 Tian Wu
Tian Wu Zipeng Liu
Zipeng Liu Timothy Shin Heng Mak
Timothy Shin Heng Mak Pak Chung Sham
Pak Chung Sham
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 10 October 2022
Sec. Statistical Genetics and Methodology
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.989639
This article is part of the Research TopicInsights in Statistical Genetics and Methodology: 2022View all 14 articles
Power calculation is a necessary step when planning genome-wide association studies (GWAS) to ensure meaningful findings. Statistical power of GWAS depends on the genetic architecture of phenotype, sample size, and study design. While several computer programs have been developed to perform power calculation for single SNP association testing, it might be more appropriate for GWAS power calculation to address the probability of detecting any number of associated SNPs. In this paper, we derive the statistical power distribution across causal SNPs under the assumption of a point-normal effect size distribution. We demonstrate how key outcome indices of GWAS are related to the genetic architecture (heritability and polygenicity) of the phenotype through the power distribution. We also provide a fast, flexible and interactive power calculation tool which generates predictions for key GWAS outcomes including the number of independent significant SNPs, the phenotypic variance explained by these SNPs, and the predictive accuracy of resulting polygenic scores. These results could also be used to explore the future behaviour of GWAS as sample sizes increase further. Moreover, we present results from simulation studies to validate our derivation and evaluate the agreement between our predictions and reported GWAS results.
Genome-wide association studies (GWAS) aim to systematically identify single-nucleotide polymorphisms (SNPs) associated with complex phenotypes. Though not necessarily causal, associated SNPs are good starting points for elucidating biological mechanisms of diseases and related phenotypes. GWAS on a wide range of phenotypes have confirmed the polygenic nature of most common traits, with thousands of SNPs each making a small contribution to individual differences in the population (Visscher et al., 2017). The recent increase in the sample size of GWAS and meta-GWAS has resulted in more of these SNPs to be identified, leading not only to more comprehensive understanding of disease etiology (Cano-Gamez and Trynka, 2020), but also greater accuracy in the calculation of polygenic scores to predict individual genetic liability to develop disease (Vilhjalmsson et al., 2015; Mak et al., 2017; Torkamani et al., 2018).
Adequate statistical power is necessary to both detect enough SNPs to inform etiology and to obtain accurate effect size estimate for polygenic score calculations (Dudbridge, 2013). Several computer programs have been developed to perform power calculation for single SNP association testing. For example, Genetic Power Calculator (GPC) (Purcell et al., 2003) used closed-form analytic results (Sham and Purcell, 2014) to perform power calculations for linkage and association studies. Genetic Association Study Power Calculator (GAS) (Johnson and Abecasis, 2017) performs power calculation for genetic association studies under case-control design. However, these tools perform power calculation for single SNPs, ignoring the polygenic nature of complex diseases, and the simultaneous testing of millions of SNPs that is now standard in GWAS (Sham and Purcell, 2014). Meta-GWAS Accuracy and Power (MetaGAP) (de Vlaming et al., 2017) performs GWAS power calculations and introduces genetic correlation parameters to account for effect size heterogeneity between studies. However, it is restricted to quantitative phenotype and random samples.
Since the goal of GWAS is to detect any truly associated SNPs, power calculation might more appropriately address the probability of detecting any number of associated SNPs, than the probability of detecting a specific associated SNP. Such a calculation would require specification of the entire distribution of effect size of all analysed SNPs, rather than the effect size of a single SNP. Several methods have been proposed to infer the underlying genetic effect size distribution based on significant GWAS hits or GWAS summary statistics (Park et al., 2010; So et al., 2010; Chatterjee et al., 2013; Moser et al., 2015; Zhang et al., 2018). Evidence shows that a point-normal distribution is adequate to fit the distribution of true effects of common variants for some complex traits (Zhang et al., 2018) and it is more practical than the infinitesimal model (Visscher et al., 2017).
This report describes a fast, flexible and interactive power calculation tool for GWAS under the assumption of a point-normal distribution of standardized effect sizes. The program generates predictions for the key outcomes of GWAS, including the distribution of statistical power across all independent causal SNPs, the expected number of independent genome-wide significant SNPs, total phenotypic variance explained by these SNPs, and the predictive accuracy of optimally weighted polygenic scores (PGS). It allows the user to specify the nature of the phenotype under consideration (quantitative or dichotomous), its epidemiological features (e.g., disease prevalence) and genetic architecture (e.g., SNP-heritability), and the study design (e.g., case-control).
The input parameters and the output indices of the program are summarized in Table 1.

TABLE 1. Key input parameters and output indices.
