 Kala F. Schilter1†Brandon A. Smith1†Qian Nie1†
Kala F. Schilter1†Brandon A. Smith1†Qian Nie1† Kathryn Stoll1Juan C. Felix1,2
Kathryn Stoll1Juan C. Felix1,2 Jason A. Jarzembowski2
Jason A. Jarzembowski2 Honey V. Reddi1*
Honey V. Reddi1*- 1Precision Medicine Laboratory, Department of Pathology, Medical College of Wisconsin, Milwaukee, WI, United States
- 2Pathology and Laboratory Medicine, Medical College of Wisconsin, Milwaukee, WI, United States
Next-Generation Sequencing (NGS) allows rapid analysis of multiple genes for the detection of clinically actionable variants. This study reports the analytical validation of a targeted pan cancer NGS panel CANSeqTMKids for molecular profiling of childhood malignancies. Analytical validation included DNA and RNA extracted from de-identified clinical specimens including formalin fixed paraffin embedded (FFPE) tissue, bone marrow and whole blood as well as commercially available reference materials. The DNA component of the panel evaluates 130 genes for the detection of single nucleotide variants (SNVs), Insertion and Deletions (INDELs), and 91 genes for fusion variants associated with childhood malignancies. Conditions were optimized to use as low as 20% neoplastic content with 5 ng of nucleic acid input. Evaluation of the data determined greater than 99% accuracy, sensitivity, repeatability, and reproducibility. The limit of detection was established to be 5% allele fraction for SNVs and INDELs, 5 copies for gene amplifications and 1,100 reads for gene fusions. Assay efficiency was improved by automation of library preparation. In conclusion, the CANSeqTMKids allows for the comprehensive molecular profiling of childhood malignancies from different specimen sources with high quality and fast turnaround time.
1 Introduction
Childhood cancers including leukemias, and tumors of the central nervous system and renal tumors are the leading disease-related causes of death in children in the United States (Siegel et al., 2018). General treatment of childhood malignancies is a combination of surgery, cytotoxic chemotherapy, and radiotherapy, with long term side effects (Kopp et al., 2012). The discovery of more personalized and less harmful therapies is a rising need, however, childhood cancers currently represent less than 1% of new cancer diagnosis (Siegel et al., 2014). Evidence demonstrates that the frequency, distribution, and types of genetic alterations of childhood cancer may differ from adult tumors (Vogelstein et al., 2013), demanding the need for a better understanding of the molecular landscape of childhood malignancies.
Molecular profiling for childhood cancer usually comes into play after diagnosis or failure to respond to standard therapy. Profiling studies using next-generation sequencing (NGS) have facilitated widespread investigation of the molecular landscape of childhood cancers in the recent years leading to the identification of a large number of biomarkers across multiple childhood cancers with both small mutations and copy number variants (Grobner et al., 2018; Ma et al., 2018). Specifically, 17% of driver genes were mutated in both leukemias and solid tumors. CDKN2A, IKZF1, ETV6, RUNX1, and FLT3 were the top genes mutated in leukemias, while somatic alterations in ALK, NF1, and PTEN primarily occurred in solid tumors, suggesting that the driver alterations are either common to cancer (e.g., cell cycle) or specific to pediatric cancer histotype (Ma et al., 2018). Given the uniqueness of childhood cancers, it is important to have a molecular profiling assay that is comprehensive and applicable across most if not all childhood malignancies.
In this study, we report the analytical validation of the CANSeqTMKids assay which uses a targeted NGS panel that interrogates both DNA and RNA to provide comprehensive genomic information across 203 unique genes known to be associated with childhood malignancies. The assay was validated across multiple specimen types including fixed paraffin embedded (FFPE) tissue, cell blocks, blood, and bone marrow prior to clinical implementation for the evaluation of pediatric tumors.
2 Materials and methods
2.1 Panel content
CANSeqTMKids is a comprehensive molecular profiling assay that evaluates relevant DNA mutations (SNVs, indels and CNVs) across 130 key genes and RNA fusions across 91 fusion transcript driver genes associated with pediatric cancer, in a single NGS assay (Table 1).
