 Muhammad Umair
Muhammad Umair Muhammad Younus
Muhammad Younus Sarfraz Shafiq3
Sarfraz Shafiq3 Anam Nayab
Anam Nayab- 1Medical Genomics Research Department, Ministry of National Guard Health Affairs (MNGH), King Abdullah International Medical Research Center (KAIMRC), King Saud Bin Abdulaziz University for Health Sciences, Riyadh, Saudi Arabia
- 2State Key Laboratory of Membrane Biology and Beijing Key Laboratory of Cardiometabolic Molecular Medicine, Institute of Molecular Medicine, College of Future Technology and Peking-Tsinghua Center for Life Sciences and PKU-IDG/McGovern Institute for Brain Research, Peking University, Beijing, China
- 3Department of Anatomy and Cell Biology, University of Western Ontario, London, ON, Canada
- 4Department of Biotechnology, Fatima Jinnah Women University, Rawalpindi, Pakistan
- 5Genetics and Precision Medicine Department, King Abdullah Specialized Children Hospital (KASCH), King Abdulaziz Medical City, Ministry of National Guard Health Affairs (MNG-HA), Riyadh, Saudi Arabia
Spondylocostal dysostosis is a genetic defect associated with severe rib and vertebrae malformations. In recent years, extensive clinical and molecular diagnosis advancements enabled us to identify disease-causing variants in different genes for such severe conditions. The identification of novel candidate genes enabled us to understand the developmental biology and molecular and cellular mechanisms involved in the etiology of these rare diseases. Here, we discuss the clinical and molecular targets associated with spondylocostal dysostosis, including clinical evaluation, genes, and pathways involved. This review might help us understand the basics of such a severe disorder, which might help in proper clinical characterization and help in future therapeutic strategies.
1 Introduction
The spondylocostal dysostosis (SCDO), a subclass of a Jarcho–Levin syndrome, represents a rare heritable group of genetic disorders. SCDO is characterized by congenital defective vertebral segmentation and rib deformation due to imperfect alignments, fusion, or reduction in numbers. The patient often presents with a short trunk and scoliosis (Solomon et al., 1978; Mortier et al., 1996; Gucev et al., 2010). SCDO can be characterized as familial or sporadic because of its autosomal recessive and dominant inheritance pattern (Turnpenny et al., 1993). SCDO is generally inherited as an autosomal recessive trait due to mutations of the genes that are involved in the Notch-signaling pathway such as Delta-like canonical Notch ligand 3 (DLL3) (Bulman et al., 2000), mesoderm posterior protein 2 (MESP2) (Whittock et al., 2004a; Whittock et al., 2004b), LFNG (Sparrow et al., 2006), and HES7 genes (Sparrow et al., 2008). Patients with familial SCDO clinically present with the most common symptoms such as anal and urogenital anomalies, congenital heart disease, limb abnormalities, plagiocephaly-torticollis deformity sequence, diaphragmatic hernia, and neuronal tube defects (in males) (Cetinkaya et al., 2008), while patients with sporadic SCDO present with a wide variety of symptoms such as asymmetrical thoracic region and scoliosis (Mortier et al., 1996). Numerous sporadic SCDO cases have been reported with VATER, VACTERL, and MURCS associations along with DiGeorge, Alagille, and Robinow syndromes. Associations with an intellectual disability and other abnormal neurological syndromes are unusual (Turnpenny et al., 2009; Turnpenny et al., 2017).
In the present review, we have classified spondylocostal dysostosis based on gene identification and associated clinical phenotypes. A total of 185 and 284 entries were obtained using the mesh “spondylocostal dysostosis” in the OMIM and PubMed (NCBI). They include both syndromic and non-syndromic spondylocostal dysostosis. Autosomal recessive spondylocostal dysostoses have been classified into seven types and are presented in Table 1, which might be helpful for researchers and clinicians to have a quick overview of the disorder, help in molecular diagnosis, and further management plans.
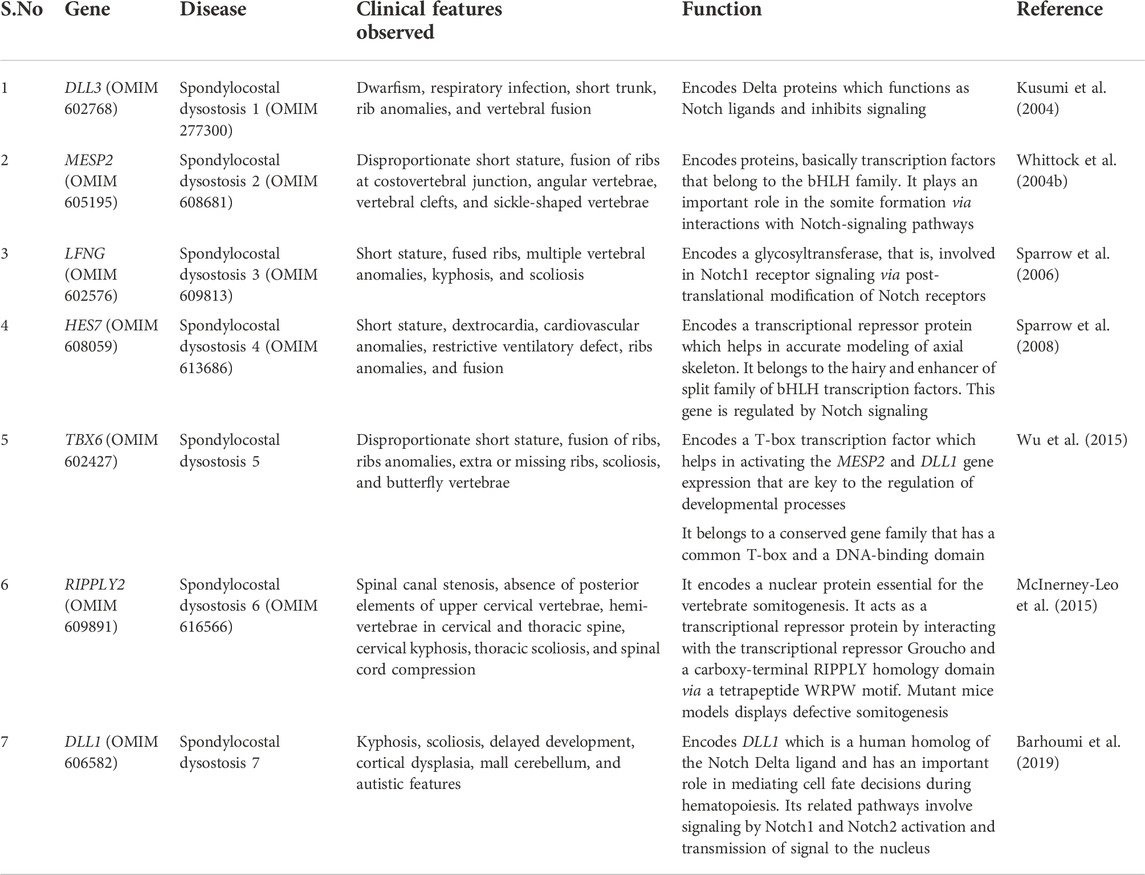
TABLE 1. Spondylocostal dysostosis-associated genes, associated clinical representation, and function in the Notch-signaling pathway.
1.1 Clinical description
Patients with SCDO have short trunks relative to their height, a short neck, and often present with thoracic insufficiency and mild to severe non-progressive scoliosis. Neonates often clinically present with mild to moderate respiratory insufficiency due to a smaller thorax. Neonatal lung growth may improve by the age of 2 years, which supports their normal growth and development. Such patients can survive but are at a higher risk of life-threatening complications such as pulmonary hypertension. Male patients with SCDO may be at a higher risk of inguinal hernia (Turnpenny et al., 2017).
1.2 Classification
SCDO is classified into seven types based on pathogenic mutations in different genes (Table 1). Autosomal recessive SCDO is isolated in nature, and is confined to the vertebrae and ribs. However, additional syndromic types have been reported in some of the subtypes.