The phenotype is either an observed quantitative trait or a disease determined by a latent continuous liability (Falconer, 1965). For simplicity, SNPs are assumed to have been made nearly independent by clumping or pruning; the total number of SNPs (m) is the effective number of independent SNPs in the entire genome. A proportion
where
For disease phenotypes, standardised log-odds ratios (
where
For quantitative traits, the regression coefficient estimate
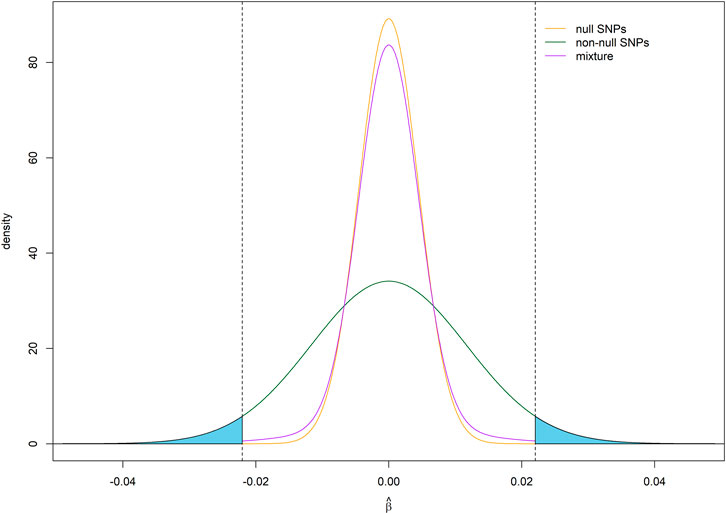
FIGURE 1. Assumed distribution of effect size estimates under a point-normal model. For illustration, the critical values for statistical significance are shown as vertical dotted lines, while average statistical power for detecting non-null SNPs is given by the shaded areas under density curve for non-null SNPs. Parameter values
For binary traits, the sampling variance of the per-standard deviation effect estimate on the liability scale depends on the disease prevalence (K) in the population and the proportion of cases (
The sample size n can be rescaled by a factor
The statistical power for an individual SNP is determined by its effect size, the sample size, and the desired significance level. In a random sample of size
From the expectation and variance of statistical power, we derived formulae for the expectation and variance of the number of independent significant SNPs, as well as the proportion of phenotypic variance explained by these SNPs. These formulae were validated by simulation studies. For SNP
The polygenic model specifies that the phenotypic value is related to SNP genotypes by
A number of different methods to determine the weights
We enabled the above framework to be used for power calculation in other study designs, including phenotypic selection of continuous traits (e.g., extreme phenotype design), and case-control studies of binary traits, by deriving the equivalent sample size
We applied our method to four phenotypes including height, body mass index (BMI), major depressive disorder (MDD) and schizophrenia (SCZ) to evaluate how well the predicted GWAS outcomes match up with the reported GWAS outcomes (Wray et al., 2018; Yengo et al., 2018; The Schizophrenia Working Group of the Psychiatric Genomics Consortium Ripke et al., 2020). We selected these four phenotypes because at least three sizeable GWAS or meta-GWAS had been conducted, so that earlier GWAS outcomes could be used to set a reasonable range for
For SNP heritability, we assumed the latest estimated value reported in literature; when several SNP heritability estimates were reported at about the same time, their average value was used. Specifically, we assumed the SNP heritabilities of height, BMI, MDD, and SCZ were 0.483 (Yengo et al., 2018), 0.249 (see Web resources), 0.089 (Howard et al., 2019) and 0.23 (Lam et al., 2019; Lee et al., 2019), respectively. In all of our applications, we set m as 60,000 (Wray et al., 2013), assuming meta-analysis samples are from European ancestry. For quantitative trait GWAS using a population cohort, the parameter n was simply the sample size of GWAS or meta-GWAS, whereas for binary phenotypes, we used the equivalent sample size described above. If earlier study was a meta-analysis, we calculated the equivalent sample size for each cohort in the meta-analysis, and used the sum of equivalent sample sizes as our model parameter n (Supplementary Tables S2, S3). We set the genome-wide significant level α as 5 × 10−8 except when predicting GWAS key outcomes for height and BMI. For these two studies, α was set as 1 × 10−8 to be consistent with the literature.