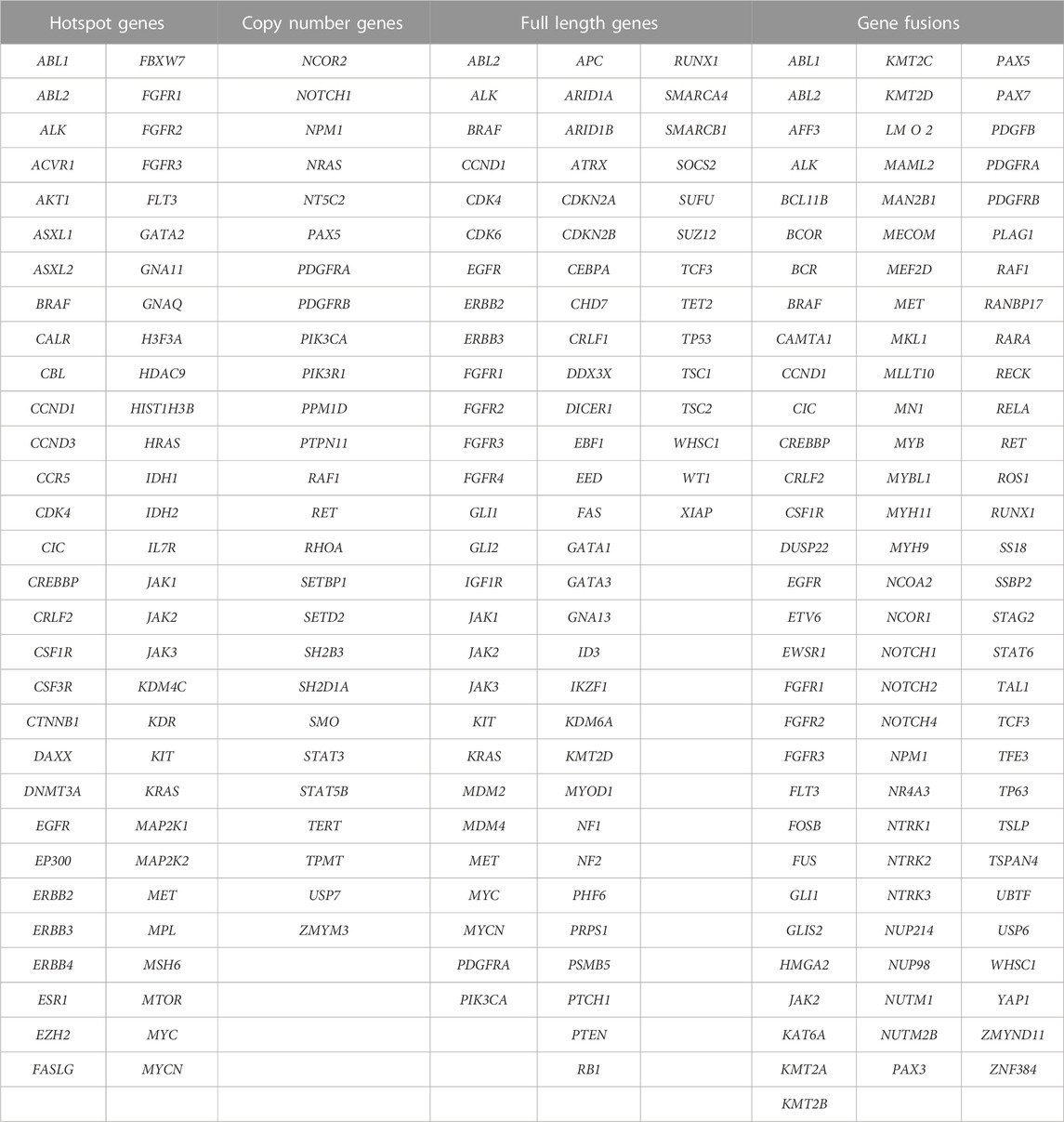
TABLE 1. Panel content (203 unique genes).
2.2 Sample cohort
A total of 65 samples including FFPE tissue (n = 32), cell blocks (n = 2), whole blood (n = 8), bone marrow (n = 4), cell lines (n = 7) and commercial controls (n = 12) were used in the validation (Table 2). The size of the sample cohort was established based on recommended guidelines (Jennings et al., 2017). This study was performed using retrospective specimens with known molecular profiling results, known diagnoses and represented different tumor types (Table 3). Specimens were de-identified per IRB guidelines prior to inclusion in the study. Due to the diverse nature of childhood cancers, the CANSeqTMKids panel has been designed to evaluate both solid tumors and hematological tissues. An analytical validation plan outlining sample cohort, validation strategy and processes involved, was reviewed and approved prior to study start. This study was approved by the Medical College of Wisconsin Institutional Review Board.
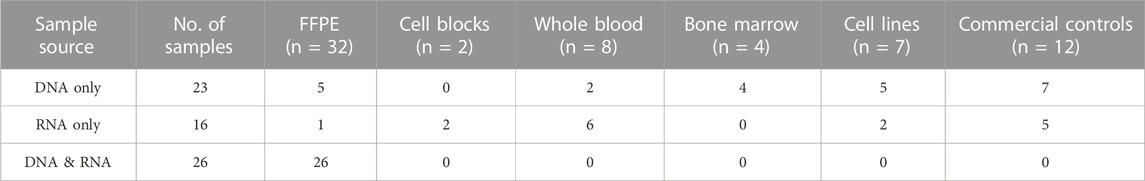
TABLE 2. Study Cohort. Summary of clinical specimens and commercial controls used in study (n = 65).
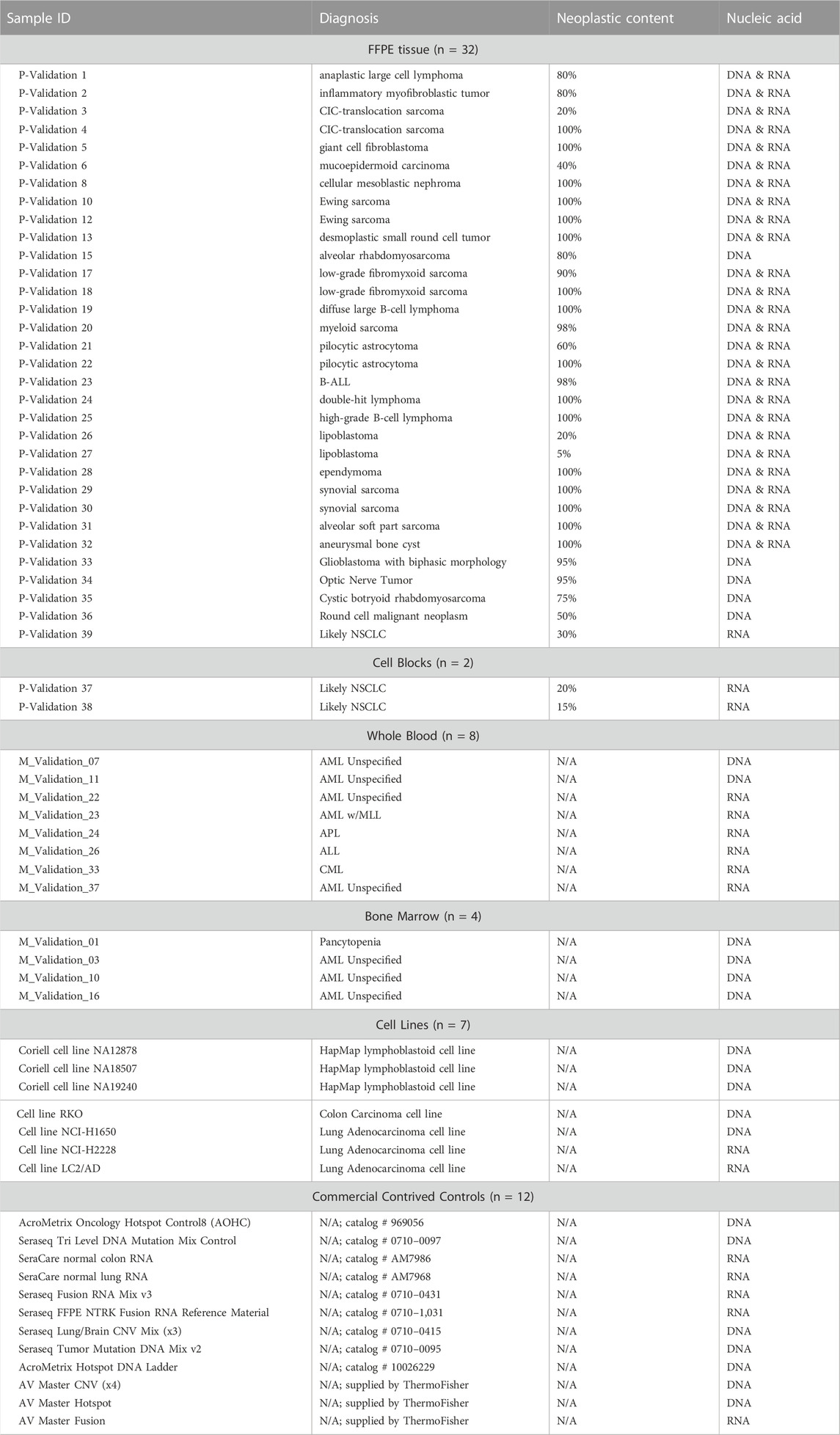
TABLE 3. Study Cohort. Details of specimens used in study.
2.3 DNA and RNA extraction
DNA and RNA from all specimens was extracted per established protocols. FFPE specimens were macro dissection-enriched prior to extraction. DNA quantification and quality was evaluated using the NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA) and considered acceptable if the resultant A260/A280 absorbance ratio was between 1.8 and 2.1. RNA quantification was evaluated using the Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA) and was considered acceptable if sufficient quantity of RNA to ensure a 10 ng input was obtained for downstream processing.
2.4 Library preparation, templating and sequencing
Libraries were prepared by both manual and automated Ion Chef process. For the DNA portion of library preparation, the manual library preparation requires 8 µL with a concentration of 2.5 ng/μL whereas the automated library preparation requires 15 µL with a concentration of 0.7 ng/μL. The RNA requirements are slightly less with 5 µL with a concentration of 2 ng/μL for manual prep and 10 µL with a concentration of 1 ng/μL for the automated process. The manual process followed the Oncomine™ Childhood Cancer Research