 Chunchun Hu
Chunchun Hu Yi Wang
Yi Wang Chunyang Li2
Chunyang Li2 Bingrui Zhou
Bingrui Zhou Qiong Xu
Qiong Xu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 28 February 2023
Sec. Genetics of Common and Rare Diseases
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1083779
This article is part of the Research Topic Genetic and Environmental Factors in the Occurrence of Paediatric Disorders – Volume II View all 10 articles
Objectives: Autism spectrum disorder (ASD) is a neurodevelopmental disorder with genetic and clinical heterogeneity. Owing to the advancement of sequencing technologies, an increasing number of ASD-related genes have been reported. We designed a targeted sequencing panel (TSP) for ASD based on next-generation sequencing (NGS) to provide clinical strategies for genetic testing of ASD and its subgroups.
Methods: TSP comprised 568 ASD-related genes and analyzed both single nucleotide variations (SNVs) and copy number variations (CNVs). The Autism Diagnostic Observation Schedule (ADOS) and the Griffiths Mental Development Scales (GMDS) were performed with the consent of ASD parents. Additional medical information of the selected cases was recorded.
Results: A total of 160 ASD children were enrolled in the cohort (male to female ratio 3.6:1). The total detection yield was 51.3% for TSP (82/160), among which SNVs and CNVs accounted for 45.6% (73/160) and 8.1% (13/160), respectively, with 4 children having both SNVs and CNV variants (2.5%). The detection rate of disease-associated variants in females (71.4%) was significantly higher than that in males (45.6%, p = 0.007). Pathogenic and likely pathogenic variants were detected in 16.9% (27/160) of the cases. SHANK3, KMT2A, and DLGAP2 were the most frequent variants among these patients. Eleven children had de novo SNVs, 2 of whom had de novo ASXL3 variants with mild global developmental delay (DD) and minor dysmorphic facial features besides autistic symptoms. Seventy-one children completed both ADOS and GMDS, of whom 51 had DD/intellectual disability (ID). In this subgroup of ASD children with DD/ID, we found that children with genetic abnormalities had lower language competence than those without positive genetic findings (p = 0.028). There was no correlation between the severity of ASD and positive genetic findings.
Conclusion: Our study revealed the potential of TSP, with lower cost and more efficient genetic diagnosis. We recommended that ASD children with DD or ID, especially those with lower language competence, undergo genetic testing. More precise clinical phenotypes may help in the decision-making of patients with genetic testing.
Autism spectrum disorder (ASD) is a highly heterogeneous neurodevelopmental disorder characterized by social deficits and restricted, repetitive patterns of behavior and interests (Lord et al., 2020). The ASD occurrence in the United States is estimated to be approximately 1 in 44, with an overall male-to-female prevalence ratio of 3.4:12). As one of the most heritable medical conditions, ASD is associated with over a thousand risk genes (He et al., 2013), of which more than 100 genes and genomic regions meet rigorous statistical thresholds for the correlation with ASD phenotype (Satterstrom et al., 2020). Models of genetic risk for ASD tend to favor complex inheritance; nevertheless, rare inherited and de novo variants contribute to a substantial risk of individuals with ASD (Iossifov et al., 2014; Sanders et al., 2015). According to recently published large case‒control studies (Satterstrom et al., 2020; Fu et al., 2022; Zhou et al., 2022), the genetic contribution to ASD continues to increase. Children with a diagnosis of ASD are recommended for etiological assessments. Chromosomal microarray analysis (CMA), detecting large duplications or deletions, was used as first-tier genetic testing for children with ASD, multiple congenital anomalies (MCA) and developmental delay (DD)/intellectual disability (ID) (Miller et al., 2010), in addition to fragile X analysis and MECP2 testing. Many physician organizations recommend next-generation sequencing (NGS) testing when CMA-based evaluation has no positive identifications. In recent years, with the remarkable maturity of technical aspects of NGS variant discovery, it has been reported that rare genetic variants can be found in up to 30% of the ASD population (Vorstman et al., 2017).
ASD is always accompanied by cooccurring conditions, such as DD/ID, language disorders, motor difficulties, attention deficit hyperactivity disorder (ADHD), and epilepsy. It is generally acknowledged that established ASD risk variants are associated with these comorbidities (Vorstman et al., 2017). Approximately 50% of children diagnosed with ASD will have ID (Shaw et al., 2021). The presence of ID and dysmorphic features are considered to account for a higher detection rate of genetic susceptibility factors contributing to ASD etiology (Tammimies et al., 2015; Husson et al., 2020). Likewise, finding genetic abnormalities may facilitate a better understanding of the pathophysiology of ASD, lead to early detection of cooccurring conditions and develop preventative guidance for children and families.
Here, we report the detection yields of the designed targeted sequencing panel (TSP) containing 568 ASD-related genes. ASD children were divided into subgroups according to clinical assessments, hoping to find the value of guidance for genetic testing and facilitate effective intervention based on pathological pathways inferred from the genetic information.
