 Jing Chen
Jing Chen Renhua Li2
Renhua Li2 Robert L. Whitener
Robert L. Whitener Edward H. Leiter
Edward H. Leiter Clayton E. Mathews
Clayton E. Mathews- 1Department of Pathology, Immunology, and Laboratory Medicine, University of Florida, Gainesville, FL, United States
- 2Henry M Jackson Foundation for the Advancement of Military Medicine (HJF), Bethesda, MD, United States
- 3The Jackson Laboratory, Bar Harbor, ME, United States
Alloxan (AL)-generated Reactive Oxygen Species (ROS) selectively destroy insulin-producing pancreatic β-cells. A previous genome-wide scan (GWS) using a cohort of 296 F2 hybrids between NOD (AL-sensitive) and ALR (AL-resistant) mice identified linkages contributing to β-cell susceptibility or resistance to AL-induced diabetes on Chromosomes (Chr) 2, 3, 8, and a single nucleotide polymorphism in mt-Nd2 of the mitochondrial genome (mtDNA). AL treatment of congenic and consomic NOD mouse stocks confirmed resistance linked to both the mtDNA and the Chr 8 locus from ALR [NOD.mtALR.ALR-(D8Mit293-D8Mit137)]. To identify possible epistatic interactions, the GWS analysis was expanded to 678 F2 mice. ALR-derived diabetes-resistance linkages on Chr 8 as well as the mt-Nd2a allele were confirmed and novel additional linkages on Chr 4, 5, 6, 7, and 13 were identified. Epistasis was observed between the linkages on Chr 8 and 2 and Chr 8 and 6. Furthermore, the mt-Nd2 genotype affected the epistatic interactions between Chr 8 and 2. These results demonstrate that a combination of nuclear-cytoplasmic genome interactions regulates β-cell sensitivity to ROS-mediated ALD.
Highlights
• Reactive oxygen species play important role in the beta cell death that accompanies in both Type 1 and Type 2 diabetes
• We utilized an alloxan-induced diabetes mouse model to determine the genetic contributions to beta cell death
• Beta cell sensitivity to free radicals is controlled by both the nuclear genome and a polymorphism in the mitochondrial gene, mt-Nd2
• Epistatic and intergenomic interactions regulate beta cell resistance to free radicals
Introduction
Alloxan (AL) is a potent free radical/reactive oxygen species (ROS) generator and selective pancreatic β-cell toxin in mice and rats. In these species β-cells are especially sensitive to ROS because of their low levels of antioxidative capabilities (Lenzen et al., 1996; Tiedge et al., 1997; Mathews et al., 2001); however, the genetic determinants of β-cell sensitivity to ROS are not well understood. Young prediabetic NOD/ShiLtJ (NOD) mice are sensitive to a threshold AL dose whereas mice of the closely related ALR/LtJ (ALR) strain, selected for AL resistance, maintain strong resistance (Ino et al., 1991). Indeed, pancreatic islet cells of the ALR strain, unlike those of autoimmune diabetes-prone NOD strain, are remarkable in their resistance to a broad variety of diabetogenic stresses, including ROS, proinflammatory cytokines, and immune cell-mediated destruction (Ino et al., 1991; Mathews et al., 2001; Mathews et al., 2003). The differential strain sensitivity to AL-mediated diabetes (ALD) has previously been employed to probe the underlying genetic control of β-cell susceptibility/resistance to ROS-mediated damage. A preliminary analysis using an F2 cohort of 296 mice linked β-cell ALD resistance to loci in both the nuclear and mitochondrial (mtDNA) genomes (Chen et al., 2008b). Nuclear loci contributing to susceptibility or resistance were identified on Chromosomes (Chr) 2, 3, and 8.
Mitochondria (mt) are important sources of ROS generation (Mathews et al., 2005; Gusdon et al., 2007; Chen et al., 2008b; Gusdon et al., 2008). The mt electron transport chain proteins are encoded by genes in both nuclear and mt genomes therefore, normal mt function requires the activity of both genomes. Although mtDNA mutations and/or polymorphisms have been attributed to many human diseases including diabetes (Mathews et al., 1999; Mathews et al., 2005; Wallace, 2010), the interaction between mtDNA and nuclear genome in the pathogenesis of diabetes has not been extensively studied. The only polymorphism distinguishing the mtDNA of ALR from NOD is a single nucleotide polymorphism (SNP) at mtDNA position 4,738 (C4738A) in mt-Nd2, NADH dehydrogenase subunit 2, resulting in a leucine to methionine amino acid substitution at residue 276 (L276M) in ALR (Mathews et al., 2005). The NOD allele is denoted as mt-Nd2c and the ALR allele as mt-Nd2a (Mathews et al., 2005). This unique difference in the mtDNA allows the analysis of mt-nuclear interactions in a manner linking nuclear loci to mt-Nd2. To define additional linkages as well as explore epistasis and intergenomic nuclear-mt interactions, we expanded the GWS study cohort to 678 F2 mice. Here we demonstrate that ALD susceptibility is a polygenic trait entailing epistatic interaction between the nuclear and mt genomes.
Methods
Mice
NOD/ShiLtJ, NOD.129S7(B6)-Rag1tm1Mom/J (NOD.Rag1−/−), and ALR/LtJ (ALR) mice were bred and maintained in our specific pathogen-free animal facility at the University of Florida. Progeny of reciprocal F1 matings between the NOD and ALR strains were intercrossed in 4 parental combinations, as described previously (Chen et al., 2008b). In the current study, the total number of F2 individuals used for the genome-wide scan (GWS) was 678, including the 296 previously reported (Chen et al., 2008b). NOD. ALRc8 congenic mice, with a long congenic segment from D8Mit293 (36.2 Mb) to D8Mit137 (103.9 Mb) were created as previously described (Chen et al., 2008a). Interval specific Chr 8 congenic mice were generated by outcrossing NOD. ALRc8 (D8Mit293-D8Mit137) mice to the parental NOD strain, typing the progeny with microsatellite markers throughout this interval, and selecting recombinants for ALR markers between D8Mit293 and D8Mit137. This resulted in the establishment of two sub-congenic lines: NOD. ALRc8 (D8Mit293-D8Mit74) and NOD. ALRc8 (D8Mit268-D8Mit137). These shortened congenic regions flank 36.2 Mb–77.8 Mb and 88.3 Mb–103.9 Mb on Chr 8, respectively. All procedures described within this manuscript were approved by the University of Florida’s Institutional Animal Care and Use Committee (UF-IACUC 201905476) and carried out in accordance with the National Institutes of Health guide for the care and use of Laboratory animals.
Alloxan injection and diagnosis of diabetes
Freshly prepared alloxan monohydrate (Sigma, St. Loius, MO) was administered i.v. to both male and female F2 mice as well as male congenic mice through the tail vein at 6 weeks of age (Chen et al., 2008b). All F2 mice in the GWS received the dose of 52 mg/kg. Congenic mice were subjected to a dose response where AL was administered at the escalating doses of 45 mg/kg, 50 mg/kg, or 55 mg/kg. Blood from non-fasted mice was drawn immediately prior to the injection of AL, and then at 7 and 14 days post-AL administration. Glucose levels were measured with a One Touch Ultra 2 glucometer (Life Scan, Milpitas, CA). Alloxan-induced diabetes (ALD) was diagnosed in mice with blood glucose of ≥ 250 mg/dl (Chen et al., 2008b).
DNA extraction and genotyping
DNA was extracted from kidney and a GWS performed using a total of 80 microsatellite and 81 single nucleotide polymorphism (SNP) markers, as described previously (Chen et al., 2008b). Physical positions of markers were based on Mouse Genome Informatics at The Jackson Laboratory (http://www.informatics.jax.org/) and NCBI build 37 of the mouse genome (http://www.ncbi.nlm.nih.gov/projects/genome/guide/mouse/).
Quantitative trait loci and statistical analyses
Quantitative Trait Loci (QTL) linkage analysis was performed using R/qtl (The Jackson Laboratory, Bar Harbor, ME. http://www.rqtl.org/) (Broman et al., 2003). Thresholds for LOD scores are based on 1,000 permutation tests (Churchill and Doerge, 1994). In this intercross the values of LOD score thresholds were 2.23 and 3.27 for suggestive (p < 0.63) and significant (p < 0.05) levels, respectively, when sex was included as an additive covariate. Confidence intervals of QTLs were determined by using the posterior probability density method (Chen and Kendziorski, 2007). Chi-squares and p values were calculated using JMP software (SAS Institute Inc.).
