 Mila Mirceta1,2
Mila Mirceta1,2 Natalie Shum
Natalie Shum Monika H. M. Schmidt
Monika H. M. Schmidt
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 17 November 2022
Sec. Neurogenomics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.985975
This article is part of the Research Topic Microsatellite expansion disorders: Molecular mechanisms and therapeutic targets View all 6 articles
Expanded tandem repeat DNAs are associated with various unusual chromosomal lesions, despiralizations, multi-branched inter-chromosomal associations, and fragile sites. Fragile sites cytogenetically manifest as localized gaps or discontinuities in chromosome structure and are an important genetic, biological, and health-related phenomena. Common fragile sites (∼230), present in most individuals, are induced by aphidicolin and can be associated with cancer; of the 27 molecularly-mapped common sites, none are associated with a particular DNA sequence motif. Rare fragile sites (
The terms “fragility” and “fragile site,” coined in 1969–70, refer to unusual secondary constrictions in chromosomes, that are distinct from the primary constrictions of the centromeres (Schmid and Vischer, 1969; Magenis et al., 1970). Under specific conditions of replicative stress, they can also manifest as chromatin gaps, breaks, or failed chromatin compaction on metaphase chromosomes. Fragile sites are found across the genome, such as in the heterochromatic regions harboring classical satellite repeats on chromosomes 1, 9, 15, 16, and Y, as well as the common and rare fragile sites (Figure 1). Fragile sites can also arise at telomeres, at telomere fusions, and at other specific genetic loci. Due to their genome-wide prevalence, fragile sites have been found to be associated with genetic and genomic instability, and are extensively linked to many disease phenotypes, including neurological disorders (sections 2.1, 2.2), immunodeficiency–centromeric instability–facial anomalies (ICF) syndrome (section 2.3), and cancer progression.
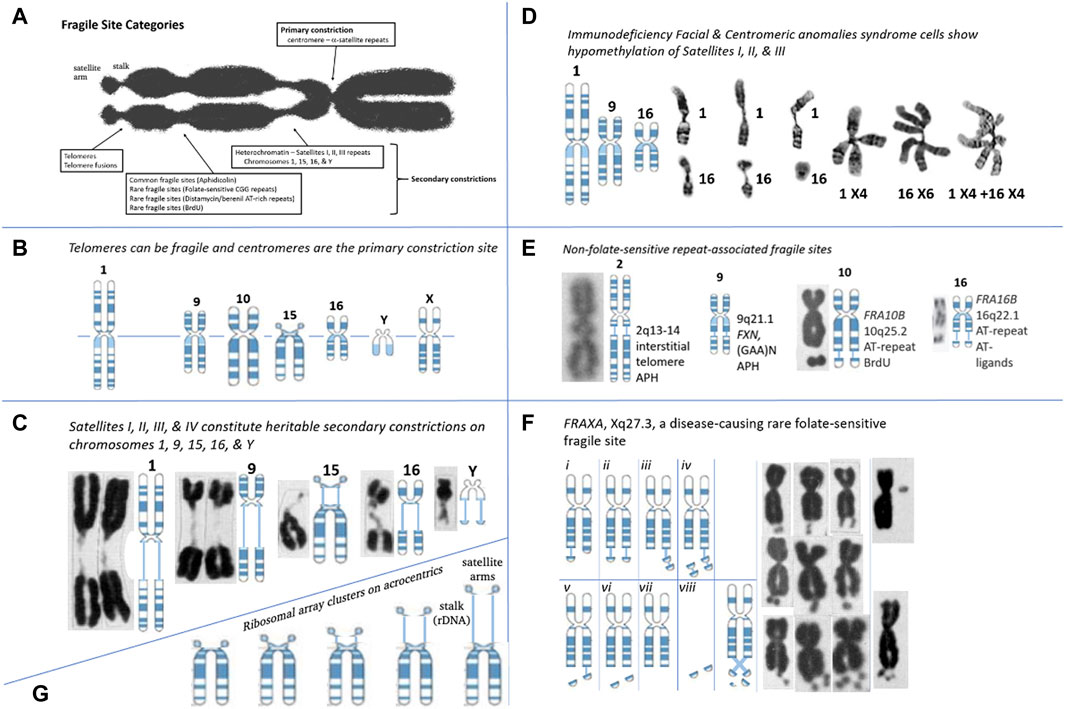
FIGURE 1. Repeat Tracts, fragile sites, and disease. (A) Categories of fragile site constrictions observable on human chromosomes. (B) Fragile sites can occur at the telomere or centromere, observed on chromosomes 1, 2, 9, 10, 15, 16, Y, and X. Telomere ends display “fragile-site like” appearances that are TRF1-dependant and APH inducible (Sfeir et al., 2009). The centromere of every chromosome is the “primary constriction,” composed of repeats. (C) Secondary constrictions are present on chromosomes 1, 9, 16, 15, and Y. Cytogeneticists were aware of these gaps prior to FRAXA and modeled it after the secondary constrictions (Lubs, 1969). These are composed of the classical satellite repeat DNAs, where four types (I-IV) of satellite DNA are located in the heterochromatic regions of chromosomes 1, 9, 15, 16, and Y, the total amount on these chromosomes and the proportion of the types being different (Vogt, 1990). Satellite regions of chromosomes 9 and Y, whose composition is the most complicated, and chromosome 15 is less complex, but like 9 and Y, it comprises all four types of satellite DNA. Chromosome 1 has type II satellite DNA, with the proportion of the remaining types being less. The C segment of chromosome 16 comprises only type II. The size/length of the secondary constrictions is highly-polymorphic amongst individuals, and segregates as a heritable state (Chromosomes: Guttenbach and Schmid, 1994). (D) In Immunodeficiency, Centromeric instability & Facial anomalies (ICF) syndrome, satellites I, II, and III of chromosomes 1, 9, and 16 are hypomethylated and show secondary constrictions within these regions. ICF syndrome was recently shown to be caused by four mutations in four genes: ICF1/DNMT3B, ICF2/ZBTB24, ICF3/CDCA7, and ICF4/HELLS (van den Boogaard et al., 2017). Satellite-containing regions on chromosomes 1, 9, and 16 are hypomethylated in individuals affected by ICF syndrome, and these show a variety of aberrant chromosomes: secondary constrictions, multibranched chromosome arms, whole arm deletions, duplications, isochromosomes, and centromeric fragility. As with