Assay (OCCRA) (Thermo Scientific, Waltham, MA) and the Ion AmpliSeq™ Library Preparation user guide. The Automated library preparation used the Oncomine™ Childhood Cancer Research Assay, Chef-Ready kit on the Ion Chef (Thermo Fisher Scientific). Libraries were barcoded with IonCode™ Barcode Adapters 1–384 Kit and normalized to 100 pmol/L by the Equalizer kit (Thermo Scientific, Waltham, MA). DNA and RNA libraries were then combined and diluted at an 80:20 DNA:RNA ratio at ∼50p.m. and templated overnight on the Ion 540 chip using Ion 540™ Kit—Chef (Thermo Scientific, Waltham, MA).
Sequencing was performed using 540 chips on the Ion GeneStudio™ S5 Prime Sequencer (Thermo Fisher Scientific, Waltham, MA). Raw reads from sequencing were processed and aligned to the reference genome hg19 on Ion Torrent Suite Software versions 5.12 and 5.14 (Thermo Fisher Scientific, Waltham, MA) and the run metrics of the Ion Torrent Suite used to determine quality control of sequencing runs. The minimum cutoff of ISP (Ion Sphere™ Particle) loading was 80% and the maximum of polyclonal ISPs was 50%, with threshold for total reads at 60M. The minimum percent usable reads were set to be 30%, and the minimum raw accuracy was 99%.
Variant calling and fusion detection was performed on Ion Reporter™ versions 5.14 and 5.16 server system by the OCCRA - w2.5 - IR workflow. The quality control and variant calling analysis were performed on the Ion Reporter™ (IR) software package. Tertiary analysis and report generation was established using the GO Pathology Workbench (GenomOncology, Cleveland, OH).
2.5 Analytical validation
Analytical validation studies were carried out per guidelines from the Association for Molecular Pathology (AMP) and College of American Pathologists for the validation of Next-Generation Sequencing–Based Oncology Panels (Jennings et al., 2017). Details of the validation addressing STARD guidelines is presented in Supplementary Table S1.
2.5.1 Specificity
Three Coriell HapMap DNA samples NA12878, NA18507, NA19240 and two normal colon and lung RNA samples (SeraCare Life Sciences, Milford, MA) were used to determine assay specificity by evaluating positive and negative variant calls of SNV/MNV, INDELs across all targeted hotspots and fusions covered by the assay. The hotspot and fusion design files (Thermo Scientific, Waltham, MA) were used to extract variants from VCF outputs followed by manual variant review.
