
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 08 August 2023
Sec. Epigenomics and Epigenetics
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1037406
This article is part of the Research Topic DNA & RNA Methylation: Impact on Cancer Progression View all 5 articles
 Melva Gutierrez-Angulo1,2*
Melva Gutierrez-Angulo1,2* Maria de la Luz Ayala-Madrigal2
Maria de la Luz Ayala-Madrigal2 Jose Miguel Moreno-Ortiz2
Jose Miguel Moreno-Ortiz2 Jorge Peregrina-Sandoval3
Jorge Peregrina-Sandoval3 Fernando Daniel Garcia-Ayala2
Fernando Daniel Garcia-Ayala2Colorectal cancer is a complex disease resulting from the interaction of genetics, epigenetics, and environmental factors. DNA methylation is frequently found in tumor suppressor genes to promote cancer development. Several factors are associated with changes in the DNA methylation pattern, and recently, the gastrointestinal microbiota could be associated with this epigenetic change. The predominant phyla in gut microbiota are Firmicutes and Bacteroidetes; however, an enrichment of Bacteroides fragilis, Fusobacterium nucleatum, and Streptococcus bovis, among others, has been reported in colorectal cancer, although the composition could be influenced by several factors, including diet, age, sex, and cancer stage. Fusobacterium nucleatum, a gram-negative anaerobic bacillus, is mainly associated with colorectal cancer patients positive for the CpG island methylator phenotype, although hypermethylation in genes such as MLH1, CDKN2A, MTSS1, RBM38, PKD1, PTPRT, and EYA4 has also been described. Moreover, Hungatella hathewayi, a gram-positive, rod-shaped bacterium, is related to hypermethylation in SOX11, THBD, SFRP2, GATA5, ESR1, EYA4, CDX2, and APC genes. The underlying epigenetic mechanism is unclear, although it could be implicated in the regulation of DNA methyltransferases, enzymes that catalyze the transfer of a methyl group on cytosine of CpG sites. Since DNA methylation is a reversible event, changes in gut microbiota could modulate the gene expression through DNA methylation and improve the colorectal cancer prognosis.
Colorectal cancer (CRC) is a complex disease caused by interactions among genetic, epigenetic, and environmental factors. DNA methylation is frequently observed in tumor suppressor genes that promote cancer development. Several factors are associated with changes in DNA methylation patterns. Recently, the gastrointestinal microbiota have been associated with this epigenetic change. The predominant phyla in the gut microbiota are Firmicutes and Bacteroidetes. However, an enrichment of Bacteroides fragilis, Fusobacterium nucleatum, and Streptococcus bovis, among others, has been reported in CRC. Nevertheless, diet, age, sex, and cancer stage could influence the composition. Fusobaterium nucleatum, a gram-negative anaerobic bacillus, is mainly associated with CRC patients positive for the CpG island methylator phenotype. Notwithstanding, hypermethylation in genes such as MLH1, CDKN2A, MTSS1, RBM38, PKD1, PTPRT, and EYA4 has also been described. Moreover, Hungatella hathewayi, a Gram-positive, rod-shaped bacterium, is related to hypermethylation in SOX11, THBD, SFRP2, GATA5, ESR1, EYA4, CDX2, and APC genes. The underlying epigenetic mechanism is unclear, although it could be implicated in regulation of DNA methyltransferases (DNMTs) , enzymes that catalyze the transfer of a methyl group on the cytosine of CpG sites. Since DNA methylation is a reversible event, changes in gut microbiota can modulate gene expression through DNA methylation and improve CRC prognosis.
CRC is the second leading cause of death and ranks fourth in incidence (Ferlay et al., 2020). CRC is a multi-step process characterized by the sequential accumulation of genetic and epigenetic changes that transform the normal epithelium into metastatic carcinoma (Li et al., 2021). Additionally, environmental factors have been found to be associated with this transformation, including consuming red and processed meat, alcohol, and tobacco; lack of physical activity; and microbiome composition (Katsaounou et al., 2022). Based on the morphological changes in colorectal tissue, CRC is divided into two categories: classical or adenoma–carcinoma sequences and alternative or serrated. In the adenoma–carcinoma sequence, the chromosomal (CIN) and microsatellite instability (MSI) has been associated with transforming normal tissue into adenocarcinoma tissue. The CIN pathway is found in 65%–70% of sporadic CRC cases and is characterized by mutations in oncogenes (KRAS) and tumor suppressor genes (APC and TP53), in addition to chromosomal aberrations (18q deletion). Furthermore, MSI is caused by defects in MLH1, MSH2, MSH6, and PMS2, which encode for mismatch DNA repair (MMR) proteins, and the MSI pathway is found in 15% of patients with sporadic CRC (Nguyen et al., 2020). In the alternative category, the serrated or sawtooth lesions include polyps, sessile, and traditional adenomas found in approximately 15% of CRC cases. The serrated pathway promotes these lesions, and the associated molecular changes include BRAF or KRAS mutations, MSI, microsatellite stability, and hypomethylation or hypermethylation in MLH1 (De Palma et al., 2019; Nguyen et al., 2020). The CpG methylator island phenotype (CIMP) has been described in both categories (De Palma et al., 2019; Nguyen et al., 2020; Huang and Yang, 2022). This pathway is characterized by promoter hypermethylation of tumor suppressor genes. CIMP-positive tumors are identified by evaluating a panel comprising up to 16 genes (Jung et al., 2020). Frequently methylated markers used were CACNA1G, IGF2, NEUROG1, RUNX3, SOCS1, CRABP1, MINT1, MINT2, and MINT31 (Jia et al., 2016). The CIMP pathway is associated with MSI through MLH1 hypermethylation, and a high frequency is observed in serrated tumors and the proximal colon of older patients (Harada and Morlote, 2020).