Our model is based on the assumption that the effect size follows a point-normal distribution. Accordingly, the effect size estimate follows a normal mixture distribution (Figure 1). Figure 2A shows the relationship between statistical power and sample size for different effect sizes for a single SNP. We define SNP explaining 0.01%, 0.1%, and 1% of SNP heritability as having small, moderate and large effect, respectively. When the effect size is large, power curve increased rapidly and saturated soon. The proportion of SNPs with at least that level of statistical power on the x-axis is shown in Figure 2B. This proportion is equivalent to one minus the cumulative probability of power. With the increase of sample size, larger proportions of SNPs remain high statistical power. The expectation and variance of power, given different levels of heritability,
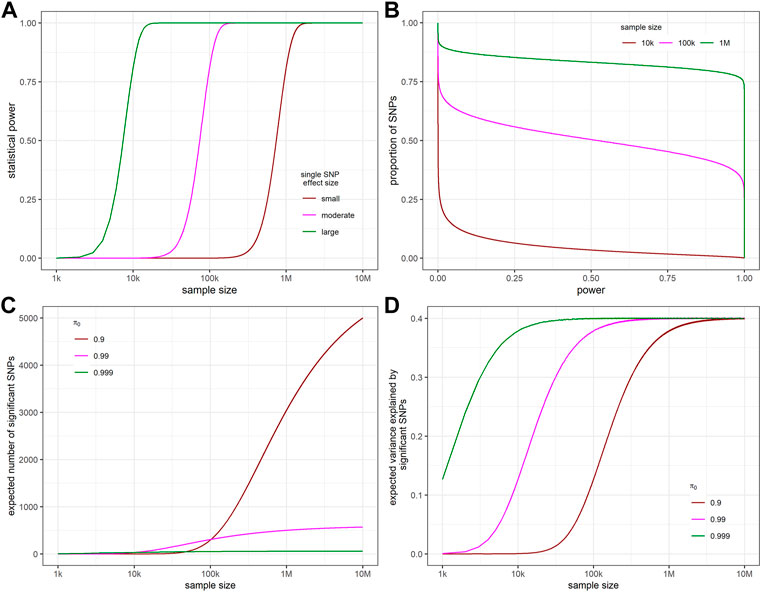
FIGURE 2. The relationship between statistical power, sample size, expected number of significant SNPs, and apparent variance explained by significant SNPs. (A) The relationship between sample size and the statistical power to detect a single SNP with different effect sizes “small”, “moderate”, and “large” representing SNPs that explain 0.01%, 0.1%, and 1% of SNP heritability. (B) Proportion of SNPs with at least that level of statistical power on the x-axis for different sample sizes. (C) Relationship between expected number of significant SNPs and sample sizes. (D) Relationship between the expected variance explained by the significant SNPs and sample sizes. For all figures,
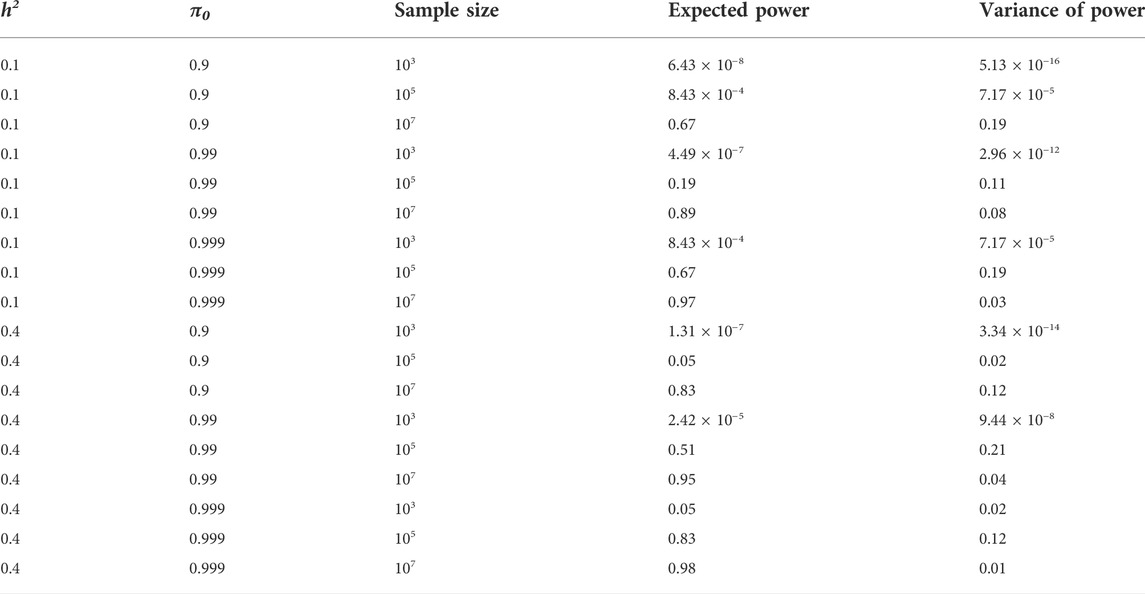
TABLE 2. The expectation and variance of statistical power across causal SNPs for different SNP heritability, polygenicity, and sample sizes. m = 60,000,
The number of independent significant SNPs is a function of statistical power across all causal SNPs. Testing the significance of each independent SNP could be regarded as a Bernoulli trial
When calculating the variance of the number of significant SNPs, null and non-null SNPs are also considered separately. For null SNPs, the number of significant SNPs is binomial with mean
In our model, sample size and
The phenotypic variance explained by independent significant SNPs in a GWAS is
When effect size estimates are calculated in different samples, the number of significant SNPs and
T is the critical value given the significance level.
The variance of variance explained by the significant SNPs is obtained using the law of total variance.
Similarly, the variance of corrected variance explained by significant SNPs can also be calculated.
The relationship between the expected apparent variance explained and sample size shows consistent pattern with that of expected number of significant SNPs and sample size (Figure 2D).