2.5.2 Sensitivity
Sensitivity was assessed using DNA and RNA from FFPE tissue, cell lines and contrived samples (Table 5). The true positive and false negative variants were determined by multiple commercial controls. Mean raw base calling accuracy was calculated for each of the samples with a target error rate <2%. The Coriell HapMap sample NA12878 is a well characterized benchmark sample for NGS validation studies. The AcroMetrix Oncology Hotspot Control (AOHC, Thermo Scientific, Waltham, MA) is a synthetic control consisting of 555 variants, with 198 covered by the OCCRA. The Seraseq Tri Level DNA Mutation Mix (SeraCare Life Sciences, Milford, MA) is a comprehensive synthetic control consisting of 40 mutations at target allele frequencies of 10, 7% and 4%, with 29 covered by the OCCRA. This control was sequenced 14 times during the validation to assess the assays’ ability of detecting variants at different allele frequencies. The Seraseq Fusion RNA Mix v4 (SeraCare Life Sciences, Milford, MA) is a reference standard containing a total of 16 fusions (14 gene fusions and 2 oncogenic isoforms). Fourteen of the 16 fusions are targeted by the OCCRA. The variant calling PPA (TP/(TP + FP) and PPV (TP/(TP + FN) was established for all variant types with IR default setting of ≥5% allele frequency (AF) for SNVs and INDELs, ≥4 copies for CNVs and ≥20 reads for fusion detection.
Limit of Detection (LOD) was determined for each variant type (SNV/MNV, INDELs, CNV and Fusions) using the contrived AOHC DNA Ladder, Seraseq Lung/Brain CNV and Seraseq RNA control titrated in a background of normal RNA (Placenta RNA Thermo Fisher). Limit of Input (LOI) was determined by diluting FFPE DNA and RNA in nuclease-free water. Nucleic acid concentration was measured using the Qubit™ dsDNA HS Assay Kit (Thermo Scientific, Waltham, MA) and Qubit™ RNA HS Assay Kit (Thermo Scientific, Waltham, MA) and two input concentrations (5ng and 1 ng) were used for downstream processing.
2.5.3 Precision (repeatability and reproducibility)
Inter-assay repeatability was evaluated using three independent DNA and RNA libraries prepped from FFPE tissue and sequenced in triplicate on the same day, chips, and system. Two of the RNA samples were pooled from two different samples to increase the number of fusions assessed. To evaluate for inter-assay reproducibility, libraries from FFPE tissue and contrived controls were prepared for DNA (n = 5) and RNA (n = 4) and sequenced 2–5 times on multiple days, chips, and systems.
3 Results
3.1 Established thresholds and quality metrics
The run metrics of the Ion Torrent Suite were used to determine quality control of sequencing runs which included base score, average sequencing depth, fusion panel control reads, minimum sequencing depth for variant calls, uniformity of coverage (ISP Loading), and strand bias of SNV and INDEL (Table 4; Figure 1). The thresholds of DNA mapped reads were 3M with mean depth ≥800x. The minimum mean read length was 75bp with uniformity ≥80% and mean raw accuracy ≥99%. The minimum RNA mapped reads was 20,000 with mean read length of 60bp.
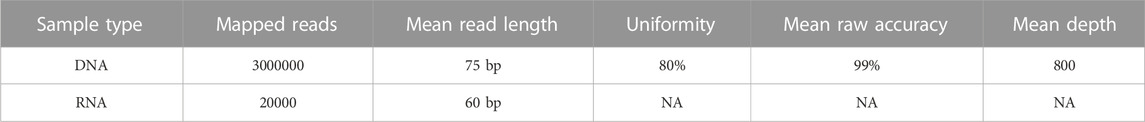
TABLE 4. Quality metrics and thresholds.
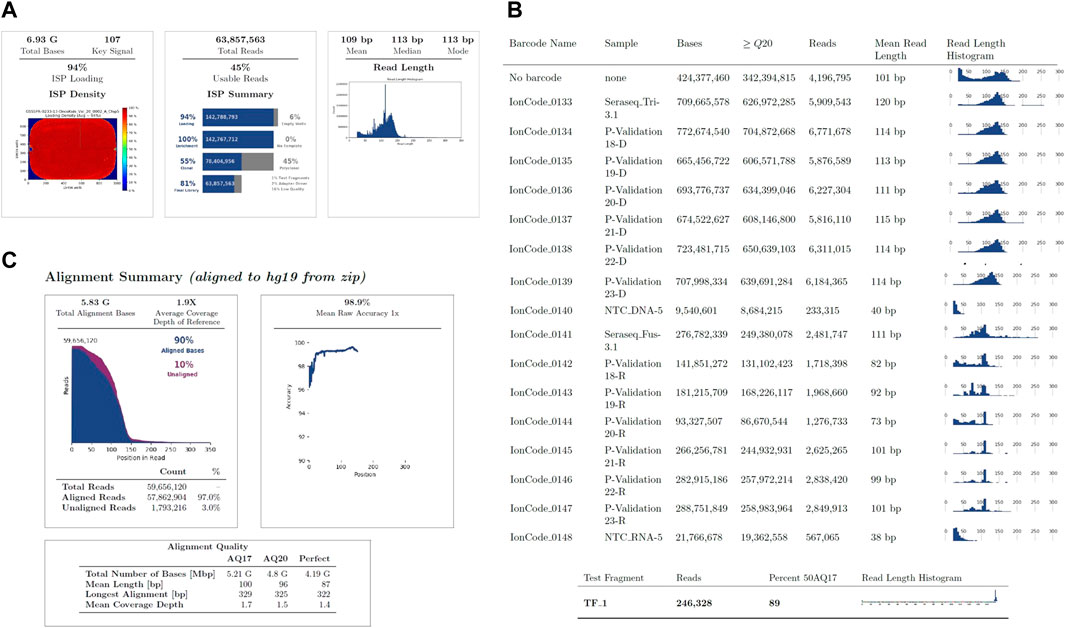
FIGURE 1. Run Summary Metrics obtained post sequencing. (A). Summary of metrics across the chip with loading density which is expected to be at ≥85% (left panel), total number of reads being ≥60M and usable reads being ≥35% (middle panel) and the average read length evaluated (right panel). (B). Run metrics for each sample on the chip. (C). Sequence alignment summary.