The term “microbiota” refers to the composition and abundance of microorganisms. In contrast, the term “microbiome” is used to describe the whole habitat, including microorganisms, their genome, and environment, or is defined as a collection of genes and genomes of the microbiota (Marchesi and Ravel, 2015). Microbiota analysis has limitations owing to the difficulty in culturing microorganisms; however, the development of high-throughput sequencing of the 16S rRNA gene and shotgun metagenomics facilitates the identification of uncultured members of the gut microbiota (Milani et al., 2017; Kumar et al., 2023). The human gastrointestinal system contains the highest density of microbiota (1011–1012 per milliliter (Rinninella et al., 2019). The Unified Human Gastrointestinal Genome (UHGG) v2.0 estimates a total of 4,744 prokaryotic species (4,716 bacteria and 28 archaea) in the gut microbiome (Mitchell et al., 2020). Although everyone contains a unique gut microbiota, the predominant phyla are Firmicutes and Bacteroidetes, accounting for 90% of the total microbiota. Clostridium is the most frequent genus of Firmicutes, while Bacteroides and Prevotella are the most frequent genera of Bacteroidetes (Rinninella et al., 2019). Nevertheless, a large diversity of genera, such as Pseudomonas, Streptococcus, Fusobacterium, Veillonella, Haemophilus, Neisseria, Porphyromonas, Collinsella, Faecalibacterium, Eubacterium, Ruminococcus, Peptococcus, Peptostreptococcus, Lactobacillus, Streptomyces, and Bifidobacterium, has been reported in the gut microbiota (Sheng et al., 2019; Peery et al., 2021). Aruguman et al. (2011) analyzed 33 samples from European individuals using multidimensional cluster and principal component analyses. They identified different enterotypes based on the abundance of three principal genera: enterotype 1 (Bacteroides), enterotype 2 (Prevotella), and enterotype 3 (Ruminococcus). The function of Bacteroides and co-occurring Parabacteroides is protein or carbohydrate fermentation. In contrast, the function of Prevotella and co-occurring Desulfovibrio similar to that of Ruminococcus and co-occurring Akkermansia is mucin degradation. Almeida et al. (2021) compiled 204,938 genomes and 170,602,708 genes from the human gut microbiome to generate a UHGG catalog from prokaryote-isolated genomes and metagenome-assembled genomes (MAGs). The last method is used to infer a new genome from de novo-assembled contigs. They reported that the most representative bacterial species were Agathobacter rectalis, Escherichia coli D, Bacteroides uniformis, and the archaeal species, Methanobrevibacter smithii. Although a MAG was used, only two of the 25 most abundant bacteria were represented by the MAG. The microbiome is dynamic and influenced by age, anatomical region, diet, antibiotics, genetics, and others (Gomaa, 2020; Ruan et al., 2020). Different microbiome compositions through life stages have been reported and associated with diet diversity and inflammatory processes. Older adults (>65 years) have shown an abundance of Enterobacteriaceae compared to adults with mainly Bacteroidetes and Firmicutes. Moreover, a study of DNA of fecal microbiota of 69 samples (24 from individuals aged 105–109 years, 15 from individuals aged 99 to 104 years, 15 from individuals aged 65 to 75 years, and 15 from individuals aged 22 to 48 years) sequenced by Illumina revealed that the oldest individuals (105–109 years old) had enrichment of Akkermansia, Bifidobacterium, and Christensenellaceae bacteria (Biagi et al., 2016; Gomaa, 2020). Regarding the anatomical location, a transcriptome analysis of 20 samples collected from healthy people reported 20%–70% of similarity in samples from the upper gastrointestinal tract, including saliva, with predominant genera Gemella, Veillonella, Neisseria, Fusobacterium, Streptococcus, Prevotella, Pseudomonas, and Actinomyces, while 20%–90% of similarity was reported for the lower gastrointestinal tract with predominant genera Faecalibacterium, Ruminococcus, and Bacteroides (Vasapolli et al., 2019).
Pan et al. (2018) conducted a genome-wide analysis of intestinal epithelial cells in the small intestine of mice at different developmental stages. They found changes in microbiota-dependent methylation patterns early after birth, and the differentially methylated positions increased according to mouse development with 1,492 in 1-week-old mice, while in 4-week and 12/16-week were 132 and 217, respectively. Moreover, they found that the expression of Dnmt3a and Tet3, which are involved in DNA methylation and demethylation, respectively, was altered in 1- and 12/16-week-old mice. Analysis of differentially methylated genes revealed enrichment of genes associated with cellular proliferation, regeneration, and immune responses. The normal function of the gut microbiota is implicated in the metabolism of nutrients, xenobiotics, and drugs; protection against pathogens; structural support of the gastrointestinal tract; and immunomodulation (Jandhyala et al., 2015; Gomaa, 2020; Rebersek, 2021).
Controversial results regarding microbiota composition have been reported in patients with CRC. Higher abundances of Butyrivibrio, Gemella, Akkermansia, Fusobacterium nucleatum, H. hathewayi, Parvimonas spp., Desulfovibrio, Streptococcus bovis, Bacteroides fragilis, and Bilophila wadsworthia have been described in CRC (Dahmus et al., 2018; Sobhani et al., 2019; Wang, et al., 2020; Xia et al., 2020). Moreover, lower proportions of Ruminococcus, Bifidobacterium, Eubacteria, and Lachnospira have been reported (Sobhani et al., 2019). Different microbiota compositions have been described depending on the anatomical segment or cancer stage. The study conducted by Suga et al. (2022) found a significant reduction in Clostridial cluster XIVa and Clostridial cluster IX in sigmoid and right-sided colon cancers, respectively, as determined by the terminal restriction fragment length polymorphism analysis. Moreover, 16S rRNA gene sequencing revealed that Firmicutes were significantly dominant in right-sided colon cancer and Verrucomicrobia in sigmoid colon cancer. In proximal and distal segments, Veillonella and Coprobacter were more abundant in distal segments (Sheng et al., 2019). In CRC staging, Alistipes were abundant in patients with stage III compared with stage IV CRC (Sheng et al., 2019). Moreover, Bacteroides fragilis is associated with 3-year survival, whereas high levels of F. nucleatum are associated with poor survival in patients with metastatic CRC (Dahmus et al., 2018; Lee et al., 2018). An analysis of 118 patients with CRC, 140 with adenomas, and 128 healthy participants revealed enrichment of Peptostreptococcus stomatis, Fusobacterium nucleatum, Parvimonas micra, Peptostreptococcus anaerobius, and Bacteroides fragilis and depletion of Coprobacter fastidiosus, Eubacterium ventriosum, Roseburia intestinalis, and Roseburia inulinivorans in CRC compared with other groups. Moreover, the quantification of 97 metabolites showed an increase in 16 metabolites (L-alanine, glycine, L-proline, L-aspartic acid, L-valine, L-leucine, L-serine, myristic acid, phenyl lactic acid, oxoglutaric acid, L-phenylalanine, L-alpha-aminobutyric acid, phenylacetic acid, palmitoleic acid, 3-aminoisobutanoic acid, and norvaline), and one metabolite (butyric acid) was depleted in patients with CRC compared to healthy individuals (Coker et al., 2022).
Inflammation, production of mutagenic biomolecules, and, recently, a dysregulation of epigenetic mechanisms have been described as possible mechanisms of microbiota-induced colorectal carcinogenesis (Dahmus et al., 2018; Peery et al., 2021).
Epigenetics regulates gene expression without modifications in the nucleotide sequence composition, which can be inherited and reversible. Epigenetic mechanisms include DNA methylation, histone modification, and regulation by non-coding RNA (Portela and Esteller, 2010). DNA methylation is the most studied mechanism and is essential for tissue- or cell-specific gene expression regulation, silencing of retroviral elements, embryogenesis, genomic imprinting, and X chromosome inactivation (Portela and Esteller, 2010; Moore et al., 2013; Edwards et al., 2017). DNA methylation is a chemical modification in the fifth carbon of cytosine in CpG dinucleotides catalyzed by DNMTs and S-adenosylmethionine (SAM) as a cofactor. Three DNMTs are involved in DNA methylation: DNMT1 maintains the methylated state immediately after the DNA strand is replicated, and DNMT3A and DNMT3B promote de novo methylation, mainly during embryogenesis. Methylation is a reversible process controlled by DNMTs and demethylases. The demethylation process can be passive due to the lack of methyl addition during replication or active by TET proteins. In contrast, the last mechanism produces the intermediate metabolites 5-hydroxymethyl cytosine, 5-formylcytosine, and 5-carboxylcytosine, which are removed by base-excision repair (Dai et al., 2021). The methyl donor SAM, which is required for methylation reactions, is involved in folate metabolism. Folate metabolism is a multi-step process that produces 10-formyl tetrahydrofolate (THF), which is involved in purine synthesis, and 5–10 methylene THFs are required for thymidylate synthesis. Moreover, in one-carbon metabolism is generated 5-methylTHF, a metabolite that acts as a methyl donor for the conversion of homocysteine to methionine, which is subsequently metabolized to SAM, a cofactor of DNMT in the DNA methylation process (Liu and Ward, 2010).
