 Woojong Jang
Woojong Jang Ji-Nam Kang
Ji-Nam Kang Ick-Hyun Jo
Ick-Hyun Jo Si-Myung Lee2
Si-Myung Lee2 Chang-Kug Kim
Chang-Kug Kim- 1Department of Herbal Crop Research, National Institute of Horticultural and Herbal Science (NIHHS), Rural Development Administration (RDA), Eumseong, South Korea
- 2Genomics Division, National Institute of Agricultural Sciences (NAS), Rural Development Administration, Jeonju, South Korea
Codonopsis lanceolata (2n = 2x = 16) belongs to the Campanulaceae family and is a valuable medicinal and vegetable plant primarily found in East Asia. Several studies have demonstrated its excellent pharmacological effects, for example in bronchial treatment. However, genomic information of C. lanceolata is scarce, hindering studies on crop improvement of the species. Here, we report a high-quality chromosome-level genome assembly of C. lanceolata based on a hybrid method using Nanopore long-read, Illumina short-read, and Hi-C data. The assembled genome was completed as 1,273 Mb (84.5% of the estimated genome size), containing eight pseudo-chromosomes, ranging from 101.3 to 184.3 Mb. The genome comprised of 71.3% repeat sequences and 46,005 protein-coding genes, of which 85.7% genes were functionally annotated. Completeness of the assembled genome and genes was assessed to be 97.5% and 90.4%, respectively, by Benchmarking Universal Single-Copy Orthologs analysis. Phylogenetic and synteny analysis revealed that C. lanceolata was closely related to Platycodon grandiflorus in the Campanulaceae family. Gene family evolution revealed significant expansion of related genes involved in saponin biosynthesis in the C. lanceolata genome. This is the first reference genome reported for C. lanceolata. The genomic data produced in this study will provide essential information for further research to improve this medicinal plant and will broaden the understanding of the Campanulaceae family.
1 Introduction
Codonopsis lanceolata (lance asiabell or bonnet bellflower) belongs to the Campanulaceae family which consists of about 2,400 species (Lammers, 2007), and is a perennial vine plant distributed primarily in East Asia. The plant grows up to 1.5 m in moist low mountain or hilly areas (Liu et al., 2019), and has been used as a valuable medicinal and vegetable plant (Lim, 2015). However, climate change and indiscriminate harvesting have resulted in the plant becoming increasingly rare in its natural habitat. This valuable plant exhibits excellent pharmacological properties due to its inherent diverse secondary metabolites such as triterpenoid saponins, phenylpropanoids, alkaloids, polyacetylenes, and other compounds (Hossen et al., 2016; Du et al., 2018). These properties include antioxidant (Jeon et al., 2013), antimicrobial (He et al., 2010), anti-inflammatory (Li et al., 2007), and immune-modulatory (Lee et al., 2007) effects, making the plant highly valuable for commercial use. Moreover, C. lanceolata is considered a substitute for Panax ginseng, commonly treated as a panacea in Korea.
Several studies have reported the pharmacological effects of C. lanceolata, however only a few genetic and genomic studies have been reported. Moreover, limited genomic information on this species is available to guide breeding strategies for crop improvement and to study the conservation of natural populations. The recent development of high-throughput sequencing technologies has reduced the burden on genomic research, making it easily accessible (Pareek et al., 2011; Park and Kim, 2016). The hybrid of Third-Generation Sequencing (TGS) and Next-Generation Sequencing (NGS) technologies such as Oxford Nanopore Technologies (ONT) and short-read sequencing from Illumina have enabled rapid and accurate genome assembly (Lu et al., 2016; Dumschott et al., 2020). These developments provide a suitable opportunity to accumulate genomic information, which is essential for performing various studies related to the minor plants that lack basic research foundations.
Here, we present the first high-quality chromosome-level genome assembly of C. lanceolata (2n = 2x = 16) using hybrid methods including NGS, TGS, and high-throughput chromosome conformation capture (Hi-C) technologies. This study provides valuable genomic resources that will enable further research into this medicinal plant and expand our understanding of the Campanulaceae family.