1.3 Spondylocostal dysostosis type 1 (SCDO1; DLL3)
SCDO1 is caused by pathogenic biallelic mutations in the Delta-like canonical Notch ligand 3 (DLL3) gene (MIM 602768) located on chromosome 19q13.2 with autosomal recessive mode of inheritance. The clinical features, reported in the affected individuals having DLL3 mutations, are the irregularity of the vertebral column on spinal radiographs in early childhood and the prenatal stage. “Pebble beach sign” of each vertebral body in the fetus is a characteristic feature, which is observed having an ovoid or round shape with smooth boundaries (Turnpenny et al., 2003). As a result, the fetus develops the pebble beach appearance of the vertebrae and gives rise to multiple irregularities to the vertebral column. In such a situation, an MRI is more feasible to view the irregularities as compared to X-rays.
The DLL3 gene (NM_016941.4) consists of eight exons encoding a 618-amino acid (NP_058637.1) long protein. The long encoded DLL3 protein is composed of different domains such as a Delta–Serrate-Lag2 region (DSL), six epidermal growth factor-like domains (EGF), and a transmembrane (TM) domain. DLL3 has been shown to be involved in the somite boundary formation and cell-signaling mechanism as it has shown a spatially restricted expression pattern when studied in different animal models (Dunwoodie et al., 2002; Kusumi et al., 2004; Dunwoodie S. L., 2009). To date, only 31 disease-causing mutations have been reported in the DLL3 (HGMD, 2021). Out of these, 28 homozygous mutations have been identified in the DLL3 gene associated with SCDO1. These mutations included seven nonsense mutations, six missense mutations, ten small deletions, and five small insertions (Table 2).
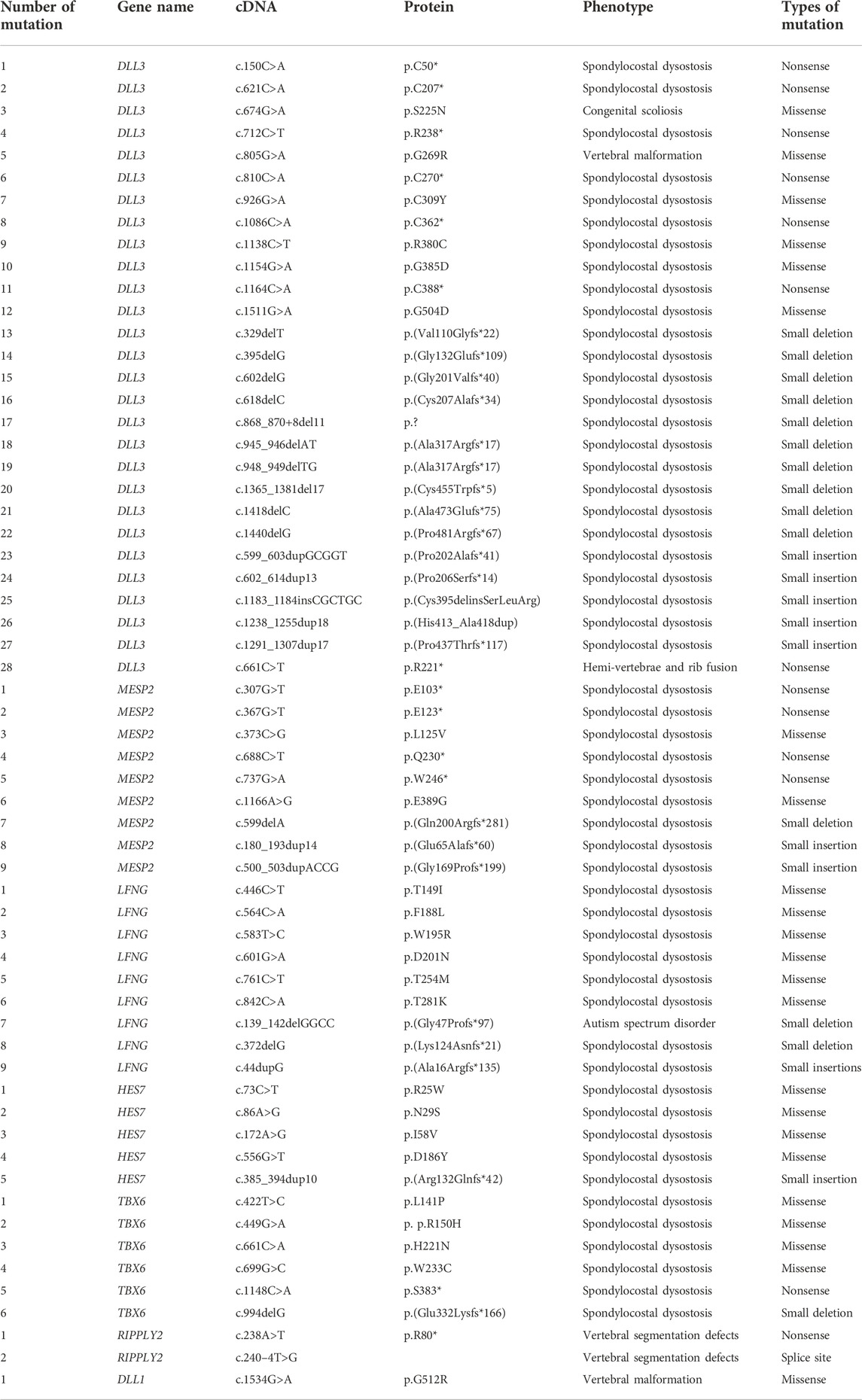
TABLE 2. Mutations reported to date in genes associated with spondylocostal dysostosis (HGMD® Professional 2022.2).
1.4 Spondylocostal dysostosis type 2 (SCDO2; MESP2)
SCDO2 is reported to be inherited as an autosomal recessive disorder, and is caused due to the mutation in the MESP2 gene which is located on chromosome 15q26.1. The mutation in mesoderm posterior basic helix–loop–helix transcription factor 2 (MESP2) (MIM 605195) may be a homozygous or a compound heterozygous mutation. SCDO2 reported cases that exhibit clinical features such as truncal shortening, short necks, and spondylocostal dysostosis. As compared to SCDO1 (DLL3), the affected individuals have relatively mildly affected lumbar vertebrae as compared to the thoracic region (Whittock et al., 2004b).
In an Arab-Lebanese family, reported by Whittock et al. (2004a), two affected individuals exhibited spondylocostal dysostosis features, such as short necks and truncal shortening without any other abnormalities. In the same family, a biallelic 4-bp duplication (c.500_503dup) has been identified in the MESP2 (NM_001039958.2, NP_001035047.1) gene as the main cause of the disease. It has been found that the mutation occurs in a sequence following the basic helix–loop–helix (bHLH) domain which leads to a frameshift, hence resulting in premature truncation (Whittock et al., 2004b).
MESP2 (MIM 605195; NM_001039958.2) consists of two exons (1614 bp) and encodes a protein comprising 397 amino acids (NP_001035047.1). The full-length MESP2 protein consists of an N-terminal domain succeeded by a basic helix–loop–helix (bHLH) domain, a GQ (glycine and glutamine repeats)-rich region, and finally, a C-terminal domain. MESP2 belongs to a family of transcriptional regulatory proteins that have a core helix–loop–helix (bHLH) domain, and plays a key role in rostro-caudal polarization and somite formation (Figure 1) (Mitsuru Morimoto et al., 2005) (Morimoto et al., 2007). To date, only nine mutations have been reported in the MESP2 gene and only nine have been associated with SCDO2, which include four nonsense mutations, two missense mutations, one small deletion, and two small insertions (Table 2).
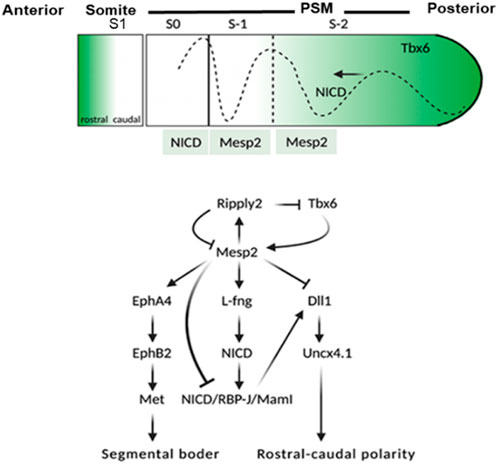
FIGURE 1. Schematic representation of events involved in the activation of MESP2. TBX6 along with Notch1 signaling leads to the expression of MESP2 in the anterior presomitic mesoderm (NICD dotted line). The dotted line toward the anterior along with the black arrow shows the Notch1 signal wavers in the presomitic mesoderm. As MESP2 protein is formed within S1, NICD is suppressed indirectly via activation of LFNG, and directly via inhibition of DLL1. The expression of MESP2 protein inhibits the expression of DLL1 and Uncx4.1 caudal genes; however, it induces the expression of rostral genes EphA4, EphB2, and followed by mesenchymal-to-epithelial transition (MET) within somites leading to segmentation of the rostral-caudal polarity within the somites is generated via a RIPPLY2–MESP2 negative feedback loop. S1 = formed somite; S0 = somite under formation; S1 and S2 = somite primordia.
