
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 10 October 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.999442
This article is part of the Research Topic RNA-Seq and transcriptome-based approaches to aid in the diagnosis and understanding of genetic diseases View all 4 articles
 Yixi Sun1,2,3
Yixi Sun1,2,3 Yangwen Qian1,2,3
Yangwen Qian1,2,3 Hai-Xi Sun4Min Chen1,2,3
Hai-Xi Sun4Min Chen1,2,3 Yuqin Luo1,2,3
Yuqin Luo1,2,3 Xiaojing Xu4
Xiaojing Xu4 Kai Yan1,2,3
Kai Yan1,2,3 Liya Wang1,2,3
Liya Wang1,2,3 Junjie Hu1,2,3
Junjie Hu1,2,3 Minyue Dong1,2,3*
Minyue Dong1,2,3*Skewed XCI plays an important role in the phenotypic heterogeneities of many X-linked disorders, even involving in diseases caused by XCI-escaping genes. DDX3X-related intellectual disability is more common in females and less common in males, who usually inherit from unaffected heterozygous mothers. As an X inactivation (XCI) escaping gene, the role of skewed XCI in the phenotype of DDX3X mutant female is unknown. Here we reported a DDX3X: c.694_711dup18 de novo heterozygous mutation in a female with intellectual disability on the maternal X chromosome on the basis of SNPs detected by PCR-sanger sequencing. AR assay revealed that the maternal mutant X chromosome was extremely inactivated in the proband. Using RNA sequencing and whole-exome sequencing, we quantified allelic read counts and allele-specific expression, and confirmed that the mutant X chromosome was inactive. Further, we verified that the mutant DDX3X allele had a lower expression level by RNA sequencing and RT-PCR, and the normal and mutated DDX3X expression accounted for respectively 70% and 30% of total. In conclusion, we found a symptomatic female with extreme skewing XCI in the DDX3X mutant allele. It was discovered that XCI in the mutant allele was insufficient to reverse the phenotype of DDX3X-related neurodevelopmental disorder. It contributed to a better understanding of the role of skewed XCI in phenotypic differences, which can aid in the genetic counseling and prenatal diagnosis of disorders in females with DDX3X defects.
Mutations in X-linked genes account for approximately 5%–10% of the causes of intellectual disability (ID) in males, whereas females with X-linked ID have much lower penetrance and phenotypic severity than hemizygous males (Dobyns et al., 2004; Lubs et al., 2012; Dikow et al., 2017). Female carriers with X-linked variants are generally thought to be protected by functional mosaicism caused by random X chromosome inactivation (XCI) or skewed XCI that favors the wild-type allele (Cotton et al., 2015; Kellaris et al., 2018).
DDX3X maps to Xp11.3–11.23 and is an XCI-escaping gene (Lahn and Page, 1997; Cotton et al., 2015). It encodes an ATP-dependent “DEAD-box” RNA helicase involved in numerous cellular processes; it is essential for cortical development, which is required in neural progenitors to produce cortical neurons during development (Linder et al., 1989; Kellaris et al., 2018; Lennox et al., 2020; Mo et al., 2021). DDX3X variants are one of the most common causes of ID, accounting for 1%–3% of females with unexplained ID (Snijders Blok et al., 2015). Around 848 individuals have been identified (https://ddx3x.org/). In general, DDX3X-related ID is more common in females with de novo variants and less common in males, who usually inherited from unaffected heterozygous mothers except few cases of de novo male patients (Snijders Blok et al., 2015; Wang et al., 2018; Nicola et al., 2019; Tang et al., 2021). Moreover, there are phenotypic differences in females with DDX3X variants.
Skewed XCI plays an important role in the phenotypic heterogeneities of many X-linked disorders, even involving in diseases caused by XCI-escaping genes (Sun et al., 2019). Although XCI-escaping genes are expressed from the inactive X chromosome (Xi) in all cells, their expression levels relative to active X chromosome (Xa) levels are usually reduced (Carrel and Willard, 1999). As similar with most of XCI-escaping genes, transcripts of DDX3X are not fully expressed from the Xi, escaping ratios of DDX3X are ranging from 29% to 61% (Garieri et al., 2018). Expressions of wild-type and mutant DDX3X are affected by skewed XCI, so we tried to explore if skewed XCI might dictate the clinical phenotypes of females with DDX3X mutations, which could aid in genetic counseling for the prenatal diagnosis of DDX3X-related intellectual disability. Here we investigated an intellectual disability girl with a de novo DDX3X mutation. By PCR-sanger sequencing, we defined that the mutation occurred from which X chromosome, and further detected the methylation and transcription levels of the X chromosome by AR assay and RNA sequencing.