Transcriptome analysis
Transcriptome analysis was performed after isolation of RNA from isolated islets using Affymetrix Genechip Mouse Gene 2.0 ST array. Pancreatic islets from 10–12-week-old NOD.ALRc8 (D8Mit293-D8Mit137) and NOD.Rag1−/− mice were isolated as previously described (Whitener et al., 2017). As a broad range of insulitis severity is seen at this age range in NOD mice, immunodeficient NOD-Rag1−/− mice that do not develop insulitis or T1D were used in lieu of NOD mice. Pancreata from two mice were used to isolate pools of 200 and 250 islets per strain. At least three pools of islets were generated for each mouse strain. RNA was extracted for islets using the Qiagen RNeasy kit according to the manufacturer’s instructions (Qiagen, Hilden, Germany). All extractions were performed inside a benchtop hood to minimize the potential for RNAse contamination and included on-column DNA digestion. Quality of the extracted RNA was verified using the Bio-Rad Experion StdSense RNA kit (Bio-Rad, Hercules, CA). All of the samples had RQI values above 6.5. As we used whole tissue comprised of multiple cell types, significance thresholds for differential expression were initially conservatively set at |log2(FC)|> 0.5 with a p-value of ≤ 0.05.
Results
The maternally-derived ALR mtDNA Nd2a allele contributes to ALD resistance
Our previous results have demonstrated that ALR females and males resist ALD while NOD females and males are susceptible to ALD development at a dose of 52 mg/kg (Mathews et al., 2005; Chen et al., 2008b). At this dose, F1 hybrid progeny of both sexes from crosses of ALR females to NOD males [(RxD)F1] resist ALD while their counterpart (DxR)F1 mice do not. As previously reported, spontaneous diabetes did not occur in any of the reciprocal F1 hybrids (Mathews et al., 2005; Chen et al., 2008b). The incidence of ALD was 23.2% (157 out of 678) in the total cohort of reciprocal F2 mice (Table 1: both sexes and all cross directions combined) and remaining close to our reported results using a smaller F2 population [62 out of 296 (20.9%) (Chen et al., 2008b)]. Males exhibited a two-fold higher incidence of ALD compared to females (30.9% versus 15.4%). Stratification of the results by cross direction and sex demonstrated that F2 male mice with both the mtDNA and Chr Y derived from NOD had the highest ALD incidence (47.8%) whereas males with the mtDNA and Chr Y from ALR associated with the lowest ALD incidence (17.5%). The SNP at mtDNA position 4,738 in mt-Nd2, the only sequence difference in the mtDNA of ALR versus NOD (Mathews et al., 2005), remained a significant linkage where the ALR-derived allele, mt-Nd2a, provided protection against ALD (Chi-square of 21.2; p < 0.0001). All F2 mice with mt-Nd2c had twice the risk of developing ALD [30.4% incidence (107 out of 353)] compared to a 15.4% (50 out of 325) incidence in mice inheriting mt-Nd2a. This confirmed our previous findings (Mathews et al., 2005) that the SNP variation resulting in a leucine to methionine amino acid substitution in ND2 is an important mediator of β-cell sensitivity to ALD (Chen et al., 2008b).

TABLE 1. Incidence of alloxan-induced diabetes in each direction of the F2 cohort.
ALD is a polygenic phenotype under control of multiple nuclear loci
The present study utilizing a larger segregating population provided additional power to confirm previous contributions to ALD from the nuclear genome and to detect additional linkages (Figures 1,2; Table 2). In this larger cohort, GWS revealed suggestive linkages (with a LOD score threshold of ≥ 2.2) where ALR genome contributed resistance on Chr 4 (dominant, χ2 = 10.9, p = 0.0123), Chr 6 (co-dominant, χ2 = 12.7, p = 0.0017), Chr 7 (dominant, χ2 = 11.9, p = 0.0078), Chr 8 (dominant, χ2 = 12.1, p = 0.0023), and Chr 13 (co-dominant, χ2 = 12.4, p = 0.0021). Interestingly, the ALD-susceptible NOD genome provided a resistance locus on Chr 5 (recessive, χ2 = 11.9, p = 0.0026). Among these 5 loci, only the linkage on Chr 8 was previously reported as associated with ALD using a smaller cohort (Chen et al., 2008b). GWS with the larger cohort failed to confirm a locus on Chr 3 where a susceptibility allele was unexpectedly contributed by ALR (Chen et al., 2008b). Additionally, whereas rs3681744 on Chr 2 had contributed negative heterosis in our previous study (Chen et al., 2008b), it was not confirmed in the larger cohort as an independent linkage. However, this region was identified (as described below) as an interacting locus with Chr. 8 and mt-Nd2.
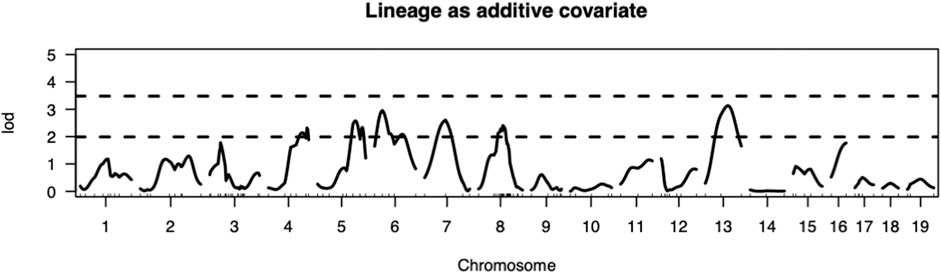
FIGURE 1. Genome-wide analysis revealed QTLs on Chromosomes 4, 5, 6, 7, 8, and 13. Thresholds for LOD scores are based on 1,000 permutation tests. In this intercross the values of LOD score thresholds are 2.23 and 3.27 for suggestive (p < 0.63) and significant (p < 0.05) levels, respectively, when sex is included as an additive covariate and mitochondrial lineage added as additive covariate.
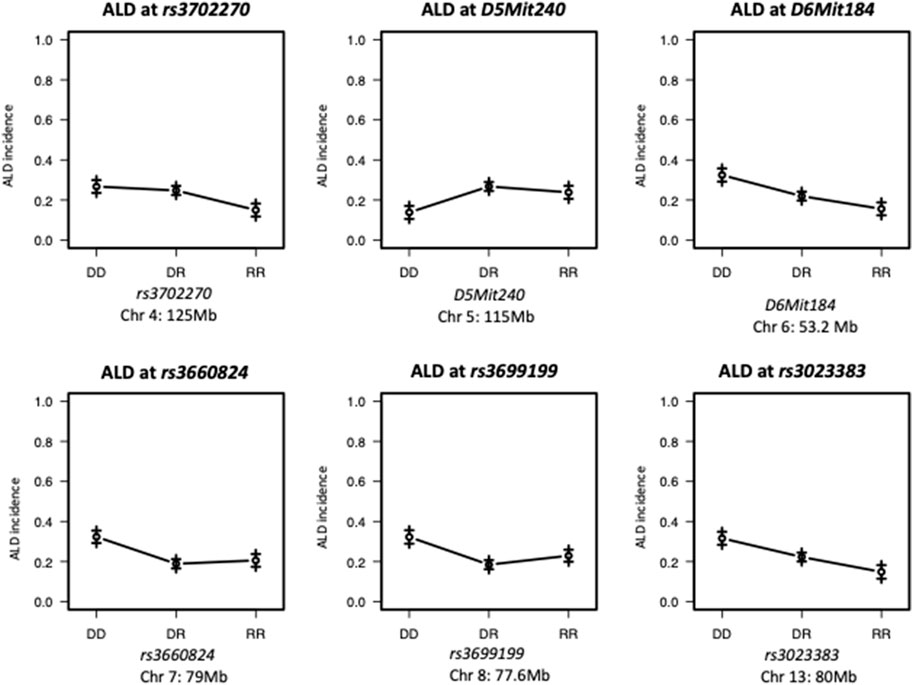
FIGURE 2. Effect Plots of markers on the peak of each chromosome. ALR allele on Chromosome 4 and 7 recessively contributes to resistance; ALR allele on Chromosomes 8 dominantly contributes to resistance. ALR allele on Chromosome 5 dominantly contributes to susceptibility; On Chromosomes 6 and 13, the susceptibility contributed by NOD allele and resistance contributed by ALR allele co-dominantly exist. DD: homozygous for NOD; DR: heterozygous for NOD and ALR; RR: homozygous for ALR.