MESP2 is integral to somite boundary formation, and is important for the development of anterior somatic compartment via suppression of Notch signaling (Figure 1). MESP2 expression was analyzed by Cornier et al. (2008) and Saga et al. (1997) in mouse models. They argued that MESP2 expressed as stripe in the presomitic mesoderm which is indicative of the future somite. It is these stripes that form a support for the vertebral development in the spine of adult mice. MESP2 expression is localized in the anterior somatic compartment. MESP2 gene knockout mice show severe vertebral segmentation defects along with proximal rib fusion which is parallel to that observed in STD patients.
1.5 Spondylocostal dysostosis type 3 (SCDO3; LFNG)
SCDO3 is clinically characterized by having long slender fingers, camptodactyly, and congenital vertebral anomalies. Radiological scans have revealed multiple vertebral ossification centers in the thoracic region of the vertebral column (Sparrow et al., 2006). SCDO3-carrying individuals exhibit more severe spine shortening as compared to SCDO1 and SCDO2 individuals, although ribs deformities have been reported to be similar to SCDO1 and SCDO2 cases. In 2006, Sparrow et al reported a biallelic missense mutation (c.564C>A; p.F188L) in the Leng O-fucosylpeptide 3-beta-N-acetylglucosaminyltransferase (LFNG) gene located on chromosome 7p22.3 as a candidate gene in a Lebanese proband. Recently, a proband from a Japanese family has been reported having features of severe multiple vertebral deformities starting from the cervical to sacral spine. Novel compound heterozygous variants [c.372delG (p.K124Nfs*) and c.601G>A (p.D201N)] in the LFNG gene have been identified which further validates the role of this gene in causing spondylocostal dysostosis type 3 in humans (Otomo et al., 2019).
LFNG (NM_001040167.2) is composed of a total of eight exons (2377bp) located on chromosome 7p22.3, encoding a 379-aa long protein (NP_001035257.1) To date, fourteen mutations have been reported in the LFNG gene responsible for causing SCDO3 phenotypes (HGMD; 2018). To date, only 14 mutations have been reported in the LFNG gene and only nine have been associated with SCDO3, including two small deletions, one small insertion, and six missense variants (Table 2). The other phenotypes reported include autism spectrum disorder (ASD), scoliosis, Asperger’s syndrome, and tetralogy of Fallot.
Apart from vertebral defects, the proband reported by Sparrow et al. (2006) had camptodactyly of the left index finger. The authors suggested that these features might not be due to the LFNG mutation, since according to one study, lack of LFNG expression in mouse models did not show limb defects (Zhang et al., 2002). It has been shown that the LFNG expression is restricted to the hemangioblasts (precursors of blood vessels) in a developing vertebrate limb. However, the expression of LFNG in the human embryo is still elusive, and human limb development might be different from that in the mouse (Sparrow et al., 2006).
1.6 Spondylocostal dysostosis type 4 (SCDO4; HES7)
SCDO4 is a severe autosomal recessive disorder triggered by the HES7 gene mutation located on chromosome 17p13.1. A proband in a Caucasian Mediterranean origin consanguineous family had features such as lumbosacral myelomeningocele; hydrocephalus; myelomeningocele; bell-shaped, shortened thorax; stenotic anus; and talipes. The radiological examination of the proband also revealed the shortening of the spine and contiguous and multiple vertebral defects. The parents were normal, second cousins, having two healthy kids (Sparrow et al., 2008). Molecular analysis of the proband led to the identification of a homozygous missense mutation (c.73C>T; p. Arg25Trp) in the HES7 gene.
Later, two affected cases from a non-consanguineous Italian family were reported to have spondylocostal dysostosis. The affected individuals showed multiple anomalies, multiple rib fusions with 11 pairs of bilateral ribs, short trunk, and short stature. MRI results revealed multiple segmentation anomalies in the cervical region of the spine (Sparrow et al., 2010). In 2013, a large consanguineous Arab family was reported to have seven SCDO4 cases. The affected individuals were reported to have consistent features of SCDO4. Three of the affected individuals were presented with dextrocardia along with situs inversus. Two of the affected individuals also exhibited secondary features such as neural tube defects (spina bifida occulta, thoracic myelomeningocele, and Chiari II) (Sparrow et al., 2013).
The HES7 gene (MIM608059; NM_001165967.2) covers 1674 bp of the genomic region, has four exons, and encodes HES7 protein. An HES7 protein is a transcription factor that belongs to the hairy and enhancer of split family of bHLH transcription factors (NP_001159439.1). To date, only seven disease-causing homozygous mutations have been identified in the HES7 gene (HGMD, 2022). Only five mutations including four missense mutations and one small insertion have been associated with SCDO4 (Table 2).
Gene expression analysis of various transcription factors in an embryonic mouse brain have suggested the low level expression of HES7 in the mid-brain and thalamus and thus, is limited to PSM (Gray et al., 2004). The expression of other members of the HES family (HES1, 3, 5, and 6) in the embryonic mouse brain has also been reported (Gray et al., 2004). Furthermore, severe neural tube defects and premature neurogenesis have been reported for HES1-null mutants (Ishibashi et al., 1995). Severe somite segmentation defects have been reported in homozygous HES7-null mouse embryos; laterality defects, however, have not been described (Bessho et al., 2001).
1.7 Spondylocostal dysostosis type 5 (SCDO5; TBX6)
SCDO5 was reported for the first time by Gucev et al. (2010) in a Macedonian descendant family segregating autosomal dominant spondylocostal dysostosis. The affected individuals had several phenotypes, such as disproportionate short trunks, short necks, mild scoliosis with hemi-vertebrae, and vertebral blocks. These affected individuals did not reveal any other abnormalities, dysmorphic features, or neurodevelopment issues. Gucev et al. (2010) identified a pathogenic heterozygous variant in the TBX6 gene. In total, twenty-three individuals from the Chinese Han population with congenital scoliosis-related compound heterozygous mutations in the TBX6 gene were identified. The affected individuals revealed hemi-vertebrae and other shared rib abnormalities (Wu et al., 2015). More recently (Lefebvre et al., 2017), identified homozygous disease-causing mutations in the TBX6 gene justified the recessive inheritance of SCDO5. The affected individuals presented features such as disproportionate short stature, short necks, rib abnormalities, missing ribs, scoliosis, syringomyelia, and butterfly vertebrae.
The TBX6 gene is located on chromosome 16p11.2 (NM_004608.3, NP_004599.2), and is composed of nine exons encoding a 463-amino acid protein, TBX6. TBX6 belongs to a family that is phylogenetically conserved and shares a common DNA-binding domain, the T-box. The T-box genes encode transcription factors that play an important role in the regulation of developmental processes. The knockout studies in mice have suggested the role of this gene in the specification of paraxial mesoderm structures (Zhao et al., 2018). To date, only 66 disease-causing homozygous mutations have been identified in the HES7 gene (HGMD, 2022). Only six mutations in the TBX6 gene have been associated with SCDO5, including four missense mutations, one nonsense mutation, and one small deletion (Table 2). Other phenotypes associated with TBX6 pathogenesis include scoliosis, Müllerian aplasia, Mayer–Rokitansky–Küster–Hauser syndrome, congenital anomalies of the kidney, tetralogy of Fallot, and autism.
The TBX6-null/mh mice exhibited vertebral malformations, similar to those in humans; the lower part of the spine is affected by the vertebral malformations (Liu et al., 2019).