The possible role of the microbiota in DNA methylation has been studied mainly in CIMP-positive tumors and F. nucleatum. CIMP is related to the hypermethylation of CpG islands found primarily in the promoters of suppressor genes, and a panel described previously was used for pathway identification. The CIMP status can be categorized into two groups: CIMP-positive or CIMP-negative, or into three groups: CIMP-high, CIMP-low, and CIMP-negative, depending on the number of methylated markers found in the colorectal tumor tissue (Jia et al., 2016). However, the microbiome and its association with CIMP have shown discordant results (Tahara et al., 2014; Ito et al., 2015; Mima et al., 2015; Park et al., 2017; Lee et al., 2018; Li J. et al., 2019). Tahara et al. (2014) identified high numbers of F. nucleatum and pan-Fusobacterium associated with CIMP positivity, and the phenotype was analyzed using seven markers (ER, SFRP1, MYOD1, MGMT, SLC16A2, SPOCK2, and N33) in CRC tissues. Moreover, concordant results have been reported by other authors who employed different markers for CIMP identification (Ito et al., 2015; Mima et al., 2015; Lee et al., 2018; Li J. et al., 2019; Ono et al., 2022), but no significant association was reported by Park et al., 2017. They analyzed eight markers for CIMP diagnosis (MLH1, NEUROG1, CRABP1, CACNA1G, CDKN2A, IGF2, SOCS1, and RUNX3), and only high F. nucleatum was significantly associated with CDKN2A methylation in CRC tissues with high MSI. Additionally, MLH1 hypermethylation is associated with an abundance of F. nucleatum (Tahara et al., 2014; Ito et al., 2015; Mima et al., 2015). Mima et al. (2015) found no significance between LINE-1 (long interspersed nucleotide element-1) methylation and F. nucleatum. Moreover, a similar distribution of LINE-1 methylation levels and F. nucleatum was observed in negative and positive tumors for MSI-high (Hamada et al., 2018). Other species, such as B. fragilis, Faecalibacterium prausnitzii, and E. coli, have been analyzed, but no relationship with CIMP has been described (Li J. et al., 2019).
Few studies have explored the influence of the microbiota on gene-specific methylation. Sobhani et al. (2019) realized transferred human fecal samples from normal and CRC donors to germ-free mice, promoting aberrant crypt foci, microbiota dysbiosis, and DNA alterations in the murine colonic mucosa. In tissues, they reported hypermethylation in SFRP1,2,3, PENK, NPY, ALX4, SEPT9, and WIF1 genes. However, only three genes (WIF1, PENK, and NPY) were selected for validation in the tissue, serum, and stool of patients with CRC, and hypermethylation in the blood was associated with microbiota. Moreover, hypermethylation in the SFRP2 gene in tissues and blood has been associated with Bilophila. The protein encoded by SFRP2 is a WNT pathway modulator that directly interacts with WNT ligands. Furthermore, the cumulative methyl index (CMI) was measured, and they found an abundance of P. micra in patients with a higher CMI in the blood (Sobhani et al., 2019). Xia et al. (2020) analyzed colonic mucosa, adenoma, and CRC tissues. They found promoter-wide methylation in MTSS1, RBM38, PKD1, and PTPRT related to F. nucleatum and SOX11, THBD, SFRP2, GATA5, and ESR1 related to H. hathewayi, whereas EYA4 was associated with both bacteria. Additionally, they analyzed MLH1, APC, PTEN, P16 (CDKN1A), and CDX1/2, which are known driver genes of CRC, and found a correlation between CpG site methylation of CDX2 and MLH1 in H. hathewayi and Streptococcus spp., respectively. Contrastingly, hypermethylation in APC was associated with both bacteria. Additionally, global DNA methylation (5-mC) was evaluated in the colonic cell lines NCM460, HCT116, and HT29 incubated with F. nucleatum and H. hathewayi, and a significant increase in global DNA methylation was reported in all of them (Xia et al., 2020).
Hypermethylated genes associated with F. nucleatum are MLH1, CDKN2A, MTSS1, RBM38, PKD1, PTPRT, and EYA4. MLH1 is a tumor suppressor gene implicated in MMR and is frequently inactivated in CRC. The primary loss-of-function mechanism occurs through mutations, mainly in inherited CRC (Lynch syndrome); however, in sporadic CRC, approximately 19% of cases show hypermethylation of its promoter (Li et al., 2013). CDKN2A has shown a methylation frequency of 15% in CRC patients. The protein encoded by this gene is a cyclin-dependent kinase inhibitor; therefore, its hypermethylation increases cell proliferation (Bagci et al., 2016). MTSS1 is a metastasis suppressor gene that is frequently hypermethylated in leukemia and prostate, gastric, and bladder cancers, excluding colorectal cancer, where there are no reports (Utikal et al., 2006; Yamashita et al., 2006; Xie et al., 2011; Chen et al., 2020; Grandits et al., 2021). Yu et al. (2010) analyzed 105 primary colon tumors and found hypermethylation in TBX5 in 68% of cases. The transcription factor encoded by this gene upregulates to MTSS1; therefore, a TBX5 hypermethylation also contributes to the inactivation of MTSS1 (Yu et al., 2010). Hypermethylation induced by F. nucleatum could only be one of the mechanisms related to the inactivation of MTSS1 since another process could be implicated. RBM38 encodes for the mRNA 3′-untranslated region-binding protein to stabilize the transcripts, and hypermethylation has only been associated significantly in breast cancer with the TP53 wild type (Léveillé et al., 2011). However, overexpression of RBM38 has been found in several cancers, including CRC (Tate et al., 2019). The protein encoded by PKD1 belongs to the PKD family, which comprises PKD1, PKD2, and PKD3 members that regulate essential processes involved in the initiation and progression of cancer (Azoitei et al., 2018). Wei et al. (2014) analyzed expression in colon cancer tissues and found increased levels of PKD2 and a lower proportion of PKD3. However, PKD1 was not detected in either mRNA or protein but only in normal colon cells. They concluded that the loss of expression could result from epigenetic mechanisms, although they could not prove this. The PTPRT gene codes for phosphatase and is involved in cancer progression and hypermethylation in colorectal, lung, and head and neck cancers (Laczmanska et al., 2013; Peyser et al., 2016; Sen et al., 2020). Methylation analysis of The Cancer Genome Atlas data revealed that colon adenocarcinoma, head and neck squamous cell carcinoma, lung adenocarcinoma, and invasive breast carcinoma had PTPRT hypermethylation that was significantly correlated with the downregulation of mRNA expression, with CRC being the most frequently hypermethylated (Peyser et al., 2016). Finally, EYA4 encodes for a phosphatase protein that functions in the DNA repair process, and the analysis has shown hypermethylation in CRC cell lines and approximately 90% of CRC patients (Kim et al., 2015; McInnes et al., 2017; Azuara et al., 2018). An analysis of epigenome-wide methylome of open chromatin in 12 CRC tissues revealed 2,187 differentially methylated regions, of which 66% located in 1,025 genes, with EY4 being the most significant (Ishak et al., 2020).