To validate the derived formula, we performed simulation studies using specific genetic architecture parameters (Figure 3). For both continuous and binary phenotypes, the 95% probability intervals of the theoretical number of significant SNPs and variance explained covers the mean of 100-time simulation results, which supports our analytic derivation. In addition, In Table 3, we listed necessary sample sizes to detect 5%, 50%, and 95% of causal SNPs for traits with different levels of
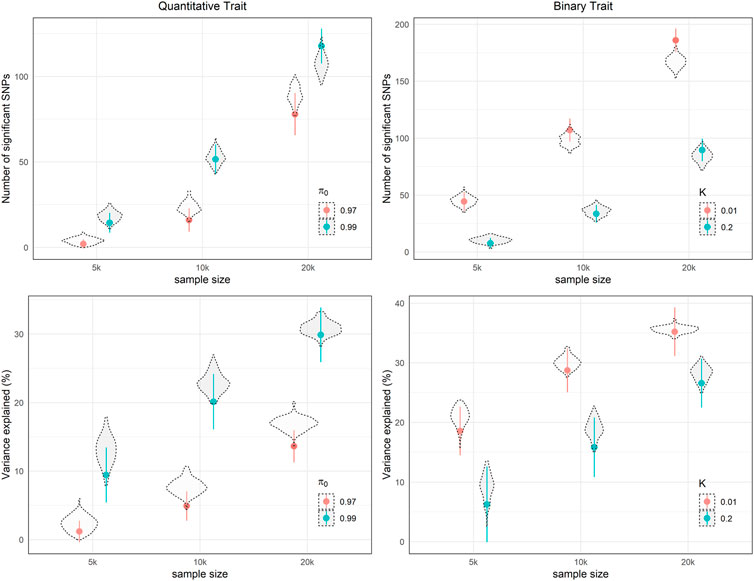
FIGURE 3. Theoretical expected number of independent significant SNPs and variance explained with 95% probability intervals i.e., dots and whiskers, with different parameters settings in 100 simulations.
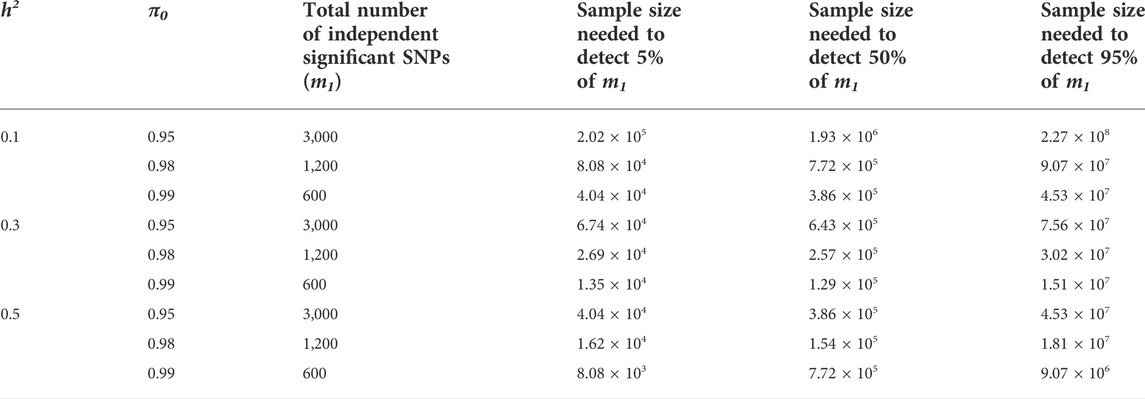
TABLE 3. The sample sizes needed to detect 5%, 50%, and 95% of independent significant SNPs for phenotypes with different levels of polygenicity, assuming the effect size following point-normal distribution, m = 60,000. m1 is the total number of causal SNPs.
For study design with phenotypic selection of continuous traits, we first consider the extreme phenotype (EP) study design (Barnett et al., 2013), which recruits subjects with extreme phenotypic values from both tail regions of truncated normal distribution (
The relationship between sample regression coefficient
Similarly, to calculate the equivalent sample size for case-control study, the key is to build up the relationship between the estimated log odds ratio based on standardised genotype, i.e.,
Under the assumption of point-normal genetic effect distribution, we also compared the efficacy of PGS constructed by the ordinary least square estimate (OLSE), p-value thresholding method and the aforementioned posterior expectation shrinkage relative to the true additive genetic value (Figure 4). In this figure, the p-value threshold is chosen to maximize the
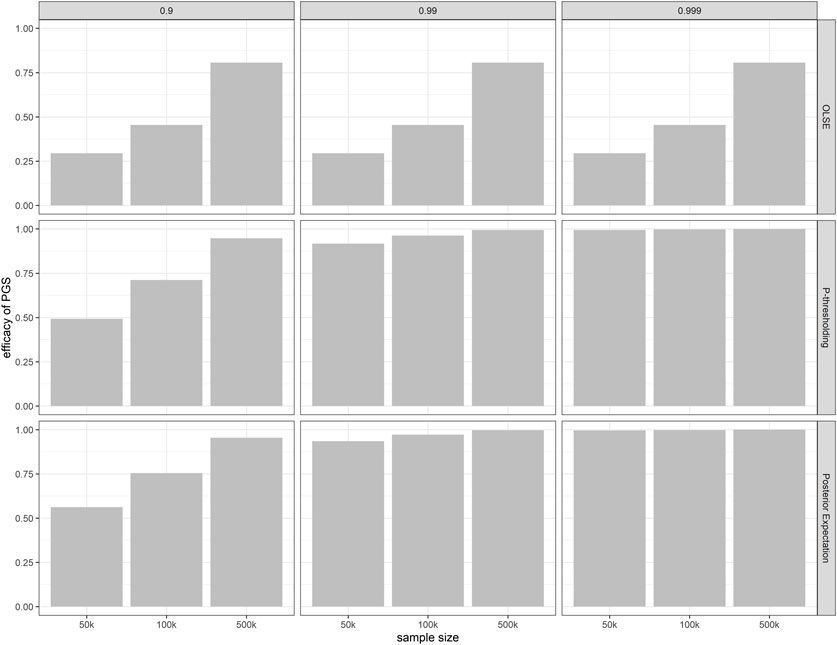
FIGURE 4. Efficacy of PGS constructed under different
We compared the predicted results with the reported meta-GWAS outcomes (Table 4). The predicted number of independent significant SNPs, the apparent and corrected variance explained are calculated based on
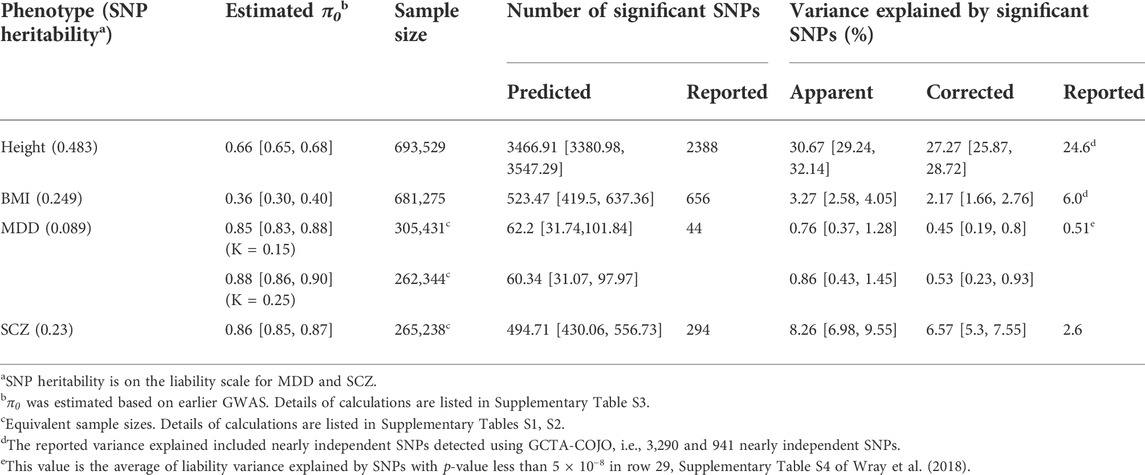