1.8 Spondylocostal dysostosis type 6 (SCDO6; RIPPLY2)
SCDO6, an autosomal recessive disorder, is caused by homozygous variants in the RIPPLY2 gene (NM_001009994.2), which is located on chromosome 6q14.2. SCDO6 exhibits severe phenotypes such as spinal canal stenosis, the descent of an occipital bone, absence of posterior elements of upper cervical vertebrae, hemi-vertebrae, butterfly vertebrae, cervical kyphosis, and thoracic scoliosis (Menger et al., 2021). McInerney-Leo et al. (2015) reported two affected individuals born to non-consanguineous parents having phenotypes such as affected posterior C1–C4 elements, butterfly vertebrae of T2–T7, cervical kyphosis, spinal canal stenosis, mild thoracic scoliosis, and hemi-vertebrae.
The RIPPLY2 gene consists of four exons (664 bp) that encode a 128-aa long nuclear protein, RIPPLY2 (RIPPLY transcriptional repressor 2 protein), that belongs to a novel family of proteins required for vertebrate somitogenesis. The members of this family of proteins are characterized by having a tetrapeptide WRPW motif at N-terminal and a RIPPLY homology domain at C-terminal (composed of a Bowline-DSCR-Ledgerline conserved region) The RIPPLY family of proteins has been reported to be the transcriptional repressors as they negatively regulate the T-box proteins, including TBX6, coded by SCDO gene 10 (Kawamura et al., 2008). The transcriptional repression occurs by the interaction of RIPPLY proteins with the DNA-binding domain of T-box proteins, via their RIPPLY homology domain, and with the transcriptional co-repressor Groucho/TLE proteins via their WRPW motif (Kawamura et al., 2005). To date, only three disease-causing mutations have been identified in the RIPPLY2 gene, including one nonsense, one splice site mutation, and one small deletion (Table 2; HGMD, 2022).
Studies involving mice, which are kept homozygous null for RIPPLY2, have been reported to have severe vertebral and ribs malformations. Such mice also displayed defects in axial skeleton segmentations due to defective somitogenesis and finally, died in their prenatal stage. The vertebral defects that have been observed in RIPPLY2-null mice had a metameric pattern of vertebral bodies, intervertebral discs, and severely disrupted neural arches (Chan et al., 2007). Such conditions are similar to those of the mice, which are homozygous null for the autosomal recessive SCDO genes DLL3, MESP2, LFNG, and HES7 (Sparrow et al., 2011).
1.9 Spondylocostal dysostosis type 7 (SCDO7; DLL1)
SCDO type 7 is a recently identified type of spondylocostal dysostosis inherited in an autosomal recessive manner. Barhoumi et al. (2019) reported two affected individuals (boys) showing features such as scoliosis, fused thoracic spines (T4–T5, T6–T8, and T11–T12), and multiple spine deformities. The whole-exome sequence analysis of these patients revealed a homozygous missense mutation (c.1534G>A; p. Gly512Arg) in exon 9 of the DLL1 gene. In addition, 14 patients from unrelated families had neurodevelopmental disorders along with other brain abnormalities. The sequence analysis of these patients revealed the heterozygous mutations in the DLL1 gene (Fischer-Zirnsak et al., 2019). All these reported patients had kyphosis/scoliosis, hyperextensible joints, hypotonia, and ataxia, as common features.
DLL1, located on chromosome 6q27, comprise a total of 11 exons (3174 bp) encoding a 723-amino acid long protein (NM_005618.4, NP_005609.3, 11). DLL1 encodes for the protein Delta-like canonical Notch ligand 1 (DLL1). DLL1 belongs to the Delta/Serrate/Jagged Family and is a human homolog of the Drosophila Delta ligand. It is important for hematopoiesis as it plays an important role in mediating cell fate decisions via cell-to-cell communication (Barhoumi et al., 2019). To date, 38 disease-causing mutations have been identified in the DLL1 gene associated with different disorders such as neurodevelopmental disorder, autism, hearing loss, congenital heart disease, and cleft lip/or palate. However, only one mutation has been associated in DLLI causing vertebral malformations (SCDO7) (Barhoumi et al., 2019).
DLL1 is composed of three domains, mainly intracellular, transmembrane, and an extracellular domain [containing eight epidermal growth factor-like (EGF-like) domains]. The mutation is mainly located in the eight EGF-like domains at a conserved nucleotide region. The mutation resulted in a change of amino acid at 512 position (from glycine to arginine), consequently affecting its binding properties with the target proteins.
Knockout mutations of DLL1 in the mouse model generated a phenotype that differed significantly from the wild-type mouse in reference to the body size. The KO mouse had a small size and suffered from osteosclerosis because of the loss of function of osteoblasts and osteoclasts. According to the histopathological study, the affected mice had a reduced bone formation rate, decreased osteoblast surface area, and a compromised metabolic bone turnover (Muguruma et al., 2017).
1.10 Molecular genetics of SCDO and involvement of the Notch-signaling pathway
In humans, vertebrae are formed during embryogenesis from somites which are the embryonic segments produced from the presomitic mesoderm (PSM) in approx. 4–5 h. These somites are produced in a rhythmic sequence following an anterior to posterior direction. This periodic series of somite formation involves a molecular segmentation clock which basically operates through a Notch-signaling pathway (Wahi et al., 2016). Most of the genes belonging to the Notch-signaling pathway are involved in the somitogenesis and the molecular oscillation generated by them is synchronized with coupling genes of Wnt and FGF pathways in the PSM. These pathways are vital for normal vertebral development and somitogenesis as the coupled oscillations of the signaling genes under these signaling pathways govern the accurate segmentation in the process of somite formation. The mutations in these genes such as LFNG, DLL3, and MESP2 have been shown to be associated with the abnormal molecular oscillations leading to the development of congenital vertebral malformations, that is, recessive spondylocostal dysostosis (SCDO) (Dequéant et al., 2006; Goldbeter and Pourquié, 2008).
The Notch-signaling pathway was identified for the first time in Drosophila. It is highly conserved through evolution from invertebrates to vertebrates (Bray, 2006). The Notch-signaling pathway is a very diverse and important pathway, which is known to interact with several other pathways such as hedgehog (Hh), fibroblast growth factor (FGF), Janus kinase/signal transducers, transcription activation (Jak/STAT), transforming growth factor-β (TGF-β), receptor tyrosine kinase (RTK), hypoxia pathways, and Wnt pathways (Gustafsson et al., 2005; Hurlbut et al., 2007). These signaling pathways have been extensively studied, and the future research might unravel the complex interactions and pathophysiology involved.
Notch receptors bind the Delta/Serrate/Lag2 ligands, also known as DSL ligands. In mammals, the DSL ligands are sub-divided into five types, which are grouped into two classes, that is, the Delta-like (homology with Drosophila Delta), which includes the DLL1, DLL3, and DLL4, as well as Serrate-like and the Serrate, which included Jagged1 and Jagged2 (Figure 2). Both the Delta-like and Serrate-like ligands have an extracellular domain having a signal peptide, an amino-terminal domain, a DSL domain, and several EGF-like repeats (Nye and Kopan, 1995).
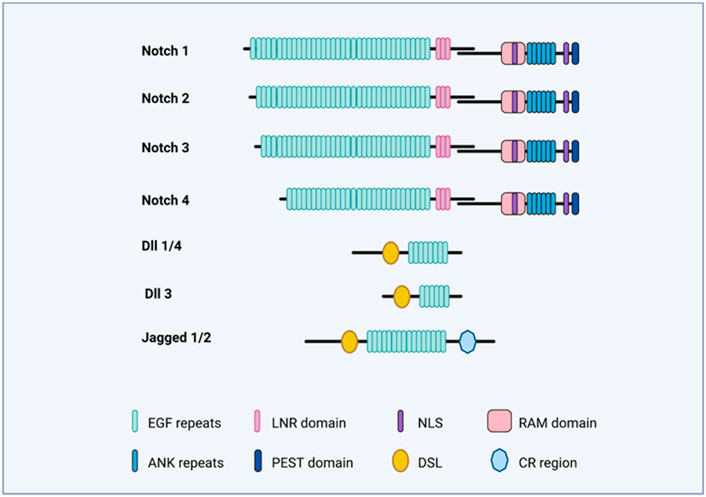
FIGURE 2. Notch-signaling illustration. The figure illustrates the domains of Notch receptors (Notch 1–4) and Notch ligands (DLL1,3,4; Jag1 and Jag2).