3.2 Analytical accuracy
Analytical accuracy was established using the reference Coriell cell line NA12878 with a mean raw accuracy of 99.8% (Table 5). The Seraseq Tri-Level mix control targets variants at different allelic frequencies (4%, 7% and 10%) establishing the limit of detection to be ≥5% allele frequency (AF) for SNVs and INDELs since variants in the 3%–5% allele frequency range are detectable but display variable reproducibility. The minimum AF for small deletions (6–15 nt) was 3.4% and for small insertions (3-4 nt) was 3.8%. and SNVs were detected at 3.5% AF (Table 6). CNVs were detected at about 4.86–6.64 copies, depending on the cancer type (Table 7). All 14 fusions of the Seraseq Fusion v3 Mix control covered by the OCCRA, were detected at 43 reads (Table 8), establishing the cutoff to be 45 for clinical implementation. Automation of library prep resulted in the fusion detection cut-off being increased to 1,100 fusion spanning reads reducing the sensitivity for fusion detection, no impact was observed on the detection of DNA variants. SNVs, INDELs and fusions were able to be detected with 1 ng DNA and RNA input respectively. Gene amplifications were only detected with 5 ng of DNA (Table 9). Results from the AOHC established a PPA of 97% and a PPV of 100% (Table 10), with the combined PPA and PPV of all variants type at 97.2% and >99% with a 95% CI of 93.3%–99%, respectively (Table 11).
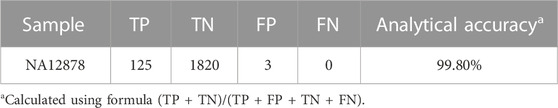
TABLE 5. Accuracy. Analytical Accuracy.
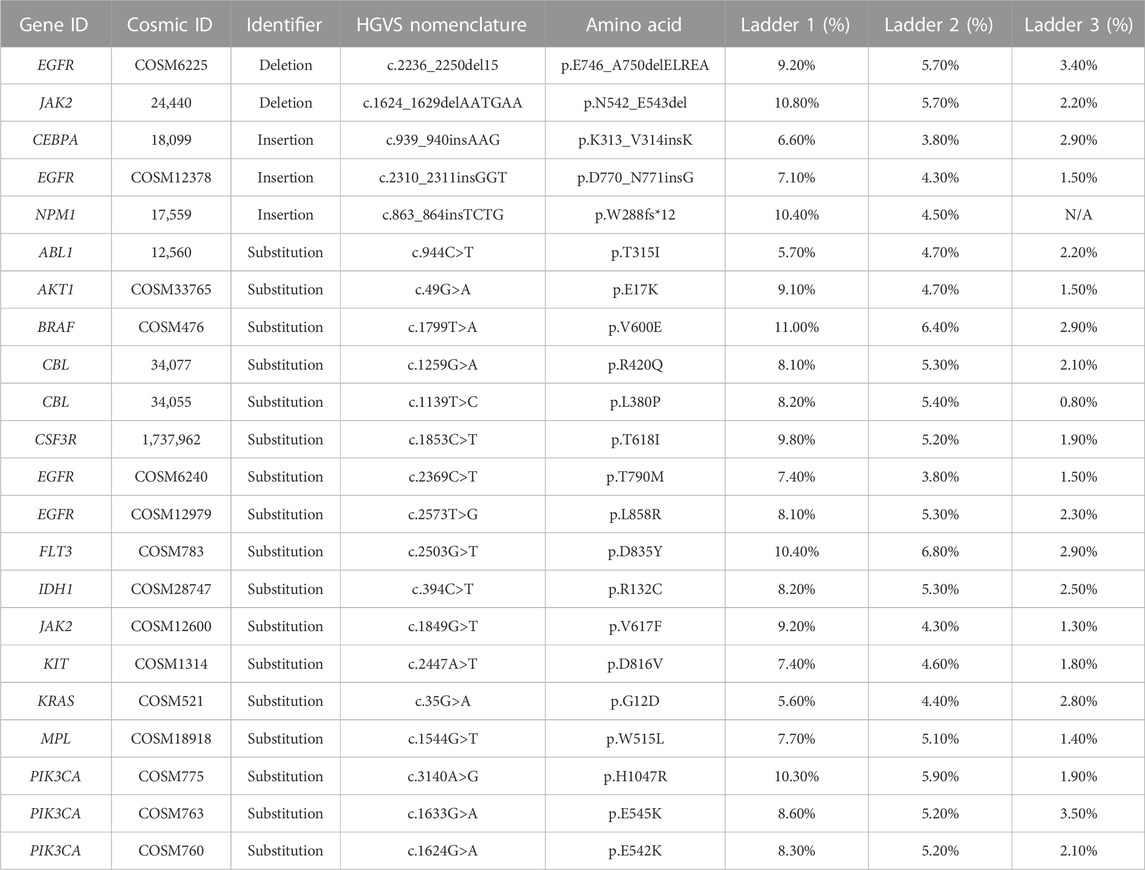
TABLE 6. Accuracy. The LOD of variant AF (SNVs and INDELs).