The proband was a 10-year-old girl without family history. The symptoms of the proband included mild to moderate intellectual disability, delayed psychomotor development, poor speech development, infantile hypotonia, autism, and behavior problems. The patient was born after a normal pregnancy and delivery. She could not sit without support at 18 months. When seeing a doctor at the age of 19 months, her gross motor and fine motor skills were obviously delayed, her abilities of grasp, visual movement, stationary and locomotivity were respectively equivalent to 12, 13, 11, and 9 months of age. She began to walk at 2 years. At the age of 26 months, her language level was equivalent to the normal 12.7-month-old child (Sym score: 5). At the age of 7 years, she was diagnosed as autism spectrum disorder, the scores of childhood autism rating scale and autistic behavior checklist were 32 and 75. Spike and slow wave complex sporadically showed in video electroencephalogram (VEEG). Brain magnetic resonance imaging (MRI) was normal. By color doppler ultrasound, abnormal cerebral blood flows were detected in middle cerebral artery (left: +3SD, right: +3SD) and anterior cerebral artery (left: +3SD, right: +2-3SD) at 7 years.
Genomic DNA and total RNA of peripheral blood mononuclear cells (PMBCs) were respectively extracted with the Gentra Puregene Kit (Qiagen, Germany) and RNAiso Plus (Takara, Japan). Extracted total RNA was reverse-transcribed using PrimeScript™ RT Master Mix (Takara, Japan). PCR was performed using GoldStar Best Master Mix (CWBIO, Beijing). Primer sequences were listed below. DDX3X-DNA-F: AGACTTGATGGCTTGTGC; DDX3X-DNA-R: TTTCTGGCTTCCTCGTAG; DDX3X-RNA-F: CAGTGATGTTGAGATGGGAGAA; DDX3X-RNA-R: AAACCACGCAAGGACGAA. T-A cloning was performed by Hieff Clone™ Zero TOPO-TA Cloning Kit (Yeasen, Shanghai) according to the manufacturer’s instruction. Sanger sequencing were performed by ABI 3730 DNA analyser.
To analyze XCI in DNA methylation level, XCI in the family was analyzed using PCR amplification of the AR gene in blood, as previously described (Allen et al., 1992). Primers sequences were listed below. AR-F: TCCAGAATCTGTTCCAGAGCGTGC (labeled by 5′6-FAM); AR-R: GCTGTGAAGGTTGCTGTTCCTCAT. Amplified products were run on an ABI 3500 genetic analyzer.
Whole-exome sequencing (WES) and RNA-sequencing were performed by Biomarker Technologies Corporation (Beijing, China). The sequencing libraries were generated using the NimbleGen SeqCap EZ Human Exome V3 (Roche, Basel, Swiss) and NEBNextR Ultra™ Directional RNA Library Prep Kit for Illumina (NEB, United States). Clustering of the index-coded samples was performed on a cBot Cluster Generation System (Illumina, United States). After cluster generation, the library preparations were sequenced on an Illumina HiSeq X Ten platform.
Burrows-Wheeler Aligner v0.7.13-r1126 was used to align each sample’s clean reads with the reference genome using default parameters. Alignment files were converted to BAM files using SAMtools software. Variant calling was performed for all samples by using the Haplotype Caller in GATK software.
To calculate the proportion of expressed wild type allele and mutant allele of DDX3X at the RNA level, we mapped the RNA-seq reads to a modified cDNA sequence of DDX3X gene (NM_001193416.2), in which the 18-bp wild type allele sequences on exon 8 were removed, using BWA (Version: 0.7.17) (Li and Durbin, 2009) with parameters “aln -t 12 -o 3 -e 35 -d 0 -i 0 -l 100 -k 0 -M 1 -O 1 -E 1 -N -n 0.0001.” Then an in-house Perl script was used to extract the wild type allele sequence or mutant allele sequence from the mapped reads.
To distinguish the transcription of alleles on the two X chromosomes, we explored the exome sequencing data for heterozygous informative SNPs on the X chromosome in II.1 to assess the parental origin. Combining RNA sequencing, informative SNPs were quantified allelic read counts and allele-specific expression (ASE).
Trio-based exome sequencing revealed a de novo heterozygous variant of NM_001193416.3: c.694_711dup18 (NC_000023.10: g.41203004_41203021dup18) in DDX3X gene of the female proband. Sanger sequencing verified the de novo variant in the proband, the parents were wild-type (Figure 1), both paternity and maternity were confirmed (PS2). This variant was absent from gnomAD, ESP and 1,000 Genomes databases (PM2). It had not been reported in the HGMD and ClinVar databases. It caused protein length changes as a result of six amino acids insertions (Ala232_Pro237dup) on the non-repeated region (PM4). Based on the American College of Medical Genetics (ACMG) guidelines (Richards et al., 2015), the variant was classified as likely pathogenic (1PS + 2PM).