In mammals, Notch receptors (Notch1–4) have four types (Figure 2), and it is structurally divided into an extracellular and intracellular domain. The extracellular domain (ECD) has EGF-like 29–36 repeats and three Lin-12/Notch (LIN) repeats. The EGF-like repeats binds the ligand, while the LIN repeats prevent ligand-independent signaling. The intracellular domain (ICD) contains the RBPj-associated molecule (RAM) domain, and a six-ankyrin (ANK) repeats domain. Both of these are protein-interacting domains. Apart from these, the intracellular domain also contains a transactivation domain (TAD), a PEST sequence, and nuclear localization signals (NLSs).
The Notch-signaling pathway is very simple, as the intracellular domain (ICD) of the Notch receptor lacks the second messengers and is thus directly involved in the target gene transcription (Figure 3). However, Notch-signaling modifiers have been involved in the complex signaling specificity (Bray, 2006). Once synthesized as a single polypeptide, Notch protein travels through the Golgi apparatus where it may be further modified (Panin et al., 1997) and is cleaved by a furin-like convertase via S1 cleavage in the trans-Golgi network convertase (Logeat et al., 1998). The associated specific epidermal growth factor (EGF)-like repeats are modified in the endoplasmic reticulum (ER) by O-fucosylation via O-fucosyltransferase 1 (Pofut1).
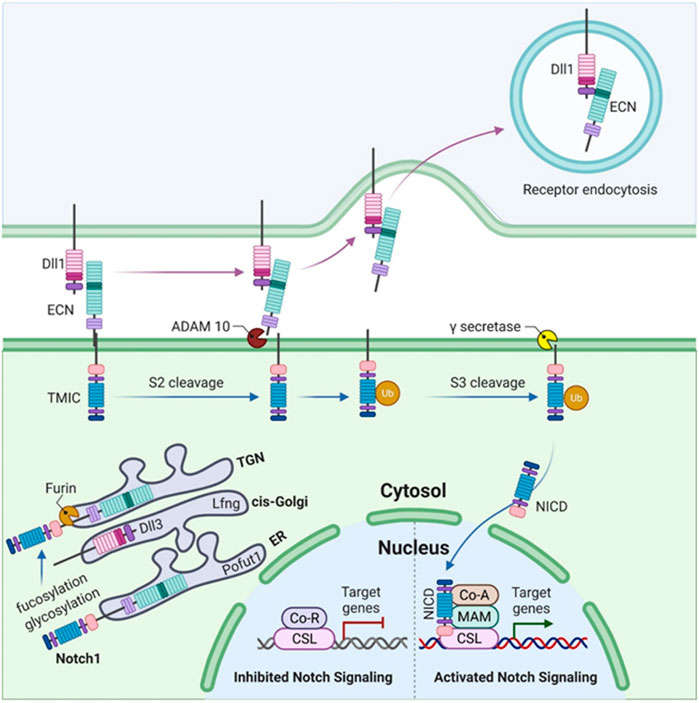
FIGURE 3. Somitogenesis in mammals illustrating Notch1 signaling. The figure illustrates the synthesis of Notch1 as a single polypeptide. O-fucosylation of specific EGF-like repeats occurs in the endoplasmic reticulum (ER) by O-fucosyltransferase 1 (Pofut1), followed by the elongation of these O-fucosylated EGF-like repeats (upon addition of GlcNAc by lunatic fringe, Lfng) as the Notch1 polypeptide passes through the cis-Golgi. S1 cleavage occurs in the trans-Golgi network (TGN) by a furin-like convertase. The Notch1 heterodimer (composed of an N-terminal extracellular truncation, ECN, and a C-terminal transmembrane and intracellular domain, TMIC) moves toward the cell surface where it binds DLL1 via ECN in trans. This binding leads to the activation of S2 cleavage by the disintegrin and metallopeptidase domain (ADAM) proteases, ADAM10 or ADAM17. This liberates the Notch1 ECN which then undergoes ubiquitylation (Ub) and is endocytosed. S3 cleavage of Notch1 ECN occurs in the transmembrane domain via γ-secretase which releases the Notch1 intracellular domain (NICD). NICD binds CSL which is a DNA-binding protein. Upon binding to CSL, co-repressor (CoR) proteins and histone deacetylases (HDACs) are released while the binding of coactivators (CoAs) occurs simultaneously which results in the activation of Notch signaling, leading to the transcription of target genes such as MESP2, LFNG, and HES7.
The C-terminal transmembrane, N-terminal extracellular truncation (ECN), and intracellular domain (TMIC) of the Notch receptor fragments form a heterodimer that proceeds toward the cell surface (Rand et al., 2000; Sanchez-Irizarry et al., 2004). Once it has reached the cell surface, it is cleaved by disintegrin (S2 cleavage) and proteases (metalloprotease domain, ADAM10 and ADAM17) due to the interaction of the ECN with the respective ligand (in the trans), resulting in the separation of Notch heterodimer separation through transendocytosis of the ECN into the signal-sending cell (Pan and Rubin, 1997; Brou et al., 2000; Hartmann et al., 2002). The Notch-signaling pathway in Figure 3 summarizes the function and involvement of each gene.
1.11 Diagnosis and genetic counseling
SCDO is mostly diagnosed by the radiological features in association with NGS (WES/WGS). The identification of the culprit gene defines the type of SCDO. It is characterized into different subtypes as a result of homozygous mutations in several genes causing autosomal recessive SCDO, such as DLL3, DLL1, LFNG, RIPPLY2, MESP2, HES7, and TBX6. Once the mutation in the specific gene is identified, genetic counseling and family carrier testing can be performed. Carrier testing will help to reduce the occurrence of diseases in coming generations (Umair et al., 2017).
2 Discussion
Genetic deformities of the skeletal system are characterized by malformation, inconsistent growth, and distortion of individual bones or groups of bones, which can cause syndromic and non-syndromic forms (Umair et al., 2019). Genetic skeletal deformities can occur due to disruption in the intricate processes of skeletal development, growth, and homeostasis. The inheritance pattern of genetic skeletal deformities can be either autosomal (dominant or recessive) or X-linked (dominant or recessive) (Umair et al., 2018b; Umair and Hayat, 2020; Ahmad et al., 2022). Similarly, spondylocostal dysostosis is a severe form of skeletal deformity that leads to severe ribs and vertebrae malformations. With the advent of NGS technology, molecular diagnosis of rare skeletal deformities such as spondylocostal dysostosis is now cost effective and quick (Umair et al., 2018a). Screening the associated seven genes in patients suffering from disorders such as spondylocostal dysostosis will enable the researchers and clinicians to treat the patient at an early stage. In addition, techniques such as Noninvasive prenatal testing (NIPT), Pre-implantation genetic testing for aneuploidy (PGT-A) and Pre-implantation genetic testing for monogenic disorders (PGT-M) have been employed to reduce the occurrence of severe genetic disorders (Alyafee et al., 2021a; Alyafee et al., 2021b; Alyafee et al., 2022). Therapeutic interventions are only possible if we know the exact molecular origin of a disease that might help the researchers to identify the exact biomarkers. In such a scenario, knowledge about the molecular etiology and pathophysiology of such a rare genetic disease is a must to implement and draw future therapeutic interventions.
The Notch-signaling pathway plays a major role in numerous cell developmental processes, the major one being the skeletal system development and bone homeostasis. The Notch-signaling system utilizes Notch receptors as a key component in the regulation of cell differentiation and function. The four Notch receptors carry out cell-specific functions in the skeletal system and any mutations in them cause alterations in the Notch signaling. Several skeletal malignancies and development of various congenital bone disorders have been reported to be associated with the variations in the Notch-signaling pathway (Canalis, 2018).
The Notch-signaling pathway is an absolute requirement for normal somitogenesis in vertebrates which has been confirmed by mouse models and cell culture studies. The somite boundary formation and its patterning largely depend on the Notch1 activity. The Notch pathway genes are largely involved in the rostral-caudal patterning, synchronized oscillations in the segmentation clock in PSM, and the segmental border formation. Disease causing genetic mutations encoding the protein involved in these essential processes has been reported to be the source of severe vertebral deformities in humans Notch pathway components (Dunwoodie SL., 2009).
Investigation of the genetic causes of abnormal vertebral segmentation (AVS), using mouse models, has reported certain genes that are important for normal vertebral development as they are the activators of Notch, FGF, and Wnt signaling pathways. These signaling pathways are pivotal for normal somite production and bones development in vertebrates. Mutations in these genes, that is, DLL3, MESP2, LFNG, and HES7, cause congenital AVS disorder, SCDO. DLL3 and DLL1 are worth mentioning as these genes serve as the DSL ligands of Notch and are likely to inhibit the pathway by acting as an inhibitor of signaling (Dunwoodie S. L., 2009).
