., Drott, M. T., and Jiménez-Díaz, R. M. (2014). Recombination between clonal lineages of the asexual fungus Verticillium dahliae detected by genotyping by sequencing. PloS one 9, e106740. doi:10.1371/journal.pone.0106740
Mullett, M., Adamson, K., Bragança, H., Bulgakov, T., Georgieva, M., Henriques, J., et al. (2018). New country and regional records of the pine needle blight pathogens Lecanosticta acicola, Dothistroma septosporum and Dothistroma pini. For. Pathol. 48, e12440. doi:10.1111/efp.12440
Mullett, M. S., Brown, A. V., and Barnes, I. (2015). Population structure and reproductive mode of Dothistroma septosporum in the Brittany peninsula of France. Eur. J. Plant Pathology 143, 261–275. doi:10.1007/s10658-015-0678-8
Mullett, M. S., Drenkhan, R., Adamson, K., Boroń, P., Lenart-Boroń, A., Barnes, I., et al. (2021). Worldwide genetic structure elucidates the Eurasian origin and invasion pathways of Dothistroma septosporum, causal agent of Dothistroma needle blight. J. fungi 7, 111. doi:10.3390/jof7020111
Myrholm, C. L., Tomm, B. D., Heinzelmann, R., Feau, N., Hamelin, R. C., Mcdougal, R., et al. (2021). Development of a rapid loop-mediated isothermal amplification assay for the detection of Dothistroma septosporum. Forests 12, 362. doi:10.3390/f12030362
Nei, M. (1978). Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89, 583–590. doi:10.1093/genetics/89.3.583
Ondrušková, E., Hečková, Z., Kádasi Horáková, M., Koltay, A., Ostrovský, R., Pažitný, J., et al. (2017). Distribution and characterization of Dothistroma needle blight pathogens on Pinus mugo in Slovakia. Eur. J. Plant Pathology 148, 283–294. doi:10.1007/s10658-016-1088-2
Ondrušková, E., Hečková-Jánošíková, Z., Adamčík, S., Kádasi Horáková, M., Rakúsová-Sládková, D., and Adamčíková, K. (2018). Needle blight caused by Dothistroma pini in Slovakia: Distribution, host range and mating types. Scand. J. For. Res. 33, 650–656. doi:10.1080/02827581.2018.1482954
Ortíz De Urbina, E., Mesanza, N., Aragonés, A., Raposo, R., Elvira-Recuenco, M., Boqué, R., et al. (2017). Emerging needle blight diseases in Atlantic Pinus ecosystems of Spain. Forests 8, 18. doi:10.3390/f8010018
Oskay, F., Tunalı, Z., Lehtijärvi, A. T., Doğmuş-Lehtijärvi, H. T., Woodward, S., and Mullett, M. (2020). Distribution and genetic diversity of Dothistroma septosporum in Pinus brutia forests of south-Western Turkey. Plant Pathol. 69, 1551–1564. doi:10.1111/ppa.13242
Pap, P., Drekić, M., Poljaković-Pajnik, L., Marković, M., and Vasić, V. (2015). Forest health monitoring in Vojvodina in 2015. Topola: Serbian, 117–133.
Pautasso, M., and Jeger, M. J. (2014). Network epidemiology and plant trade networks. AoB PLANTS 6, plu007. doi:10.1093/aobpla/plu007
Peakall, R., and Smouse, P. E. (2012). GenAlEx 6.5: Genetic analysis in excel. Population genetic software for teaching and research—an update. Bioinformatics 28, 2537–2539. doi:10.1093/bioinformatics/bts460
Piškur, B., Hauptman, T., and Jurc, D. (2013). Dothistroma needle blight in Slovenia is caused by two cryptic species: Dothistroma pini and Dothistroma septosporum. For. Pathol. 43, 518–521. doi:10.1111/efp.12059
Puechmaille, S. (2016). The program STRUCTURE does not reliably recover the correct population structure when sampling is uneven: Subsampling and new estimators alleviate the problem. Mol. Ecol. Resour. 16, 608–627. doi:10.1111/1755-0998.12512
Queloz, V., Wey, T., and Holdenrieder, O. (2014). First record of Dothistroma pini on Pinus nigra in Switzerland. Plant Dis. 98, 1744. doi:10.1094/PDIS-06-14-0630-PDN
Rodas, C. A., Wingfield, M. J., Granados, G. M., and Barnes, I. (2016). Dothistroma needle blight: An emerging epidemic caused by Dothistroma septosporum in Colombia. Plant Pathol. 65, 53–63. doi:10.1111/ppa.12389
Rosenberg, N. (2004). Distruct: A program for the graphical display of population structure. Mol. Ecol. Notes 4, 137–138. doi:10.1046/j.1471-8286.2003.00566.x
Schmitz, S., Gischer, F., and Chandelier, A. (2013). First detection of Dothistroma pini in Belgium. Poster presentation at COST Action FP1102 meeting, 23–24.