TABLE 4. Predicted versus reported numbers of independent significant SNPs and variance explained by these SNPs with 95% probability intervals (PIs) based on the range of estimated π0 for height, body mass index (BMI), major depressive disorder (MDD), and schizophrenia (SCZ).
For BMI and MDD, the predicted key GWAS outcomes are close to the reported values. However, our model over-estimated the results for height and SCZ. For height, one of the possible reasons is that the effect size distribution is not as simple as a point-normal, which is supported by other reference (Zhang et al., 2018). For schizophrenia, mixed population in discovery samples, for example, Asian samples are included in Ripke et al. (2014) and PGC3—SCZ (The Schizophrenia Working Group of the Psychiatric Genomics Consortium Ripke et al., 2020), may lead to the phenomenon that the reported number of significant SNPs is less than expected and it is out of the scope of our model. For different populations, m would be different, but how exactly the mixed population in discovery sample would affect the detected number of significant SNPs needs further study.
In this paper, we derived theoretical results and provided computational algorithms for predicting the key outcomes of GWAS or meta-GWAS using parameters regarding the genetic architecture of phenotype and sample size, under the assumption that the standardised effect sizes of all SNPs in the genome follow a point-normal distribution. We conducted simulation studies to validate our theoretical results, and applied our model to GWAS data on four example complex traits.
Our results show that the density function of statistical power across causal SNPs under the assumed effect size distribution is bimodal with peaks near 0 and 1 (a variation of Figure 2B; Supplementary Figure S1). In other words, most causal SNPs have statistical power close to either zero or one, because of “floor” and “ceiling” effects. The relative heights of the two peaks are influenced by sample size; increasing sample size will increase the statistical power of all causal SNPs and thus reduce the height of the peak near zero and increase the height near one. From the distribution of statistical power, the expectations and variances of key GWAS outcomes, such as the number of independent genome-wide significant SNPs and the phenotypic variance explained by these SNPs, can be calculated. These calculations have been implemented in an online interactive tool named Polygenic Power Calculator.