2 Materials and methods
2.1 Sampling, library construction, and sequencing
Whole plant body of C. lanceolata was collected from the National Institute of Horticultural and Herbal Science research field in Eumseong, Korea, and was registered to the National Agrobiodiversity Center (http://genebank.rda.go.kr/) under the voucher number IT239928. The fresh leaves were ground in liquid nitrogen using a mortar and pestle, and genomic DNA was extracted using Exgene Plant SV midi kit (GeneAll Biotechnology, Korea) according to the manufacturer’s instructions. The genomic DNA was purified using ×0.5 AMPure XP bead (Beckman Coulter, United States) according to the manufacturer’s instructions. The quality and quantity of genomic DNA were examined using the Qubit fluorometer (Invitrogen, United States) and Agilent 2200 TapeStation (Agilent Technologies, United States).
An ONT sequencing library was prepared using the ONT 1D ligation sequencing kit SQK-LSK109 (ONT, UK). ONT sequencing was performed using the 1D flowcell vR9.4 and GridION platform operated with MinKNOW software v3.1.20 according to the manufacturer’s instructions. Raw ONT sequencing data (FAST5 files) were converted to FASTQ format using Guppy v2.0.10 (Wick et al., 2019) using default parameters. All Nanopore sequencing procedures were serviced by Phyzen Co. (www.phyzen.com, Korea). An NGS sequencing library was constructed according to the standard Illumina paired-end (PE) library protocol and sequenced using the Illumina HiSeq X platform, all of which were serviced by Macrogen Co. (www.macrogen.com, Korea).
2.2 Data trimming and genome size estimation
The ONT sequencing data were trimmed using Porechop v0.2.3 (https://github.com/rrwick/Porechop) using default parameters to remove adaptor and chimeric sequences. Raw Illumina PE data were trimmed using Trimmomatic v0.38 (Bolger et al., 2014) with default parameters. The genome size of C. lanceolata was estimated using k-mer frequency analyses based on the high-quality Illumina PE data. An optimal k-mer value was obtained by Jellyfish v2.0 (Marcais and Kingsford, 2011), and genome size was estimated using GenomeScope v2.0 (Ranallo-Benavidez et al., 2020) based on the 17-mer frequency distribution data.
2.3 Genome assembly
The trimmed ONT data were self-corrected using the Canu assembler v1.71 (Koren et al., 2017) with default parameters, and the corrected ONT data were de novo assembled using SMARTdenovo (https://github.com/ruanjue/smartdenovo) with a minimum read length of 1,000 bp and other default parameters. The assembled contig sequences were polished twice based on the trimmed PE data using Pilon v1.23 (Walker et al., 2014). An additional polishing process was performed using mapping information of the PE data to improve the assembly quality. The trimmed PE data were mapped to the assembled contig sequences using BWA-MEM v0.7.17 (Li and Durbin, 2009) and Samtools v1.9 (Li et al., 2009) with default parameters. Variant calling was performed using GATK v4.1.4 (https://software.broadinstitute.org/gatk) and Picard v2.20.4 (http://broadinstitute.github.io/picard). Consensus sequence generation through variant substitutions was performed using VCFtools v0.1.13 (https://vcftools.github.io/index.html). Haplotigs in the assembled contig sequences were removed using Purge haplotigs (Roach et al., 2018) with default parameters.
A Hi-C library of C. lanceolata was constructed for chromosome-level assembly using Proximo Hi-C Plant Kit (Phase Genomics, United States) according to the manufacturer’s instructions. Crosslinked DNA was digested using the Sau3A I restriction enzyme, and proximity ligated with biotinylated nucleotides. The molecules were pulled down with streptavidin beads and processed into an Illumina-compatible sequencing library. Sequencing was performed using the Illumina HiSeq X platform. The generated Hi-C PE data were aligned to the assembled contigs using BWA-MEM v0.7.17 (Li and Durbin, 2009) with -5SP option, and unique mapped reads were detected using SAMBLASTER v0.1.26 (Faust and Hall, 2014) and Samtools v1.9 (Li et al., 2009). A chromosome-level assembly was performed using LACHESIS methods (Burton et al., 2013). All Hi-C assembly procedures were serviced by the Phase Genomics Co. (www.phasegenomics.com, United States). Completeness of the assembled draft genome sequence was validated by NGS data mapping using BWA-MEM v0.7.17 (Li and Durbin, 2009) with default parameters and Benchmarking Universal Single-Copy Orthologs (BUSCO) v5.0.0 (Simao et al., 2015) using the embryophyta_odb10 lineage dataset. LRT Assembly Index (LAI) was also used to assess the genome assembly quality (Qu et al., 2018).