The study included 160 patients who were diagnosed with ASD in the Department of Child Healthcare, Children’s Hospital of Fudan University, from June 2017 to March 2019 for genetic testing. The inclusion criteria for the cases were as follows: children met the criteria of ASD diagnosed by experienced pediatricians according to the Diagnostic and Statistical Manual of Mental Disorders, fifth edition (DSM-V) (American Psychiatric Association, 2013). Patients were also recommended to complete the Autism Diagnostic Observation Schedule, second edition (ADOS-2) (Lord C et al., 2012). The results of the ADOS included two subdomains: social affect (SA) and restricted and repetitive behavior (RRB). The total raw score was converted into the ADOS calibrated severity score, from 1 (none) to 10 (severe). The Griffiths Mental Development Scales (GMDS) (Griffiths, 1984; Huntley, 1996) were also performed with the consent of ASD parents. The raw scores of the 5 subscales (Locomotor (Lm), Personal and Social (P/S), Hearing and Speech (H/Sp), Eye and Hand (E/Hd), and Performance (Pf)) of the GMDS were transformed into developmental quotients (DQ). A DQ lower than 70 was considered delayed. For the selected cases, additional medical information was recorded.
We selected 568 candidate genes in TSP as follows: genes marked from 1 to 4 in the ranking categories of the Gene-Scoring (2017) in SFARI Gene (https://gene.sfari.org/), genes predicted by the TADA model with a False discovery rate (FDR) value less than 0.3 (6, 17), and genes reported in large-scale studies (Vorstman et al., 2017; O'Roak et al., 2012; Wang et al., 2016). Genes were grouped and classified into 3 groups: “Group1-Definitive”, 66 genes ranked 1 or 2 in SFARI Gene and their FDR value < 0.05; “Group2-Probably”, 157 genes ranked 3 in SFARI Gene and their FDR value < 0.3 with more than one genetic study identified loss-of-function mutations that related to ASD; “Group3-Possible”, 345 genes ranked 4 in SFARI Gene and their FDR value < 0.3 with no loss-of-function mutation founding that had possible relationship with ASD. There were 110 genes in TSP correlated with ID (from JuniorDoc Database, http://drwang. top/) and 51 genes correlated with epilepsy (from EpilepsyGene Database, http://www.wzgenomics.cn/EpilepsyGene/).
Genomic DNA of participants was isolated from blood samples according to standard procedures by a QIAamp DNA Blood Midi Kit. Two hundred nanograms of genomic DNA from each individual was sheared by a Biorupter (Diagenode, Belgium) to acquire 150–200 bp fragments. The ends of the DNA fragments were repaired, and Illumina Adaptor was added (Fast Library Prep Kit, iGeneTech, Beijing, China). After the sequencing library was constructed, the whole exons were hybridized with costumed probes designed and synthesized by iGeneTech as mentioned above. Captured libraries were mixed in equal molar amounts and sequenced on an Illumina HiSeq2000 platform (Illumina, San Diego, CA) with 150 base paired-end reads. The average on-target sequencing rate was 98.7%, and the target bases covered at >=20X and >=10X were 97.7% and 97.6%, respectively. Raw reads were filtered to remove low-quality reads by using FastQC. Then, clean reads were mapped to the reference genome GRCh37/Hg19 by using BWA. After removing duplications, SNVs and InDels were called and annotated by using GATK. The variants were interpreted according to ACMG guidelines (Richards, et al. Genetics in Medicine (2015) 17, 405) and patient phenotypes and were classified as pathogenic (P), likely pathogenic (LP), variants of unknown significance (VUS), likely benign (LB) or benign (B). A CNV kit was used to call the large copy number variations (CNVs), and the default parameters were used. To identify CNVs, part of the sequencing library was sequenced directly, and each sample yielded 1G raw data. CNVs were called by using CNVseq, and the controls were the healthy parents. For the diagnostic SNVs of patients and parents, Sanger sequencing was used for variant confirmation. For diagnostic CNVs, qPCR/MLPA was performed.
Conventional descriptive statistical methods were used for presenting characteristics of the study cohort. We used unpaired t-test or Mann–Whitney U test depending on normality for the comparisons of ADOS scores and DQs of the GMDS in subgroups with positive and negative genetic findings in DD/ID subgroup. For categorical variables, the sex and subgroup differences of detection yields were compared by chi-squared tests. Data were presented as the mean ± standard deviation (SD) or medians and interquartile ranges (IQR) for continuous variables according to whether the data were normally distributed. Data were presented as percentages for categorical variables. Data analysis was performed using SPSS 22.0 (IBM, Armonk, NY, United States).
A total of 160 children who were diagnosed with ASD were included in the cohort (125 males and 35 females). The mean age of the patients was 3.24 ± 1.27 years. Ninety-four children completed the ADOS-2, and 75 children were assessed using the GMDS, with 71 children having both ADOS-2 and GMDS assessments (Table 1).