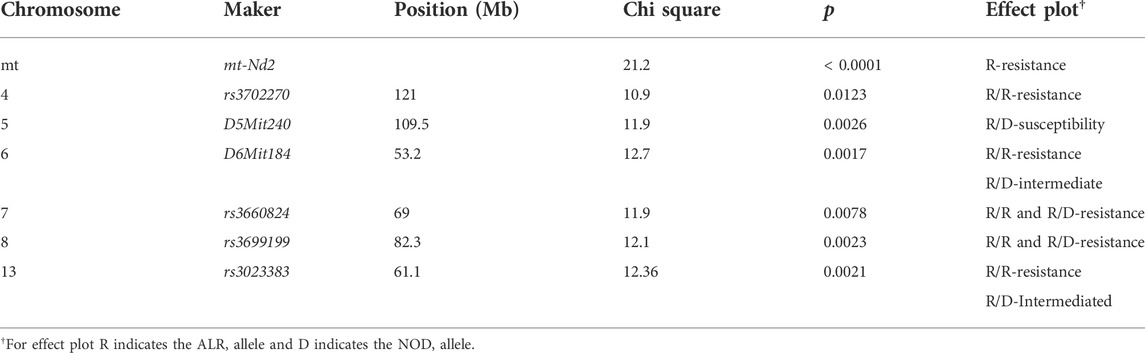
TABLE 2. Loci that are significantly linked to ALD resistance or susceptibility.
Sub-congenic analysis reduced the region of Chromosome 8 providing resistance to ALD
We have previously reported that a Chr 8 congenic line, designated NOD. ALRc8 (D8Mit205-D8Mit137) and generated by introgressing ALR Chr 8 genetic markers into the NOD genome between 50.8 Mb and 103.8 Mb, demonstrated resistance to ALD (Chen et al., 2008b). Further backcrossing to NOD of this long congenic line allowed generation of a shorter sub-congenic strain, NOD. ALRc8 (D8Mit293-D8Mit74) (36.2 Mb–77.8 Mb) that partially overlaps with the long congenic line to reduce the size of this ALR-derived protective genetic region. When challenged with different doses of AL, this short congenic line demonstrated the same resistance as the long congenic line (Figure 3). Thus, the protective locus is located in the proximal region between 50.8 Mb and 77.8 Mb. Consistently, the peak for linkage on Chr 8 is in this region at 77 Mb (Figures 1, 4, 5).
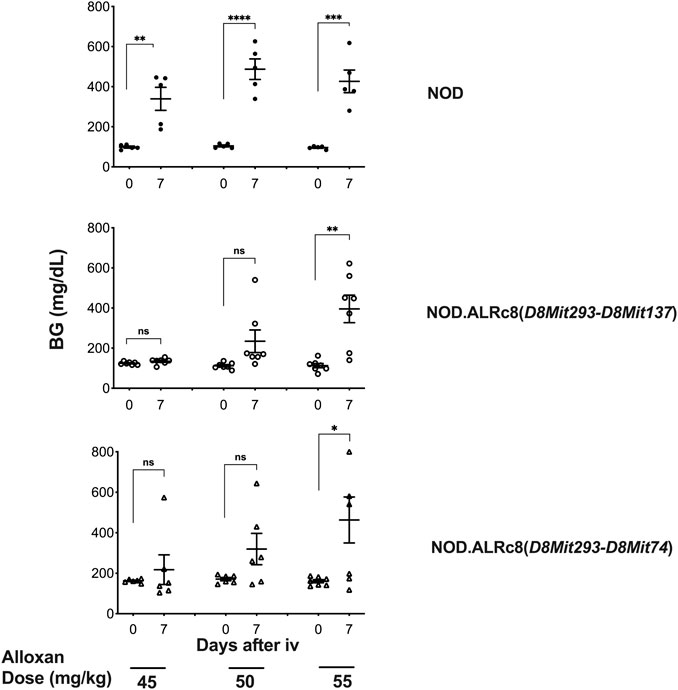
FIGURE 3. Mice congenic for ALR on chromosome 8 with short and long congenic regions show the same resistance to alloxan. Six-week-old male mice were challenged with different doses of alloxan as indicated. Blood glucose were measured before and 7 days after alloxan treatment. Upper panel: NOD control; middle panel: NOD. ALRc8 (D8Mit293-D8Mit137); lower panel: NOD. ALRc8 (D8Mit293-D8Mit74). n = 5–7. Unpaired t test. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
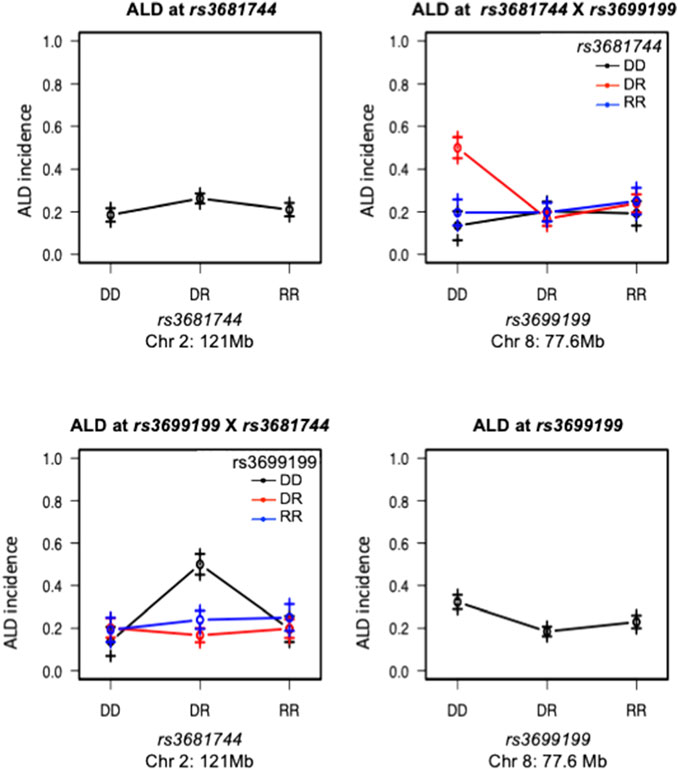
FIGURE 4. Interaction between Chromosomes 2 and 8 was plotted for ALD incidence. Tope left and lower right panels are effect plot of Chr 2 and Chr 8 respectively, same as in Figure 2. Top right panel: Chr 8 genotype is on the x-axis, Chr 2 genotype is shown as group points. ALR allele on Chr 8 dominantly suppresses the sensitivity, therefore the highest ALD incidence is seen in the co-existence of homozygous NOD genotype on Chr 8 and heterozygosity on Chromosome 2; Bottom left panel, showing the same interaction result with Chr 2 genotype on the x-axis and Chr 8 genotype as group points. The interaction is significant as shown in Table 3. DD: homozygous for NOD; DR: heterozygous for NOD/ALR; RR: homozygous for ALR.
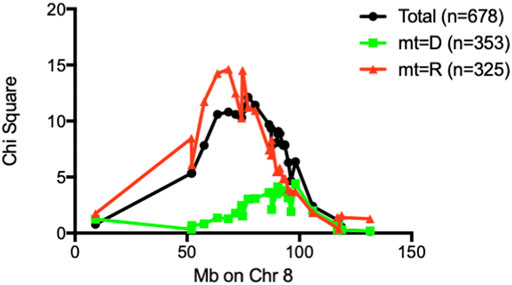
FIGURE 5. Chi-square values of each marker on Chr 8 were plotted against marker position in Mb. When stratified by mt-genome, the linkage existed only when mt-genome is ALR derived mt-Nd2a. Black line: Whole population of F2 (n = 678); Red line: population of F2 mice with ALR-derived mt-Nd2a (n = 325); Green line: population of F2 mice with NOD-derived mt-Nd2c (n = 353).
Epistatic interactions contribute to sensitivity of pancreatic β-cells to free radical induced damage
Interaction between unlinked loci was tested using R/qtl, fitting multiple regression models and dropping one QTL at a time. In addition to confirmation of linkages to mt-Nd2 (C4738A genotyping), Chr 4, 5, 6, 7, 8, and 13 (Figure 1) that were detected in the single QTL scan, a significant interaction between Chr 2 and 8 was detected (Table 3; Figure 4). The highest ALD incidence was observed in mice with the combination of DD genotype at marker rs3699199 on Chr 8 and a heterozygous genotype (DR) at marker rs3681744 on Chr 2 (Figure 4). While this Chr 2 linkage was not detected as a single QTL as it did not reach significance (Figure 1), the current GWS does confirm that rs3681744 contributes overdominance to ALD but only when interacting with homozygous NOD genome marked by rs3699199 on Chr 8.