2.4 Genome annotation
Initial prediction of repeat sequences in the assembled genome was performed using RepeatModeler v1.0.9 (http://www.repeatmasker.org/RepeatModeler.html), which were merged with previously reported repeat sequences deposited in RepBase v28.04 (https://www.girinst.org/repbase/) to use as a reference repeat database. Consensus repeat sequences in the C. lanceolata genome were identified and characterized using RepeatMasker v4.0.9 (http://www.repeatmasker.org) with the custom database.
Gene prediction was carried out based on repeat-masked assembly sequences using an evidence-based annotation method. The protein sequences of four species including Platycodon grandiflorus (Jia et al., 2022), Helianthus annuus (Badouin et al., 2017), P. ginseng (Wang et al., 2022), and Arabidopsis thaliana (Cheng et al., 2017) were downloaded from each genome database for homology-based prediction. The transcriptome evidence of two Codonopsis species were also collected. The unigene sequences of C. tangshen were obtained from a previous study (He et al., 2019). RNA-seq data of C. pilosula (Gao et al., 2015) were retrieved from the GenBank Sequence Rad Archive (SRA) database and de novo assembled using Trinity v 2.9.1 (Grabherr et al., 2011) with default parameters. Initial gene prediction was performed based on these evidences using MAKER3 v3.01.03 (Holt and Yandell, 2011). The ab initio data for final gene prediction was generated using GeneMark-ES v4.38 (Lomsadze et al., 2005), SNAP v2006-07-28 (Zaharia et al., 2011), and AUGUSTUS v3.3.2 (Stanke et al., 2006). The final gene set for C. lanceolata was confirmed based on the ab initio data using MAKER3 v3.01.03 (Holt and Yandell, 2011) and EvidenceModeler v1.1.1 (Haas et al., 2008).
Functional annotation of the predicted genes was performed by similarities analysis against the NCBI non-redundant (nr) protein database using DIAMOND v0.9.30.131 (Buchfink et al., 2015) with an E-value cutoff of 1e-5. Gene Ontology (GO) terms were assigned to genes using Blast2GO Command Line v1.4.4 (Gotz et al., 2008) with default parameters based on the similarity results. A metabolic pathway was also assigned to genes by searching against the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database (Du et al., 2014) using the KEGG Automatic Annotation Server (KAAS) v2.1 (Moriya et al., 2007) with the single-directional best hit (SBH) method and representative gene sets for eukaryotes. Conserved domains within the protein-coding genes were determined using InterProScan v5.34-73.0 (Jones et al., 2014) with default parameters.
2.5 Comparative genomic analyses
The collinear blocks within C. lanceolata chromosomes and the synteny blocks between C. lanceolata and P. grandiflorus were identified using MCScanX (Wang et al., 2012) with default parameters. Each block was visualized using Circos (Krzywinski et al., 2009) and SynVisio (https://synvisio.github.io), respectively.
To investigate the phylogenetic status and gene family evolution of C. lanceolata, single-copy orthologous genes were searched with eight other species including Arctium lappa (Fan et al., 2022), A. thaliana (Cheng et al., 2017), Daucus carota (Iorizzo et al., 2016), H. annuus (Badouin et al., 2017), Oryza sativa (Sakai et al., 2013), P. grandiflorus (Jia et al., 2022), Solanum lycopersicum (Hosmani et al., 2019), and Vitis vinifera (Jaillon et al., 2007) using OrthoFinder v2.5.4 (Emms and Kelly, 2019). The divergence time and phylogenetic tree were constructed based on the extracted single-copy orthologous genes among the nine species using BEAST. Analysis of the likelihood for gene family gain and loss of C. lanceolata and eight related species was performed using CAFÉ5 (Han et al., 2013).
The total gene set of C. lanceolata was compared to those of four other related species including P. grandiflorus (Jia et al., 2022), H. annuus (Badouin et al., 2017), P. ginseng (Wang et al., 2022), and A. thaliana (Cheng et al., 2017). Homologous protein sequences were identified using BLASTP analysis with an E-value cutoff of 1e-5. The unique and shared genes among the five species were classified based on the sequence similarity using OrthoVenn2 (Xu et al., 2019) with plants group parameters. GO enrichment analysis was performed on the shared genes among the five species and the unique genes in C. lanceolata. Candidate genes encoding enzymes involved in saponin biosynthesis were identified by searching genes assigned to the sesquiterpenoid and triterpenoid biosynthesis pathway (https://www.genome.jp/dbget-bin/www_bget?ko00909) using KAAS analysis. Phylogenetic analysis based on the candidate genes was performed using the maximum likelihood (ML) method with 1,000 bootstraps using MEGA v11 (Tamura et al., 2021) with default parameters after aligning predicted amino acid sequences using MUSCLE (Edgar, 2004) with default parameters.