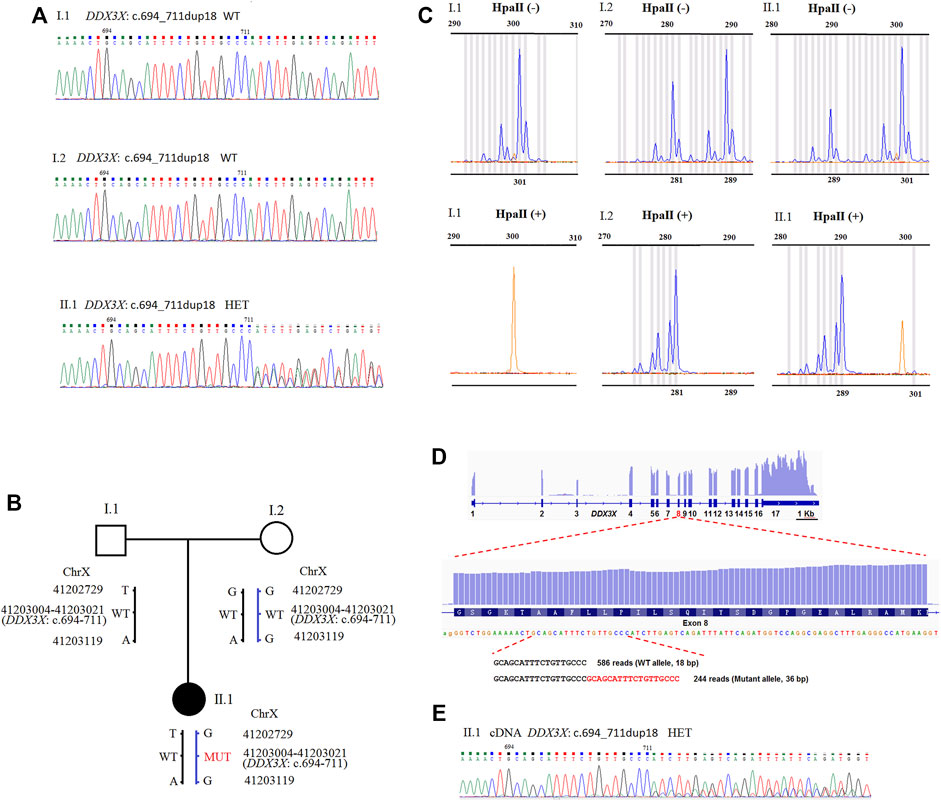
FIGURE 1. (A) Sequence analysis of genomic DNA from family members. The genotypes of DDX3X were wild type, wild type, and c.694_711dup18 HET, in I.1 (father), I.2 (mother), and II.1 (proband). (B) Schematic representation of the DDX3X mutation and two SNPs in the family. Besides the DDX3X c.694_711dup18, G > T and G > A variant were also detected at the positions NC_000023.10: g.41202729 and g.41203119 in II.1 by PCR-sanger sequencing. DDX3X c.694_711dup18 was confirmed to inherit from mother I.2 by the two SNPs. HET, heterozygote; WT, wild type; MUT, mutation type. (C) XCI pattern was detected by AR assay. A peak of 301 bp for AR was observed by assaying the undigested PCR product of I.1 and no peaks were observed for the HpaII digested product. The undigested PCR product of I.2 gave two peaks of 281 and 289 bp, and major peak of 281 bp was observed with the HpaII digested product. The undigested PCR product of the proband II.1 gave two peaks of 289 and 301 bp. One X-chromosome linked with the 289 bp peak of AR was inherited from the mother I.2 and the other from the father I1. The product of HpaII digestion gave only one major peak of 289 bp. II.1 exhibited extreme skewing of XCI. The inactivated X-chromosome was linked with the 289 bp peak of AR, which inherited from the mother I.2. (D) The IGV Genome Browser view showing RNA-seq reads mapped to DDX3X locus. The numbers represent exon number of DDX3X gene. Read number of wild type allele and mutant allele detected by RNA-seq is shown on the bottom. (E) Sequence analysis of the RT-PCR product from PMBCs of II.1. The mutant peaks were lower than the wild. HET, heterozygote.
Despite the fact that the variant was de novo, we were able to trace its origins back to the SNPs of PCR-sanger sequencing. Using a pair of PCR primers, we not only verified the variant, but also found two SNPs in the family. As presented in Supplementary Figure S1A, a heterozygous variant NC_000023.10: g.41202729G>T was detected in the proband II.1 by forward sequencing. As reverse sequencing showed that the base in this position was T, which was inherited from the father I.1. The in-frame duplication originated from the mother I.2. It indicated that the variant of NM_001193416.3: c.694_711dup18 occurred on the maternal X chromosome. The result was also confirmed by another SNP NC_000023.10: g.41203119G>A (Supplementary Figure S1B). The results were further confirmed by T-A cloning, sanger sequence chromatogram of single allele showed that the bases in two SNPs (NC_000023.10: g.41202729 and g.41203119) linked with the duplication were all G, which was inherited from the mother I.2. It revealed that the duplication existed on maternal allele (Supplementary Figure S2).