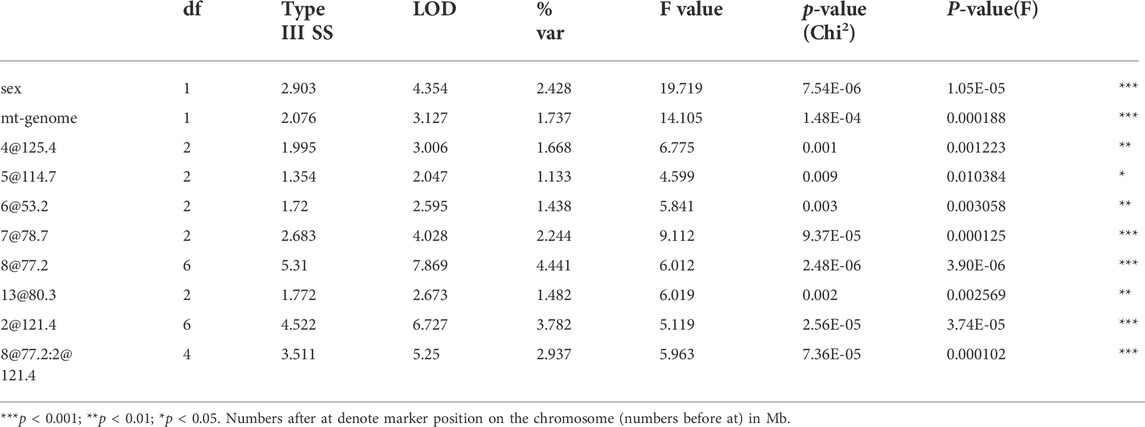
TABLE 3. Multiple regression model, drop one QTL at a time ANOVA table.
Intergenomic interactions affect sensitivity to ALD
An analysis was performed to calculate Chi-squares for each locus tested based on mt genotype. This investigation identified an interaction between mt-Nd2a and rs3699199 on Chr 8 [Figure 2 (effect plot) and Table 2] that provides dominant ALD resistance. In fact, when stratified by mt-genome, the linkage on Chr 8 exists only when mt-genome is ALR derived mt-Nd2a (Figure 5). Further, the intragenomic epistasis between QTL on Chr 8 and 2 was affected by the mt-Nd2c genotype (Figure 6). Here it was identified that mice with mt-Nd2c exhibited increased ALD susceptibility with an overall incidence of 57% when heterozygous for rs3681744 on Chr 2 in combination with homozygosity for the D genotype at rs3699199 on Chr 8 (Figure 6A). In contrast, ALR alleles on Chr 8 dominantly contribute to resistance in F2 mice with the ALR mtDNA, thus reducing ALD incidence in all genotype combinations, including when rs3681744 is heterozygous and rs3699199 is DD, where incidence decreased to 40% (Figure 6B). The different degree of the Chr 8-2 interaction in the two cohorts with different allotypes of mt-Nd2 implicates the important contribution of intergenomic interactions to ALD susceptibility.
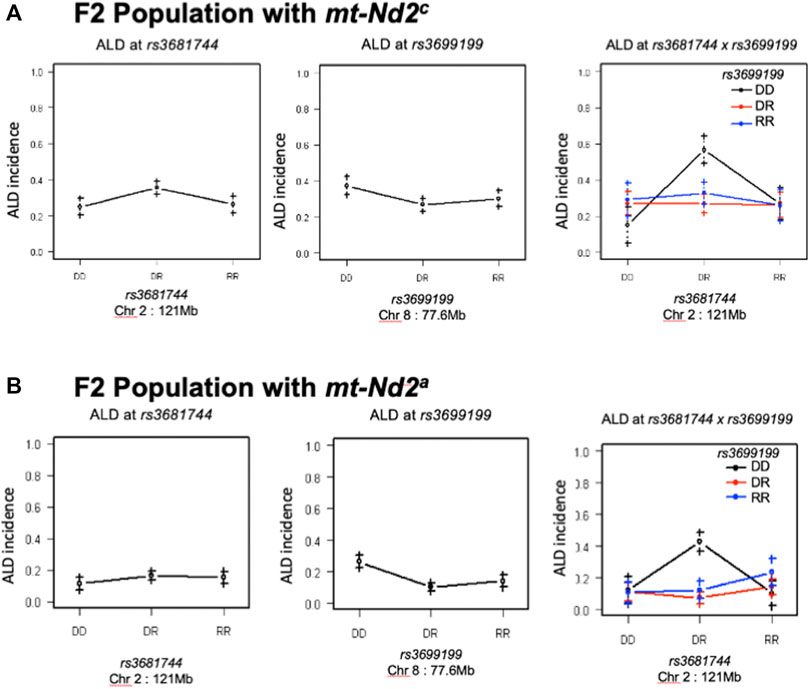
FIGURE 6. The interaction between Chr 8 and 2 were analyzed in 2 subgroups according to mt-genotype. Panel (A), interaction in the subgroup of mice with mt-Nd2c type. Left and middle are the effects of QTL on Chr 2 and 8 respectively. Right plot shows their joint effects with chr 2 genotypes on the x-axis and chr 8 genotypes indicated by grouping of points (DD, black; DR, red; and RR, blue). Panel (B), the same analysis as in Panel A was done in the group of mice with mt-Nd2a type.
Likewise, the interaction between QTLs on Chrs 8 and 6 was highly dependent on the genotype at mtDNA nt 4,738 (Figure 7). Figure 7A elaborates the interactions of Chr 8 and Chr 6 in the full population of F2 mice (both mt populations present). Here the absence of ALR genome on Chr 8 allows the penetrance of the ALD susceptibility phenotype regardless of Chr 6 genotype. In the group of mice with mt-Nd2c allele (Figure 7B) the co-existence of DD on both Chromosomes 6 and 8 contributes to the highest incidence of ALD. Stratification for the ALR-derived mt-Nd2a allele, in comparison to the NOD-derived mt-Nd2c allele, showed suppressed action of homozygous NOD alleles on Chr 6 and Chr 8 respectively (Figure 7B,C). However, in the ALR-derived mt-Nd2a group, combination of NOD homozygosity (DD) on Chr 8 and ALR homozygosity (RR) on Chr 6 (Figure 7C) unexpectedly showed enhanced sensitivity.
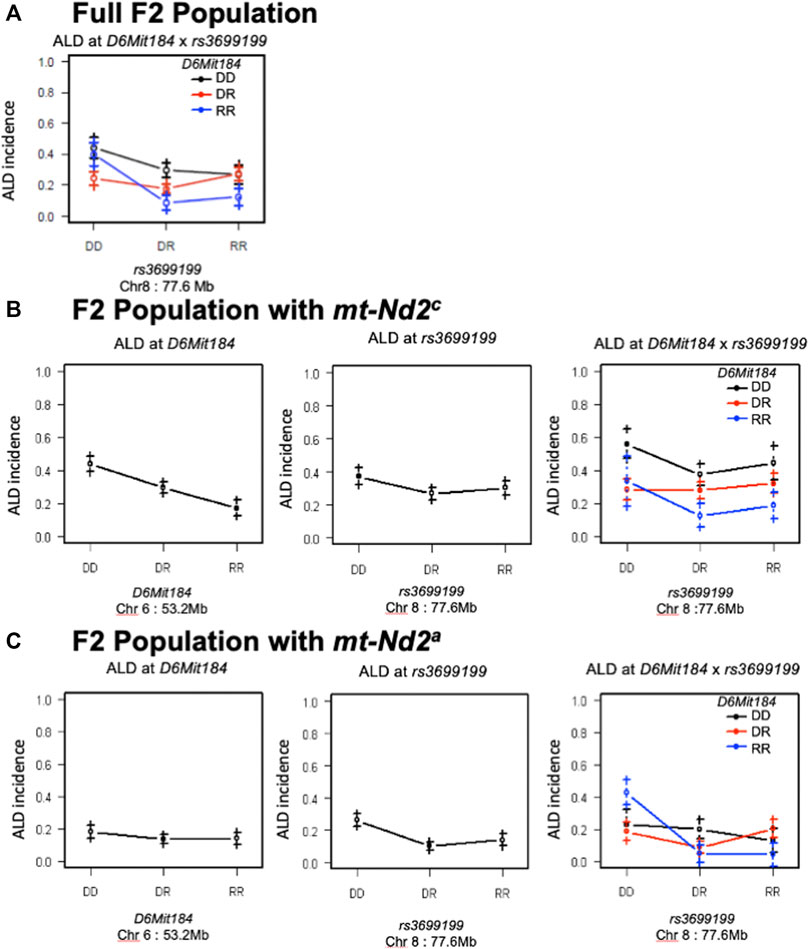
FIGURE 7. The interaction between Chr 8 and 6 were analyzed in 2 subgroups according to mt-genotype. Panel (A) shows the interactions of Chr 8 and Chr 6 in the full population of F2 mice. Panel (B), interaction in the subgroup of mice with NOD derived mt-Nd2c type. In this subgroup, coexistence of homozygous DD type on both chromosomes denotes the highest susceptibility to ALD; panel (C), interaction in the subgroup of mice with mt-Nd2a type. In this group, homozygous DD on Chr 8 and RR on Chr 6 confer the highest susceptibility to ALD.
