
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
DATA REPORT article
Front. Genet., 20 February 2023
Sec. Livestock Genomics
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1091544
 Qing Wang1,2†
Qing Wang1,2† Fengping He3†
Fengping He3† Ru-Yi Huang4†
Ru-Yi Huang4† Xueke Yang1Diancheng Yang5Jacob Njaramba Ngatia6,7Yanan Gong5
Xueke Yang1Diancheng Yang5Jacob Njaramba Ngatia6,7Yanan Gong5 Yanchun Xu6Song Huang5*Hui Liu1*
Yanchun Xu6Song Huang5*Hui Liu1*The oriental garden lizard Calotes versicolor (Daudin, 1802) is a medium to large-sized lizard characterized by the posterodorsal orientation of lateral scales in genus Calotes, subfamily Draconinae and family Agamidae (http://www.reptile-database.org/). Calotes is the most widely geographically distributed genus with more than 27 species being found in southern Iran, Afghanistan, Nepal, India, Sri Lanka, Brunei, Indonesia, the Malay Peninsula, Sumatra and southern China (Boulenger and Robinson, 1912; Smith, 1943; Erdelen, 1986; Zhao, 1993). Moreover, the oriental garden lizard has been introduced into Kenya, Borneo, Sulawesi, Seychelles, Mauritius, Oman and Florida (United States) (Gowande et al., 2016). The oriental garden lizard is a diurnal lizard, semi-arboreal to arboreal in habit and has been recorded mostly in scrub, deciduous forests and plantations. They usually bask in the Sun on tree trunks or branches with their heads up or down to regulate their body temperature (Diong et al., 1994). Male and female are identical in general morphology and scalation. However, females have much smaller nuchal and dorsal spines compared to the males and lack a gular sac (Pal et al., 2018). During breeding, adult males have bright body colors which can be quickly changed, and it is also called ‘chameleon tree lizard’ (Shanbhag, 2003).
Calotes versicolor is known to play a role in controlling insect populations, and also serves as a prey item of snakes and birds (Matyot, 2004; Sudasinghe and Somaweera, 2015). Studies on the mitochondrial DNA of C. versicolor conducted in Hainan island and adjacent mainland China found high genetic variation between two major lineages (Huang et al., 2013). Many researchers have mentioned the high level of morphological variation in this species across different populations, and some have considered it to be a species complex (Zug et al., 2006; Gowande et al., 2016). Recent studies have mainly focused on taxology, morphology, physiology and ecology of the C. versicolor (Gowande et al., 2016; Gowande et al., 2021; Tantrawatpan et al., 2021). However, the genomic background of C. versicolor is poorly characterized.
Rapid development of high-throughput sequencing technologies during the last decade has opened new avenues to address the genetic basis of adaptation and speciation in natural populations (Vijay et al., 2016). The use of genetic data has proven to be valuable in delimiting taxa that would not have been recognized based on morphology alone (Spinks and Shaffer, 2005; Wenner et al., 2012; Rodríguez et al., 2020). A high-quality genome is a valuable genetic resource to explore the possible genetic basis for the biological features of lizards (Alföldi et al., 2011; Lind et al., 2019; Gemmell et al., 2020). Therefore, obtaining the high-quality C. versicolor genome will be important for elucidating the genetic mechanisms underlying the species’ distinct biological characteristics and complexity.
Here, we generated the first high-quality genome of the oriental garden lizard using the oxford nanopore technologies (ONT) and DNBSEQ sequencing technology. This is the first and only high-quality genome of subfamily Draconinae. This high-quality C. versicolor genome with high contiguity and completeness become the genomic basis for the molecular studies in the subfamily Draconinae. It could be a valuable resource to conduct future research on the ecology, evolution and genetic mechanisms of biological characteristics of C. versicolor and the subfamily Draconinae.