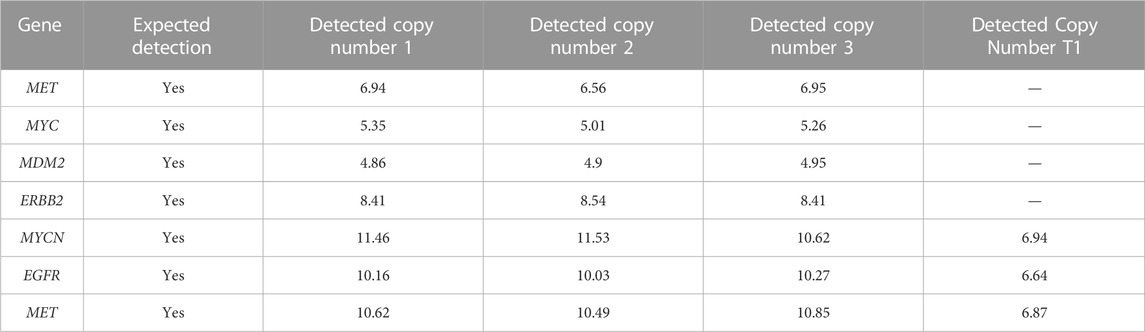
TABLE 7. Accuracy. The LOD of CNV detection.
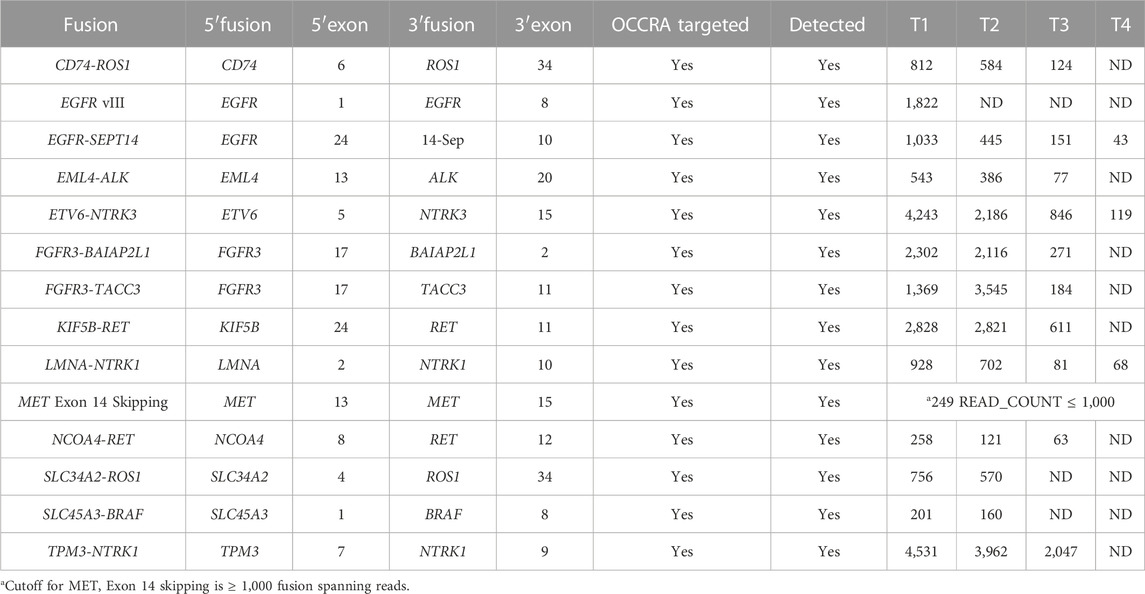
TABLE 8. Accuracy. The LOD of fusion detection.

TABLE 9. Accuracy. The LOI of DNA and RNA.
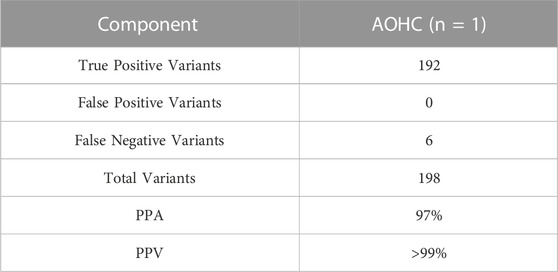
TABLE 10. Accuracy. PPA and PPV established by AcroMetrix Oncology Hotspot control.
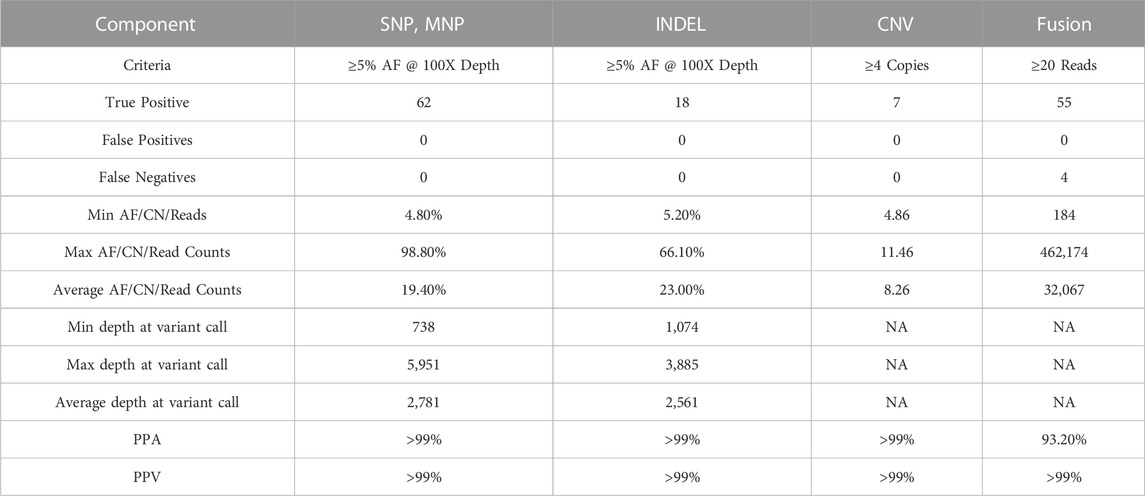
TABLE 11. Accuracy. Overall PPA and PPV of different variant types.