Genes differentially expressed in islets from insulitis-free NOD-Rag1−/− and NOD.ALRc8(D8Mit293-D8Mit137) mice may confer inter-genomic interaction
Transcriptome analysis revealed over 3,000 transcripts were differentially expressed in the islets of these two mouse strains (threshold values of |log2(FC)|>0.5, p-value <0.05), of which 164 were located on Chr 8. Figure 8 shows some of these genes that are of interest (discussed below). The NOD. ALRc8 (D8Mit293-D8Mit137) and NOD-Rag1−/− islet gene expression array data are available at https://www.ncbi.nlm.nih.gov/geo/(Accession number: GSE206705).
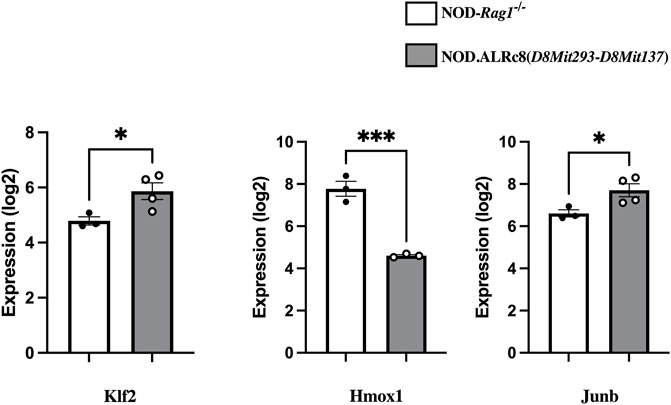
FIGURE 8. Transcriptome analysis identified genes differentially expressed in islets from insulitis-free NOD-Rag1−/− and NOD. ALRc8 (D8Mit293-D8Mit137) mice. Shown here are the expression of Klf2, Hmox1 and Junb in islets from NOD-Rag1−/− (open bars, n = 3 pools) and NOD. ALRc8 (D8Mit293-D8Mit137) (Grey bars, n = 4 pools), respectively compared using an unpaired t test. *p < 0.05. ***p < 0.001.
Discussion
AL is a toxic glucose analogue that is transported into and accumulates in rodent pancreatic β-cells by glucose transporter 2 (GLUT2). The β-cytotoxic action of this compound is mediated by ROS. Once inside β-cells, AL forms a conjugate with glutathione and is then reduced by thiol reductases (e.g., glutaredoxins) to form dialuric acid (Washburn and Wells, 1997). Autoxidation of dialuric acid generates superoxide radicals, hydrogen peroxide and, in a final iron-catalyzed reaction step, hydroxyl radicals. Hydroxyl radicals are ultimately responsible for death of the β-cells and the ensuing state of insulin-requiring, ALD. Due to the particularly low antioxidative defense capacity of rodent β-cells and the proposed role of ROS in the development of both Type 1 and Type 2 diabetes, ALD has been a useful model to understand mechanisms of β-cell failure. Indeed, the ALR mouse strain that was developed to resist ALD is also resistant to induction of T2D (Sekiguchi et al., 1991; Mathews et al., 2004), as well as spontaneous autoimmune diabetes (Mathews et al., 2003; Mathews et al., 2005; Chen et al., 2011a; Chen et al., 2011b). Earlier genetic analyses for ALR-derived resistance to T1D identified mt-Nd2 of the mtDNA as well as loci on Chr 17 (Idd16), Chr 8 (Idd22), and Chr 3 (Susp) (Mathews et al., 2003; Mathews et al., 2005; Pomerleau et al., 2005).
Our previous study utilized a cohort of 296 reciprocal F2 hybrids from outcrosses of ALR and NOD mice. This population was used to identify the unique genetic resistance of the ALR strain to ALD. Four loci were linked to ALD, including three in the nuclear genome (Chr 2, 3, and 8) and one in the mtDNA (mt-Nd2). The ALR genome provided diabetes resistance at both mt-Nd2 and a defined region on Chr 8 that has an overlapping confidence interval with Idd22 (Chen et al., 2008b). In the current study with a larger F2 cohort (n = 678) we confirmed both of these linkages as well as a Y-linked trait. However, the results here with a larger cohort of F2 mice failed to replicate the independent linkage on Chr 2 or the linkage to Chr 3 while providing suggestive evidence for ALR-derived protective loci on Chr 4, 6, 7, 8, and 13. Interestingly, one protective linkage on Chr 5 of the NOD genome was identified. Further, both intragenomic and intergenomic epistasis were uncovered. The intergenomic epistasis for ALD resistance between ALR alleles at mt-Nd2 and Chr 8 confirmed our previous observation using a congenic and conplastic mouse approach that NOD. mtALR-Idd22 mice were more resistant to ALD than either NOD. mtALR or NOD. Idd22 (Chen et al., 2008a).
The unique SNP and resulting allele/allotype in the mtDNA of ALR, mt-Nd2a, was previously associated with resistance against ALD (Chen et al., 2008b) and confirmed in the current study (Table 2). In contrast, the NOD mt-Nd2c allele has been associated with elevated mt ROS production (Gusdon et al., 2007; Gusdon et al., 2008) and β-cell sensitivity to free radical mediated (hydrogen peroxide and alloxan) as well as autoimmune destruction (Chen et al., 2008a; Chen et al., 2011b). The resistance contributed by mt-Nd2a to the potent free radicals generated by AL may be the result of suppressed endogenous mt free radical production (Mathews et al., 2005; Gusdon et al., 2007; Gusdon et al., 2008) and/or a higher level of antioxidant defenses unique to ALR pancreatic islets (Mathews et al., 2001; Mathews et al., 2005). Indeed, mt-Nd2a was also mapped to be protective against spontaneous T1D in mouse models (Mathews et al., 2005), and a corresponding human mt-ND2a (mtDNA D haplotype) was associated with resistance to T1D in humans (Uchigata et al., 2002) as well as to T2D in certain human populations (Liou et al., 2012). Therefore, the mechanisms underlying mt-Nd2a-associated β-cell resistance are a possible new target to preserve β-cell survival in T1D and potentially in T2D.
The Chr 8 ALD-resistance locus was mapped previously using a smaller F2 population of 296 mice to have an ALR-derived AL-resistance locus (Chen et al., 2008b). In a separate study using (NODxALR)F1 x NOD backcross 1 mice, the ALR allele on Chr 8, denoted as Idd22, was mapped to be protective against spontaneous T1D (Mathews et al., 2003) and later confirmed using congenic analysis (Whitener et al., 2017). These two Chr 8 linkages are overlapping (Chen et al., 2008b). With the current larger F2 sample size, the ALD-resistant linkage on Chr 8 is further confirmed (Figure 1). We have previously created congenic mice to examine the increased threshold for AL when ALR alleles on Chr 8 were introgressed onto the NOD background (Chen et al., 2008b). In the current study, we further shortened the congenic interval to a proximal region between 36.2 Mb and 77.8 Mb. The proximal short congenic mice show the same elevated threshold for AL resistance as the previous longer congenic (Figure 3), suggesting the gene or genes that contribute to resistance are in this proximal region. Consistent with this result, the peak of Chi-squares on Chr 8 is located within this region at 77 Mb (Figure 5), with 95% confidence interval between markers D8Mit205 (51.9 Mb) and D8Mit252 (93.48 Mb). Previously we determined, using congenic mice, that the mechanism of Idd22-mediated autoimmune diabetes resistance is through inhibition of autoreactive T cell extravasation through the vascular endothelium (Whitener et al., 2017). This was performed in NOD with the ALR allele of Idd22 and the NOD allele of mt-Nd2 excluding the possibility of observing the important intergenomic interaction between Idd22 and mt-Nd2 that provides protection to beta cells [Figure 5 and (Chen et al., 2008b)]. The fact that ALR and NOD mtDNA differ only at the mt-Nd2 SNP allowed us to analyze the interaction of mt-Nd2 with ALD-linked nuclear loci by stratification of the 678 F2 cohort by mt-Nd2 genotype. The Chr 8 ALD-linkage was mapped only in the group of mice with mt-Nd2a, ergo, the effect of the allele inherited from ALR (R) on Chr 8 requires the co-existence of mt-Nd2a to provide protection against ALD (Figure 5). Conplastic and congenic NOD. mtALR-Idd22 mice exhibited enhanced resistance to ALD, further supporting the intergenomic epistasis (Chen et al., 2008a).
Disease endotypes have been attributed to interactions between mitochondria and the nucleus. Proper mitochondrial function necessitates expression of genes from the nuclear and mitochondrial genomes. An important factor coordinating these two compartments is retrogenic signals that radiate from the mitochondria in response to metabolism or stress to impact gene expression and influence pathology (Cannino et al., 2007; Kim et al., 2018; Walker and Moraes, 2022). Genetic cooperation between the nucleus and mitochondria has been demonstrated to impact diabetes related phenotypes such as glucose metabolism and insulin sensitivity using mouse models (Sammy et al., 2021). The data presented here demonstrate directly that intergenomic epistasis controls beta cell sensitivity to β-cell death and diabetes onset.