In total, we generated ∼103.75 gigabases (Gb) of ONT long reads (×63 depth) for genome assembly. The average length of long reads was 15,716 bp, and the N50 of long reads was 27,282 bp. We also generated ∼222.61 Gb of clean whole genome sequencing (WGS) data (×138 depth) for genome assembly and ∼30.56 Gb of RNA-seq data for gene annotation (Table 1). Based on the prediction, the total number of 17-mer present in this subset was 186,766,705,020 and the peak depth was ×110 (Supplementary Figure S1). The C. versicolor genome is estimated to be 1.70 Gb in size. We yielded a draft genome assembly with 104 contigs, a total length of 1.61 Gb, and an N50 contig size of 91.60 Mb (Supplementary Table S1). Benchmarking universal Single-Copy Ortholog (BUSCO) analysis showed that 98% of 3354 BUSCO genes (vertebrata_odb10) were identified, with 97.2% single and 0.8% duplicated copy. The remaining 0.7% and 1.3% were fragmented and missing, respectively (Supplementary Table S2). This was significantly improved when compared with the published assemblies (Wilson et al., 2019) (ASM2071127v1, genome size: 0.91 Gb, contig N50: 1.62 kb and BUSCO: 6.8%, Supplementary Table S1). The C. versicolor genome (43.79% GC content) has a much more homogenous GC distribution in 500 kb windows than either the green anole lizard (Anolis carolinensis), human (Homo sapiens) or chicken (Gallus Gallus) genome (Supplementary Figure S2).
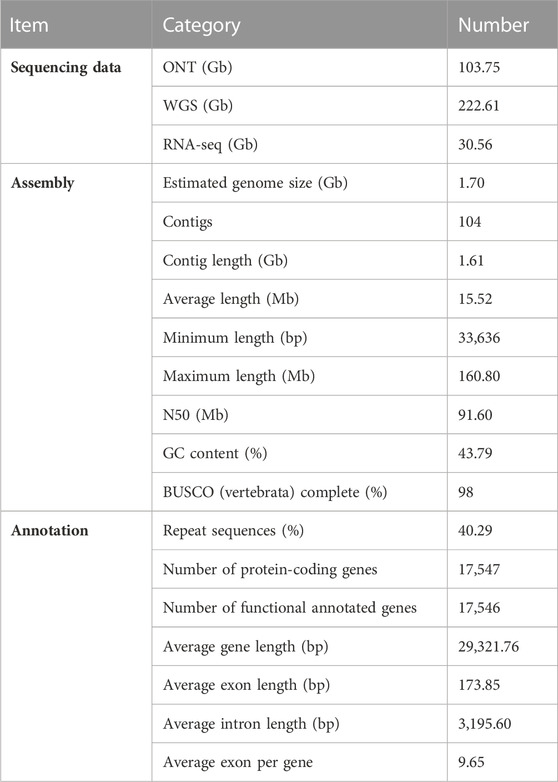
TABLE 1. Summary of genome assemblies and gene annotations of Calotes versicolor draft genome.
In total, we identified 650.26 Mb repetitive elements representing 40.29% of our assembled C. versicolor genome size (Supplementary Table S3). The repeat category with the highest proportion in the genome was LTRs (22.53%) followed by LINEs (6.21%), DNA elements (6.07%), and SINEs (0.88%) (Supplementary Tables S4–S5). The final 17,547 protein-coding genes was generated by combining high-quality homology-based, de novo, and RNA-seq supported genes. The average gene length, exon length, and intron length were 29,321.76 bp, 173.85 bp and 3,195.60 bp, respectively (Supplementary Figure S3), which is consistent with other animals used in annotation (Table 1, Supplementary Figure S4). BUSCO analysis showed that 95.1% and 2.1% of complete and fragmented BUSCO were identified, respectively, indicative of a high-quality gene set (Supplementary Table S2). Finally, 17,546 (99.99%) protein-coding genes were functionally annotated in at least one of the five databases that were used (Supplementary Table S6). In addition, we predicted 195 miRNA, 744 tRNA, 404 rRNA and 384 snRNA in the C. versicolor genome, respectively (Supplementary Table S7).