The main hypermethylated genes associated with H. hathewayi were SOX11, THBD, SFRP2, APC, GATA5, CDX2, ESR1 and EYA4. SOX11 encodes a transcription factor, and its hypermethylation has been found in various malignancies (excluding CRC) and in the inflammatory rectal mucosa of ulcerative patients (Pugongchai et al., 2017; Tahara et al., 2017; Li X. et al., 2019; Shan et al., 2019). THBD encodes a receptor with an affinity for thrombin, and its hypermethylation is significant in gastric cancer-positive for Helicobacter pylori and CRC (Shin et al., 2010; Lange et al., 2012). Moreover, hypermethylation in ESR1, CDX2, GATA5, and APC has been reported in CRC patients (Dawson et al., 2014; Sahnane et al., 2015; Liang et al., 2017; Liu et al., 2017; Zhu et al., 2021). ESR1 codes for estrogen receptor alpha, a transcription factor that regulates genes involved in differentiation and cell proliferation. However, the predominant form in colon tissue is estrogen receptor beta; therefore, its implication in CRC is unclear (Das et al., 2023). CDX2 encodes a transcription factor that regulates genes in the intestinal epithelium (UniProt Consortium, 2021), and gene hypermethylation is associated with BRAF mutations and CIMP-high (Dawson et al., 2014). The protein produced by the GATA5 gene is a transcription factor required for cardiovascular development. Nonetheless, in CRC, GATA5 has been included in the CIMP panel because it is frequently methylated (Jia et al., 2016). Moreover, the protein encoded for the APC gene is involved in the WNT signaling pathway affected in 90% of sporadic CRC cases (Zhao et al., 2022).
Recently, the role of epigenetic changes in tumor tissue induced by bacteria has been described, although there are few studies in this field. DNA methylation is associated with F. nucleatum, Bilophila, P. micra, H. hathewayi, and Streptococcus spp.
The most studied opportunistic pathogen is F. nucleatum which is found in several reservoirs and patients with CRC, and high levels have been associated with development, prognosis, and treatment response (Brennan and Garret, 2019). Fusobacterium is hypothesized to be associated with CRC through chronic infection and dysregulation of the expression of genes involved in the WNT pathway, immune system, and cell cycle (Gholizadeh et al., 2017; Brennan and Garret, 2019; Stokowa-Sołtys et al., 2021). F. nucleatum has been associated with CIMP+ and the promoter methylation of MLH1, CDKN2A, MTSS1, RBM38, PKD1, PTPRT, and EYA4, which are mutated and dysregulated in CRC according to the COSMIC database (Tate et al., 2019). CIMP+ tumors are characterized by hypermethylation in promoters of suppressor tumor genes, and sporadic CRC is positive for this phenotype up to 15%. Different molecular markers have been used for diagnosis, and MLH1, GATA5, and CDKN2A are occasionally included in the diagnosis panel (Jia et al., 2016; Park et al., 2017). The mechanisms of F. nucleatum that explain its role in DNA methylation are linked to inflammation and the generation of reactive oxygen species (ROS) (Koi et al., 2018). Although the mechanism of inflammation remains unclear, there is evidence of aberrant methylation associated with inflammatory cells in ulcerative colitis and gastric cancer, which are positive for Fusobacterium and H. pylori, respectively (Tahara et al., 2017; Koi et al., 2018). Regarding ROS, guanine is damaged to produce 7-8-dihydro-8-oxo-guanine, and CpG islands are highly dense in this nucleotide; therefore, the affected region recruits DNMTs, which silence the gene through hypermethylation (Koi et al., 2018). Moreover, F. nucleatum promotes the expression and activity of DNMT1 and DNMT3A in two CRC cell lines (HT29 and HCT116) and DNMT3B in the normal cell line, NCM460 (Xia et al., 2020). Fusobacterium and its relationship with DNA methylation could be due to butyrate fatty acids because high Fusobacterium abundance was significantly correlated with a decrease in 4-hydroxybutyric acid in CRC patients (Wang et al., 2020). 4-Hydroxybutyric acid is an intermediate in butyrate synthesis that regulates enzymes involved in DNA demethylation, methylation, histone acetylation, and methylation (Wang et al., 2022).
Only one study on Bilophila and SFRP2 methylation reported a methylation frequency of 66% in CRC patients associated with poorly differentiated tissues (Bagci et al., 2016; Sobhani et al., 2019; Yu et al., 2019). Bilophila and Fusobacterium are sulfate-reducing bacteria that produce hydrogen sulfide, a genotoxic compound that causes DNA damage (Dahmus et al., 2018). However, there is no evidence linking the alterations in DNA methylation to sulfide.
Hungatella hathewayi, a gram-positive, rod-shaped bacterium, is associated with hypermethylation in SOX11, THBD, SFRP2, GATA5, ESR1, EYA4, CDX2, and APC genes. H. hathewayi seems to increase the expression and nuclear activity of DNMT1 and DNMT3A in the CRC cell lines HT29 and HCT116 and the normal cell line NCM460 (Xia et al., 2020). Therefore, DNA methylation induced by this microorganism could occur through this pathway.
Streptococcus spp. are associated with MLH1 and APC hypermethylation, genes frequently associated with CRC (Li et al., 2013; Liang et al., 2017; Zhu et al., 2021). Streptococcus species, such as Streptococcus bovis and Streptococcus gallolyticus, have been described in CRC, and their carcinogenic effect could be linked to increased production of inflammatory molecules; the subspecies gallolyticus upregulates β-catenin, a transcription factor involved in cell proliferation (Karpiński et al., 2022).
In addition, WIF1, PENK, and NPY hypermethylation has been associated with the microbiota; although no specific microorganism has been reported, a CMI was associated with P. micra (Sobhani et al., 2019). Hypermethylation in WIF1, PENK, and NPY is found in CRC. However, the roles of NPY and PENK in tumorigenesis are not clear, whereas that of WIF1 is related to the inhibition of the WNT pathway (Hu et al., 2018; Overs et al., 2021; Pulverer et al., 2021). Parvimonas micra is associated with high-grade tumors and the consensus molecular subtype 1 (defined by increased immune infiltration) in CRC patients (Löwenmark et al., 2022). Therefore, the possible mechanism of DNA methylation could be described for Fusobacterium.
Furthermore, the influence of the microbiota on the methylation of transcripts has been tested in animal models. Jabs et al. (2020) compared N6-methyladenosine mRNA in the cecal segment of conventional and germ-free mice and found 312 differentially methylated transcripts; moreover, when they transplanted the microbiota (derived from conventional mice) in germ-free mice, after 4 weeks of follow-up, they did not find differences in N6-methyladenosine compared to both groups of the study (Jabs et al., 2020).
The main bacteria associated with DNA methylation and their possible mechanisms are shown in Figure 1.