We examined XCI patterns using PCR-based AR (androgen receptor) assay in the family. After digestion with the methylation-sensitive restriction enzyme HpaII, only the allele on the inactive X chromosome could be amplified by PCR. Meanwhile, segregation analysis was used to determine the origin of the inactivated X chromosome. The undigested PCR product of II.1 yielded two peaks of 289 bp and 301 bp, respectively. For the HpaII-digested product, a major peak representing 289 bp was observed (Figure 1C), indicating the inactivated X chromosome inherited from the mother I.2. It was discovered that the X chromosome with the DDX3X variant of II.1 is inherited from the mother. It revealed that the X chromosome with the DDX3X variant from the mother had extreme skewing of XCI.
To distinguish whole transcription on two X chromosomes of the proband II.1, we explored the exome sequencing data for heterozygous informative SNPs on the X chromosome of the proband to assess the parental origin. Combining RNA sequencing, informative SNPs were quantified allelic read counts. For the proband II.1, we found that most genes on the maternal X-chromosome were lower transcription, especially the XCI genes were almost no expression on the maternal X-chromosome (Supplementary Table S1). It was consistent with the result by AR assay. It revealed that the maternal mutant X chromosome was extremely inactivated in the proband.
In peripheral blood mononuclear cells (PMBCs), RNA sequencing revealed no differences in DDX3X total expressions between the proband II.1 and the mother I.2 (data not shown). We found DDX3X expressions in both wild and mutant alleles. The read counts for the DDX3X wild and mutant alleles were 586 and 244 (71:29), respectively, indicating that the mutant allele had lower expression (Figure 1D). We also identified the results using RT-PCR and Sanger sequence from PMBCs, and the mutant peaks were lower than the wild ones (Figure 1E). Meanwhile, it showed that the duplication and base G in NC_000023.10: g.41203119 were all in the same RNA-seq reads, further confirming the duplication located on maternal allele (Supplementary Figure S3).
DDX3X encodes a 662-amino-acid protein, its functional core contains highly conserved helicase core composed of two RecA-like domains (D1, D2) and N- and C-terminal extensions. As one of the most intolerant genes, tolerated variations in DDX3X are extremely rare (Snijders Blok et al., 2015). The mutation of DDX3X: c.694_711dup18 (p.Ala232_Pro237dup) locates on the communication between RNA and ATP binding sites of D1 domain. Three mutations (c.698C > T/p.A233V, c.704T > A/p.L235Q, c.704T > C/p.L235P) have been reported in the duplicated region, and A233V, as a recurrent mutation, was confirmed to impair helicase activity by functional studies (Snijders Blok et al., 2015; Turner et al., 2019; Lennox et al., 2020). In our study, DDX3X: c.694_711dup18 caused DDX3X protein length changes as a result of six amino acids insertions of the highly conserved region (Supplementary Figure S4), and altered protein topology (Supplementary Figure S5), which might affect its function.
DDX3X is one of the most common X-linked genes linked to intellectual disability ranging from mild to severe in females, but some females with DDX3X variants have no symptoms. Many studies have tried to explore genotype-phenotype correlations. Functional studies in zebrafish have shown that mutant DDX3X alleles, which found in male patients inherited from the normal mothers, had wild-type or near wild-type activity, suggesting that they are hypomorphic alleles, and both DDX3X missense and nonsense mutations function in a haploinsufficient manner (Snijders Blok et al., 2015; Kellaris et al., 2018).
In addition to loss of function (LoF), Lennox et al. (2020) recently elucidated that the same recurrent de novo variants (R326, I415, T532) had similar phenotypes, and missense variants were more likely to have abnormal brain structural, such as polymicrogyria (PMG). Clinical severity of DDX3X-related neurodevelopmental disorder was linked to reduced helicase activity and RNA-protein granules, missense variants could function in a dominant-negative manner (Lennox et al., 2020; Dai et al., 2022).
DDX3X is an XCI–escaping gene, the role of skewed XCI in phenotypic differences of DDX3X-related ID is unclear. Research in this field is limited. As Table 1, initially, Snijders Blok et al. (2015) reported that 6 out of 15 females with DDX3X mutations and ID had almost complete skewing of X-inactivation (>95%), which was higher than typical female populations, but it was unknown which X-chromosome was inactivated due to most mutations were de novo. Subsequently, Fieremans et al. (2016) reported two females with DDX3X mutations and ID were extreme skewing XCI, and only the reference allele was present at cDNA level in a de novo stop variant (NM001193417: p.G177X). At the transcriptional level, the mutant allele was not detected, but nonsense mediated mRNA decay could not be ruled out, so they could not conclude the mutant allele must be inactive. Meanwhile, the parental origin of the variant was not definite, so it could not distinguish which X-chromosome was inactivated by AR assay in DNA methylation level.