Disease-causing variants in Notch-signaling pathway members cause different types of developmental disorders that affect different parts of the body, including the skeleton, heart, liver, kidneys, eye, face, and vasculature. Notch-associated disorders include different types of severe disorders such as Alagille syndrome caused by mutations in both ligand JAG1 and receptor Notch2 and autosomal recessive spondylocostal dysostosis, caused by mutations in ligand DLL3, and many other members of the Notch-signaling pathway (Penton et al., 2012). Similarly, the other genes associated with recessive spondylocostal dysostosis (MESP2, LFNG, and HES7) have been associated with regulating Notch-signaling activity during somitogenesis (Penton et al., 2012).
It has been observed that DLL3 interacts with full-length Notch1 in the late endocytic compartment and that high levels of DLL3 expression are correlated with low levels of Notch1, suggesting that DLL3 targets Notch1 for lysosomal degradation (Chapman et al., 2010). The LFNG is an N-acetylglucosaminyltransferase that adds N-acetylglucosamine to fucose residues on Notch receptors and acts to suppress Notch signaling during somitogenesis in mice (Sparrow et al., 2006). HES7 acts to downregulate LFNG expression. LFNG, DLL3, and MESP2 mutant embryos display a broadening of Notch-signaling activity during somitogenesis, detected by antibodies to NICD (Chapman et al., 2010; Feller et al., 2008) (Figures 3, 4). DLL3 is predicted to participate in lysosomal degradation of Notch1, MESP2 destabilizes MAML, and LFNG down-regulates Notch signaling immediately posterior to the forming somite (Chapman et al., 2010; Sasaki et al., 2011) (Figures 2, 4). Not surprisingly, ubiquitous activation of Notch in the presomitic mesoderm results in abnormal somite formation (Feller et al., 2008).
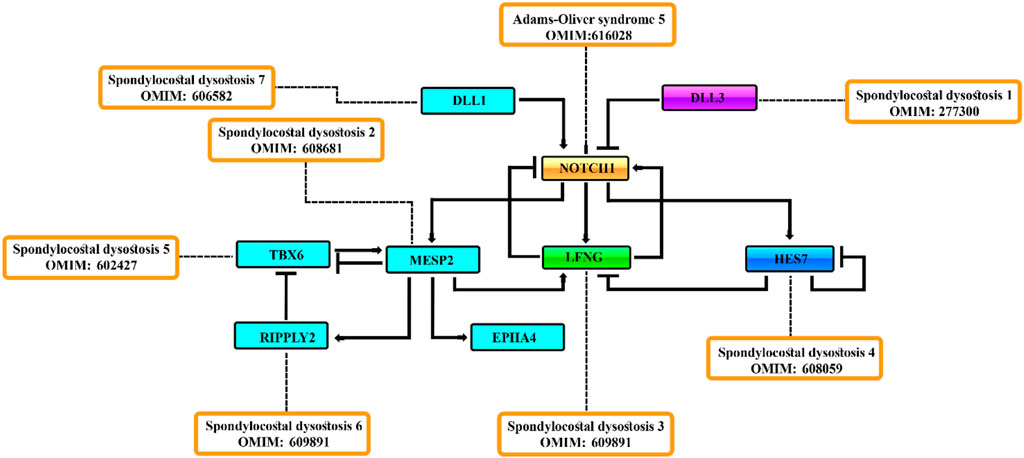
FIGURE 4. Negative feedback [act as repressors] loop consisting of the MESP2, LFNG, HES7, and DLL3 genes regulate the Notch-signaling activity during somitogenesis.
3 Conclusion
In conclusion, proper somite formation is dependent on the Notch-signaling pathway which regulates the skeletal homeostasis. SCDO is an abnormal congenital disorder of vertebral and rib deformation which occurs due to the mutations of the genetic components of the Notch-signaling pathway. SCDO is diagnosed based on characteristic symptoms, family history, an extensive clinical evaluation (radiographs of the spine), and molecular diagnosis (WGS/WES). No specific treatment is available; however, surgery can be performed to repair an inguinal hernia and scoliosis, and antibiotics can be suggested for recurrent respiratory infections. As a rare genetic disorder, the exact incidence or prevalence of SCDO is unknown, and a proper genetic and molecular analysis is required to identify the culprit gene, which might help in proper diagnosis and timely treatments.
Author contributions
MU: idea and wrote the first draft; MY, SS, and AN: edited and approved the final draft. MA: edited the final version.
Funding
This study was funded by King Abdullah International Medical Research Center (KAIMRC) (RC19/352/R).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alyafee, Y., Alam, Q., Altuwaijri, A., Umair, M., Haddad, S., Alharbi, M., et al. (2021a). Next-generation sequencing-based pre-implantation genetic testing for aneuploidy (PGT-A): First report from Saudi arabia. Genes (Basel) 12 (4), 461. doi:10.3390/genes12040461
Alyafee, Y., Al Tuwaijri, A., Alam, Q., Umair, M., Haddad, S., Alharbi, M., et al. (2021b). Next generation sequencing based non-invasive prenatal testing (nipt): First report from Saudi Arabia. Front. Genet. 12, 630787. doi:10.3389/fgene.2021.630787
Alyafee, Y., Al Tuwaijri, A., Umair, M., Alharbi, M., Haddad, S., Ballow, M., et al. (2022). Non-invasive prenatal testing for autosomal recessive disorders: A new promising approach. Front. Genet. doi:10.3389/fgene.2022.1047474
Ahmad, Z., Liaqat, R., Palander, O., Bilal, M., Zeb, S., Ahmad, F., et al. (2022). Genetic overview of postaxial polydactyly (PAP): Updated classification. Clin. Genet. doi:10.1111/cge.14224
Barhoumi, T., Nashabat, M., Alghanem, B., Alhallaj, A., Boudjelal, M., Umair, M., et al. (2019). Delta like-1 gene mutation: A novel cause of congenital vertebral malformation. Front. Genet. 510, 534. doi:10.3389/fgene.2019.00534
Bessho, Y., Sakata, R., Komatsu, S., Shiota, K., Yamada, S., and Kageyama, R. (2001). Dynamic expression and essential functions of Hes7 in somite segmentation. Genes Dev. 15, 2642–2647. doi:10.1101/gad.930601
Bray, S. J. (2006). Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 7, 678–689. doi:10.1038/nrm2009
Brou, C., Logeat, F., Gupta, N., Bessia, C., LeBail, O., Doedens, J. R., et al. (2000). A novel proteolytic cleavage involved in notch signaling: The role of the disintegrin-metalloprotease TACE. Mol. Cell 5, 207–216. doi:10.1016/s1097-2765(00)80417-7
Bulman, M. P., Kusumi, K., Frayling, T. M., McKeown, C., Garrett, C., Lander, E. S., et al. (2000). Mutations in the human delta homologue, DLL3, cause axial skeletal defects in spondylocostal dysostosis. Nat. Genet. 24, 438–441. doi:10.1038/74307
Canalis, E. (2018). Notch in skeletal physiology and disease. Osteoporos. Int. 29 (12), 2611–2621. doi:10.1007/s00198-018-4694-3
Cetinkaya, M., Ozkan, H., Köksal, N., Yazici, Z., and Yalçinkaya, U. (2008). Spondylocostal dysostosis associated with diaphragmatic hernia and neural tube defects. Clin. Dysmorphol. 17, 151–154. doi:10.1097/MCD.0b013e3282f2699c
Chan, T., Kondow, A., Hosoya, A., Hitachi, K., Yukita, A., Okabayashi, K., et al. (2007). Ripply2 is essential for precise somite formation during mouse early development. FEBS Lett. 581, 2691–2696. doi:10.1016/j.febslet.2007.05.017
Chapman, G., Sparrow, D. B., Kremmer, E., and Dunwoodie, S. L. (2010). Notch inhibition by the ligand DELTA-LIKE 3 defines the mechanism of abnormal vertebral segmentation in spondylocostal dysostosis. Hum. Mol. Genet. 20 (5), 905–916. doi:10.1093/hmg/ddq529
Cornier, A. S., Staehling-Hampton, K., Delventhal, K. M., Saga, Y., Caubet, J. F., Sasaki, N., et al. (2008). Mutations in the MESP2 gene cause spondylothoracic dysostosis/Jarcho-Levin syndrome. Am. J. Hum. Genet. 82, 1334–1341. doi:10.1016/j.ajhg.2008.04.014
Dequéant, M. L., Glynn, E., Gaudenz, K., Wahl, M., Chen, J., Mushegian, A., et al. (2006). A complex oscillating network of signaling genes underlies the mouse segmentation clock. Science 314, 1595–1598. doi:10.1126/science.1133141
Dunwoodie, S. L. (2009b). Mutation of the fucose-specific beta1, 3 N-acetylglucosaminyltransferase LFNG results in abnormal formation of the spine. Biochim. Biophys. Acta 1792 (2), 100–111. doi:10.1016/j.bbadis.2008.11.003
Dunwoodie, S. L., Clements, M., Sparrow, D. B., Sa, X., Conlon, R. A., and Beddington, R. S. (2002). Axial skeletal defects caused by mutation in the spondylocostal dysplasia/pudgy gene DLL3 are associated with disruption of the segmentation clock within the presomitic mesoderm. Development 129, 1795–1806. doi:10.1242/dev.129.7.1795
Dunwoodie, S. L. (2009a). The role of Notch in patterning the human vertebral column. Curr. Opin. Genet. Dev. 19 (4), 329–337. doi:10.1016/j.gde.2009.06.005