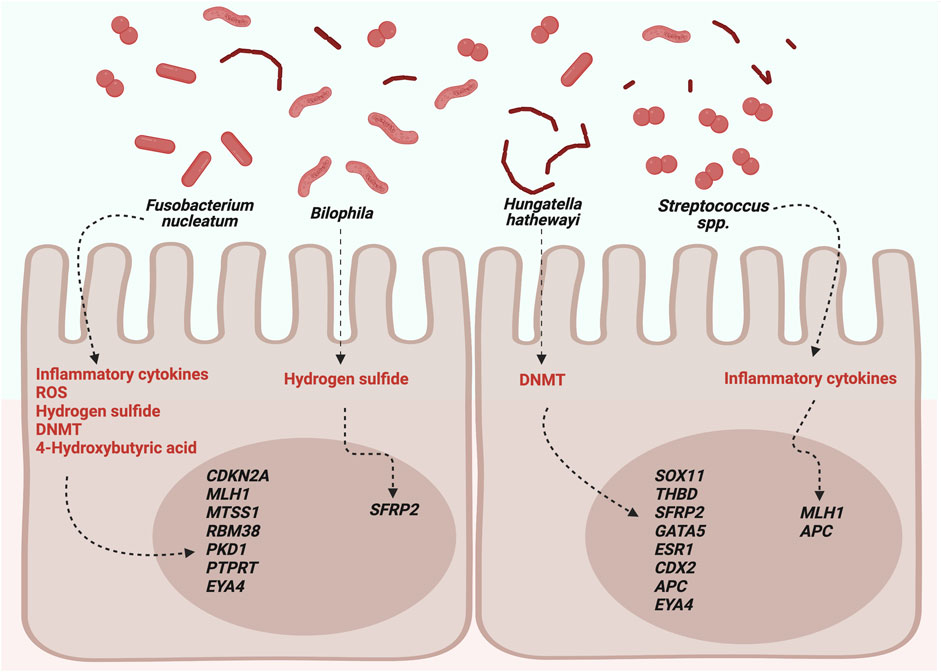
FIGURE 1. Possible roles of microbiota in DNA methylation. DNMTs, DNA methyltransferases; ROS, reactive oxygen species. Created with BioRender.com.
MG-A conceived, designed, and supervised the study. MG-A, MA-M, JM-O, JP-S, and FG-A collected the data and critically reviewed the content. All authors contributed to the article and approved the submitted version.
FG-A is a student of the PhD program in Human Genetics, and he was supported by the CONACyT scholarship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Almeida, A., Nayfach, S., Boland, M., Strozzi, F., Beracochea, M., Shi, Z. J., et al. (2021). A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol. 39, 105–114. doi:10.1038/s41587-020-0603-3
Arumugam, M., Raes, J., Pelletier, E., Le Paslier, D., Yamada, T., Mende, D. R., et al. (2011). Enterotypes of the human gut microbiome. Nat 473, 174–180. doi:10.1038/nature09944
Azoitei, N., Cobbaut, M., Becher, A., Van Lint, J., and Seufferlein, T. (2018). Protein kinase D2: A versatile player in cancer biology. Oncogene 37, 1263–1278. doi:10.1038/s41388-017-0052-8
Azuara, D., Aussó, S., Rodriguez-Moranta, F., Guardiola, J., Sanjuan, X., Lobaton, T., et al. (2018). New methylation biomarker panel for early diagnosis of dysplasia or cancer in high-risk inflammatory bowel disease patients. Inflamm. Bowel. Dis. 24, 2555–2564. doi:10.1093/ibd/izy255
Bagci, B., Sari, M., Karadayi, K., Turan, M., Ozdemir, O., and Bagci, G. (2016). KRAS, BRAF oncogene mutations and tissue specific promoter hypermethylation of tumor suppressor SFRP2, DAPK1, MGMT, HIC1 and p16 genes in colorectal cancer patients. Cancer. Biomark. 17, 133–143. doi:10.3233/CBM-160624
Biagi, E., Franceschi, C., Rampelli, S., Severgnini, M., Ostan, R., Turroni, S., et al. (2016). Gut microbiota and extreme longevity. Curr. Biol. 26, 1480–1485. doi:10.1016/j.cub.2016.04.016
Brennan, C. A., and Garrett, W. S. (2019). Fusobacterium nucleatum - symbiont, opportunist and oncobacterium. Nat. Rev. Microbiol. 17, 156–166. doi:10.1038/s41579-018-0129-6
Chen, J., Huang, L., Zhu, Q., Wang, Z., and Tang, Z. (2020). MTSS1 hypermethylation is associated with prostate cancer progression. J. Cell. Physiol. 235, 2687–2697. doi:10.1002/jcp.29172
Coker, O. O., Liu, C., Wu, W. K. K., Wong, S. H., Jia, W., Sung, J. J. Y., et al. (2022). Altered gut metabolites and microbiota interactions are implicated in colorectal carcinogenesis and can be non-invasive diagnostic biomarkers. Microbiome 10, 35. doi:10.1186/s40168-021-01208-5
Dahmus, J. D., Kotler, D. L., Kastenberg, D. M., and Kistler, C. A. (2018). The gut microbiome and colorectal cancer: A review of bacterial pathogenesis. J. Gastrointes. Oncol. 9, 769–777. doi:10.21037/jgo.2018.04.07
Dai, X., Ren, T., Zhang, Y., and Nan, N. (2021). Methylation multiplicity and its clinical values in cancer. Expert. Rev. Mol. Med. 23, e2. doi:10.1017/erm.2021.4
Das, P. K., Saha, J., Pillai, S., Lam, A. K., Gopalan, V., and Islam, F. (2023). Implications of estrogen and its receptors in colorectal carcinoma. Cancer Med. 12, 4367–4379. doi:10.1002/cam4.5242
Dawson, H., Galván, J. A., Helbling, M., Muller, D. E., Karamitopoulou, E., Koelzer, V. H., et al. (2014). Possible role of Cdx2 in the serrated pathway of colorectal cancer characterized by BRAF mutation, high-level CpG Island methylator phenotype and mismatch repair-deficiency. Int. J. Cancer. 134, 2342–2351. doi:10.1002/ijc.28564
De Palma, F., D'Argenio, V., Pol, J., Kroemer, G., Maiuri, M. C., and Salvatore, F. (2019). The molecular hallmarks of the serrated pathway in colorectal cancer. Cancers 11, 7. doi:10.3390/cancers11071017
Edwards, J. R., Yarychkivska, O., Boulard, M., and Bestor, T. H. (2017). DNA methylation and DNA methyltransferases. Epigenetics. Chromatin. 10, 23. doi:10.1186/s13072-017-0130-8
Ferlay, J., Ervik, M., Lam, F., Colombet, M., Mery, L., Piñeros, M., et al. (2020). Global cancer observatory: Cancer today. Lyon, France: International Agency for Research on Cancer.