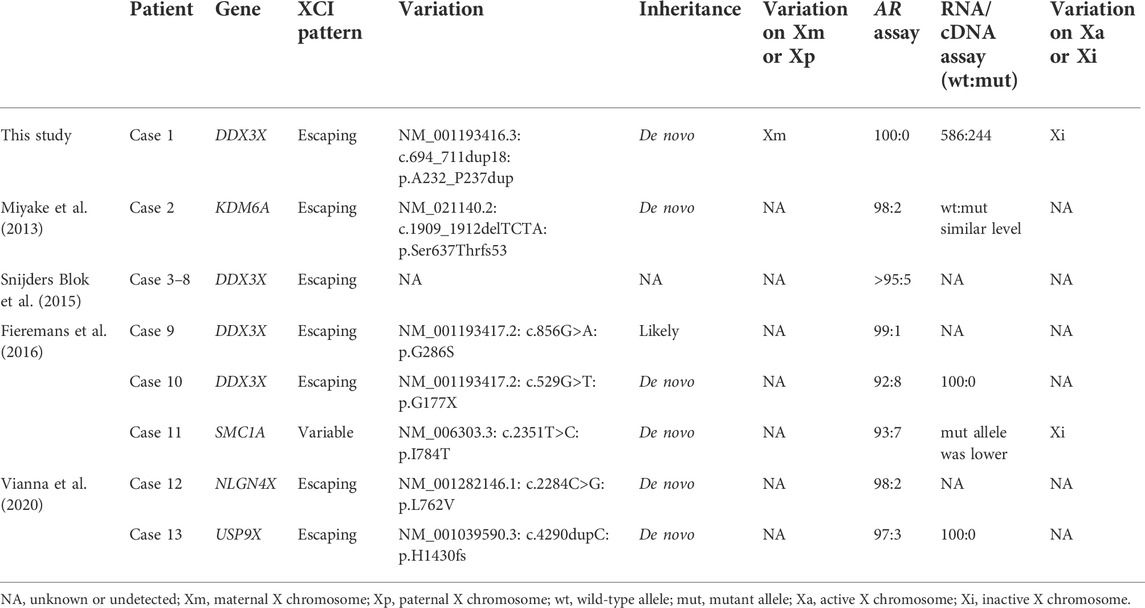
TABLE 1. Skewed XCI of female intellectual disability (ID) patients with variations in the escaping or variable XCI genes.
In our study, we reported a de novo DDX3X mutation in a female with intellectual disability, but we were able to find the mutation occurred on the maternal X chromosome based on SNPs detected by PCR-sanger sequencing, and we confirmed that the mutant X chromosome was inactive detected by AR assay and RNA sequencing both in DNA methylation and transcriptional levels. Females with DDX3X mutations on the inactive X chromosome were still affected, according to the findings. Skewed XCI was insufficient to reverse the phenotype of XCI-escaping gene DDX3X deficiency.
As most XCI-escaping genes, including transcripts of DDX3X, are not fully expressed from the Xi, escaping ratios of DDX3X range from 29% to 61% (Garieri et al., 2018). Using RNA sequence, escaping ratio in our study is approximately 42% (244/586). If we only considered escaping XCI and skewed XCI, we would assume that DDX3X expression on the active X chromosome (Xa) was 1 in one cell, and according to our data, its expression on the inactive X chromosome (Xi) was 0.42 (1*42%). As shown in Figure 2, if we just calculated DDX3X expression from two cells. DDX3X expressions of wild type (wt) in the normal female with random XCI, paternal and maternal skewed XCI all were 2.84 (1 + 0.42 + 1 + 0.42), whereas DDX3X expressions of wild type and mutant type in maternal skewed XCI pattern of were 2 (1 + 1) and 0.84 (0.42 + 0.42). The proband with about 70% (2/2.84) normal and 30% (0.84/2.84) mutated DDX3X expression was still affected.