Comparative genomic analyses were performed between the C. versicolor and 16 other species and identified 6,121 single-copy orthologs, 2,224 multiple-copy orthologs, 8,737 other orthologs and 18 unclustered genes (Figure 1B). We further identified 4,357 single-copy genes shared by these species (Supplementary Table S8). A phylogenetic tree was constructed using these genes, with divergence times being calculated between each pair of species. It was found that the C. versicolor and Pogona vitticeps is in a clade with a divergence time of 82 (35.7–122.5) million years ago (MYA), which is much later than the divergence time between the C. versicolor and snake (Figure 1B, Supplementary Figure S5). The gene family expansion and contraction analysis showed that 545 gene families were expanded and 1,394 gene families were contracted (Figure 1B). We performed Gene Ontology (GO) enrichment analysis of 177 genes (Supplementary Table S9) in significantly expanded gene families (N = 22), which showed that they were significantly enriched in 81 GO terms (Supplementary Table S10), especially those related to sensory perception and biological regulation, including sensory perception of taste (GO:0050909, p = 1.43E-77), chemical stimulus (GO:0007606, p = 1.43E-77), and sensory perception (GO:0007600, p = 6.93E-53). We further performed Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of these significantly expanded genes. The analysis showed that they were significantly enriched in 59 KEGG pathways (Supplementary Table S11, Figure 1C), especially those related to the immune system, including NF-kappa B signaling pathway (map04064, p = 1.20E-49), B cell receptor signaling pathway (map04662, p = 6.68E-42), Natural killer cell mediated cytotoxicity (map04650, p = 2.29E-40) and T cell receptor signaling pathway (map04660, p = 1.14E-4). The biological characteristics, included Taste transduction (map04742, p = 3.06E-46), Retinol metabolism (map00830, p = 4.36E-08) and Melanogenesis (map04916, p = 1.87E-4).
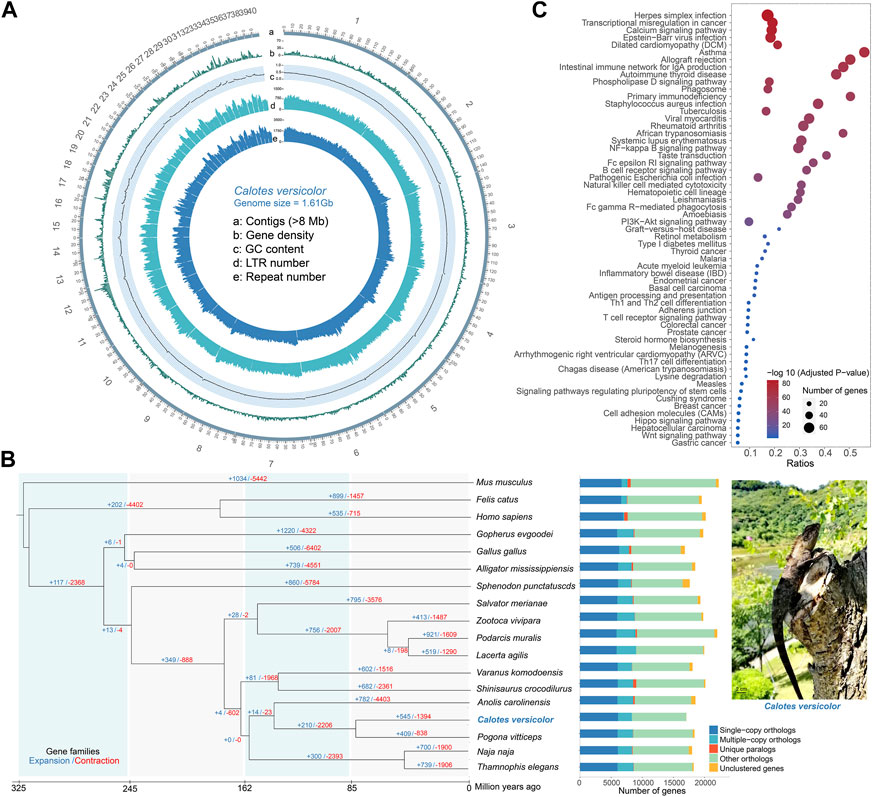
FIGURE 1. Genome landscape of the Calotes versicolor genome, comparative genomics analysis and enrichment analysis of expanded gene families. (A) Genomic features landscape of the C. versicolor genome (500 kb window). a: 40 contigs (92.63% of the genome) with a length range from 8.67 Mb to 160.80 Mb. b: gene count (0–61). c: GC content density (0.0011–0.6632). d: Long Terminal Repeats (LTR) count (0–1464). e: repeat density (2–3482). (B) The phylogenetic relationship of 17 species and the estimated divergence time. Gene family expansions (+) and contractions (−) are indicated by blue and red colors, respectively. The stacked bar plot presents the number of orthologous for 17 species. The right figure is adult of C. versicolor. (C) Significantly enriched 59 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways in the C. versicolor genome compared with the other 16 species.