3.3 Specificity
A total of 3,640 negative variants were identified in both NA12878 and NA19240 samples with 772 INDELs and 2,869 SNVs/MNVs. Total 1820 negative variants were identified in NA18507 sample with 386 INDELs and 1,434 SNVs/MNVs. There were no positive variants detected across all hotspots, giving a specificity of ≥99% for all HapMap DNA samples (Table 12). Two normal colon and lung RNA samples were used to establish specificity of fusion detection of the assay. There was one false positive non-targeted fusion FHIT-TIRAP. F8T4 detected at 2,827 reads in one of the normal colon RNA replicates, resulting in the specificity greater than 99% in fusion detection (Table 13).
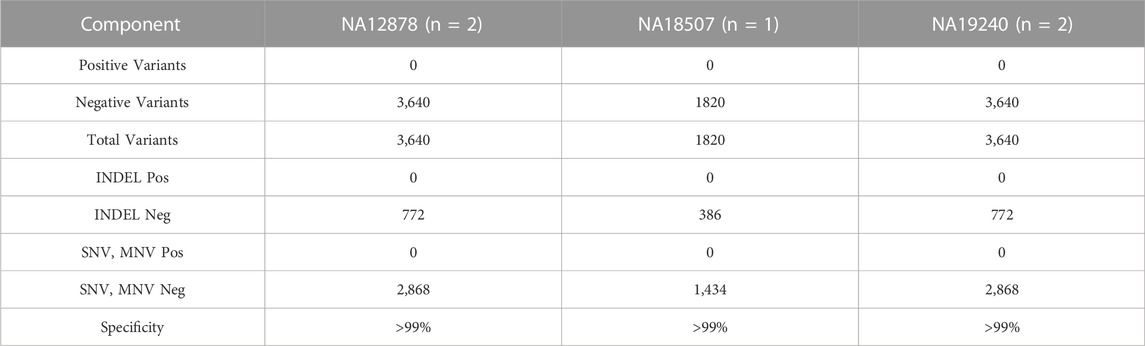
TABLE 12. Specificity. Analytical specificity of DNA samples.
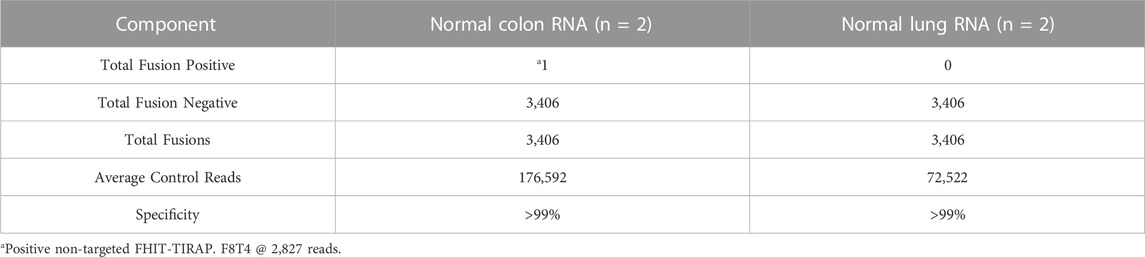
TABLE 13. Specificity. Analytical specificity of RNA samples.
3.4 Repeatability and reproducibility
A total of 39 true positive variants of SNV/MNV, INDEL, CNV and fusions were detected across the samples and all replicates, resulting in an overall combined variant repeatability of >99% (95% CI of 91.0%–100%) (Table 14). A total of 73 true positive variants of SNV/MNV, INDEL, CNV and fusions were detected in the combined samples and all replicates resulting in an overall combined variant reproducibility of >99% (Table 15).
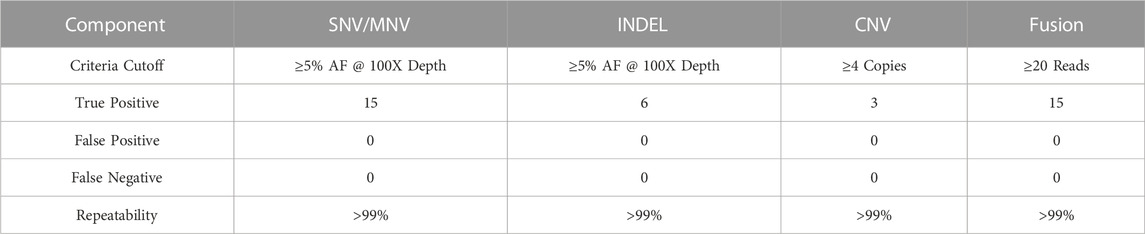
TABLE 14. Repeatability and Reproducibility. Intra-assay Repeatability.
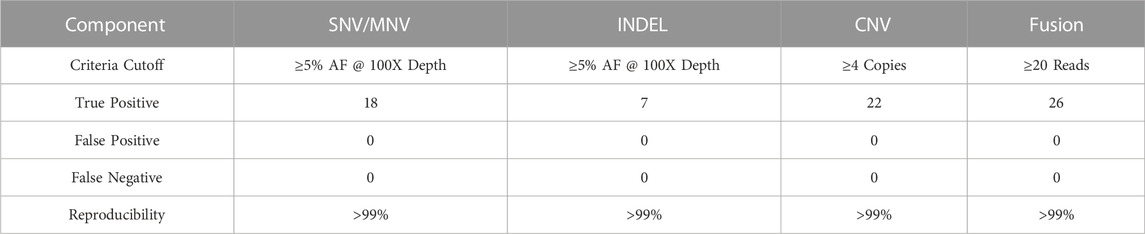
TABLE 15. Repeatability and Reproducibility. Inter-assay Reproducibility.