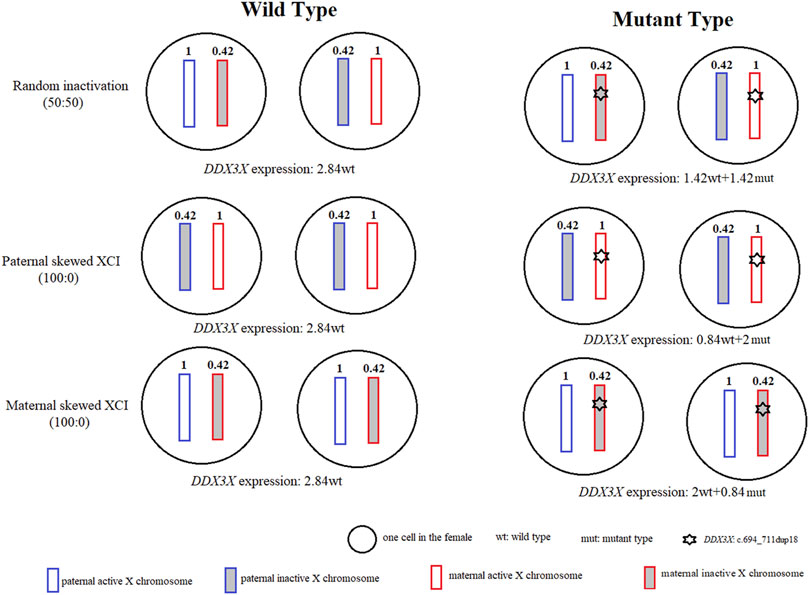
FIGURE 2. Modeled DDX3X expression levels in normal females and females with DDX3X mutation. For two normal female cells, there were no differences for DDX3X expressions in random XCI, paternal and maternal skewed XCI, all were 2.84. For two female cells with DDX3X mutation. The wild type of DDX3X expressions respectively were 1.42, 0.84, 2 in random XCI, paternal and maternal skewed XCI, which were affected by the XCI pattern. The white and gray boxes represented the active X chromosome (Xa) and inactive X chromosome (Xi). The blue and red showed the paternal and maternal X chromosome. The number inside the X chromosome indicated the proportion of DDX3X expression according to our data. wt, wild type; mut, mutation type.
For the DDX3X in-frame insertion, we did not confirm if LoF or dominant-negative manner, play a key role in the pathogenic mechanism, and did not know the degree of LoF or dominant-negative caused by the mutation. If the mechanism was LoF, it indicated that DDX3X was very sensitive to gene dosage. If we assumed the function of mutated DDX3X completely lost. It showed that not only 50% normal DDX3X product by the single wild-type X chromosome was not sufficient for complete function, even though normal DDX3X dosage increased up to 70% by skewed XCI on the mutant X chromosome, the female with DDX3X heterozygous mutation still affected. In mice, Lennox et al. (2020) also reinforced the exquisite dosage sensitivity of the developing brain to DDX3X, as even a 25% reduction in DDX3X levels strongly perturbed neurogenesis.
In consideration of no differences in DDX3X total expressions between the proband II.1 and the mother I.2. This variant was more likely to behave like a missense than a loss-of-function, the pathogenic mechanism was more likely to dominant-negative manner. If we assumed all mutated DDX3X proteins were detrimental, it suggested that only 30% mutated DDX3X expression was enough to cause abnormal clinical phenotype, the expression of mutation did not have to come to 50%. No matter which mechanism acts, it does not change the conclusion, the female with DDX3X mutation in inactive X chromosome was still affected.
Other escaping or variable XCI genes associated with intellectual disability (ID) (Table 1), such as SMC1A and USP9X, are in a similar situation (Limongelli et al., 2010; Gervasini et al., 2013; Fieremans et al., 2016; Vianna et al., 2020). Variants in these genes were found to cause both X-linked ID and skewing of XCI, with the variant allele expressing less than the wild-type allele. However, the exact mechanisms underlying XCI skewing in females harboring variants in escape XCI genes remain unknown. Perhaps these genes were X-Y homologous genes, which were too important for life, as females with mutations on Xa were difficult to survive.
Of course, there are some limitations in our study. Most DDX3X variants are de novo, tracing their origins of parents are usually ignored, and then it is unable to assess the XCI status on the methylation level. Our research is just a case report, providing a new idea to detect XCI status, further studies in a larger cohort with different variation types are needed. In addition, we just detected the XCI status of blood, the status of brain was unknown. Although Werner et al. (2022) recently modeled variability in human XCI ratios across tissues and individuals using the GTEx dataset, determining that XCI ratios are consistent across all tissue lineages. Due to no sufficient evidence, we could not confirm that XCI status must be consistent between blood and brain in patients with DDX3X variants. It remains to be proved by further experiments.