3 Results and discussion
Approximately 61.2 Gb Nanopore long-reads with an average read length of 4,423 bp and 104.9 Gb Illumina short-reads were generated after the trimming process from raw sequencing data for genome assembly of C. lanceolata (Supplementary Table S1). The C. lanceolata genome was estimated to be about 1,507 Mb, with 1.74% heterozygosity and 82.03% repeat sequences, based on optimal 17-mer analysis using high-quality Illumina short-reads (Supplementary Figure S1; Supplementary Table S2). Initial draft sequences of 1,272 Mb, consisting of 19,667 contigs showing N50 value of 88.7 kb (Supplementary Table S3), were assembled based on Nanopore long-reads used as the seed sequences through a polishing process using Illumina short-reads. Finally, a chromosome-level genome assembly for C. lanceolata, that was 1,273 Mb (84.5% of estimated genome size) and composed of 4,828 scaffolds with N50 value of 154.4 Mb, was completed through a scaffolding process using 47.1 Gb Illumina data produced from Hi-C library (Table 1; Supplementary Table S1). The longest eight scaffolds, ranging in length from 101.3 to 184.3 Mb, included 90.1% (1,147 Mb) of the completed assembled genome sequence (Figure 1A; Supplementary Table S4). The Hi-C interaction heatmap showed distinct interaction signals that distinguished eight pseudo-chromosomes within each pseudo-chromosome (Supplementary Figure S2). BUSCO analysis assessed that the assembled draft genome sequence captured 1,574 (97.5%) complete BUSCOs including 1,476 (91.4%) single-copy BUSCOs, 98 (6.1%) duplicated BUSCOs, and 21 (1.3%) fragmented BUSCOs (Table 1). LAI for assembly quality assessment of repetitive sequences in the draft genome sequence was calculated as 9.08. These results demonstrated that the C. lanceolata genome sequence completed in this study was assembled with a high-quality of completeness.

TABLE 1. Summary statistics of genome assembly and gene prediction of C. lanceolata.
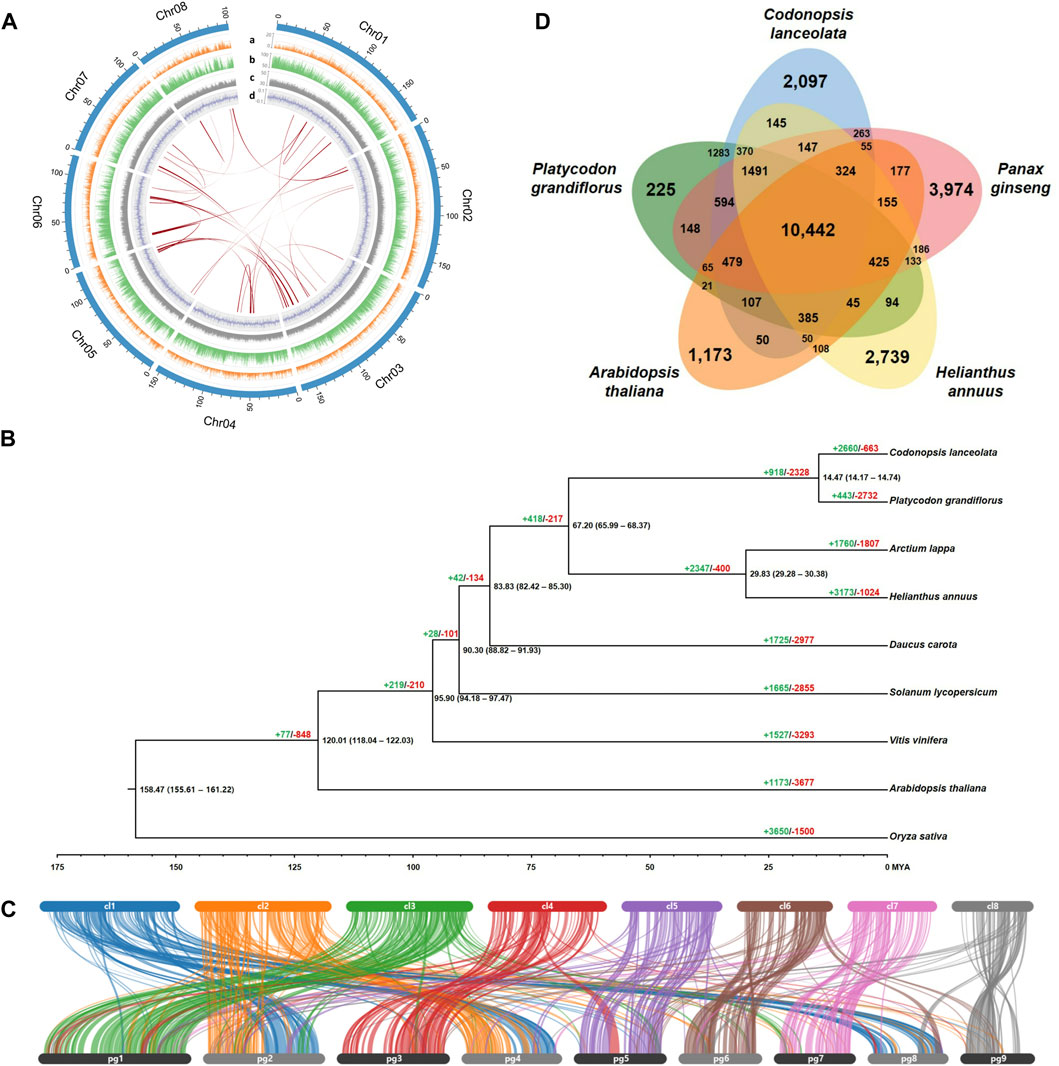