Feller, J., Schneider, A., Schuster-Gossler, K., and Gossler, A. (2008). Noncyclic Notch activity in the presomitic mesoderm demonstrates uncoupling of somite compartmentalization and boundary formation. Genes Dev. 22 (16), 2166–2171. doi:10.1101/gad.480408
Fischer-Zirnsak, B., Segebrecht, L., Schubach, M., Charles, P., Alderman, E., Brown, K., et al. (2019). Haploinsufficiency of the notch ligand DLL1 causes variable neurodevelopmental disorders. Am. J. Hum. Genet. 105 (3), 631–639. doi:10.1016/j.ajhg.2019.07.002
Goldbeter, A., and Pourquié, O. (2008). Modeling the segmentation clock as a network of coupled oscillations in the Notch, Wnt and FGF signaling pathways. J. Theor. Biol. 252, 574–585. doi:10.1016/j.jtbi.2008.01.006
Gray, P. A., Fu, H., Luo, P., Zhao, Q., Yu, J., Ferrari, A., et al. (2004). Mouse brain organization revealed through direct genome-scale TF expression analysis. Science 306, 2255–2257. doi:10.1126/science.1104935
Gucev, Z. S., Tasic, V., Pop-Jordanova, N., Sparrow, D. B., Dunwoodie, S. L., Ellard, S., et al. (2010). Autosomal dominant spondylocostal dysostosis in three generations of a Macedonian family: Negative mutation analysis of DLL3, MESP2, HES7, and LFNG. Am. J. Med. Genet. A 152a, 1378–1382. doi:10.1002/ajmg.a.33471
Gustafsson, M. V., Zheng, X., Pereira, T., Gradin, K., Jin, S., Lundkvist, J., et al. (2005). Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev. Cell 9, 617–628. doi:10.1016/j.devcel.2005.09.010
Hartmann, D., de Strooper, B., Serneels, L., Craessaerts, K., Herreman, A., Annaert, W., et al. (2002). The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum. Mol. Genet. 11, 2615–2624. doi:10.1093/hmg/11.21.2615
Hurlbut, G. D., Kankel, M. W., Lake, R. J., and Artavanis-Tsakonas, S. (2007). Crossing paths with Notch in the hyper-network. Curr. Opin. Cell Biol. 19, 166–175. doi:10.1016/j.ceb.2007.02.012
Ishibashi, M., Ang, S. L., Shiota, K., Nakanishi, S., Kageyama, R., and Guillemot, F. (1995). Targeted disruption of mammalian hairy and Enhancer of split homolog-1 (HES-1) leads to up-regulation of neural helix-loop-helix factors, premature neurogenesis, and severe neural tube defects. Genes Dev. 9, 3136–3148. doi:10.1101/gad.9.24.3136
Kawamura, A., Koshida, S., Hijikata, H., Ohbayashi, A., Kondoh, H., and Takada, S. (2005). Groucho-associated transcriptional repressor ripply1 is required for proper transition from the presomitic mesoderm to somites. Dev. Cell 9, 735–744. doi:10.1016/j.devcel.2005.09.021
Kawamura, A., Koshida, S., and Takada, S. (2008). Activator-to-repressor conversion of T-box transcription factors by the Ripply family of Groucho/TLE-associated mediators. Mol. Cell. Biol. 28, 3236–3244. doi:10.1128/MCB.01754-07
Kusumi, K., Mimoto, M. S., Covello, K. L., Beddington, R. S., Krumlauf, R., and Dunwoodie, S. L. (2004). DLL3 pudgy mutation differentially disrupts dynamic expression of somite genes. Genesis 39, 115–121. doi:10.1002/gene.20034
Lefebvre, M., Duffourd, Y., Jouan, T., Poe, C., Jean-Marçais, N., Verloes, A., et al. (2017). Autosomal recessive variations of TBX6, from congenital scoliosis to spondylocostal dysostosis. Clin. Genet. 91, 908–912. doi:10.1111/cge.12918
Liu, J., Wu, N., Yang, N., Takeda, K., Chen, W., Li, W., et al. (2019). TBX6-associated congenital scoliosis (TACS) as a clinically distinguishable subtype of congenital scoliosis: Further evidence supporting the compound inheritance and TBX6 gene dosage model. Genet. Med. 21, 1548–1558. doi:10.1038/s41436-018-0377-x
Logeat, F., Bessia, C., Brou, C., LeBail, O., Jarriault, S., Seidah, N. G., et al. (1998). The Notch1 receptor is cleaved constitutively by a furin-like convertase. Proc. Natl. Acad. Sci. U. S. A. 95, 8108–8112. doi:10.1073/pnas.95.14.8108
McInerney-Leo, A. M., Harris, J. E., Leo, P. J., Marshall, M. S., Gardiner, B., Kinning, E., et al. (2015). Whole exome sequencing is an efficient, sensitive and specific method for determining the genetic cause of short-rib thoracic dystrophies. Clin. Genet. 88, 550–557. doi:10.1111/cge.12550
Mitsuru Morimoto, M., Takahashi, Y., Endo, M., and Saga, Y. (2005). The Mesp2 transcription factor establishes segmental borders by suppressing Notch activity. Nature 19435 (7040), 354–359. doi:10.1038/nature03591
Morimoto, M., Sasaki, N., Oginuma, M., Kiso, M., Igarashi, K., Aizaki, K., et al. (2007). The negative regulation of Mesp2 by mouse Ripply2 is required to establish the rostro-caudal patterning within a somite. Development 134 (8), 1561–1569. doi:10.1242/dev.000836
Mortier, G. R., Lachman, R. S., Bocian, M., and Rimoin, D. L. (1996). Multiple vertebral segmentation defects: Analysis of 26 new patients and review of the literature. Am. J. Med. Genet. 61, 310–319. doi:10.1002/(SICI)1096-8628(19960202)61:4<310::AID-AJMG3>3.0.CO;2-Y
Muguruma, Y., Hozumi, K., Warita, H., Yahata, T., Uno, T., Ito, M., et al. (2017). Maintenance of bone homeostasis by DLL1 -mediated notch signaling. J. Cell. Physiol. 232, 2569–2580. doi:10.1002/jcp.25647
Nye, J. S., and Kopan, R. (1995). Developmental signaling. Vertebrate ligands for Notch. Curr. Biol. 5, 966–969. doi:10.1016/s0960-9822(95)00189-8
Otomo, N., Mizumoto, S., Lu, H. F., Takeda, K., Campos-Xavier, B., Mittaz-Crettol, L., et al. (2019). Identification of novel LFNG mutations in spondylocostal dysostosis. J. Hum. Genet. 64, 261–264. doi:10.1038/s10038-018-0548-2
Pan, D., and Rubin, G. M. (1997). Kuzbanian controls proteolytic processing of Notch and mediates lateral inhibition during Drosophila and vertebrate neurogenesis. Cell 90, 271–280. doi:10.1016/s0092-8674(00)80335-9
Panin, V. M., Papayannopoulos, V., Wilson, R., and Irvine, K. D. (1997). Fringe modulates Notch-ligand interactions. Nature 387, 908–912. doi:10.1038/43191
Penton, A. L., Leonard, L. D., and Spinner, N. B. (2012). Notch signaling in human development and disease. Semin. Cell Dev. Biol. 23 (4), 450–457. doi:10.1016/j.semcdb.2012.01.010
Rand, M. D., Grimm, L. M., Artavanis-Tsakonas, S., Patriub, V., Blacklow, S. C., Sklar, J., et al. (2000). Calcium depletion dissociates and activates heterodimeric notch receptors. Mol. Cell. Biol. 20, 1825–1835. doi:10.1128/mcb.20.5.1825-1835.2000
Saga, Y., Hata, N., Koseki, H., and Taketo, M. M. (1997). Mesp2: A novel mouse gene expressed in the presegmented mesoderm and essential for segmentation initiation. Genes Dev. 11, 1827–1839. doi:10.1101/gad.11.14.1827
Sanchez-Irizarry, C., Carpenter, A. C., Weng, A. P., Pear, W. S., Aster, J. C., and Blacklow, S. C. (2004). Notch subunit heterodimerization and prevention of ligand-independent proteolytic activation depend, respectively, on a novel domain and the LNR repeats. Mol. Cell. Biol. 24, 9265–9273. doi:10.1128/MCB.24.21.9265-9273.2004
Sasaki, N., Kiso, M., Kitagawa, M., and Saga, Y. (2011). The repression of Notch signaling occurs via the destabilization of mastermind-like 1 by Mesp2 and is essential for somitogenesis. Development 138 (1), 55–64. [PubMed] [Google Scholar]. doi:10.1242/dev.055533
Solomon, L., Jimenez, R. B., and Reiner, L. (1978). Spondylothoracic dysostosis: Report of two cases and review of the literature. Arch. Pathol. Lab. Med. 102, 201–205.