In conclusion, we described a de novo DDX3X mutation in a female with intellectual disability. Using the AR assay and RNA sequencing, we verified that the X chromosome with the DDX3X mutation was extreme skewing inactivation. However, it was insufficient to reverse the phenotype of DDX3X-related neurodevelopmental disorder. It contributed to our understanding of the role of skewed XCI in the phenotypic differences, which can aid in the genetic counseling and prenatal diagnosis of disorders in females with DDX3X defect.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the Ethics Committee of Women’s Hospital’s recommendations, School of Medicine at Zhejiang University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
MD designed the study. PCR and RT-PCR was carried out by YS, KY, and JH; YS and MC contributed the AR assay detection. YL and LW performed WES and ASE analysis. RNA sequence analysis was performed by H-XS and XX. YS and YQ wrote the draft manuscript. All co-authors provided feedback on the estimates and contributed to the subsequent versions of the manuscript. All authors read and approved the final version of the manuscript.
This study was supported by the National Natural Science Foundation of China (Grant Nos. 81801441, 81901382), the Key Projects Jointly Constructed by the Ministry and the Province of Zhejiang Medical and Health Science and Technology Project (Grant No. WKJ-ZJ-2127), the Key Research and Development Program of the Zhejiang Province (Grant No. 2019C03025), the Medical Health Science and Technology Project of Zhejiang Provincial Health Commission (Grant No. 2021KY772), the Public Welfare Technology Research Program of Zhejiang Province (Grant No. LGC20H200003).
We appreciate all the participants involved in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.999442/full#supplementary-material
Allen, R. C., Zoghbi, H. Y., Moseley, A. B., Rosenblatt, H. M., and Belmont, J. W. (1992). Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 51 (6), 1229–1239.
Carrel, L., and Willard, H. F. (1999). Heterogeneous gene expression from the inactive X chromosome: An X-linked gene that escapes X inactivation in some human cell lines but is inactivated in others. Proc. Natl. Acad. Sci. U.S.A. 96 (13), 7364–7369. doi:10.1073/pnas.96.13.7364
Cotton, A. M., Price, E. M., Jones, M. J., Balaton, B. P., Kobor, M. S., and Brown, C. J. (2015). Landscape of DNA methylation on the X chromosome reflects CpG density, functional chromatin state and X-chromosome inactivation. Hum. Mol. Genet. 24 (6), 1528–1539. doi:10.1093/hmg/ddu564
Dai, Y., Yang, Z., Guo, J., Li, H., Gong, J., Xie, Y., et al. (2022). Expansion of clinical and genetic spectrum of DDX3X neurodevelopmental disorder in 23 Chinese patients. Front. Mol. Neurosci. 15, 793001. doi:10.3389/fnmol.2022.793001
Dikow, N., Granzow, M., Graul‐Neumann, L. M., Karch, S., Hinderhofer, K., Paramasivam, N., et al. (2017). DDX3X mutations in two girls with a phenotype overlapping Toriello-Carey syndrome. Am. J. Med. Genet. 173 (5), 1369–1373. doi:10.1002/ajmg.a.38164
Dobyns, W. B., Filauro, A., Tomson, B. N., Chan, A. S., Ho, A. W., Ting, N. T., et al. (2004). Inheritance of most X-linked traits is not dominant or recessive, just X-linked. Am. J. Med. Genet. 129A (2), 136–143. doi:10.1002/ajmg.a.30123
Fieremans, N., Van Esch, H., Holvoet, M., Van Goethem, G., Devriendt, K., Rosello, M., et al. (2016). Identification of intellectual disability genes in female patients with a skewed X-inactivation pattern. Hum. Mutat. 37 (8), 804–811. doi:10.1002/humu.23012
Garieri, M., Stamoulis, G., Blanc, X., Falconnet, E., Ribaux, P., Borel, C., et al. (2018). Extensive cellular heterogeneity of X inactivation revealed by single-cell allele-specific expression in human fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 115 (51), 13015–13020. doi:10.1073/pnas.1806811115
Gervasini, C., Russo, S., Cereda, A., Parenti, I., Masciadri, M., Azzollini, J., et al. (2013). Cornelia de Lange individuals with new and recurrentSMC1Amutations enhance delineation of mutation repertoire and phenotypic spectrum. Am. J. Med. Genet. 161 (11), 2909–2919. doi:10.1002/ajmg.a.36252
Kellaris, G., Khan, K., Baig, S. M., Tsai, I. C., Zamora, F. M., Ruggieri, P., et al. (2018). A hypomorphic inherited pathogenic variant in DDX3X causes male intellectual disability with additional neurodevelopmental and neurodegenerative features. Hum. Genomics 12 (1), 11. doi:10.1186/s40246-018-0141-y
Lahn, B. T., and Page, D. C. (1997). Functional coherence of the human Y chromosome. Science 278 (5338), 675–680. doi:10.1126/science.278.5338.675
Lennox, A. L., Hoye, M. L., Jiang, R., Johnson-Kerner, B. L., Suit, L. A., Venkataramanan, S., et al. (2020). Pathogenic DDX3X mutations impair RNA metabolism and neurogenesis during fetal cortical development. Neuron 106 (3), 404–420. e408. doi:10.1016/j.neuron.2020.01.042
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 (14), 1754–1760. doi:10.1093/bioinformatics/btp324
Limongelli, G., Russo, S., Digilio, M. C., Masciadri, M., Pacileo, G., Fratta, F., et al. (2010). Hypertrophic cardiomyopathy in a girl with Cornelia de Lange syndrome due to mutation in SMC1A. Am. J. Med. Genet. 152A (8), 2127–2129. doi:10.1002/ajmg.a.33486
Linder, P., Lasko, P. F., Ashburner, M., Leroy, P., Nielsen, P. J., Nishi, K., et al. (1989). Birth of the D-E-A-D box. Nature 337 (6203), 121–122. doi:10.1038/337121a0
Lubs, H. A., Stevenson, R. E., and Schwartz, C. E. (2012). Fragile X and X-linked intellectual disability: Four decades of discovery. Am. J. Hum. Genet. 90 (4), 579–590. doi:10.1016/j.ajhg.2012.02.018
Miyake, N., Koshimizu, E., and Okamoto, N. (2013). MLL2 and KDM6A mutations in patients with Kabuki syndrome. Am. J. Med. Genet. 161A, 2234–2243.