FIGURE 1. C. lanceolata genome characteristics, phylogenomics, synteny structure, and gene clusters. (A) Circos plot representing the eight pseudo-chromosomes in the C. lanceolata assembly and their features. The outermost track (light blue) indicates eight pseudo-chromosomes. Tracks a–d depict the distribution of genes (orange), repeat elements (green), GC contents (gray), and GC skew (blue), respectively. The innermost lines (red) represent collinear blocks identified within the C. lanceolata genome. (B) Phylogenetic tree and gene family evolution of nine plant species. The black numbers above each node indicate divergence times between groups. The green (+) and red (−) numbers indicate the values of expanded and contracted gene families, respectively. (C) Synteny blocks between C. lanceolata and P. grandiflorus genomes. The upper and lower bars indicate each pseudo-chromosomes of C. lanceolata and P. grandifloras, respectively. (D) Shared and unique gene clusters in C. lanceolata and four related species.
The genome annotation characterized 908.3 Mb repeat sequences in the C. lanceolata genome, accounting for 71.3% of the genome (Supplementary Table S5). Among the various repeat elements, long terminal repeats (LTRs), especially Gypsy (17.0%) and Copia (11.5%) type, were remarkably prevalent in the genome. A total of 46,005 genes were predicted based on protein and transcriptome evidence in the C. lanceolata genome (Table 1). The total length of the gene set was 42.41 Mb with an average length of 3,568 bp, and GC content of 44.04%. Average exon and intron length of the gene set were calculated as 922 bp and 2,646 bp, respectively. Among them, 39,435 genes (85.7%) could be functionally annotated by comparing their homology against libraries of known proteins (Supplementary Table S6).
In order to detect the degree of duplication, collinear blocks within the C. lanceolata chromosomes were searched using the annotated gene information. A total of 27 collinear blocks were identified, indicating that there were few duplication events in the entire C. lanceolata genome (Figure 1A; Supplementary Table S7).
Phylogenetic analysis based on 844 single-copy orthologous genes showed that C. lanceolata was closely related to P. grandiflorus in the Campanulaceae family (Figure 1B). The 14.47 MYA of divergence time between two species corresponded with the synteny result indicating that the gene structures and contents were highly conserved each other (Figures 1B, C). Gene family evolution among the nine species by CAFÉ analysis revealed that 2660 and 663 gene families were significantly expanded and contracted in the C. lanceolata genome, respectively.