An adult male oriental garden lizard was collected from car crash in Haikou, Hainan Province, China. The muscle sample was collected, transferred to liquid nitrogen immediately and stored at −80°C for long fragment DNA isolation. Procedures were approved by the College of Forestry, Hainan University (HNUAUCC-2022–000151). For the Nanopore library, 8–10 µg of genomic DNA was purified (>50 kb) with the SageHLS HMW library system (Sage Science). DNA libraries (∼800 ng) were constructed and sequenced on the PromethION (Oxford Nanopore Technologies, ONT) at the NOVOGENE (Beijing, China). Additionally, total RNA from a pooling sample of three different tissues (blood, muscle and skin) was extracted using TRlzol reagent (Invitrogen, United States) according to the manufacturer’s instructions. RNA integrity and purity was measured by a Qubit 3.0 Fluorometer (Life Technologies, United States). cDNA libraries were reverse transcribed from 200 to 400 bp RNA fragments. For whole genome sequence (WGS) library, total genomic DNA was extracted using a DNeasy Blood and Tissue Kit (Qiagen, United States). Both RNA ang WGS were subjected to paired-end sequencing using a DNBSEQ-T1 sequencer (MGI tech, Shenzhen, Guangdong, China).
To estimate the genome size of the C. versicolor, we generated the k-mer depth distribution with a 17 bp k-mer size using DNBSEQ short reads (∼222.61 Gb) were analyzed by KmerFreq v1.0, (Liu et al., 2013). The K-mer frequency distribution was plotted in R v3.5.1, ‘ggplot2’ package (Villanueva and Chen, 2019). The genome size of C. versicolor was estimated using the following formula: estimated genome size = kmer_num/pkdepth, where the kmer_num is the total number of k-mers, and pkdepth refers to the most frequent peak. According to that prediction, the total number of 17-mer present in this subset was 186,766,705,020 and the peak depth was ×110. The size of the C. versicolor genome was estimated to be 1.70 Gb.
Assembly was performed using the NextDenovo v 2.5.0, (https://github.com/Nextomics/NextDenovo). Meanwhile, NextDenovo contains two core modules: NextCorrect and NextGraph. The NextCorrect module was used for raw Nanopore long-reads correction and consensus sequence extraction. The NextGraph module was used for preliminary assembly (Hu et al., 2020). The read cutoff was set at 1 kb for the NextDenovo genome assembly, while default parameters were used for other settings. The primary assembly further improved the single base accuracy using NextPolish v1.4.0, (Hu et al., 2020), with all parameters set to default. At the genome polishing stage, Nanopore long-reads were used repetitively two times, and DNBSEQ short reads were used four times for genome correction. The completeness of the genome and gene set were evaluated by Benchmarking universal Single-Copy Orthologs (BUSCO, v3.1.0) (Simão et al., 2015) analysis using the database of vertebrata_odb10. GC content was measured in non-overlapping 500 kb windows in C. versicolor, A. carolinensis, H. sapiens and G. Gallus genome. In the window, GC% was (G + C)/(A + C + G + T).