Sparrow, D. B., Chapman, G., and Dunwoodie, S. L. (2011). The mouse notches up another success: Understanding the causes of human vertebral malformation. Mamm. Genome 22, 362–376. doi:10.1007/s00335-011-9335-5
Sparrow, D. B., Chapman, G., Wouters, M. A., Whittock, N. V., Ellard, S., Fatkin, D., et al. (2006). Mutation of the LUNATIC FRINGE gene in humans causes spondylocostal dysostosis with a severe vertebral phenotype. Am. J. Hum. Genet. 78, 28–37. doi:10.1086/498879
Sparrow, D. B., Faqeih, E. A., Sallout, B., Alswaid, A., Ababneh, F., Al-Sayed, M., et al. (2013). Mutation of HES7 in a large extended family with spondylocostal dysostosis and dextrocardia with situs inversus. Am. J. Med. Genet. A 161a, 2244–2249. doi:10.1002/ajmg.a.36073
Sparrow, D. B., Guillén-Navarro, E., Fatkin, D., and Dunwoodie, S. L. (2008). Mutation of Hairy-and-Enhancer-of-Split-7 in humans causes spondylocostal dysostosis. Hum. Mol. Genet. 17, 3761–3766. doi:10.1093/hmg/ddn272
Sparrow, D. B., Sillence, D., Wouters, M. A., Turnpenny, P. D., and Dunwoodie, S. L. (2010). Two novel missense mutations in HAIRY-AND-ENHANCER-OF-SPLIT-7 in a family with spondylocostal dysostosis. Eur. J. Hum. Genet. 18, 674–679. doi:10.1038/ejhg.2009.241
Turnpenny, P. D., Sloman, M., and Dunwoodie, S.ICVS (international consortium for vertebral anomalies and scoliosis) (2009). "Spondylocostal dysostosis, autosomal recessive," in GeneReviews® [internet]. M. P. Adam, H. H. Ardinger, R. A. Pagonet al. (Seattle (WA): University of Washington, Seattle). [Updated 2017 Dec 21]Available from: https://www.ncbi.nlm.nih.gov/books/NBK8828/.
Turnpenny, P. D., Sloman, M., and Dunwoodie, S.ICVS (International Consortium for Vertebral Anomalies and Scoliosis) (1993). “Spondylocostal dysostosis, autosomal recessive,” in GeneReviews(®). Editors M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, G. Mirzaaet al. (Seattle (WA): University of Washington, Seattle Copyright © 1993-2021University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.).
Turnpenny, P. D., Whittock, N., Duncan, J., Dunwoodie, S., Kusumi, K., and Ellard, S. (2003). Novel mutations in DLL3, a somitogenesis gene encoding a ligand for the Notch signalling pathway, cause a consistent pattern of abnormal vertebral segmentation in spondylocostal dysostosis. J. Med. Genet. 40, 333–339. doi:10.1136/jmg.40.5.333
Umair, M., Ahamd, F., Bilal, M., Asiri, A., Younus, M., and Khan, A. (2019). A comprehensive review of genetic skeletal disorders reported from Pakistan: A brief commentary. Meta Gene 20, 100559. doi:10.1016/j.mgene.2019.100559
Umair, M., Ahmad, F., and Ullah, A. (2018a). Whole exome sequencing as a diagnostic tool for genetic disorders in Pakistan. PJMR 57 (2), 90–91.
Umair, M., Ahmad, F., Bilal, M., and Arshad, M. (2018b). Frontonasal dysplasia: A review. J. Biochem. Clin. Gene. 1 (2), 1–14.
Umair, M., and Hayat, A. (2020). Nonsyndromic split-hand/foot malformation: Recent classification. Mol. Syndromol. 10 (5), 243–254. doi:10.1159/000502784
Umair, M., Rafique, A., Ullah, A., Ahmad, F., Ali, R. H., Nasir, A., et al. (2017). Novel homozygous sequence variants in the GDF5 gene underlie acromesomelic dysplasia type-grebe in consanguineous families. Congenit. Anom. 57 (2), 45–51. doi:10.1111/cga.12187
Wahi, K., Bochter, M. S., and Cole, S. E. (2016). The many roles of Notch signaling during vertebrate somitogenesis. Semin. Cell Dev. Biol. 49, 68–75. doi:10.1016/j.semcdb.2014.11.010
Whittock, N. V., Ellard, S., Duncan, J., de Die-Smulders, C. E., Vles, J. S., and Turnpenny, P. D. (2004a). Pseudodominant inheritance of spondylocostal dysostosis type 1 caused by two familial delta-like 3 mutations. Clin. Genet. 66, 67–72. doi:10.1111/j.0009-9163.2004.00272.x
Whittock, N. V., Sparrow, D. B., Wouters, M. A., Sillence, D., Ellard, S., Dunwoodie, S. L., et al. (2004b). Mutated MESP2 causes spondylocostal dysostosis in humans. Am. J. Hum. Genet. 74, 1249–1254. doi:10.1086/421053
Wu, N., Ming, X., Xiao, J., Wu, Z., Chen, X., Shinawi, M., et al. (2015). TBX6 null variants and a common hypomorphic allele in congenital scoliosis. N. Engl. J. Med. 372, 341–350. doi:10.1056/NEJMoa1406829
Zhang, N., Norton, C. R., and Gridley, T. (2002). Segmentation defects of Notch pathway mutants and absence of a synergistic phenotype in lunatic fringe/radical fringe double mutant mice. Genesis 33, 21–28. doi:10.1002/gene.10081
Keywords: Spondylocostal dysostosis, SCDO, Genetic skeletal disorders, SCDO1-7, Notch-signaling pathway
Citation: Umair M, Younus M, Shafiq S, Nayab A and Alfadhel M (2022) Clinical genetics of spondylocostal dysostosis: A mini review. Front. Genet. 13:996364. doi: 10.3389/fgene.2022.996364
Received: 17 July 2022; Accepted: 31 October 2022;
Published: 25 November 2022.
Edited by:
Yi Zhang, Xiangya Hospital, Central South University, ChinaReviewed by:
Lianlei Wang, Peking Union Medical College Hospital (CAMS), ChinaYunjia Wang, Xiangya Hospital, Central South University, China
Copyright © 2022 Umair, Younus, Shafiq, Nayab and Alfadhel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Muhammad Umair, a2h1Z29vNHVAeWFob28uY29t, dW1haXJtdUBuZ2hhLm1lZC5zYQ==