We have previously associated mt-Nd2c with increased mtROS production (Gusdon et al., 2007; Gusdon et al., 2008). The interaction between Chr 8 and mt-Nd2 might localize to a gene or genes on Chr 8 that control the dissipation of ROS or mitochondrial function to suppress mtROS production. When endogenous ROS produced at mitochondria is suppressed with mt-Nd2a, we propose that the free radical dissipation capacity of the ALR-allele on Chr 8 will be set free to extinguish the exogenous free radicals induced by AL. Since the ALD-linked Chr 8 locus overlaps with T1D-protective Idd22 (Mathews et al., 2003), and mt-Nd2a was also associated with T1D (Mathews et al., 2005), the interaction between mt-Nd2 and Chr 8 appears to regulate β-cell survival during development of diabetes. We have compared gene expression of islets from NOD. Rag1−/− and NOD.ALRc8 (D8Mit293-D8Mit137) mice using transcriptome arrays. Among 164 differentially expressed Chr 8 transcripts between these two strains there are some candidates linked to mitochondrial function, ROS levels, or cell death. A few examples (Figure 8): Klf2 (73 Mb) encodes Kruppel-like factor 2, a transcription factor involved in endothelium function, T cell development and immune activation and is closely related with mitochondrial function (Coon et al., 2022). Hmox1 (75.1 Mb) encodes Heme Oxygenase 1, a known antioxidative enzyme exhibited higher expression in NOD islets compared to NOD. ALRc8(D8Mit293-D8Mit137). Induced expression of Hmox1 protects NOD mice from T1D and islets form destruction (Pileggi et al., 2001; Pogu et al., 2019). However, the increased expression is likely an indicator of increased mitochondrial oxidative stress in NOD compared to the Idd22 congenic strain (Johnson and DeMars, 2004) as opposed to Hmox1 being an ideal candidate gene. Junb (84.98 Mb) encodes a subunit of AP-1 transcription factor. JunB has been shown to be protective against mitochondrial stress in human lymphoma cells (Son et al., 2010), and structurally, JunB can localize in mitochondria (Liu et al., 2018). These possible candidate genes locate close to the peak linkage (77 Mb) on Chr 8 (Figure 5) and are within the 95% confidence interval. These may contribute to ALD resistance by impacting the interactions between Chr 8 and mt-Nd2. Further studies are needed to develop β-cell lines with both mt-Nd2a and Chr 8 (ALD-linkage/Idd22) to isolate the effect of this interaction at β-cell level.
The mtDNA haplotype also affects interactions of Chr 8 with Chr 2 (Figure 6). Although the linkage on Chr 2 was not alone significant as an independent linkage in the larger cohort studied here, a strong epistatic interaction between Chr 2 and Chr 8 was detected in the full population of F2 mice (Figures 4, 6; Table 3). The interaction between Chr 2 and Chr 8 appeared using multiple regression modeling [drop one QTL at a time ANOVA analysis (Figure 4; Table 3)]. This epistatic interaction is affected by the mt-Nd2 allele (Figure 6). Dominant protection of ALR genome on Chr 8 suppresses the ALD sensitivity linked to the heterozygous genotype on Chr 2. Therefore, the negative heterosis conferred by the linkage on Chr 2 is only observed when Chr 8 is homozygous for the NOD genotype resulting in heightened ALD incidence, and is at the highest when mt-Nd2 is also NOD type. As the Chr 2 linkage alone is not significant, it is likely that the ALR allele on Chr 8 provides too powerful a resistance and outplays sensitivity conferred by the Chr 2 linkage. The peak on Chr 2 is denoted by marker rs3681744 at 121 Mb. An interesting candidate gene near rs3681744 is 28S ribosomal protein S5, mitochondrial (Mrps5). Expression analysis demonstrates that Mrps5 transcripts are at a significantly lower level in isolated NOD. Rag1−/− islets compared to NOD.ALRc8(D8Mit293-D8Mit137) islets. As this regulates mitochondrial protein it may affect the Chr2-Chr8-mt-Nd2 interaction to prevent AL-induced mitochondrial damage through control of translation of genes encoded on the mitochondrial genome, including mt-Nd2 (Titova et al., 2020).
The NOD linkage (D) on Chr 6 contributes dominant AL sensitivity whereas the NOD contribution on Chr 8 is recessive (Figure 2). A Chr 8-Chr 6 was not detected in the full population of 678 F2 mice not stratified by mt-ND2 genotype (Figure 7A). The NOD allele (D) on Chr 6 contributes dominant AL sensitivity but the NOD susceptibility contribution on Chr 8 appears to act recessively (Figure 2). However, a distinct interaction was revealed when the population of F2 mice was stratified by mt genotype. In segregants expressing the NOD mt-Nd2c, homozygous D type on both Chr 8 and 6 synergistically contributed to ALD susceptibility (Figure 7B). On the contrary, in segregants expressing the ALR mt-Nd2a, homozygosity for the NOD Chr 8 linkage (DD) exhibited maximum susceptibility in combination with homozygosity for the ALR linkage (RR) on Chr 6 (Figure 7C). The peak linkage on Chr 6 is at marker D6Mit184 (53.2 Mb), with 95% confidence interval between markers D6Mit48 (31.9 Mb) and D6Mit323 (86.85 Mb). Analysis of differentially expressed genes in the islets from NOD. Rag1−/− compared to NOD. ALRc8 (D8Mit293-D8Mit137) identified Hdac11 (histone deacetylase 11) at 91 Mb, as expressed at lower level in the islets of ALD-susceptible NOD. Rag1−/− mice. Hdac11 has been reported to affect mitochondrial function (Hurtado et al., 2021). At 98.9 Mb, a gene Foxp1, encodes forkhead box P1 is a transcription factor for mtDNA binding protein (Wang et al., 2019) and has reduced expression in ALD-resistant NOD.ALRc8(D8Mit293-D8Mit137) islets. As AL can induce oxidative stress-mediated mtDNA damage (Driggers et al., 1997), a reduction in Foxp1 may protect against β-cell death by reducing replication of damaged mtDNA allowing for repair to occur. Although these genes are outside of the 95% confidence interval on Chr 6, given their association with mitochondria, these remain interesting candidates.
In autoimmune T1D genes may impact both the immune system and non-immune cells (i.e., β-cells) (Barrett et al., 2009; Inshaw et al., 2020). These results therefore do not exclude the possibility that Idd22 affects both β-cells and immune cells either by different genes in this region or by a single common gene. In fact, Idd22 alone confers resistance to spontaneous T1D in NOD (Mathews et al., 2005) by hampering autoreactive T cell trafficking to islets (Whitener et al., 2017), while islet susceptibility to in vitro autoimmune attack is not affected (Whitener et al., 2017). This is another example of T1D as a complex and multifactorial disease (Driver et al., 2012; Chen Y. G. et al., 2018).
With the emerging data from genetic studies since tools like GWAS became available, genes and loci involved in T1D and T2D have been identified. Genes identified by GWAS to be involved in diabetes have been linked with mitochondria or mitochondrial function as well as β-cell death. An example is GLIS Family Zinc Finger 3 (GLIS3), which is associated with both T1D and T2D (Wen and Yang, 2017). Knockdown of GLIS3 induced BAX translocation to mitochondria, increasing cleaved caspase 3 and 9, and cytochrome c release, therefore potentiates cytokine-induced β-cell death via the mitochondrial pathway of apoptosis (Nogueira et al., 2013). PDX1, another gene associated with both T2D (Hani et al., 1999) and monogenic diabetes of the young (MODY) (Stoffers et al., 1997), is thought to control β-cell function by regulation of mitophagy, a key mechanism for mitochondrial turnover and quality control (Ding and Yin, 2012), through controlling of the T1D-associated gene CLEC16A (Soleimanpour et al., 2014; Soleimanpour et al., 2015). The mt-ND2 SNP rs28357984 (5178 A/C) in the human mitochondrial genome that corresponds to the mouse mt-Nd2, was also associated with both human T1D (Uchigata et al., 2002) and T2D (Liou et al., 2012). The intergenomic epistasis was studied in mt-exchange mouse model of fatty liver disease (Betancourt et al., 2014). In human T1D, mt-nuclear epistasis is associated with patients’ BMI (Ludwig-Slomczynska et al., 2020). Interestingly, in this study, the mtDNA SNP that interacts with nuclear genes was also in mt-ND2 (Ludwig-Slomczynska et al., 2020). Both fatty liver disease and BMI are factors associated with T2D. Therefore, mitochondria might be a common factor in the pathogenesis of both T1D and T2D through affecting β-cell fitness. Since both types of diabetes are multifactorial diseases, mt-nuclear interactions may play pathogenic or protective roles in both diseases. To the best of our knowledge, the current study is the first to investigate the role of mt-nuclear interactions in the control of β-cell sensitivity to ROS and development of diabetes.