Refrom each species was used to performpetitive elements were identified using a combination of homology-based and de novo approaches. For the homology-based approach at both the DNA and protein levels, the genome assembly was aligned to the known repeat database RepBbase using RepeatMasker v4.0.5, (Chen, 2004), RepeatProteinMask (Chen, 2004) and Tandem Repeats Finder v4.07b, (Ou and Jiang, 2018). For the de novo-based approach, RepeatModeler v2.0, (Flynn et al., 2020), and LTR_retriever (Ou and Jiang, 2018) were used to construct a de novo repeat library. All repetitive elements were masked for gene annotation. We also mapped the gene density, GC content, LTR density and repeat density onto 40 contigs (length >8.67 Mb and 92.63% of the C. versicolor genome) using the CIRCOS v0.69–8, (Krzywinski et al., 2009).
We predict assembled gene set combined homology-based, de novo and transcriptome-based methods. In the homology-based method, protein sequences of G. Gallus, H. sapiens, A. carolinensis, P. vitticeps, Varanus komodoensis and Podarcis muralis available in the NCBI database were mapped to the C. versicolor genome using GeneWise v2.4.1, (Birney et al., 2004), with an E-value cutoff of 1e-5. In the de novo method, we ran the repeat-masked genome using Augustus v3.0.3, (Stanke et al., 2004). In the transcriptome-based method, transcripts were assembled using StringTie v1.3.3b, (Pertea et al., 2015), based on clean RNA-seq data. The final protein-coding gene set was generated using the MAKER pipeline v3.01.03, (Campbell et al., 2014), by combining high-quality homology-based, de novo and RNA-seq supported genes. The completeness of the genome and gene set were evaluated through BUSCO analysis using the database of vertebrata_odb10. Functional annotations of protein-coding genes were carried out using BLAST (e-value cut-off of 1e-5) against publicly available databases including the Swiss-Prot, TrEMBL and KEGG database. InterProScan v5.52–86.0, (Jones et al., 2014), was used to predict motifs and domains, as well as Gene ontology (GO) terms. In addition, non-coding RNA genes, including tRNA, miRNA, snRNA and rRNA, were predicted in the assembled genome. tRNA genes were identified using tRNAscan-SE v1.3.1, (Lowe and Eddy, 1997). SnRNA and miRNA genes were detected by searching the reference genome sequences against the content of the Rfam database (Release 12.0) using BLAST. The rRNA genes were detected by alignment with BLASTN against known human rRNA sequences, with an e-value of 1e-5.
We performed a comparative genomic analysis between the C. versicolor and 16 other species with an outgroup Mus musculus (Supplementary Table S8). First, the longest transcript of each gene from each species was used to perform all-to-all BLAST v2.2.26, (Mount, 2007), analysis with the parameter “-p blastp -m8 -e 1e-5 -F F”. Then, genes were clustered using Treefam v1.4, (Li et al., 2006), pipeline with hierarchically clustering on a sparse graph. Finally, 21,771 gene families were identified in all 17 reference genomes, with 4,357 single-copy genes being shared by these 17 species. These single-copy genes were used to construct a Maximum-Likelihood (ML) phylogenetic tree using IQTREE v1.6.12, (Nguyen et al., 2015), with the best-fit evolutionary substitution model (GTR + F + R4) that was evaluated using ModelFinder (Kalyaanamoorthy et al., 2017). To estimate the divergence time between C. versicolor and other 16 species, we used MCMC Tree v4.5, (Yang, 2007), implemented in the PAML package. Sequences for 4,357 single-copy genes were used as the input file for MCMC Tree, and multiple fossil times (G. gallus-H. sapiens: 312.3–330.4, G. gallus-A. carolinensis: 259.7–299.8, G. gallus-P. vitticeps: 276.0–286.8, G. gallus-Alligator mississippiensis: 240.9–247.0, Naja naja-A. carolinensis: 148.5–166.4 and A. carolinensis-P. vitticeps:135.0–160.5) were used for time calibrations from Timetree (http://www.timetree.org/). The Markov chain Monte Carlo (MCMC) process was run for 1,500,000 iterations with a sampling frequency of 150 after aburn-in of 500,000 iterations.