For many phenotypes, meta-GWAS sample sizes have not reached the halfway point of the desired level to detect most of the contributing SNPs. Taking MDD as an example, we estimate that 7.36 × 10 (de Vlaming et al., 2017) equivalent total samples are needed to detect 95% of all causal SNPs when MDD prevalence is 15% whereas the existing equivalent sample size only reaches 3.05 × 10 (Torkamani et al., 2018). On the other hand, it takes a much smaller sample size to capture most of the genetic variance. Figures 2C,D shows that when
In genetic association studies, the most common definition of effect size is the per-allele effect
The parameter
In the early days of GWAS, only a few independent significant SNPs were observed from GWAS and meta-GWAS due to limited sample size. Visscher et al. (Visscher et al., 2012) made the empirical observation of a roughly linear relationship between discovery sample size and the number of genome-wide significant hits, once the sample size reached a level sufficient to detect a few SNPs. This pattern matches the linear part of the S-shape in Figure 2C. In this study, we further extended the range of sample size to that needed to detect nearly all
Our method has some limitations. First, we assumed the SNPs to be independent, on the basis that GWAS or meta-GWAS usually report independent SNPs after pruning or clumping. This assumption simplifies the model and bridges the relationship between genetic architecture parameters and key GWAS outcomes directly in a concise manner. We adopted 60,000 as the number of independent SNPs, but the appropriate number may depend on the population, minor allele frequency cutoff, and sample size. A more satisfactory approach in the future may be to explicitly take LD into account, expressing marginal SNP effects by weighted sums of joint effects, while making reasonable assumptions for the joint effect size distribution. Second, we adopted the per standard deviation allele effect as effect size and ignored possible differences in the relationships between allele frequency to effect size distribution for different phenotypes. Although this definition has been widely adopted (Daetwyler et al., 2008; Dudbridge, 2013), models taking allele frequency into account in effect size distribution are not uncommon (Park et al., 2010; So et al., 2010). Third, we assumed the standardised effect sizes followed a point-normal distribution but several other effect size distributions have been proposed (Zhou et al., 2013). Thus, it would be interesting to investigate how these other distributions would alter the predicted behaviour of GWAS outcomes. Fourth, our model ignores the contribution of rare variants (allele frequency < 1%). As GWAS are increasing in both sample size and number of genotyped or imputed SNPs, more rare variants with large effect size are being detected. The observed discrepancies between the predicted values from our model and the reported empirical results for height and schizophrenia also suggest possible inadequacies in our model, including misspecification of effect size distribution, inaccurate estimates of parameters such as
Heritability of BMI can be found here: http://www.nealelab.is/uk-biobank/. The online power calculator is available at https://twexperiment.shinyapps.io/PPC_v2_1/.
The original contributions presented in the study are included in the article and its Supplementary Material, further inquiries can be directed to the corresponding author.
PS conceived of the presented idea. TW, ZL, and TM developed the theory. TW performed the computations and drafted the article. ZL and PS made revision of the article. All authors discussed the results, contributed to, and approved the final manuscript.
This work was supported by Hong Kong Research Grants Council Collaborative Research Grant C7044-19G, Theme-based Research Scheme Grant T12-712/21-R, Hong Kong Innovation and Technology Bureau funding for the State Key Laboratory of Brain and Cognitive Sciences, and National Natural Science Foundation of China (32170637).
Author TM was employed by Fano Labs.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.989639/full#supplementary-material
Amanat, S., Requena, T., and Lopez-Escamez, J. A. (2020). A systematic review of extreme phenotype strategies to search for rare variants in genetic studies of complex disorders. Genes 11, 987. doi:10.3390/genes11090987
Barnett, I. J., Lee, S., and Lin, X. (2013). Detecting rare variant effects using extreme phenotype sampling in sequencing association studies. Genet. Epidemiol. 37, 142–151. doi:10.1002/gepi.21699
Bigdeli, T. B., Lee, D., Webb, B. T., Riley, B. P., Vladimirov, V. I., Fanous, A. H., et al. (2016). A simple yet accurate correction for winner's curse can predict signals discovered in much larger genome scans. Bioinformatics 32, 2598–2603. doi:10.1093/bioinformatics/btw303
Bulik-Sullivan, B. K., Loh, P. R., Finucane, H. K., Ripke, S., Yang, J., Patterson, N., et al. (2015). LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295. doi:10.1038/ng.3211
Cano-Gamez, E., and Trynka, G. (2020). From GWAS to function: Using functional genomics to identify the mechanisms underlying complex diseases. Front. Genet. 11, 424. doi:10.3389/fgene.2020.00424
Chatterjee, N., Wheeler, B., Sampson, J., Hartge, P., Chanock, S. J., and Park, J. H. (2013). Projecting the performance of risk prediction based on polygenic analyses of genome-wide association studies. Nat. Genet. 45, 400–405. doi:10.1038/ng.2579
Daetwyler, H. D., Villanueva, B., and Woolliams, J. A. (2008). Accuracy of predicting the genetic risk of disease using a genome-wide approach. PLoS One 3, e3395. doi:10.1371/journal.pone.0003395
de Vlaming, R., Okbay, A., Rietveld, C. A., Johannesson, M., Magnusson, P. K. E., Uitterlinden, A. G., et al. (2017). Meta-GWAS accuracy and power (MetaGAP) calculator shows that hiding heritability is partially due to imperfect genetic correlations across studies. PLoS Genet. 13, e1006495. doi:10.1371/journal.pgen.1006495
Dudbridge, F. (2013). Power and predictive accuracy of polygenic risk scores. PLoS Genet. 9, e1003348. doi:10.1371/journal.pgen.1003348
Euesden, J., Lewis, C. M., and O'Reilly, P. F. (2015). PRSice: Polygenic risk score software. Bioinformatics 31, 1466–1468. doi:10.1093/bioinformatics/btu848
Falconer, D. S. (1996). Introduction to quantitative genetics. Harlow, United Kingdom: Prentice-Hall.