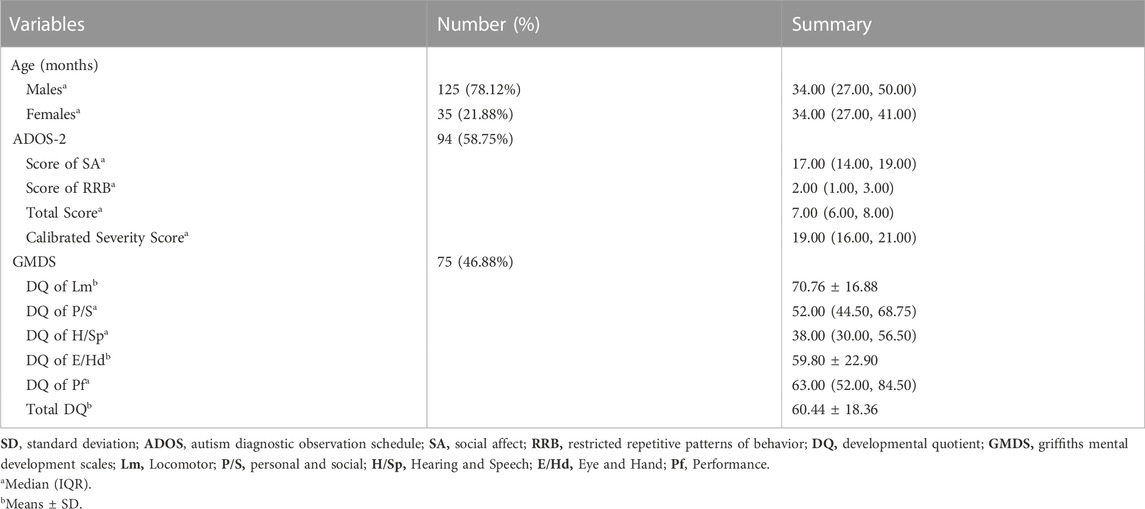
TABLE 1. Clinical characteristics and the results of ADOS/GMDS of ASD patients in the cohort.
The overall detection yield of TSP was 51.3% (82/160) for analyzing both SNVs and CNVs, of which 57 were male and 25 were female. Although the number of males was higher than that of females, the detection rate of disease-associated variants in females (71.4%) was significantly higher than that in males (45.6%, χ2 = 7.30, p = 0.007). The “pathogenic/likely pathogenic (P/LP)” rate was 16.9% (27/160), including 16 males and 11 females. For SNVs, 73 children had positive results (24 females), accounting for a detection rate of 45.6%, of which “P” variants were found in 4.4% of cases (7/160), “LP” in 8.1% of cases (13/160), and “VUS” in 33.1% of cases (53/160). We identified a total of 90 SNVs, of which 74 were missense mutations, 5 were frameshift mutations, and 11 were splicing mutations. The most common variants were DLGAP2, SHANK3, and KMT2A, which were present in 3 probands for each variant. Of 73 patients with SNVs, 68 patients underwent parental testing (both father and mother), and 3 patients had variant confirmation only by mother. We found 11 cases carried de novo variants, including ASXL3, KMT2A, and MECP2, which accounted for 16.2% (11/68) of the analyzed trios. Of the other 74 variants, 42 were of maternal origin, and 30 were of paternal origin, with 2 variants of non-maternal origin (lack of paternal samples) (Table 2). CNVs were found in 13 patients (3 females), which accounted for 8.1% (13/160), whereas 4 children had dual SNV and CNV. The percentage of pathogenic CNVs was 15.4% (2/13), “LP” was 46.2% (6/13) and “VUS” was 38.5% (5/13). A total of 23.1% (3/13) were duplication variants, and 76.9% (10/13) were heterozygous deletions. 17p11.2 had the greatest number of reportable CNVs, accounting for 46.2% (6/13) (Table 3).
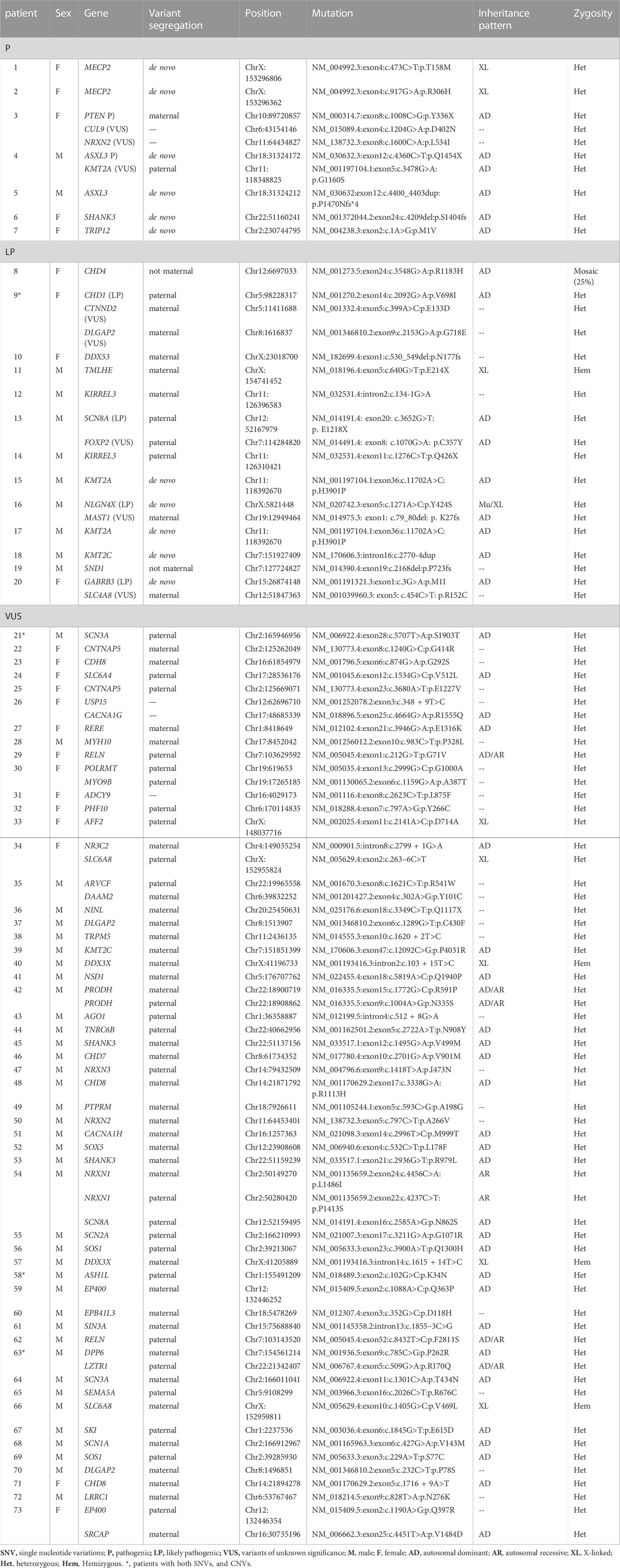
TABLE 2. SNVs identified from TSP.