Using the divergence time ranges between the 17 species and the inferred phylogenetic tree, the expanded and contracted gene families were detected using CAFÉ v4.2.1, (De Bie et al., 2006). KEGG enrichment analyses were performed on the expanded gene families with all annotated genes used as the background. Fisher’s exact test was used to improve the accuracy of the conducted χ2 tests. Finally, the Benjamini–Hochberg method (Peng et al., 2017) was used to generate adjusted p-values.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: China National GeneBank DataBase Sequence Archive (https://db.cngb.org/cnsa/) of the China National GeneBank DataBase under accession number CNP0003598.
The animal study was reviewed and approved by College of Forestry, Hainan University (HNUAUCC-2022–000151).
HL and SH designed and initiated the project. R-YH, XY collected the samples. YG, R-YH, and DY performed the DNA and RNA extraction, library preparation, and genome sequencing. QW, FH, and R-YH performed the bioinformatics analysis and visualized the results. QW wrote the manuscript with the assistance from HL, SH, JN, and YX revised and edited the manuscript. All authors have read and approved the final version of manuscript.
Our project was financially supported by the Fundamental Research Funds for the Central Universities (2572020DR10). This work was also supported by China National GeneBank (CNGB).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1091544/full#supplementary-material
Alföldi, J., Di Palma, F., Grabherr, M., Williams, C., Kong, L., Mauceli, E., et al. (2011). The genome of the green anole lizard and a comparative analysis with birds and mammals. Nature 477 (7366), 587–591. doi:10.1038/nature10390
Birney, E., Clamp, M., and Durbin, R. (2004). GeneWise and genomewise. Genome Res. 14 (5), 988–995. doi:10.1101/gr.1865504
Boulenger, G.-A., and Robinson, H. C. (1912). A vertebrate fauna of the Malay Peninsula from the Isthmus of Kra to Singapore including the adjacent islands. Nature 90, 619. doi:10.1038/090619a0
Campbell, M. S., Holt, C., Moore, B., and Yandell, M. (2014). Genome annotation and curation using MAKER and MAKER-P. Curr. Protoc. Bioinforma. 48 (1), 4.11.111–4.11.3914.11. 39. doi:10.1002/0471250953.bi0411s48
Chen, F. Z., You, L. J., Yang, F., Wang, L. N., Guo, X. Q., Gao, F., et al. (2020). CNGBdb: China national genebank database. Yi Chuan= Hered. 42 (8), 799–809. doi:10.16288/j.yczz.20-080
Chen, N. (2004). Using Repeat Masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinforma. 5 (1), 4–14.10. 14. doi:10.1002/0471250953.bi0410s05
De Bie, T., Cristianini, N., Demuth, J. P., and Hahn, M. W. (2006). Cafe: A computational tool for the study of gene family evolution. Bioinformatics 22 (10), 1269–1271. doi:10.1093/bioinformatics/btl097
Diong, C., Chou, L., and Lim, K. (1994). Calotes versicolor: The changeable lizard. Malaysia): Nature Malaysia.
Erdelen, W. (1986). The genus Calotes (Sauria: Agamidae) in Sri Lanka: Clutch sizes and reproductive seasonality of Calotes versicolor-preliminary results. Spixiana 9 (1), 111–115.