Schneider, S., Jung, E., Queloz, V., Meyer, J. B., and Rigling, D. (2019). Detection of pine needle diseases caused by Dothistroma septosporum, Dothistroma pini and Lecanosticta acicola using different methodologies. For. Pathol. 2019, e12495. doi:10.1111/efp.12495.1111/efp.12495
Shannon, C. E. (2001). A mathematical theory of communication. ACM Sigmob. Mob. Comput. Commun. Rev. 5, 3–55. doi:10.1145/584091.584093
Short, D. P. G., Gurung, S., Hu, X., Inderbitzin, P., and Subbarao, K. V. (2014). Maintenance of sex-related genes and the co-occurrence of both mating types in Verticillium dahliae. PLOS ONE 9, e112145. doi:10.1371/journal.pone.0112145
Siziba, V. I., Wingfield, M. J., Sadiković, D., Mullett, M. S., Piškur, B., and Barnes, I. (2016). Development of microsatellite markers for the pine needle blight pathogen, Dothistroma pini. For. Pathol. 46, 497–506. doi:10.1111/efp.12282
Smith, J. M., Smith, N. H., O'rourke, M., and Spratt, B. G. (1993). How clonal are bacteria? Proc. Natl. Acad. Sci. U. S. A. 90, 4384–4388. doi:10.1073/pnas.90.10.4384
Stoddart, J. A., and Taylor, J. F. (1988). Genotypic diversity: Estimation and prediction in samples. Genetics 118, 705–711. doi:10.1093/genetics/118.4.705
Szpiech, Z. A., Jakobsson, M., and Rosenberg, N. A. (2008). ADZE: A rarefaction approach for counting alleles private to combinations of populations. Bioinformatics 24, 2498–2504. doi:10.1093/bioinformatics/btn478
Van Der Nest, A., Wingfield, M. J., Janoušek, J., and Barnes, I. (2019a). Lecanosticta acicola: A growing threat to expanding global pine forests and plantations. Mol. Plant Pathol. 20, 1327–1364. doi:10.1111/mpp.12853
Van Der Nest, A., Wingfield, M. J., Ortiz, P. C., and Barnes, I. (2019b). Biodiversity of Lecanosticta pine-needle blight pathogens suggests a Mesoamerican Centre of origin. IMA Fungus 10, 2. Article 2 (2019). doi:10.1186/s43008-019-0004-8
Vuillemin, P. (1896). Les Hypostomacées, nouvelle famille de champignons parasites. Bull. Soc. Sci. Nancy 1896, 15–52.
Wartalska, P., Oszako, T., Bakier, S., Belbahri, L., Malewski, T., Hsiang, T., et al. (2021). Dothistroma septosporum not detected in Pinus sylvestris seed trees from investigated stands in southern Poland. Forests 12, 1323. doi:10.3390/f12101323
Welsh, C., Lewis, K., and Woods, A. (2009). The outbreak history of Dothistroma needle blight: An emerging forest disease in northwestern British columbia, Canada. Can. J. For. Res. 39, 2505–2519. doi:10.1139/x09-159
Keywords: Dothistroma needle blight, Dothistroma pini, microsatellites, mating types, pine needle pathogen, Mycosphaerella pini, red band needle blight
Citation: van der Nest A, Wingfield MJ, Sadiković D, Mullett MS, Marçais B, Queloz V, Adamčíková K, Davydenko K and Barnes I (2023) Population structure and diversity of the needle pathogen Dothistroma pini suggests human-mediated movement in Europe. Front. Genet. 14:1103331. doi: 10.3389/fgene.2023.1103331
Received: 20 November 2022; Accepted: 27 January 2023;
Published: 16 February 2023.
Edited by:
Hao Zhang, Institute of Plant Protection (CAAS), ChinaCopyright © 2023 van der Nest, Wingfield, Sadiković, Mullett, Marçais, Queloz, Adamčíková, Davydenko and Barnes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Irene Barnes, aXJlbmUuYmFybmVzQGZhYmkudXAuYWMuemE=; Valentin Queloz, dmFsZW50aW4ucXVlbG96QHdzbC5jaA==