In summary, using a F2 cohort generated by outcrosses of the NOD and ALR mouse strains, we detected loci that affect pancreatic β-cell sensitivity to free radical-generating chemical alloxan on Chromosomes 4, 5, 6, 7, 8, and 13, as well as mt-Nd2 of the mtDNA. Further, we identified that both intra- and intergenomic epistasis contribute to resistance or sensitivity of β-cell death in this model. This is the first report to demonstrate that interactions of the mitochondrial genome and the nuclear genome can regulate diabetes. Mitochondria are pivotal for β-cell function and insults or dysfunction of this organelle may impact pathogenesis (Uchigata et al., 2002; Chen et al., 2011b; Liou et al., 2012; Chen et al., 2017; Chen J. et al., 2018). Therefore, epigenetic interactions that affect β-cell resistance to free radical stress may alter the development of both T1D and T2D.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/geo/, GSE206705.
Ethics statement
The animal study was reviewed and approved by All procedures described within this manuscript were approved by the University of Florida’s Institutional Animal Care and Use Committee. UF-IACUC 201905476: mt-Nd2 and resistance to autoimmune diabetes.
Author contributions
JC and CM designed the research plan, performed experiments, analyzed data, and wrote the manuscript. RL performed data analysis and also contributed to the research plan. SK and GZ contributed significantly to the completion of the research. RW performed islet transcriptome analysis and participated in manuscript editing. EL participated in the experimental design and edited the manuscript. All authors have reviewed and gave final approval of the version that was submitted.
Funding
This work was supported by grants from the National Institutes of Health DK074656 and AI56374 (CEM), DK36175 and DK27722 (EHL), as well as the American Diabetes Association.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Barrett, J. C., Clayton, D. G., Concannon, P., Akolkar, B., Cooper, J. D., Erlich, H. A., et al. (2009). Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 41, 703–707. doi:10.1038/ng.381
Betancourt, A. M., King, A. L., Fetterman, J. L., Millender-Swain, T., Finley, R. D., Oliva, C. R., et al. (2014). Mitochondrial-nuclear genome interactions in non-alcoholic fatty liver disease in mice. Biochem. J. 461, 223–232. doi:10.1042/BJ20131433
Broman, K. W., Wu, H., Sen, S., and Churchill, G. A. (2003). R/qtl: QTL mapping in experimental crosses. Bioinformatics 19, 889–890. doi:10.1093/bioinformatics/btg112
Cannino, G., Di Liegro, C. M., and Rinaldi, A. M. (2007). Nuclear-mitochondrial interaction. Mitochondrion 7, 359–366. doi:10.1016/j.mito.2007.07.001
Chen, M., and Kendziorski, C. (2007). A statistical framework for expression quantitative trait loci mapping. Genetics 177, 761–771. doi:10.1534/genetics.107.071407
Chen, J., Gusdon, A. M., Thayer, T. C., and Mathews, C. E. (2008a). Role of increased ROS dissipation in prevention of T1D. Ann. N. Y. Acad. Sci. 1150, 157–166. doi:10.1196/annals.1447.045
Chen, J., Lu, Y., Lee, C. H., Li, R., Leiter, E. H., and Mathews, C. E. (2008b). Commonalities of genetic resistance to spontaneous autoimmune and free radical--mediated diabetes. Free Radic. Biol. Med. 45, 1263–1270. doi:10.1016/j.freeradbiomed.2008.07.020
Chen, J., Gusdon, A. M., and Mathews, C. E. (2011a). Role of genetics in resistance to type 1 diabetes. Diabetes. Metab. Res. Rev. 27, 849–853. doi:10.1002/dmrr.1260
Chen, J., Gusdon, A. M., Piganelli, J. D., Leiter, E. H., and Mathews, C. E. (2011b). mt-Nd2(a) Modifies resistance against autoimmune type 1 diabetes in NOD mice at the level of the pancreatic β-cell. Diabetes 60, 355–359. doi:10.2337/db10-1241
Chen, J., Chernatynskaya, A. V., Li, J. W., Kimbrell, M. R., Cassidy, R. J., Perry, D. J., et al. (2017). T cells display mitochondria hyperpolarization in human type 1 diabetes. Sci. Rep. 7, 10835. doi:10.1038/s41598-017-11056-9
Chen, J., Stimpson, S. E., Fernandez-Bueno, G. A., and Mathews, C. E. (2018). Mitochondrial reactive oxygen species and type 1 diabetes. Antioxid. Redox Signal. 29, 1361–1372. doi:10.1089/ars.2017.7346
Chen, Y. G., Mathews, C. E., and Driver, J. P. (2018). The role of NOD mice in type 1 diabetes research: Lessons from the past and recommendations for the future. Front. Endocrinol. 9, 51. doi:10.3389/fendo.2018.00051
Churchill, G. A., and Doerge, R. W. (1994). Empirical threshold values for quantitative trait mapping. Genetics 138, 963–971. doi:10.1093/genetics/138.3.963
Coon, B. G., Timalsina, S., Astone, M., Zhuang, Z. W., Fang, J., Han, J., et al. (2022). A mitochondrial contribution to anti-inflammatory shear stress signaling in vascular endothelial cells. J. Cell Biol. 221, e202109144. doi:10.1083/jcb.202109144
Ding, W. X., and Yin, X. M. (2012). Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 393, 547–564. doi:10.1515/hsz-2012-0119
Driggers, W. J., Holmquist, G. P., Ledoux, S. P., and Wilson, G. L. (1997). Mapping frequencies of endogenous oxidative damage and the kinetic response to oxidative stress in a region of rat mtDNA. Nucleic Acids Res. 25, 4362–4369. doi:10.1093/nar/25.21.4362
Driver, J. P., Chen, Y. G., and Mathews, C. E. (2012). Comparative genetics: synergizing human and NOD mouse studies for identifying genetic causation of type 1 diabetes. Rev. Diabet. Stud. 9, 169–187. doi:10.1900/RDS.2012.9.169
Gusdon, A. M., Votyakova, T. V., Reynolds, I. J., and Mathews, C. E. (2007). Nuclear and mitochondrial interaction involving mt-Nd2 leads to increased mitochondrial reactive oxygen species production. J. Biol. Chem. 282, 5171–5179. doi:10.1074/jbc.M609367200
Gusdon, A. M., Votyakova, T. V., and Mathews, C. E. (2008). mt-Nd2a suppresses reactive oxygen species production by mitochondrial complexes I and III. J. Biol. Chem. 283, 10690–10697. doi:10.1074/jbc.M708801200
Hani, E. H., Stoffers, D. A., Chevre, J. C., Durand, E., Stanojevic, V., Dina, C., et al. (1999). Defective mutations in the insulin promoter factor-1 (IPF-1) gene in late-onset type 2 diabetes mellitus. J. Clin. Invest. 104, R41–R48. doi:10.1172/JCI7469
Hurtado, E., Nunez-Alvarez, Y., Munoz, M., Gutierrez-Caballero, C., Casas, J., Pendas, A. M., et al. (2021). HDAC11 is a novel regulator of fatty acid oxidative metabolism in skeletal muscle. FEBS J. 288, 902–919. doi:10.1111/febs.15456
Ino, T., Kawamoto, Y., Sato, K., Nishikawa, K., Yamada, A., Ishibashi, K., et al. (1991). Selection of mouse strains showing high and low incidences of alloxan-induced diabetes. Jikken Dobutsu. 40, 61–67. doi:10.1538/expanim1978.40.1_61
Inshaw, J. R. J., Cutler, A. J., Crouch, D. J. M., Wicker, L. S., and Todd, J. A. (2020). Genetic variants predisposing most strongly to type 1 diabetes diagnosed under age 7 Years lie near candidate genes that function in the immune system and in pancreatic beta-cells. Diabetes Care 43, 169–177. doi:10.2337/dc19-0803
Johnson, W. T., and DeMars, L. C. (2004). Increased heme oxygenase-1 expression during copper deficiency in rats results from increased mitochondrial generation of hydrogen peroxide. J. Nutr. 134, 1328–1333. doi:10.1093/jn/134.6.1328
Kim, K. H., Son, J. M., Benayoun, B. A., and Lee, C. (2018). The mitochondrial-encoded peptide MOTS-c translocates to the nucleus to regulate nuclear gene expression in response to metabolic stress. Cell Metab. 28, 516–524. doi:10.1016/j.cmet.2018.06.008