Gene clustering analysis based on similarity among protein sequences revealed that the C. lanceolata gene products were grouped into 10,442 gene clusters with shared genes from P. grandiflorus, P. ginseng, H. annuus, and A. thaliana, as well as 2,097 clusters with genes unique to C. lanceolata (Figure 1D). GO enrichment analysis of the shared clusters identified the abundant GO terms, such as the biological process GO terms related to regulation of transcription, RNA modification, and rRNA processing, as well as molecular function GO terms related to oxidoreductase activity, oxidoreductase activity, and carboxylic ester hydrolase activity (Supplementary Table S8). In the clusters with genes unique to C. lanceolata, biological process GO terms related to terpenoid biosynthetic process were abundant (Supplementary Table S9). A total of 106 candidate genes involved in the saponin biosynthesis pathway were identified using KAAS analysis (Supplementary Table S10). Of these, putative beta-amyrin synthase genes that are important oxidosqualene cyclases for triterpenoid saponin biosynthesis were identified to be expanded and distinctly grouped in C. lanceolata compared to the other four plant species examined (Supplementary Figure S3).
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA627046; https://figshare.com/, 10.6084/m9.figshare.21507774.
Author contributions
Project design and oversight: WJ, J-NK, and C-KK; Sample collection and curation: J-NK, S-ML, G-HP, and C-KK; Experiment conduction and data analysis: WJ, J-NK, and C-KK; Figure and table preparation: WJ, J-NK, and C-KK; Result interpretation and discussion: WJ, J-NK, I-HJ, S-ML, G-HP, and C-KK; Manuscript writing and revision: WJ, J-NK, I-HJ, S-ML, G-HP, and C-KK; Funding acquisition: C-KK. All authors have read and approved the final version of this manuscript.
Funding
This work was supported by grants from the “Research Program for Agricultural Science and Technology Development” (Project No. PJ015713) in the Rural Development Administration, South Korea.
Acknowledgments
We would like to thank the Bioinformatics and the Sales team in Phyzen for their technical assistance and contribution to the management of this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1100819/full#supplementary-material
References
Badouin, H., Gouzy, J., Grassa, C. J., Murat, F., Staton, S. E., Cottret, L., et al. (2017). The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 546 (7656), 148–152. doi:10.1038/nature22380
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi:10.1093/bioinformatics/btu170
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. methods 12 (1), 59–60. doi:10.1038/nmeth.3176
Burton, J. N., Adey, A., Patwardhan, R. P., Qiu, R., Kitzman, J. O., and Shendure, J. (2013). Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31 (12), 1119–1125. doi:10.1038/nbt.2727
Cheng, C. Y., Krishnakumar, V., Chan, A. P., Thibaud-Nissen, F., Schobel, S., and Town, C. D. (2017). Araport11: A complete reannotation of the Arabidopsis thaliana reference genome. Plant J. 89 (4), 789–804. doi:10.1111/tpj.13415
Du, J., Yuan, Z., Ma, Z., Song, J., Xie, X., and Chen, Y. (2014). KEGG-PATH: Kyoto encyclopedia of genes and genomes-based pathway analysis using a path analysis model. Mol. Biosyst. 10 (9), 2441–2447. doi:10.1039/c4mb00287c
Du, Y. E., Lee, J. S., Kim, H. M., Ahn, J. H., Jung, I. H., Ryu, J. H., et al. (2018). Chemical constituents of the roots of Codonopsis lanceolata. Arch. Pharm. Res. 41 (11), 1082–1091. doi:10.1007/s12272-018-1080-9
Dumschott, K., Schmidt, M. H., Chawla, H. S., Snowdon, R., and Usadel, B. (2020). Oxford Nanopore sequencing: New opportunities for plant genomics? J. Exp. Bot. 71 (18), 5313–5322. doi:10.1093/jxb/eraa263
Edgar, R. C. (2004). Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32 (5), 1792–1797. doi:10.1093/nar/gkh340
Emms, D. M., and Kelly, S. (2019). OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 20 (1), 238. doi:10.1186/s13059-019-1832-y
Fan, W., Wang, S., Wang, H., Wang, A., Jiang, F., Liu, H., et al. (2022). The genomes of chicory, endive, great burdock and yacon provide insights into Asteraceae palaeo-polyploidization history and plant inulin production. Mol. Ecol. Resour. 22, 3124–3140. doi:10.1111/1755-0998.13675