Gholizadeh, P., Eslami, H., and Kafil, H. S. (2017). Carcinogenesis mechanisms of Fusobacterium nucleatum. Biomed. Pharmacother. 89, 918–925. doi:10.1016/j.biopha.2017.02.102
Gomaa, E. Z. (2020). Human gut microbiota/microbiome in health and diseases: A review. Ant. Van. Leeuwenhoek. 113, 2019–2040. doi:10.1007/s10482-020-01474-7
Grandits, A. M., Nguyen, C. H., Schlerka, A., Hackl, H., Sill, H., Etzler, J., et al. (2021). Downregulation of MTSS1 in acute myeloid leukemia is associated with a poor prognosis, chemotherapy resistance, and disease aggressiveness. Leukemia 35, 2827–2839. doi:10.1038/s41375-021-01224-2
Hamada, T., Zhang, X., Mima, K., Bullman, S., Sukawa, Y., Nowak, J. A., et al. (2018). Fusobacterium nucleatum in colorectal cancer relates to immune response differentially by tumor microsatellite instability status. Cancer. Immunol. Res. 6, 1327–1336. doi:10.1158/2326-6066.CIR-18-0174
Harada, S., and Morlote, D. (2020). Molecular pathology of colorectal cancer. Adv. Anat. Pathol. 27, 20–26. doi:10.1097/PAP.0000000000000247
Hu, H., Li, B., Zhou, C., Ying, X., Chen, M., Huang, T., et al. (2018). Diagnostic value of WIF1 methylation for colorectal cancer: A meta-analysis. Oncotarget 9 (4), 5378–5386. doi:10.18632/oncotarget.23870
Huang, Z., and Yang, M. (2022). Molecular network of colorectal cancer and current therapeutic options. Front. Oncol. 12, 852927. doi:10.3389/fonc.2022.852927
Ishak, M., Baharudin, R., Rose, I. M., Sagap, I., Mazlan, L., Azman, Z., et al. (2020). Genome-wide open chromatin methylome profiles in colorectal cancer. Biomolecules 10, 719. doi:10.3390/biom10050719
Ito, M., Kanno, S., Nosho, K., Sukawa, Y., Mitsuhashi, K., Kurihara, H., et al. (2015). Association of Fusobacterium nucleatum with clinical and molecular features in colorectal serrated pathway. Int. J. Cancer. 137, 1258–1268. doi:10.1002/ijc.29488
Jabs, S., Biton, A., Bécavin, C., Nahori, M. A., Ghozlane, A., Pagliuso, A., et al. (2020). Impact of the gut microbiota on the m6A epitranscriptome of mouse cecum and liver. Nat. Commun. 11, 1344. doi:10.1038/s41467-020-15126-x
Jandhyala, S. M., Talukdar, R., Subramanyam, C., Vuyyuru, H., Sasikala, M., and Nageshwar Reddy, D. (2015). Role of the normal gut microbiota. World. J. Gastroenterol. 21, 8787–8803. doi:10.3748/wjg.v21.i29.8787
Jia, M., Gao, X., Zhang, Y., Hoffmeister, M., and Brenner, H. (2016). Different definitions of CpG island methylator phenotype and outcomes of colorectal cancer: A systematic review. Clin. Epigenetics. 8, 25. doi:10.1186/s13148-016-0191-8
Jung, G., Hernández-Illán, E., Moreira, L., Balaguer, F., and Goel, A. (2020). Epigenetics of colorectal cancer: Biomarker and therapeutic potential. Nat. Rev. Gastroenterol. Hepatol. 17, 111–130. doi:10.1038/s41575-019-0230-y
Karpiński, T. M., Ożarowski, M., and Stasiewicz, M. (2022). Carcinogenic microbiota and its role in colorectal cancer development. Semin. Cancer. Biol. S1044-579X, 420–430. Online ahead of print. doi:10.1016/j.semcancer.2022.01.004
Katsaounou, K., Nicolaou, E., Vogazianos, P., Brown, C., Stavrou, M., Teloni, S., et al. (2022). Colon cancer: From epidemiology to prevention. Metabolites 12, 499. doi:10.3390/metabo12060499
Kim, S. J., Tae, C. H., Hong, S. N., Min, B. H., Chang, D. K., Rhee, P. L., et al. (2015). EYA4 acts as a new tumor suppressor gene in colorectal cancer. Mol. Carcinog. 54, 1748–1757. doi:10.1002/mc.22247
Koi, M., Okita, Y., and Carethers, J. M. (2018). Fusobacterium nucleatum infection in colorectal cancer: Linking inflammation, DNA mismatch repair and genetic and epigenetic alterations. J. Anus. Rectum. Colon. 2, 37–46. doi:10.23922/jarc.2017-055
Kumar, R., Yadav, G., Kuddus, M., Ashraf, G. M., and Singh, R. (2023). Unlocking the microbial studies through computational approaches: How far have we reached? Environ. Sci. Pollut. Res. Int. 30, 48929–48947. doi:10.1007/s11356-023-26220-0
Laczmanska, I., Karpinski, P., Bebenek, M., Sedziak, T., Ramsey, D., Szmida, E., et al. (2013). Protein tyrosine phosphatase receptor-like genes are frequently hypermethylated in sporadic colorectal cancer. J. Hum. Genet. 58, 11–15. doi:10.1038/jhg.2012.119
Lange, C. P., Campan, M., Hinoue, T., Schmitz, R. F., van der Meulen-de Jong, A. E., Slingerland, H., et al. (2012). Genome-scale discovery of DNA-methylation biomarkers for blood-based detection of colorectal cancer. PloS. One. 7, e50266. doi:10.1371/journal.pone.0050266
Lee, D. W., Han, S. W., Kang, J. K., Bae, J. M., Kim, H. P., Won, J. K., et al. (2018). Association between Fusobacterium nucleatum, pathway mutation, and patient prognosis in colorectal cancer. Ann. Surg. Oncol. 25, 3389–3395. doi:10.1245/s10434-018-6681-5
Léveillé, N., Elkon, R., Davalos, V., Manoharan, V., Hollingworth, D., Oude Vrielink, J., et al. (2011). Selective inhibition of microRNA accessibility by RBM38 is required for p53 activity. Nat. Commun. 2, 513. doi:10.1038/ncomms1519
Li, J., Ma, X., Chakravarti, D., Shalapour, S., and DePinho, R. A. (2021). Genetic and biological hallmarks of colorectal cancer. Genes. Dev. 35, 787–820. doi:10.1101/gad.348226.120
Li, J., Wei, Z., Cao, S., Tan, X., Liu, S., Yao, Z., et al. (2019a). A pilot study on clinicopathological features and intestinal microflora changes in colorectal cancer patients born over a nine-year period encompassing three years before and after the Great Chinese famine. Cancer. Epidemiol. 59, 166–172. doi:10.1016/j.canep.2019.02.001
Li, X., Wu, X., Li, Y., Cui, Y., Tian, R., Singh, N., et al. (2019b). Promoter hypermethylation of SOX11 promotes the progression of cervical cancer in vitro and in vivo. Oncol. Rep. 41, 2351–2360. doi:10.3892/or.2019.6993
Li, X., Yao, X., Wang, Y., Hu, F., Wang, F., Jiang, L., et al. (2013). MLH1 promoter methylation frequency in colorectal cancer patients and related clinicopathological and molecular features. PloS. One. 8, e59064. doi:10.1371/journal.pone.0059064