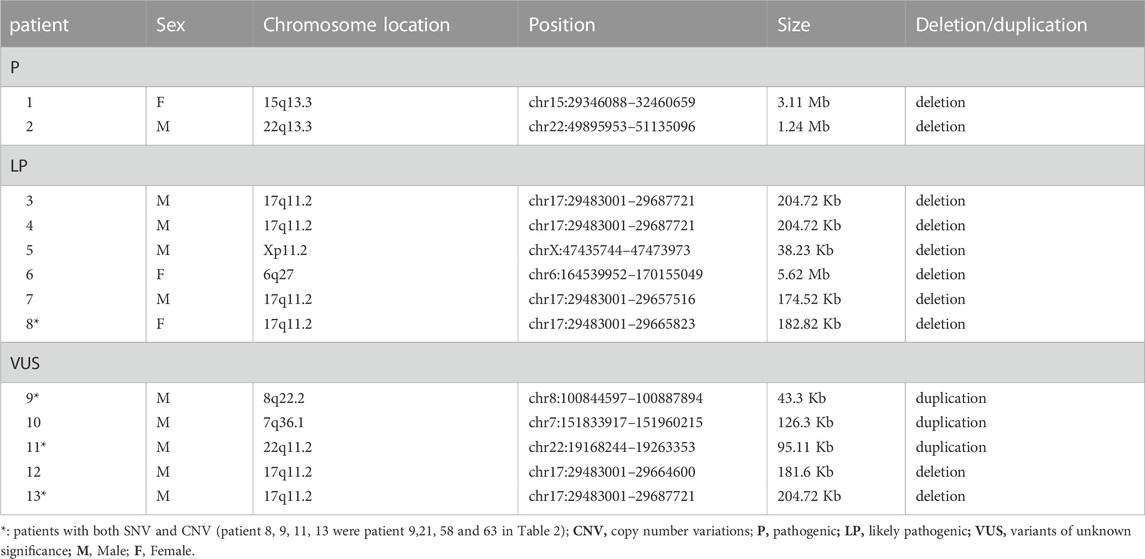
TABLE 3. CNVs in ASD patients from TSP.
Among 71 children, the average DQ of the GMDS was 61.26 ± 17.43, and the calibrated severity score of the ADOS-2 was 7.14 ± 1.45. The detection yield of these 71 children was 54.9% (39/71), with P/LP variants reached 19.7% (14/71). The detection rate of this subgroup was not statistically different from that of general ASD cohort (χ2 = 0.77, p = 0.774). There were 51 patients (71.8%, 51/71) with a total DQ under 70. In this subgroup of ASD children combined DD, 30 patients had positive genetic variants (58.8%, 30/51), with P/LP rate reaching 21.6% (11/51). Children with genetic abnormalities had lower language competence than children without positive genetic findings (Z = -2.20, p = 0.028). There were no correlations between ASD symptoms and the detection of genetic abnormalities in this DD subgroup (Table 4).