Falconer, D. S. (1965). The inheritance of liability to certain diseases estimated from the incidence among relatives. Ann. Hum. Genet. 29, 51–76. doi:10.1111/j.1469-1809.1965.tb00500.x
Genomes Project Consortium Auton, A., Brooks, L. D., Durbin, R. M., Garrison, E. P., Kang, H. M., Korbel, J. O., et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. doi:10.1038/nature15393
Holland, D., Frei, O., Desikan, R., Fan, C. C., Shadrin, A. A., Smeland, O. B., et al. (2020). Beyond SNP heritability: Polygenicity and discoverability of phenotypes estimated with a univariate Gaussian mixture model. PLoS Genet. 16, e1008612. doi:10.1371/journal.pgen.1008612
Howard, D. M., Adams, M. J., Clarke, T. K., Hafferty, J. D., Gibson, J., Shirali, M., et al. (2019). Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 22, 343–352. doi:10.1038/s41593-018-0326-7
Hyde, C. L., Nagle, M. W., Tian, C., Chen, X., Paciga, S. A., Wendland, J. R., et al. (2016). Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat. Genet. 48, 1031–1036. doi:10.1038/ng.3623
Johnson, J. L., and Abecasis, G. (2017). GAS power calculator: Web-based power calculator for genetic association studies. bioRxiv.
Lam, M., Chen, C. Y., Li, Z. Q., Martin, A. R., Bryois, J., Ma, X. X., et al. (2019). Comparative genetic architectures of schizophrenia in East Asian and European populations. Nat. Genet. 51, 1670–1678. doi:10.1038/s41588-019-0512-x
Lee, P. H., Anttila, V., Won, H., Feng, Y. A., Rosenthal, J., Zhu, Z., et al. (2019). Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell 179, 1469–1482. e1411. doi:10.1016/j.cell.2019.11.020
Lee, S. H., Goddard, M. E., Wray, N. R., and Visscher, P. M. (2012). A better coefficient of determination for genetic profile analysis. Genet. Epidemiol. 36, 214–224. doi:10.1002/gepi.21614
Lloyd-Jones, L. R., Zeng, J., Sidorenko, J., Yengo, L., Moser, G., Kemper, K. E., et al. (2019). Improved polygenic prediction by Bayesian multiple regression on summary statistics. Nat. Commun. 10, 5086. doi:10.1038/s41467-019-12653-0
Locke, A. E., Kahali, B., Berndt, S. I., Justice, A. E., Pers, T. H., Felix, R., et al. (2015). Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206. doi:10.1038/nature14177
Mak, T. S. H., Kwan, J. S., Campbell, D. D., and Sham, P. C. (2016). Local true discovery rate weighted polygenic scores using GWAS summary data. Behav. Genet. 46, 573–582. doi:10.1007/s10519-015-9770-2
Mak, T. S. H., Porsch, R. M., Choi, S. W., Zhou, X., and Sham, P. C. (2017). Polygenic scores via penalized regression on summary statistics. Genet. Epidemiol. 41, 469–480. doi:10.1002/gepi.22050
Moser, G., Lee, S. H., Hayes, B. J., Goddard, M. E., Wray, N. R., and Visscher, P. M. (2015). Simultaneous discovery, estimation and prediction analysis of complex traits using a bayesian mixture model. PLoS Genet. 11, e1004969. doi:10.1371/journal.pgen.1004969
Palmer, C., and Pe'er, I. (2017). Statistical correction of the Winner's Curse explains replication variability in quantitative trait genome-wide association studies. PLoS Genet. 13, e1006916. doi:10.1371/journal.pgen.1006916
Park, J. H., Wacholder, S., Gail, M. H., Peters, U., Jacobs, K. B., Chanock, S. J., et al. (2010). Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat. Genet. 42, 570–575. doi:10.1038/ng.610
Privé, F., Arbel, J., and Vilhjálmsson, B. J. (2020). LDpred2: Better, faster, stronger. Bioinformatics 36, 5424–5431. doi:10.1093/bioinformatics/btaa1029
Purcell, S., Cherny, S. S., and Sham, P. C. (2003). Genetic power calculator: Design of linkage and association genetic mapping studies of complex traits. Bioinformatics 19, 149–150. doi:10.1093/bioinformatics/19.1.149
Purcell, S., Wray, N. R., Stone, J. L., Visscher, P. M., O'Donovan, M. C., Sullivan, P. F., et al. International Schizophrenia Consortium (2009). Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460, 748–752. doi:10.1038/nature08185
Qian, J., Tanigawa, Y., Du, W., Aguirre, M., Chang, C., Tibshirani, R., et al. (2020). A fast and scalable framework for large-scale and ultrahigh-dimensional sparse regression with application to the UK Biobank. PLoS Genet. 16, e1009141. doi:10.1371/journal.pgen.1009141