Liang, T. J., Wang, H. X., Zheng, Y. Y., Cao, Y. Q., Wu, X., Zhou, X., et al. (2017). APC hypermethylation for early diagnosis of colorectal cancer: A meta-analysis and literature review. Oncotarget 8, 46468–46479. doi:10.18632/oncotarget.17576
Liu, J. J., and Ward, R. L. (2010). Folate and one-carbon metabolism and its impact on aberrant DNA methylation in cancer. Adv. Genet. 71, 79–121. doi:10.1016/B978-0-12-380864-6.00004-3
Liu, J., Li, H., Sun, L., Wang, Z., Xing, C., and Yuan, Y. (2017). Aberrantly methylated-differentially expressed genes and pathways in colorectal cancer. Cancer. Cell. Int. 17, 75. doi:10.1186/s12935-017-0444-4
Löwenmark, T., Li, X., Löfgren-Burström, A., Zingmark, C., Ling, A., Kellgren, T. G., et al. (2022). Parvimonas micra is associated with tumour immune profiles in molecular subtypes of colorectal cancer. Cancer. Immunol. Immunother. 71, 2565–2575. doi:10.1007/s00262-022-03179-4
Marchesi, J. R., and Ravel, J. (2015). The vocabulary of microbiome research: A proposal. Microbiome 3, 31. doi:10.1186/s40168-015-0094-5
McInnes, T., Zou, D., Rao, D. S., Munro, F. M., Phillips, V. L., McCall, J. L., et al. (2017). Genome-wide methylation analysis identifies a core set of hypermethylated genes in CIMP-H colorectal cancer. Bmc. Cancer. 17, 228. doi:10.1186/s12885-017-3226-4
Milani, C., Duranti, S., Bottacini, F., Casey, E., Turroni, F., Mahony, J., et al. (2017). The first microbial colonizers of the human gut: Composition, activities, and health implications of the infant gut microbiota. Microbiol. Mol. Biol. Rev. 81, e00036-17. doi:10.1128/MMBR.00036-17
Mima, K., Sukawa, Y., Nishihara, R., Qian, Z. R., Yamauchi, M., Inamura, K., et al. (2015). Fusobacterium nucleatum and T cells in colorectal carcinoma. JAMA. Oncol. 1, 653–661. doi:10.1001/jamaoncol.2015.1377
Mitchell, A. L., Almeida, A., Beracochea, M., Boland, M., Burgin, J., Cochrane, G., et al. (2020). MGnify: The microbiome analysis resource in 2020. Nucleic. acids. Res. 48, D570–D578. doi:10.1093/nar/gkz1035
Moore, L. D., Le, T., and Fan, G. (2013). DNA methylation and its basic function. Neuropsychopharmacology 38, 23–38. doi:10.1038/npp.2012.112
Nguyen, L. H., Goel, A., and Chung, D. C. (2020). Pathways of colorectal carcinogenesis. Gastroenterology 158, 291–302. doi:10.1053/j.gastro.2019.08.059
Ono, T., Yamaguchi, T., Takao, M., Kojika, E., Iijima, T., and Horiguchi, S. I. (2022). Fusobacterium nucleatum load in MSI colorectal cancer subtypes. Int. J. Clin. Oncol. 27, 1580–1588. doi:10.1007/s10147-022-02218-5
Overs, A., Flammang, M., Hervouet, E., Bermont, L., Pretet, J. L., Christophe, B., et al. (2021). The detection of specific hypermethylated WIF1 and NPY genes in circulating DNA by crystal digital PCR™ is a powerful new tool for colorectal cancer diagnosis and screening. Bmc. Cancer. 21, 1092. doi:10.1186/s12885-021-08816-2
Pan, W. H., Sommer, F., Falk-Paulsen, M., Ulas, T., Best, P., Fazio, A., et al. (2018). Exposure to the gut microbiota drives distinct methylome and transcriptome changes in intestinal epithelial cells during postnatal development. Genome. Med. 10, 27. doi:10.1186/s13073-018-0534-5
Park, H. E., Kim, J. H., Cho, N. Y., Lee, H. S., and Kang, G. H. (2017). Intratumoral Fusobacterium nucleatum abundance correlates with macrophage infiltration and CDKN2A methylation in microsatellite-unstable colorectal carcinoma. Virchows. Arch. 471, 329–336. doi:10.1007/s00428-017-2171-6
Peery, R. C., Pammi, M., Claud, E., and Shen, L. (2021). Epigenome - a mediator for host-microbiome crosstalk. Semin. Perinatol. 45, 151455. doi:10.1016/j.semperi.2021.151455
Peyser, N. D., Freilino, M., Wang, L., Zeng, Y., Li, H., Johnson, D. E., et al. (2016). Frequent promoter hypermethylation of PTPRT increases STAT3 activation and sensitivity to STAT3 inhibition in head and neck cancer. Oncogene 35, 1163–1169. doi:10.1038/onc.2015.171
Portela, A., and Esteller, M. (2010). Epigenetic modifications and human disease. Nat. Biotechnol. 28, 1057–1068. doi:10.1038/nbt.1685
Pugongchai, A., Bychkov, A., and Sampatanukul, P. (2017). Promoter hypermethylation of SOX11 correlates with adverse clinicopathological features of human prostate cancer. Int. J. Exp. Pathol. 98, 341–346. doi:10.1111/iep.12257
Pulverer, W., Kruusmaa, K., Schönthaler, S., Huber, J., Bitenc, M., Bachleitner-Hofmann, T., et al. (2021). Multiplexed DNA methylation analysis in colorectal cancer using liquid biopsy and its diagnostic and predictive value. Curr. Issues. Mol. Biol. 43, 1419–1435. doi:10.3390/cimb43030100
Rebersek, M. (2021). Gut microbiome and its role in colorectal cancer. Bmc. Cancer. 21, 1325. doi:10.1186/s12885-021-09054-2
Rinninella, E., Raoul, P., Cintoni, M., Franceschi, F., Miggiano, G., Gasbarrini, A., et al. (2019). What is the healthy gut microbiota composition? A changing ecosystem across age, environment, diet, and diseases. Microorganisms 7, 14. doi:10.3390/microorganisms7010014
Ruan, W., Engevik, M. A., Spinler, J. K., and Versalovic, J. (2020). Healthy human gastrointestinal microbiome: Composition and function after a decade of exploration. Dig. Dis. Sci. 65, 695–705. doi:10.1007/s10620-020-06118-4
Sahnane, N., Magnoli, F., Bernasconi, B., Tibiletti, M. G., Romualdi, C., Pedroni, M., et al. (2015). Aberrant DNA methylation profiles of inherited and sporadic colorectal cancer. Clin. Epigenetics. 7, 131. doi:10.1186/s13148-015-0165-2
Sen, M., Kindsfather, A., Danilova, L., Zhang, F., Colombo, R., LaPorte, M. G., et al. (2020). PTPRT epigenetic silencing defines lung cancer with STAT3 activation and can direct STAT3 targeted therapies. Epigenetics 15, 604–617. doi:10.1080/15592294.2019.1676597