Faust, G. G., and Hall, I. M. (2014). Samblaster: Fast duplicate marking and structural variant read extraction. Bioinformatics 30 (17), 2503–2505. doi:10.1093/bioinformatics/btu314
Gao, J. P., Wang, D., Cao, L. Y., and Sun, H. F. (2015). Transcriptome sequencing of Codonopsis pilosula and identification of candidate genes involved in polysaccharide biosynthesis. PLoS One 10 (2), e0117342. doi:10.1371/journal.pone.0117342
Gotz, S., Garcia-Gomez, J. M., Terol, J., Williams, T. D., Nagaraj, S. H., Nueda, M. J., et al. (2008). High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36 (10), 3420–3435. doi:10.1093/nar/gkn176
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29 (7), 644–652. doi:10.1038/nbt.1883
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9 (1), R7–R22. doi:10.1186/gb-2008-9-1-r7
Han, M. V., Thomas, G. W., Lugo-Martinez, J., and Hahn, M. W. (2013). Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 30 (8), 1987–1997. doi:10.1093/molbev/mst100
He, X., Kim, S. S., Park, S. J., Seong, D. H., Yoon, W. B., Lee, H. Y., et al. (2010). Combined effects of probiotic fermentation and high-pressure extraction on the antioxidant, antimicrobial, and antimutagenic activities of deodeok (Codonopsis lanceolata). J. Agric. Food Chem. 58 (3), 1719–1725. doi:10.1021/jf903493b
He, Y., Zhang, M., Zhou, W., Ai, L., You, J., Liu, H., et al. (2019). Transcriptome analysis reveals novel insights into the continuous cropping induced response in Codonopsis tangshen, a medicinal herb. Plant Physiology Biochem. 141, 279–290. doi:10.1016/j.plaphy.2019.06.001
Holt, C., and Yandell, M. (2011). MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinforma. 12, 491. doi:10.1186/1471-2105-12-491
Hosmani, P. S., Flores-Gonzalez, M., van de Geest, H., Maumus, F., Bakker, L. V., Schijlen, E., et al. (2019). “An improved de novo assembly and annotation of the tomato reference genome using single-molecule sequencing, Hi-C proximity ligation and optical maps,”. BioRxiv, 767764.
Hossen, M. J., Kim, M. Y., Kim, J. H., and Cho, J. Y. (2016). Codonopsis lanceolata: A review of its therapeutic potentials. Phytother. Res. 30 (3), 347–356. doi:10.1002/ptr.5553
Iorizzo, M., Ellison, S., Senalik, D., Zeng, P., Satapoomin, P., Huang, J., et al. (2016). A high-quality carrot genome assembly provides new insights into carotenoid accumulation and asterid genome evolution. Nat. Genet. 48 (6), 657–666. doi:10.1038/ng.3565
Jaillon, O., Aury, J. M., Noel, B., Policriti, A., Clepet, C., Casagrande, A., et al. (2007). The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449 (7161), 463–467. doi:10.1038/nature06148
Jeon, S.-M., Kim, S.-Y., Kim, I.-H., Go, J.-S., Kim, H.-R., Jeong, J.-Y., et al. (2013). Antioxidant activities of processed Deoduck (Codonopsis lanceolata) extracts. J. Korean Soc. Food Sci. Nutr. 42 (6), 924–932. doi:10.3746/jkfn.2013.42.6.924
Jia, Y., Chen, S., Chen, W., Zhang, P., Su, Z., Zhang, L., et al. (2022). A chromosome-level reference genome of Chinese balloon flower (Platycodon grandiflorus). Front. Genet. 13, 869784. doi:10.3389/fgene.2022.869784
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: Genome-scale protein function classification. Bioinformatics 30 (9), 1236–1240. doi:10.1093/bioinformatics/btu031
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27 (5), 722–736. doi:10.1101/gr.215087.116
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: An information aesthetic for comparative genomics. Genome Res. 19 (9), 1639–1645. doi:10.1101/gr.092759.109
Lee, Y. G., Kim, J. Y., Lee, J. Y., Byeon, S. E., Hong, E. K., Lee, J., et al. (2007). Regulatory effects of Codonopsis lanceolata on macrophage-mediated immune responses. J. Ethnopharmacol. 112 (1), 180–188. doi:10.1016/j.jep.2007.02.026
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25 (16), 2078–2079. doi:10.1093/bioinformatics/btp352
Li, J. P., Liang, Z. M., and Yuan, Z. (2007). Triterpenoid saponins and anti-inflammatory activity of Codonopsis lanceolata. Pharmazie 62 (6), 463–466.