Mo, J., Liang, H., Su, C., Li, P., Chen, J., and Zhang, B. (2021). DDX3X: Structure, physiologic functions and cancer. Mol. Cancer 20 (1), 38. doi:10.1186/s12943-021-01325-7
Nicola, P., Blackburn, P. R., Rasmussen, K. J., Bertsch, N. L., Klee, E. W., Hasadsri, L., et al. (2019). De novo DDX3X missense variants in males appear viable and contribute to syndromic intellectual disability. Am. J. Med. Genet. 179 (4), 570–578. doi:10.1002/ajmg.a.61061
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Snijders Blok, L., Madsen, E., Juusola, J., Gilissen, C., Baralle, D., Reijnders, M. R., et al. (2015). Mutations in DDX3X are a common cause of unexplained intellectual disability with gender-specific effects on wnt signaling. Am. J. Hum. Genet. 97 (2), 343–352. doi:10.1016/j.ajhg.2015.07.004
Sun, Y., Luo, Y., Qian, Y., Chen, M., Wang, L., Li, H., et al. (2019). Heterozygous deletion of the SHOX gene enhancer in two females with clinical heterogeneity associating with skewed XCI and escaping XCI. Front. Genet. 10, 1086. doi:10.3389/fgene.2019.01086
Tang, L., Levy, T., Guillory, S., Halpern, D., Zweifach, J., Giserman-Kiss, I., et al. (2021). Prospective and detailed behavioral phenotyping in DDX3X syndrome. Mol. Autism 12 (1), 36. doi:10.1186/s13229-021-00431-z
Turner, T. N., Wilfert, A. B., Bakken, T. E., Bernier, R. A., Pepper, M. R., Zhang, Z., et al. (2019). Sex-based analysis of de novo variants in neurodevelopmental disorders. Am. J. Hum. Genet. 105 (6), 1274–1285. doi:10.1016/j.ajhg.2019.11.003
Vianna, E. Q., Piergiorge, R. M., Gonçalves, A. P., dos Santos, J. M., Calassara, V., Rosenberg, C., et al. (2020). Understanding the landscape of X-linked variants causing intellectual disability in females through extreme X chromosome inactivation skewing. Mol. Neurobiol. 57 (9), 3671–3684. doi:10.1007/s12035-020-01981-8
Wang, X., Posey, J. E., Rosenfeld, J. A., Bacino, C. A., Scaglia, F., Immken, L., et al. (2018). Phenotypic expansion in DDX3X - a common cause of intellectual disability in females. Ann. Clin. Transl. Neurol. 5 (10), 1277–1285. doi:10.1002/acn3.622
Keywords: DDX3X, skewed X-chromosome inactivation (XCI), intellectual disability, escaping X-chromosome inactivation (XCI), RNA sequencing
Citation: Sun Y, Qian Y, Sun H-X, Chen M, Luo Y, Xu X, Yan K, Wang L, Hu J and Dong M (2022) Case Report: De novo DDX3X mutation caused intellectual disability in a female with skewed X-chromosome inactivation on the mutant allele. Front. Genet. 13:999442. doi: 10.3389/fgene.2022.999442
Received: 21 July 2022; Accepted: 26 September 2022;
Published: 10 October 2022.
Edited by:
Long Guo, RIKEN Center for Integrative Medical Sciences, JapanReviewed by:
Noriko Miyake, National Center for Global Health and Medicine, JapanCopyright © 2022 Sun, Qian, Sun, Chen, Luo, Xu, Yan, Wang, Hu and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Minyue Dong, ZG9uZ215QHpqdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.