Lenzen, S., Drinkgern, J., and Tiedge, M. (1996). Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic. Biol. Med. 20, 463–466. doi:10.1016/0891-5849(96)02051-5
Liou, C. W., Chen, J. B., Tiao, M. M., Weng, S. W., Huang, T. L., Chuang, J. H., et al. (2012). Mitochondrial DNA coding and control region variants as genetic risk factors for type 2 diabetes. Diabetes 61, 2642–2651. doi:10.2337/db11-1369
Liu, F., Lossl, P., Rabbitts, B. M., Balaban, R. S., and Heck, A. J. R. (2018). The interactome of intact mitochondria by cross-linking mass spectrometry provides evidence for coexisting respiratory supercomplexes. Mol. Cell. Proteomics 17, 216–232. doi:10.1074/mcp.RA117.000470
Ludwig-Slomczynska, A. H., Seweryn, M. T., Kapusta, P., Pitera, E., Handelman, S. K., Mantaj, U., et al. (2020). Mitochondrial GWAS and association of nuclear - mitochondrial epistasis with BMI in T1DM patients. BMC Med. Genomics 13, 97. doi:10.1186/s12920-020-00752-7
Mathews, C. E., Mcgraw, R. A., Dean, R., and Berdanier, C. D. (1999). Inheritance of a mitochondrial DNA defect and impaired glucose tolerance in BHE/Cdb rats. Diabetologia 42, 35–40. doi:10.1007/s001250051109
Mathews, C. E., Graser, R. T., Savinov, A., Serreze, D. V., and Leiter, E. H. (2001). Unusual resistance of ALR/lt mouse beta cells to autoimmune destruction: role for beta cell-expressed resistance determinants. Proc. Natl. Acad. Sci. U. S. A. 98, 235–240. doi:10.1073/pnas.98.1.235
Mathews, C. E., Graser, R. T., Bagley, R. J., Caldwell, J. W., Li, R., Churchill, G. A., et al. (2003). Genetic analysis of resistance to Type-1 Diabetes in ALR/Lt mice, a NOD-related strain with defenses against autoimmune-mediated diabetogenic stress. Immunogenetics 55, 491–496. doi:10.1007/s00251-003-0603-8
Mathews, C. E., Bagley, R., and Leiter, E. H. (2004). ALS/Lt: a new type 2 diabetes mouse model associated with low free radical scavenging potential. Diabetes 53, S125–S129. doi:10.2337/diabetes.53.2007.s125
Mathews, C. E., Leiter, E. H., Spirina, O., Bykhovskaya, Y., Gusdon, A. M., Ringquist, S., et al. (2005). mt-Nd2 Allele of the ALR/Lt mouse confers resistance against both chemically induced and autoimmune diabetes. Diabetologia 48, 261–267. doi:10.1007/s00125-004-1644-8
Nogueira, T. C., Paula, F. M., Villate, O., Colli, M. L., Moura, R. F., Cunha, D. A., et al. (2013). GLIS3, a susceptibility gene for type 1 and type 2 diabetes, modulates pancreatic beta cell apoptosis via regulation of a splice variant of the BH3-only protein Bim. PLoS Genet. 9, e1003532. doi:10.1371/journal.pgen.1003532
Pileggi, A., Molano, R. D., Berney, T., Cattan, P., Vizzardelli, C., Oliver, R., et al. (2001). Heme oxygenase-1 induction in islet cells results in protection from apoptosis and improved in vivo function after transplantation. Diabetes 50, 1983–1991. doi:10.2337/diabetes.50.9.1983
Pogu, J., Tzima, S., Kollias, G., Anegon, I., Blancou, P., and Simon, T. (2019). Genetic restoration of heme oxygenase-1 expression protects from type 1 diabetes in NOD mice. Int. J. Mol. Sci. 20, E1676. doi:10.3390/ijms20071676
Pomerleau, D. P., Bagley, R. J., Serreze, D. V., Mathews, C. E., and Leiter, E. H. (2005). Major histocompatibility complex-linked diabetes susceptibility in NOD/lt mice: subcongenic analysis localizes a component of Idd16 at the H2-D end of the diabetogenic H2(g7) complex. Diabetes 54, 1603–1606. doi:10.2337/diabetes.54.5.1603
Sammy, M. J., Connelly, A. W., Brown, J. A., Holleman, C., Habegger, K. M., and Ballinger, S. W. (2021). Mito-Mendelian interactions alter in vivo glucose metabolism and insulin sensitivity in healthy mice. Am. J. Physiol. Endocrinol. Metab. 321, E521–E529. doi:10.1152/ajpendo.00069.2021
Sekiguchi, F., Ishibashi, K., Kawamoto, Y., and Ino, T. (1991). Diabetic peculiarity of the ALS-Ay and ALR-Ay strains. Jikken Dobutsu. 40, 323–329. doi:10.1538/expanim1978.40.3_323
Soleimanpour, S. A., Gupta, A., Bakay, M., Ferrari, A. M., Groff, D. N., Fadista, J., et al. (2014). The diabetes susceptibility gene Clec16a regulates mitophagy. Cell 157, 1577–1590. doi:10.1016/j.cell.2014.05.016
Soleimanpour, S. A., Ferrari, A. M., Raum, J. C., Groff, D. N., Yang, J., Kaufman, B. A., et al. (2015). Diabetes susceptibility genes Pdx1 and Clec16a function in a pathway regulating mitophagy in beta-cells. Diabetes 64, 3475–3484. doi:10.2337/db15-0376
Son, Y. O., Heo, J. S., Kim, T. G., Jeon, Y. M., Kim, J. G., and Lee, J. C. (2010). Over-expression of JunB inhibits mitochondrial stress and cytotoxicity in human lymphoma cells exposed to chronic oxidative stress. BMB Rep. 43, 57–61. doi:10.5483/bmbrep.2010.43.1.057
Stoffers, D. A., Ferrer, J., Clarke, W. L., and Habener, J. F. (1997). Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat. Genet. 17, 138–139. doi:10.1038/ng1097-138
Tiedge, M., Lortz, S., Drinkgern, J., and Lenzen, S. (1997). Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 46, 1733–1742. doi:10.2337/diab.46.11.1733
Titova, A. A., Mavlikeev, M. O., Kaligin, M. S., Suleymanova, D. M., Chekmaryeva, I. A., Kiyasov, A. P., et al. (2020). Early ultra- and microstructural alterations in rat pancreas in alloxan-induced diabetes mellitus. Ultrastruct. Pathol. 44, 61–70. doi:10.1080/01913123.2019.1710313
Uchigata, Y., Okada, T., Gong, J. S., Yamada, Y., Iwamoto, Y., and Tanaka, M. (2002). A mitochondrial genotype associated with the development of autoimmune-related type 1 diabetes. Diabetes Care 25, 2106. doi:10.2337/diacare.25.11.2106
Walker, B. R., and Moraes, C. T. (2022). Nuclear-mitochondrial interactions. Biomolecules 25, 427. doi:10.3390/biom12030427
Wallace, D. C. (2010). Mitochondrial DNA mutations in disease and aging. Environ. Mol. Mutagen. 51, 440–450. doi:10.1002/em.20586
Wang, G., Wang, Q., Huang, Q., Chen, Y., Sun, X., He, L., et al. (2019). Upregulation of mtSSB by interleukin-6 promotes cell growth through mitochondrial biogenesis-mediated telomerase activation in colorectal cancer. Int. J. Cancer 144, 2516–2528. doi:10.1002/ijc.31978
Washburn, M. P., and Wells, W. W. (1997). Glutathione dependent reduction of alloxan to dialuric acid catalyzed by thioltransferase (glutaredoxin): a possible role for thioltransferase in alloxan toxicity. Free Radic. Biol. Med. 23, 563–570. doi:10.1016/s0891-5849(97)00001-4
Wen, X., and Yang, Y. (2017). Emerging roles of GLIS3 in neonatal diabetes, type 1 and type 2 diabetes. J. Mol. Endocrinol. 58, R73–R85. doi:10.1530/JME-16-0232
Keywords: diabetes, free radical, mouse model, epistasis, mitochondrial genome, beta cell (β cell), alloxan
Citation: Chen J, Li R, Knapp S, Zhu G, Whitener RL, Leiter EH and Mathews CE (2022) Intergenomic and epistatic interactions control free radical mediated pancreatic β-cell damage. Front. Genet. 13:994501. doi: 10.3389/fgene.2022.994501
Received: 14 July 2022; Accepted: 20 September 2022;
Published: 07 October 2022.
Edited by:
Marc S. Horwitz, University of British Columbia, CanadaReviewed by:
John Corbett, Medical College of Wisconsin, United StatesMaria Bettini, The University of Utah, United States
David Wagner, University of Colorado Anschutz Medical Campus, United States
Copyright © 2022 Chen, Li, Knapp, Zhu, Whitener, Leiter and Mathews. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Clayton E. Mathews, Y2xheXRvbi5tYXRoZXdzQHBhdGhvbG9neS51ZmwuZWR1