Ripke, S., Neale, B. M., Corvin, A., Walters, J. T. R., Farh, K., Holmans, P. A., et al. (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. doi:10.1038/nature13595
The Schizophrenia Working Group of the Psychiatric Genomics Consortium Ripke, S., Walters, J. T. R., and O'Donovan, M. C. (2020). Mapping genomic loci prioritises genes and implicates synaptic biology in schizophrenia. medRxiv. doi:10.1101/2020.09.12.20192922
Sham, P. C., and Purcell, S. M. (2014). Statistical power and significance testing in large-scale genetic studies. Nat. Rev. Genet. 15, 335–346. doi:10.1038/nrg3706
So, H. C., and Sham, P. C. (2017). Improving polygenic risk prediction from summary statistics by an empirical Bayes approach. Sci. Rep. 7, 41262. doi:10.1038/srep41262
So, H. C., Yip, B. H., and Sham, P. C. (2010). Estimating the total number of susceptibility variants underlying complex diseases from genome-wide association studies. PLoS One 5, e13898. doi:10.1371/journal.pone.0013898
Song, S., Jiang, W., Hou, L., and Zhao, H. (2020). Leveraging effect size distributions to improve polygenic risk scores derived from summary statistics of genome-wide association studies. PLoS Comput. Biol. 16, e1007565. doi:10.1371/journal.pcbi.1007565
Speed, D., Cai, N., Johnson, M. R., Nejentsev, S., Balding, D. J., and Consortium, U. (2017). Reevaluation of SNP heritability in complex human traits. Nat. Genet. 49, 986–992. doi:10.1038/ng.3865
Torkamani, A., Wineinger, N. E., and Topol, E. J. (2018). The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 19, 581–590. doi:10.1038/s41576-018-0018-x
Vilhjalmsson, B. J., Yang, J., Finucane, H. K., Gusev, A., Lindstrom, S., Ripke, S., et al. (2015). Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am. J. Hum. Genet. 97, 576–592. doi:10.1016/j.ajhg.2015.09.001
Visscher, P. M., Brown, M. A., McCarthy, M. I., and Yang, J. (2012). Five years of GWAS discovery. Am. J. Hum. Genet. 90, 7–24. doi:10.1016/j.ajhg.2011.11.029
Visscher, P. M., Wray, N. R., Zhang, Q., Sklar, P., McCarthy, M. I., Brown, M. A., et al. (2017). 10 years of GWAS discovery: Biology, function, and translation. Am. J. Hum. Genet. 101, 5–22. doi:10.1016/j.ajhg.2017.06.005
Wood, A. R., Esko, T., Yang, J., Vedantam, S., Pers, T. H., Gustafsson, S., et al. (2014). Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet. 46, 1173–1186. doi:10.1038/ng.3097
Wray, N. R., Ripke, S., Mattheisen, M., Trzaskowski, M., Byrne, E. M., Abdellaoui, A., et al. (2018). Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 50, 668–681. doi:10.1038/s41588-018-0090-3
Wray, N. R., Yang, J., Hayes, B. J., Price, A. L., Goddard, M. E., and Visscher, P. M. (2013). Pitfalls of predicting complex traits from SNPs. Nat. Rev. Genet. 14, 507–515. doi:10.1038/nrg3457
Wu, T., and Sham, P. C. (2021). On the transformation of genetic effect size from logit to liability scale. Behav. Genet. 51, 215–222. doi:10.1007/s10519-021-10042-2
Yang, J., Benyamin, B., McEvoy, B. P., Gordon, S., Henders, A. K., Nyholt, D. R., et al. (2010). Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 42, 565–569. doi:10.1038/ng.608
Yengo, L., Sidorenko, J., Kemper, K. E., Zheng, Z., Wood, A. R., Weedon, M. N., et al. (2018). Meta-analysis of genome-wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum. Mol. Genet. 27, 3641–3649. doi:10.1093/hmg/ddy271
Zhang, Y., Qi, G., Park, J. H., and Chatterjee, N. (2018). Estimation of complex effect-size distributions using summary-level statistics from genome-wide association studies across 32 complex traits. Nat. Genet. 50, 1318–1326. doi:10.1038/s41588-018-0193-x
Keywords: GWAS, polygenic model, power calculation, online tool, statistical method
Citation: Wu T, Liu Z, Mak TSH and Sham PC (2022) Polygenic power calculator: Statistical power and polygenic prediction accuracy of genome-wide association studies of complex traits. Front. Genet. 13:989639. doi: 10.3389/fgene.2022.989639
Received: 08 July 2022; Accepted: 02 September 2022;
Published: 10 October 2022.
Edited by:
Rongling Wu, The Pennsylvania State University (PSU), United StatesReviewed by:
Christoph Lippert, Hasso Plattner Institute, GermanyCopyright © 2022 Wu, Liu, Mak and Sham. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pak Chung Sham, cGNzaGFtQGhrdS5oaw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.