Lim, T. (2015). “Codonopsis lanceolata,” in Edible medicinal and non medicinal plants (Germany: Springer).
Liu, Y., Ren, X., and Jeong, B. R. (2019). Night temperature affects the growth, metabolism, and photosynthetic gene expression in Astragalus membranaceus and Codonopsis lanceolata plug seedlings. Plants (Basel) 8 (10), 407. doi:10.3390/plants8100407
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, Y. O., and Borodovsky, M. (2005). Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 33 (20), 6494–6506. doi:10.1093/nar/gki937
Lu, H., Giordano, F., and Ning, Z. (2016). Oxford Nanopore MinION sequencing and genome assembly. Genomics Proteomics Bioinforma. 14 (5), 265–279. doi:10.1016/j.gpb.2016.05.004
Marcais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27 (6), 764–770. doi:10.1093/bioinformatics/btr011
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., and Kanehisa, M. (2007). Kaas: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185. doi:10.1093/nar/gkm321
Pareek, C. S., Smoczynski, R., and Tretyn, A. (2011). Sequencing technologies and genome sequencing. J. Appl. Genet. 52 (4), 413–435. doi:10.1007/s13353-011-0057-x
Park, S. T., and Kim, J. (2016). Trends in next-generation sequencing and a new era for whole genome sequencing. Int. Neurourol. J. 20, S76–S83. doi:10.5213/inj.1632742.371
Qu, S., Chen, J., and Jiang, N. (2018). Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46 (21), e126. doi:10.1093/nar/gky730
Ranallo-Benavidez, T. R., Jaron, K. S., and Schatz, M. C. (2020). GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11 (1), 1432. doi:10.1038/s41467-020-14998-3
Roach, M. J., Schmidt, S. A., and Borneman, A. R. (2018). Purge haplotigs: Allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinforma. 19 (1), 460. doi:10.1186/s12859-018-2485-7
Sakai, H., Lee, S. S., Tanaka, T., Numa, H., Kim, J., Kawahara, Y., et al. (2013). Rice annotation project database (RAP-DB): An integrative and interactive database for rice genomics. Plant Cell Physiol. 54 (2), e6. doi:10.1093/pcp/pcs183
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31 (19), 3210–3212. doi:10.1093/bioinformatics/btv351
Stanke, M., Keller, O., Gunduz, I., Hayes, A., Waack, S., and Morgenstern, B. (2006). Augustus: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439. doi:10.1093/nar/gkl200
Tamura, K., Stecher, G., and Kumar, S. (2021). MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38 (7), 3022–3027. doi:10.1093/molbev/msab120
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9 (11), e112963. doi:10.1371/journal.pone.0112963
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40 (7), e49. doi:10.1093/nar/gkr1293
Wang, Z. H., Wang, X. F., Lu, T., Li, M. R., Jiang, P., Zhao, J., et al. (2022). Reshuffling of the ancestral core-eudicot genome shaped chromatin topology and epigenetic modification in Panax. Nat. Commun. 13 (1), 1902. doi:10.1038/s41467-022-29561-5
Wick, R. R., Judd, L. M., and Holt, K. E. (2019). Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 20 (1), 129. doi:10.1186/s13059-019-1727-y
Xu, L., Dong, Z., Fang, L., Luo, Y., Wei, Z., Guo, H., et al. (2019). OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 47 (W1), W52–W58. doi:10.1093/nar/gkz333
Keywords: Campanulaceae, chromosome-level genome assembly, Codonopsis lanceolata, comparative genomics, Hi-C
Citation: Jang W, Kang J-N, Jo I-H, Lee S-M, Park G-H and Kim C-K (2023) The chromosome-level genome assembly of lance asiabell (Codonopsis lanceolata), a medicinal and vegetable plant of the Campanulaceae family. Front. Genet. 14:1100819. doi: 10.3389/fgene.2023.1100819
Received: 17 November 2022; Accepted: 23 January 2023;
Published: 01 February 2023.
Edited by:
Mather A. Khan, Heinrich Heine University of Düsseldorf, GermanyReviewed by:
Jungeun Kim, Genome Research Foundation, South KoreaJie Qiu, Shanghai Normal University, China
Hua Jin, Beijing Institute of Technology, China
Copyright © 2023 Jang, Kang, Jo, Lee, Park and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chang-Kug Kim, Y2hhbmdAa29yZWEua3I=
†These authors have contributed equally to this work