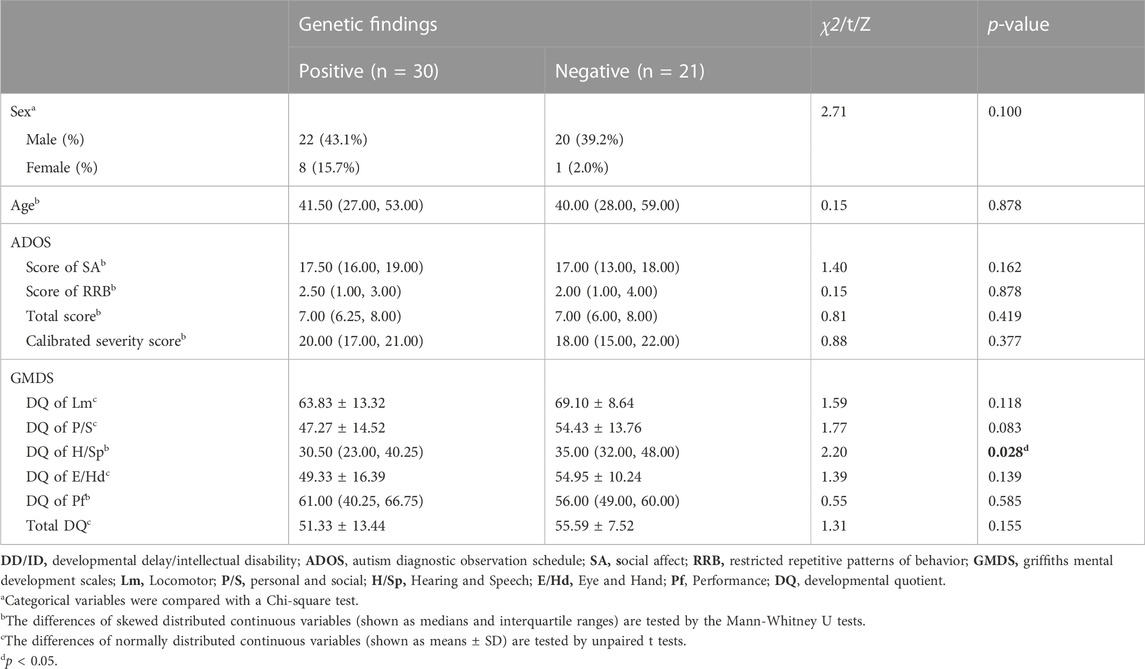
TABLE 4. Comparison of ASD symptoms and developmental scores between children with positive and negative genetic variants in DD/ID subgroup.
Patient 4 and Patient 5 had de novo variants of ASXL3 (Figure 1). Patient 4 was referred to our clinic at 19 months for delayed development. He had poor eye contact as well as response to names. Repetitive behaviors included stamping and shanking head/hands. Tracing the developmental milestones, the patient was unable to crawl and pull up to stand at that time and he learned to sit without support until the age of 10 months. He could only make repeated single-syllable sounds. His birthweight was normal, but feeding seemed very difficult in the early stage, resulting in poor postnatal growth (2 SD below the mean). Physical examination showed that he had a prominent forehead, wildly spaced eyes, strabismus and malformation of external auditory canals. When he had reexamination at 6 years old, he still had language delay. An oral examination revealed that he had dental overcrowding. The Wechsler Preschool and Primary Scale of Intelligence (WPPSI) (Wechsler, 2012) showed that his intelligence quotient (IQ) was 69. Patient 5 was a 2.7-year-old boy. He displayed repetitive behaviors such as throwing and biting objects, turning the wheels and sometimes squinting. He had obvious delayed speech and language development because he was non-verbal at the time of referral. Feeding difficulty also happened to him. Walking independently was at the age of 20 months. His total DQ of the GMDS was 55.2 (the DQs of all the subscales were less than 70). Facial dysmorphism was prominent forehead but there was no obvious deformity in other parts. He had febrile convulsions twice, while electroencephalogram (EEG) and magnetic resonance imaging (MRI) were normal. Two patients shared the common characteristics of ASXL3 variants, but they had only mild ID/DD, which was noteworthy.
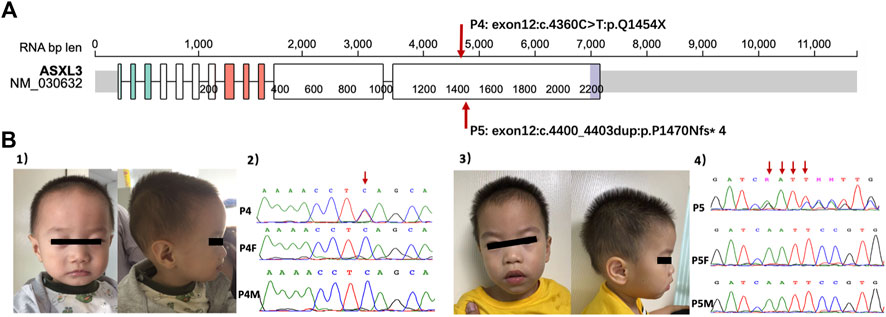
FIGURE 1. (A) The location of ASXL3 variants in patients based on Protein Paint (https://proteinpaint.stjude.org, access in September 29th). The numbers of patients correspond with Table 2. (B) (1) The facial features of P4. (2) Sanger sequence of P4 and his parents with ASXL3 genetic locus. The red arrow showed the mutation stie of P4. (3) The facial features of P5. (4) Sanger sequence of P5 and his parents with ASXL3 genetic locus. The red arrows showed the duplication sties of P5. P4, patient 4; P4F, father of patient 4; P4M, mother of patient 4; P5, patient 5; P5F, father of patient 5; P5M, mother of patient 5.