Flynn, J. M., Hubley, R., Goubert, C., Rosen, J., Clark, A. G., Feschotte, C., et al. (2020). RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117 (17), 9451–9457. doi:10.1073/pnas.1921046117
Gemmell, N. J., Rutherford, K., Prost, S., Tollis, M., Winter, D., Macey, J. R., et al. (2020). The tuatara genome reveals ancient features of amniote evolution. Nature 584 (7821), 403–409. doi:10.1038/s41586-020-2561-9
Gowande, G., Mishra, A., and Mirza, Z. A. (2016). Neotype designation for Calotes versicolor daudin, 1802 (sauria: Agamidae) with notes on its systematics. Zootaxa 4126 (2), 271–279. doi:10.11646/zootaxa.4126.2.7
Gowande, G., Pal, S., Jablonski, D., Masroor, R., Phansalkar, P. U., Dsouza, P., et al. (2021). Molecular phylogenetics and taxonomic reassessment of the widespread agamid lizard Calotes versicolor (Daudin, 1802)(Squamata, Agamidae) across South Asia. Vertebr. Zool. 71, 669–696. doi:10.3897/vz.71.e62787
Guo, X., Chen, F., Gao, F., Li, L., Liu, K., You, L., et al. (2020). Cnsa: A data repository for archiving omics data. Database 2020, baaa055. doi:10.1093/database/baaa055
Hu, J., Fan, J., Sun, Z., and Liu, S. (2020). NextPolish: A fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255. doi:10.1093/bioinformatics/btz891
Huang, Y., Guo, X., Ho, S. Y., Shi, H., Li, J., Li, J., et al. (2013). Diversification and demography of the oriental garden lizard (Calotes versicolor) on Hainan Island and the adjacent mainland. PLoS One 8 (6), e64754. doi:10.1371/journal.pone.0064754
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: Genome-scale protein function classification. Bioinformatics 30 (9), 1236–1240. doi:10.1093/bioinformatics/btu031
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K., Von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. methods 14 (6), 587–589. doi:10.1038/nmeth.4285
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: An information aesthetic for comparative genomics. Genome Res. 19 (9), 1639–1645. doi:10.1101/gr.092759.109
Li, H., Coghlan, A., Ruan, J., Coin, L. J., Heriche, J. K., Osmotherly, L., et al. (2006). TreeFam: A curated database of phylogenetic trees of animal gene families. Nucleic acids Res. 34, D572–D580. doi:10.1093/nar/gkj118
Lind, A. L., Lai, Y. Y., Mostovoy, Y., Holloway, A. K., Iannucci, A., Mak, A. C., et al. (2019). Genome of the Komodo dragon reveals adaptations in the cardiovascular and chemosensory systems of monitor lizards. Nat. Ecol. Evol. 3 (8), 1241–1252. doi:10.1038/s41559-019-0945-8
Liu, B., Shi, Y., Yuan, J., Hu, X., Zhang, H., Li, N., et al. (2013). Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv preprint arXiv:1308.2012.
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic acids Res. 25 (5), 955–964. doi:10.1093/nar/25.5.955
Matyot, P. (2004). The establishment of the crested tree lizard, Calotes versicolor (Daudin, 1802)(Squamata: Agamidae) in Seychelles. Phelsuma 12, 35–47.
Mount, D. W. (2007). Using the basic local alignment search tool (BLAST). Cold Spring Harb. Protoc. 2007 (7), top17. doi:10.1101/pdb.top17
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32 (1), 268–274. doi:10.1093/molbev/msu300
Ou, S., and Jiang, N. (2018). LTR_retriever: A highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant physiol. 176 (2), 1410–1422. doi:10.1104/pp.17.01310
Pal, S., Vijayakumar, S., Shanker, K., Jayarajan, A., and Deepak, V. (2018). A systematic revision of Calotes Cuvier, 1817 (Squamata: Agamidae) from the Western Ghats adds two genera and reveals two new species. Zootaxa 4482 (3), 401–450. doi:10.11646/zootaxa.4482.3.1
Peng, J., Liu, W., Bretz, F., and Shkedy, Z. (2017). Multiple confidence intervals for selected parameters adjusted for the false coverage rate in monotone dose-response microarray experiments. Biometrical J. 59 (4), 732–745. doi:10.1002/bimj.201500254
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33 (3), 290–295. doi:10.1038/nbt.3122
Rodríguez, A., Rodríguez, B., Montelongo, T., Garcia-Porta, J., Pipa, T., Carty, M., et al. (2020). Cryptic differentiation in the Manx shearwater hinders the identification of a new endemic subspecies. J. Avian Biol. 51 (11), jav.02633. doi:10.1111/jav.02633
Shanbhag, B. A. (2003). Reproductive strategies in the lizard, Calotes versicolor. Curr. Sci. 2003, 646–652.