4 Discussion
The present study describes the analytical validation and implementation of a pan cancer NGS panel CANSeqTMKids for the detection of clinical actionable variants in childhood malignancies. Using a total of 65 samples, the study determined that the assay performed with greater than 99% accuracy, sensitivity, repeatability, and reproducibility, across different specimen types. Assay was optimized to use low input DNA (1–5 ng) and RNA (1 ng). Limit of detection of the assay was established to be ≥5% allele fraction for SNVs and INDELs, ≥4 copies for gene amplifications and 1,100 reads for gene fusions with automated library preparation. The study is presented in line with STARD (Standards for Reporting of Diagnostic Accuracy Studies) guidelines (Cohen et al., 2016), details are provided in Supplementary Table S1. The validated assay implemented for patient testing is listed on the National Institute of Health Genetic Test Registry (https://www.ncbi.nlm.nih.gov/gtr/labs/500088/), associated with the clinical test menu of the Precision Medicine Laboratory.
Targeted sequencing of a subset of genes is the most common test in clinical molecular diagnostic laboratories. However, given the various tumor types and molecular profiles of childhood malignancies, small gene panels that only covers genes of certain tumor type cannot satisfy the needs for appropriate disease management. The validation of the CANSeqTMKids included over 30 childhood tumor types/subtypes (Table 2) and includes comprehensive screening of 230 unique genes known to be associated with childhood malignancies across FFPE, whole blood and bone marrow specimens. The CANSeqTMKids evaluates both RNA and DNA for exonic hot spot regions of 86 genes, complete exonic regions of 44 genes, copy number of 28 genes and 91 fusion genes with variant types such as SNVs, INDELs, gene amplifications and gene fusions being detected. Overall, the assay covers a wide range of clinically actionable genes for a multitude of childhood tumor types and has greater than 99% accuracy, sensitivity, repeatability, and reproducibility with lower nucleic acid input amounts.
Data availability statement
The data presented in the study are deposited in the https://submit.ncbi.nlm.nih.gov/subs/sra/SUB12695707/overview repository, accession number SUB12695707.
Ethics statement
The studies involving human participants were reviewed and approved by Institutional Review Board, Medical College of Wisconsin.
Author contributions
HR conceived the study and obtained IRB approval. KS, BS, and QN conducted the study under oversight of HR. JF was the pathologist on the study. JJ provided de-identified clinical specimens for the study. All authors reviewed, commented, and edited later drafts of the manuscript, and approved the final version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1067457/full#supplementary-material
References
Cohen, J. F., Korevaar, D. A., Altman, D. G., Bruns, D. E., Gatsonis, C. A., Hooft, L., et al. (2016). STARD 2015 guidelines for reporting diagnostic accuracy studies: Explanation and elaboration. BMJ Open 6 (11), e012799. Cited in Pubmed; PMID 28137831. doi:10.1136/bmjopen-2016-012799
Grobner, S. N., Worst, B. C., Weischenfeldt, J., Buchhalter, I., Kleinheinz, K., Rudneva, V. A., et al. (2018). The landscape of genomic alterations across childhood cancers. Nature 555 (7696), 321–327. Epub 2018/03/01. doi:10.1038/nature25480
Jennings, L. J., Arcila, M. E., Corless, C., Kamel-Reid, S., Lubin, I. M., Pfeifer, J., et al. (2017). Guidelines for validation of next-generation sequencing–based Oncology panels: A joint consensus recommendation of the association for molecular Pathology and College of American pathologists. J. Mol. Diagn 19 (3), 341–365. Cited in Pubmed; PMID 28341590. doi:10.1016/j.jmoldx.2017.01.011
Kopp, L. M., Gupta, P., Pelayo-Katsanis, L., Wittman, B., and Katsanis, E. (2012). Late effects in adult survivors of pediatric cancer: A guide for the primary care physician. Am. J. Med. 125 (7), 636–641. Epub 2012/05/09. doi:10.1016/j.amjmed.2012.01.013
Ma, X., Liu, Y., Liu, Y., Alexandrov, L. B., Edmonson, M. N., Gawad, C., et al. (2018). Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 555 (7696), 371–376. Epub 2018/03/01. doi:10.1038/nature25795
Siegel, R., Ma, J., Zou, Z., and Jemal, A. (2014). Cancer statistics. CA Cancer J. Clin. 64 (1), 9–29. Epub 2014/01/09. doi:10.3322/caac.21208
Siegel, R. L., Miller, K. D., and Jemal, A. (2018). Cancer statistics. CA Cancer J. Clin. 68 (1), 7–30. Epub 2018/01/10. doi:10.3322/caac.21442
Keywords: pan cancer assay, childhood cancer, next generation sequencing panel, molecular profiling, clinical implementation, assay validation
Citation: Schilter KF, Smith BA, Nie Q, Stoll K, Felix JC, Jarzembowski JA and Reddi HV (2023) Analytical validation and implementation of a pan cancer next-generation sequencing panel, CANSeqTMKids for molecular profiling of childhood malignancies. Front. Genet. 14:1067457. doi: 10.3389/fgene.2023.1067457
Received: 11 October 2022; Accepted: 20 January 2023;
Published: 09 February 2023.
Edited by:
Jiannis (Ioannis) Ragoussis, McGill University, CanadaReviewed by:
Parul Singh, Immunology Center, United StatesAmmar Husami, Cincinnati Children’s Hospital Medical Center, United States
Copyright © 2023 Schilter, Smith, Nie, Stoll, Felix, Jarzembowski and Reddi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Honey V. Reddi, aHJlZGRpQG1jdy5lZHU=
†These authors have contributed equally to this work and share first authorship