In this study, we investigated the detection yields and novel variants through TSP of 568 ASD-associated genes in an ASD cohort. The detection yield was 51.3% in TSP, with the rate of “P/LP” reaching 16.9%. With the falling costs of sequencing, more patients with neurodevelopmental disorders are allowed to receive genetic testing whose positive results give them better access to new treatments. CMA was considered the appropriate initial test for the etiologic evaluation of ASD children (Hyman et al., 2020). There is increasing evidence that NGS, whole-exome sequencing (WES) and whole-genome sequencing (WGS) offer diagnostic advantages over CMA (22). Sirvstava et al.‘s review (Srivastava et al., 2019) revealed a yield in the range of 30%–40% for exome sequencing, which exceeds the 10%–20% yield for CMA. Feliciano et al. (2019) conducted WES in 457 ASD families with genetic identification in 15.2% multiplex families and 10.1% simplex families. According to Ghralaigh’s study (Ni Ghralaigh et al., 2020), the diagnostic yield in ASD was 31% using WES and 42.4% using WGS, but the cost estimates were €79.33 and €1239.5 for choosing different technologies. For panel sequencing, a meta-analysis by Stefanski et al. (2021) showed that the identification of genetic defects accounted for 22.6%, compared to 27.2% for WES. Speak frankly, WES and WGS have higher diagnostic yields of ASD than panel sequencing; however, the benefits do not outweigh their drawbacks. WES and WGS offer higher costs than panel sequencing; on the other hand, due to the larger amount of data, more time is required for analysis and processing. Therefore, an affordable sequencing panel that can capture relevant genes may be a good compromise. It can not only achieve molecular diagnosis and detection efficiency in less cost and time but also avoid the waste of resources. The most important factor in ASD families’ decision about genetic testing is cost. Sequencing panel is still the most cost-effective choice. Ghralaigh et al.‘s report (Ni Ghralaigh et al., 2022) demonstrated 0.22%–10.02% diagnostic yields of gene panels to derive the conclusion that gene panels marketed for use in ASD are currently of limited clinical utility. However, gene selection and numbers for inclusion of gene panels are the key factors for results. A well-defined/comprehensive gene set is required in gene panels. We selected genes with the most promising diagnostic purpose of ASD. The most frequent variants in our cohort were SHANK3, KMT2A, and DLGAP2, which was a slightly different from the previous ASD cohort studies (Satterstrom et al., 2020; Fu et al., 2022). ASD frequent genes like SCN2A, CHD8, PTEN and so on were identified in our cohort whereas we did not find SYNGAP1, ADNP variants according to our sample size. For 72 genes associated with ASD at FDR value <=0.001 in Fu et al.’ s study (Fu et al., 2022), we have 51genes overlapped in our panel. Of identified 102 risk gene in Satterstrom’s study (Satterstrom et al., 2020), 66 of them overlapped with our TSP. Our designed TSP including most of the ASD frequent genes and whose detection yield reached 51.3%, is specialized for ASD patients, and can be considered a success for panel sequencing and potential for the clinical utility of ASD.
Although the reported prevalence sex ratio is four times higher in males than in females (Brugha et al., 2016), we observed that the detection rate of genetic variants was 1.5 times higher in females than males. Sex differences were also observed in other genetic studies (De Rubeis et al., 2014; Satterstrom et al., 2020). The possible reasons were that cognitive defects and autistic traits in females are less severe than those in males. Conversely, females may need clearer autistic characteristics and comorbid DD/ID to receive a diagnosis of ASD. It is believed that sex differences are consistent with the female protective effect model, which assumes that women need an increased genetic load to reach the threshold for ASD diagnosis (Werling, 2016). Thus, more prominent phenotypes demonstrate a higher risk for genetic variants in females than males with ASD.
Language plays a major part in the outcomes of ASD. ASD children whose language is impaired, could have a large impact on the social interaction and general wellbeing of individuals (Nudel et al., 2021). Improvements in the language of ASD children before 5 years old may result in catching up to overall average levels in developmental trajectories, whereas the remainders may develop ID (Pickles et al., 2014). Furthermore, patients who have lower cognitive abilities are more likely to obtain an identifiable genetic risk variant than those with a higher IQ (Sanders et al., 2015). Interestingly, our results showed that in the subgroup of ASD children with DD, children with genetic variants had lower language competence. In other words, children with lower language competence had a greater chance of finding genetic variants. Nudel et al. (2021) considered it as pleiotropy between language impairment and ASD. They observed a significant genetic overlap between specific language impairment and childhood autism (which excluded Asperger’s syndrome). Another hypothesis is that children with genetic conditions are more likely to display delays in early developmental milestones, especially in language and motor functions. Compared with idiopathic ASD, children with PPP2R5D, ADNP, ASXL3, DYRK1A, MED13L variants and so on were marked by extensive delays, 2.7 times for single words and 5.7 times for combined words (Wickstrom et al., 2021). Thus, ASD children with DD or ID, especially those with lower language competence, are recommended for genetic testing.
In our subjects, 2 patients had de novo AXSL3 variants. It is a transcriptional regulator that belongs to a group of vertebrate asx-like proteins. The ASXL3 gene is highly expressed in the cerebral cortex as an epigenetic regulator that plays a role in regulating and controlling gene expression through chromatin remodeling (Katoh and Katoh, 2004; Katoh, 2015). Most ASXL3 variants are de novo, placing it among the top 10 neurodevelopmental genes with the highest frequency of de novo variants (Wright et al., 2015). The characteristics of ASXL3-related syndrome (also called Bainbridge-Ropers syndrome) are DD/ID (moderate to severe), language impairment or absent speech, hypotonia and dysmorphic facial features. Our patients had typical phenotypic characteristics, such as feeding difficulties and delayed motor and language abilities. However, they had only mild developmental delay with IQ/DQ higher than 55, and no obvious signs of hypotonia or epilepsy compared with other patients with ASXL3 variants (Katoh and Katoh, 2004; Katoh, 2015). Although most ASXL3-related syndromes rely on molecular confirmation, many individuals with pathogenic variants of ASXL3 can be identified by a combination of clinical symptoms and unique phenotypes and do not omit those with mild developmental delays.