Shan, T., Uyar, D. S., Wang, L. S., Mutch, D. G., Huang, T. H., Rader, J. S., et al. (2019). SOX11 hypermethylation as a tumor biomarker in endometrial cancer. Biochimie 162, 8–14. doi:10.1016/j.biochi.2019.03.019
Sheng, Q., Du, H., Cheng, X., Cheng, X., Tang, Y., Pan, L., et al. (2019). Characteristics of fecal gut microbiota in patients with colorectal cancer at different stages and different sites. Oncol. Lett. 18, 4834–4844. doi:10.3892/ol.2019.10841
Shin, C. M., Kim, N., Jung, Y., Park, J. H., Kang, G. H., Kim, J. S., et al. (2010). Role of Helicobacter pylori infection in aberrant DNA methylation along multistep gastric carcinogenesis. Cancer. Sci. 101, 1337–1346. doi:10.1111/j.1349-7006.2010.01535.x
Sobhani, I., Bergsten, E., Couffin, S., Amiot, A., Nebbad, B., Barau, C., et al. (2019). Colorectal cancer-associated microbiota contributes to oncogenic epigenetic signatures. Proc. Nat. Acad. Sci. U. S. A. 116, 24285–24295. doi:10.1073/pnas.1912129116
Stokowa-Sołtys, K., Wojtkowiak, K., and Jagiełło, K. (2021). Fusobacterium nucleatum - friend or foe? J. Inorg. Biochem. 224, 111586. doi:10.1016/j.jinorgbio.2021.111586
Suga, D., Mizutani, H., Fukui, S., Kobayashi, M., Shimada, Y., Nakazawa, Y., et al. (2022). The gut microbiota composition in patients with right- and left-sided colorectal cancer and after curative colectomy, as analyzed by 16S rRNA gene amplicon sequencing. Bmc. Gastroenterol. 22, 313. doi:10.1186/s12876-022-02382-y
Tahara, T., Hirata, I., Nakano, N., Nagasaka, M., Nakagawa, Y., Shibata, T., et al. (2017). Comprehensive DNA methylation profiling of inflammatory mucosa in ulcerative colitis. Inflamm. Bowel. Dis. 23, 165–173. doi:10.1097/MIB.0000000000000990
Tahara, T., Yamamoto, E., Suzuki, H., Maruyama, R., Chung, W., Garriga, J., et al. (2014). Fusobacterium in colonic flora and molecular features of colorectal carcinoma. Cancer. Res. 74, 1311–1318. doi:10.1158/0008-5472.CAN-13-1865
Tate, J. G., Bamford, S., Jubb, H. C., Sondka, Z., Beare, D. M., Bindal, N., et al. (2019). Cosmic: The catalogue of somatic mutations in cancer. Nucleic. acids. Res. 47, D941–D947. doi:10.1093/nar/gky1015
UniProt Consortium (2021). UniProt: The universal protein knowledgebase in 2021. Nucleic. acids. Res. 49, D480–D489. doi:10.1093/nar/gkaa1100
Utikal, J., Gratchev, A., Muller-Molinet, I., Oerther, S., Kzhyshkowska, J., Arens, N., et al. (2006). The expression of metastasis suppressor MIM/MTSS1 is regulated by DNA methylation. Int. J. Cancer. 119, 2287–2293. doi:10.1002/ijc.22106
Vasapolli, R., Schütte, K., Schulz, C., Vital, M., Schomburg, D., Pieper, D. H., et al. (2019). Analysis of transcriptionally active bacteria throughout the gastrointestinal tract of healthy individuals. Gastroenterology 157, 1081–1092.e3. doi:10.1053/j.gastro.2019.05.068
Wang, L., Shannar, A., Wu, R., Chou, P., Sarwar, M. S., Kuo, H. C., et al. (2022). Butyrate drives metabolic rewiring and epigenetic reprogramming in human colon cancer cells. Mol. Nutr. Food. Res. 66, e2200028. doi:10.1002/mnfr.202200028
Wang, Q., Ye, J., Fang, D., Lv, L., Wu, W., Shi, D., et al. (2020). Multi-omic profiling reveals associations between the gut mucosal microbiome, the metabolome, and host DNA methylation associated gene expression in patients with colorectal cancer. Bmc. Microbiol. 20 (1), 83. doi:10.1186/s12866-020-01762-2
Wei, N., Chu, E., Wipf, P., and Schmitz, J. C. (2014). Protein kinase d as a potential chemotherapeutic target for colorectal cancer. Mol. Cancer. Ther. 13, 1130–1141. doi:10.1158/1535-7163.MCT-13-0880
Xia, X., Wu, W., Wong, S. H., Liu, D., Kwong, T., Nakatsu, G., et al. (2020). Bacteria pathogens drive host colonic epithelial cell promoter hypermethylation of tumor suppressor genes in colorectal cancer. Microbiome 8, 108. doi:10.1186/s40168-020-00847-4
Xie, F., Ye, L., Ta, M., Zhang, L., and Jiang, W. G. (2011). MTSS1: A multifunctional protein and its role in cancer invasion and metastasis. Front. Biosci. Sch. Ed. . 3, 621–631. doi:10.2741/s175
Yamashita, S., Tsujino, Y., Moriguchi, K., Tatematsu, M., and Ushijima, T. (2006). Chemical genomic screening for methylation-silenced genes in gastric cancer cell lines using 5-aza-2'-deoxycytidine treatment and oligonucleotide microarray. Cancer. Sci. 97, 64–71. doi:10.1111/j.1349-7006.2006.00136.x
Yu, J., Ma, X., Cheung, K. F., Li, X., Tian, L., Wang, S., et al. (2010). Epigenetic inactivation of T-box transcription factor 5, a novel tumor suppressor gene, is associated with colon cancer. Oncogene 29, 6464–6474. doi:10.1038/onc.2010.370
Yu, J., Xie, Y., Li, M., Zhou, F., Zhong, Z., Liu, Y., et al. (2019). Association between SFRP promoter hypermethylation and different types of cancer: A systematic review and meta-analysis. Oncol. Lett. 18, 3481–3492. doi:10.3892/ol.2019.10709
Zhao, H., Ming, T., Tang, S., Ren, S., Yang, H., Liu, M., et al. (2022). Wnt signaling in colorectal cancer: Pathogenic role and therapeutic target. Mol. Cancer. 21, 144. doi:10.1186/s12943-022-01616-7
Keywords: microbiota, DNA methylation, colorectal cancer, microbiome, DNA methyltransferase, tumor suppressor gene
Citation: Gutierrez-Angulo M, Ayala-Madrigal MdlL, Moreno-Ortiz JM, Peregrina-Sandoval J and Garcia-Ayala FD (2023) Microbiota composition and its impact on DNA methylation in colorectal cancer. Front. Genet. 14:1037406. doi: 10.3389/fgene.2023.1037406
Received: 05 September 2022; Accepted: 20 July 2023;
Published: 08 August 2023.
Edited by:
Raghavendra Upadhya, Texas A and M University, United StatesReviewed by:
Gokhan Akkoyunlu, Akdeniz University, TürkiyeCopyright © 2023 Gutierrez-Angulo, Ayala-Madrigal, Moreno-Ortiz, Peregrina-Sandoval and Garcia-Ayala. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Melva Gutierrez-Angulo, bWVsdmEuZ3V0aWVycmV6QGFjYWRlbWljb3MudWRnLm14
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.