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31 (19), 3210–3212. doi:10.1093/bioinformatics/btv351
Smith, M. A. (1943). The fauna of British India including Ceylon and Burma. Red Lion Court, Fleet St.; London: Taylor and Francis.
Spinks, P. Q., and Shaffer, H. B. (2005). Range-wide molecular analysis of the Western pond turtle (Emys marmorata): Cryptic variation, isolation by distance, and their conservation implications. Mol. Ecol. 14 (7), 2047–2064. doi:10.1111/j.1365-294X.2005.02564.x
Stanke, M., Steinkamp, R., Waack, S., and Morgenstern, B. (2004). Augustus: A web server for gene finding in eukaryotes. Nucleic acids Res. 32, W309–W312. doi:10.1093/nar/gkh379
Sudasinghe, H., and Somaweera, R. (2015). Calotes versicolor (oriental garden lizard). Diet. Herpetol. Rev. 46, 625–629.
Tantrawatpan, C., Thongnetr, W., Pilap, W., Suksavate, W., Agatsuma, T., Tawong, W., et al. (2021). Genetic diversity and population structure of the oriental garden lizard, Calotes versicolor Daudin, 1802 (Squamata: Agamidae) along the Mekong River in Thailand and Lao PDR. Asian Herpetological Res. 12 (1), 49–57.
Vijay, N., Bossu, C. M., Poelstra, J. W., Weissensteiner, M. H., Suh, A., Kryukov, A. P., et al. (2016). Evolution of heterogeneous genome differentiation across multiple contact zones in a crow species complex. Nat. Commun. 7 (1), 13195. doi:10.1038/ncomms13195
Villanueva, R. A. M., and Chen, Z. J. (2019). ggplot2: elegant graphics for data analysis. Measurement: Interdisciplinary Research and Perspectives 17 (3), 160–167. doi:10.1080/15366367.2019.1565254
Wenner, T. J., Russello, M. A., and Wright, T. F. (2012). Cryptic species in a neotropical parrot: Genetic variation within the Amazona farinosa species complex and its conservation implications. Conserv. Genet. 13 (5), 1427–1432. doi:10.1007/s10592-012-0364-8
Wilson, C. A., Titus, T., Batzel, P., Postlethwait, J. H., and Raman, R. (2019). A search for sex-linked loci in the agamid lizard, Calotes versicolor. Sex. Dev. 13 (3), 143–150. doi:10.1159/000500465
Yang, Z. (2007). Paml 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24 (8), 1586–1591. doi:10.1093/molbev/msm088
Keywords: Calotes versicolor, oxford nanopore technologies (ONT), draft genome, phylogenomic analysis, reptile
Citation: Wang Q, He F, Huang R-Y, Yang X, Yang D, Ngatia JN, Gong Y, Xu Y, Huang S and Liu H (2023) Draft genome of the oriental garden lizard (Calotes versicolor). Front. Genet. 14:1091544. doi: 10.3389/fgene.2023.1091544
Received: 07 November 2022; Accepted: 10 February 2023;
Published: 20 February 2023.
Edited by:
Shi-Yi Chen, Sichuan Agricultural University, ChinaReviewed by:
Li Hu, Institute of Zoology (CAS), ChinaCopyright © 2023 Wang, He, Huang, Yang, Yang, Ngatia, Gong, Xu, Huang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Song Huang, c25ha2VtYW5AYWhudS5lZHUuY24=; Hui Liu, bGl1aHVpQGhhaW5hbnUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.