Our work shows the utility of TSP, which has lower cost and more efficient genetic diagnosis and confirms the effectiveness of the test strategy. TSP should be offered to ASD patients in the expectation of preventative guidance and early detection of comorbidities. Subtypes of ASD children, especially those with language deficits, are recommended for testing to help families develop better intervention strategies.
The data presented in the study are deposited in the GSA-Human repository, accession number HRA003901.
The studies involving human participants were reviewed and approved by Ethics Committee of the Children’s Hospital of Fudan University. Written informed consent to participate in this study was provided by the participants and legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
QX, XX, YL, YW, and CH designed the study. CH, CL, YW, BZ, DL, HL, QX, and XX collected the study samples. YW, LM performed the sequencing experiments. QX, CH, and YW analyzed the data. CH drafted the manuscript, and XX and XQ modified the manuscript. All authors have agreed to the published version of the manuscript.
This study was supported in part by the National Natural Science Foundation of China (2021NSFC, 82171540), the National Key Research and Development Program of China (No. 2016YFC1306205), and the Key Subject Construction Project of Shanghai Municipal Health Commission (shslczdzk02903).
We thank children and their families from the Department of Child Healthcare, Children’s Hospital of Fudan University for participation. We thank Xia Wang and Yaping Yang from Ailife Diagnostics (United States) for their help. We also thank Yonghui Jiang from Yale university for his comments.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
American Psychiatric Association (2013). Diagnostic and statistical manual of mental disorders. 5th ed. Washington, DC: American Psychiatric Association.
Brugha, T. S., Spiers, N., Bankart, J., Cooper, S. A., McManus, S., Scott, F. J., et al. (2016). Epidemiology of autism in adults across age groups and ability levels. Br. J. Psychiatry 209 (6), 498–503. doi:10.1192/bjp.bp.115.174649
De Rubeis, S., He, X., Goldberg, A. P., Poultney, C. S., Samocha, K., Cicek, A. E., et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515 (7526), 209–215. doi:10.1038/nature13772
Feliciano, P., Zhou, X., Astrovskaya, I., Turner, T. N., Wang, T., Brueggeman, L., et al. (2019). Exome sequencing of 457 autism families recruited online provides evidence for autism risk genes. NPJ Genom Med. 4, 19. doi:10.1038/s41525-019-0093-8
Fu, J. M., Satterstrom, F. K., Peng, M., Brand, H., Collins, R. L., Dong, S., et al. (2022). Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat. Genet. 54, 1320–1331. doi:10.1038/s41588-022-01104-0
He, X., Sanders, S. J., Liu, L., De Rubeis, S., Lim, E. T., Sutcliffe, J. S., et al. (2013). Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet. 9 (8), e1003671. doi:10.1371/journal.pgen.1003671
Huntley, M. (1996). The Griffiths mental developmental scales manual from birth to two years. Oxford, UK: The Test Agency.
Husson, T., Lecoquierre, F., Cassinari, K., Charbonnier, C., Quenez, O., Goldenberg, A., et al. (2020). Rare genetic susceptibility variants assessment in autism spectrum disorder: Detection rate and practical use. Transl. Psychiatry 10 (1), 77. doi:10.1038/s41398-020-0760-7
Hyman, S. L., Levy, S. E., and Myers, S. M. (2020). Executive summary: Identification, evaluation, and management of children with autism spectrum disorder. Pediatrics 145 (1), e20193448. doi:10.1542/peds.2019-3448
Iossifov, I., O'Roak, B. J., Sanders, S. J., Ronemus, M., Krumm, N., Levy, D., et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515 (7526), 216–221. doi:10.1038/nature13908
Katoh, M. (2015). Functional proteomics of the epigenetic regulators ASXL1, ASXL2 and ASXL3: A convergence of proteomics and epigenetics for translational medicine. Expert Rev. Proteomics 12 (3), 317–328. doi:10.1586/14789450.2015.1033409
Katoh, M., and Katoh, M. (2004). Identification and characterization of ASXL3 gene in silico. Int. J. Oncol. 24 (6), 1617–1622.
Lord, C., Brugha, T. S., Charman, T., Cusack, J., Dumas, G., Frazier, T., et al. (2020). Autism spectrum disorder. Nat. Rev. Dis. Prim. 6 (1), 5. doi:10.1038/s41572-019-0138-4
Lord C, M., Rutter, P. D., Labore, S., Risi, K., Gotham, S., and Bishop, S. L. (2012). Autism diagnostic observation schedule. second edition. Los Angeles, CA: Western Pschological Services. ADOS-2.
Miller, D. T., Adam, M. P., Aradhya, S., Biesecker, L. G., Brothman, A. R., Carter, N. P., et al. (2010). Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 86 (5), 749–764. doi:10.1016/j.ajhg.2010.04.006
Ni Ghralaigh, F., Gallagher, L., and Lopez, L. M. (2020). Autism spectrum disorder genomics: The progress and potential of genomic technologies. Genomics 112 (6), 5136–5142. doi:10.1016/j.ygeno.2020.09.022
Ni Ghralaigh, F., McCarthy, E., Murphy, D. N., Gallagher, L., and Lopez, L. M. (2022). Brief report: Evaluating the diagnostic yield of commercial gene panels in autism. J. Autism Dev. Disord [Epub ahead of print]. doi:10.1007/s10803-021-05417-7
Nudel, R., Appadurai, V., Buil, A., Nordentoft, M., and Werge, T. (2021). Pleiotropy between language impairment and broader behavioral disorders-an investigation of both common and rare genetic variants. J. Neurodev. Disord. 13 (1), 54. doi:10.1186/s11689-021-09403-z
O'Roak, B. J., Vives, L., Fu, W., Egertson, J. D., Stanaway, I. B., Phelps, I. G., et al. (2012). Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338 (6114), 1619–1622. doi:10.1126/science.1227764
Pickles, A., Anderson, D. K., and Lord, C. (2014). Heterogeneity and plasticity in the development of language: A 17-year follow-up of children referred early for possible autism. J. Child. Psychol. Psychiatry 55 (12), 1354–1362. doi:10.1111/jcpp.12269
Sanders, S. J., He, X., Willsey, A. J., Ercan-Sencicek, A. G., Samocha, K. E., Cicek, A. E., et al. (2015). Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87 (6), 1215–1233. doi:10.1016/j.neuron.2015.09.016
Satterstrom, F. K., Kosmicki, J. A., Wang, J., Breen, M. S., De Rubeis, S., An, J. Y., et al. (2020). Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180 (3), 568–584. doi:10.1016/j.cell.2019.12.036
Shaw, K. A., Maenner, M. J., Bakian, A. V., Bilder, D. A., Durkin, M. S., Furnier, S. M., et al. (2021). Early identification of autism spectrum disorder among children aged 4 Years - autism and developmental disabilities monitoring network, 11 sites, United States, 2018. MMWR Surveill. Summ. 70 (10), 1–14. doi:10.15585/mmwr.ss7010a1
Srivastava, S., Love-Nichols, J. A., Dies, K. A., Ledbetter, D. H., Martin, C. L., Chung, W. K., et al. (2019). Meta-analysis and multidisciplinary consensus statement: Exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet. Med. 21 (11), 2413–2421. doi:10.1038/s41436-019-0554-6
Stefanski, A., Calle-Lopez, Y., Leu, C., Perez-Palma, E., Pestana-Knight, E., and Lal, D. (2021). Clinical sequencing yield in epilepsy, autism spectrum disorder, and intellectual disability: A systematic review and meta-analysis. Epilepsia 62 (1), 143–151. doi:10.1111/epi.16755
Tammimies, K., Marshall, C. R., Walker, S., Kaur, G., Thiruvahindrapuram, B., Lionel, A. C., et al. (2015). Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA 314 (9), 895–903. doi:10.1001/jama.2015.10078
Vorstman, J. A. S., Parr, J. R., Moreno-De-Luca, D., Anney, R. J. L., Nurnberger, J. I., and Hallmayer, J. F. (2017). Autism genetics: Opportunities and challenges for clinical translation. Nat. Rev. Genet. 18 (6), 362–376. doi:10.1038/nrg.2017.4
Wang, T., Guo, H., Xiong, B., Stessman, H. A., Wu, H., Coe, B. P., et al. (2016). De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat. Commun. 7, 13316. doi:10.1038/ncomms13316
Wechsler, D. (2012). Wechsler Preschool and primary scale of intelligence. Fourth Edition. San Antonio, TX: The Psychological Corporation.
Werling, D. M. (2016). The role of sex-differential biology in risk for autism spectrum disorder. Biol. Sex. Differ. 7, 58. doi:10.1186/s13293-016-0112-8
Wickstrom, J., Farmer, C., Green Snyder, L., Mitz, A. R., Sanders, S. J., Bishop, S., et al. (2021). Patterns of delay in early gross motor and expressive language milestone attainment in probands with genetic conditions versus idiopathic ASD from SFARI registries. J. Child. Psychol. Psychiatry 62 (11), 1297–1307. doi:10.1111/jcpp.13492
Wright, C. F., Fitzgerald, T. W., Jones, W. D., Clayton, S., McRae, J. F., van Kogelenberg, M., et al. (2015). Genetic diagnosis of developmental disorders in the DDD study: A scalable analysis of genome-wide research data. Lancet 385 (9975), 1305–1314. doi:10.1016/S0140-6736(14)61705-0
Keywords: targeted sequencing, NGS, autism spectrum disorder, gene, ASXL3
Citation: Hu C, Wang Y, Li C, Mei L, Zhou B, Li D, Li H, Xu Q and Xu X (2023) Targeted sequencing and clinical strategies in children with autism spectrum disorder: A cohort study. Front. Genet. 14:1083779. doi: 10.3389/fgene.2023.1083779
Received: 29 October 2022; Accepted: 05 January 2023;
Published: 28 February 2023.
Edited by:
Maria Elisabetta Baldassarre, University of Bari Aldo Moro, ItalyReviewed by:
Weizhen Ji, Yale University, United StatesCopyright © 2023 Hu, Wang, Li, Mei, Zhou, Li, Li, Xu and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiu Xu, eHV4aXVAZnVkYW4uZWR1LmNu; Qiong Xu, eHVxaW9uZ0BmdWRhbi5lZHUuY24=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.