fragile sites, these involved double-strand DNA breaks (Sawyer et al., 1995; Tuck-Muller et al., 2000). (Chromosomes: Tuck-Muller et al., 2000). (E) Many non-folate sensitive fragile sites have been mapped at repetitive regions. The interstitial telomeric repeat on chromosome 2, the AT-repeats of FRA10B and FRA16B, and the GAA repeat of FXN on chromosome 9 has “fragile-like” characteristics. Fragile site at 2q13-14 at an interstitial inverted head-to-head array of the telomeric repeat (TAGAGGG)54-(CCCTAA)104, a result of an ancient telomeric fusion, not “telomere healing” event (IJdo et al., 1991; Bosco and de Lange, 2012). Notably, there are other interstitial telomeric sequences (Wells et al., 1990). A “fragile-like” site has been reported at 9q21.1 in the expanded (GAA)N repeat in FXN, which causes Friedreich’s ataxia (Kumari et al., 2015). FRA10B at 10q25.2 is induced by BrdU and mapped to an expanded ∼42 bp AT-rich minisatellite repeat (Sutherland et al., 1980; Hewett et al., 1998) (Chromosomes: Bosco and de Lange, 2012; Sutherland et al., 1980; Felbor et al., 2003). (F) Various presentations of the FRAXA site in CGG-expanded FXS patient cells (Crippa et al., 1984; Fitchett and Seabright, 1984; Savage and Fitchett, 1988): chromatid breaks/gaps, isochromatid breaks, isolated double-minutes, deleted X’s, secondary duplications (double satellite). Satellite association and variations in length of the nucleolar constriction of normal and variant human G chromosomes. (Chromosomes: Lubs, 1969). (G) Ribosomal DNAs can vary the length of the chromosome by varying lengths of the secondary constrictions (stalks) of the acrocentrics (Chr 13, 14, 15, 21, and 22) on which they reside (Orye, 1974; Cheung et al., 1989; Heliot et al., 1997).
The first fragile site was observed in 1965 (Dekaban, 1965), followed by the discovery of the first disease-associated fragile site at the fragile X locus (Lubs, 1969), later demonstrated to be Martin-Bell syndrome (Richards et al., 1981). This initial discovery remained largely ignored until it was serendipitously induced in specific folate-deficient culture conditions, leading to the renaming of the disease to fragile X syndrome (FXS) (Sutherland, 1977) (reviewed in (Hecht and Kaiser-McCaw, 1979). Since then, discovery of these sites at specific loci has broadened.
The current classifications of fragile sites fall into two categories largely based on frequency of expression and induction method: common fragile sites (CFSs) and rare fragile sites (RFSs). The Human Genome Database documents ∼90 CFSs and ∼30 RFSs that have been cytogenetically observed and documented in previous studies (reviewed by (Feng and Chakraborty, 2017).
CFSs are present in a large proportion of the population, and are induced by aphidicolin, 5-azacytidine, and bromodeoxyuridine (BrdU) (Glover et al., 1984; Yunis and Soreng, 1984; Sutherland et al., 1985b). RFSs are observed to a maximal frequency of 5% in the population (Schmid et al., 1986) and can be induced by folate deficiency/thymidylate stress, distamycin A, and BrdU (Sutherland, 1983; Hecht and Sutherland, 1984; Sutherland et al., 1985a). Detailed protocols for the detection and analysis of both CFSs and RFSs have been published recently (Bjerregaard et al., 2018). As CFSs are linked to regions of chromosomal rearrangements in cancer, this group of fragile sites has been far more extensively studied than RFSs (reviewed in (Dillon et al., 2010; Ozeri-Galai et al., 2012; Sarni and Kerem, 2016; Glover et al., 2017; Irony-Tur Sinai and Kerem, 2019; Kaushal and Freudenreich, 2019). Harnessing knowledge about CFSs could empower the field of RFSs and provide important clues as to how fragility contributes to other disease phenotypes and genetic abnormalities (i.e., repeat instability).
The current distinction between common and rare fragile sites is problematic, being based both on the conditions that induce their expression, and the frequency with which they are present in the population (Hecht, 1986; Mrasek et al., 2010). There is no clear numerical delineation between the frequency of “common” and “rare” fragile sites. Some CFSs are rare in their manifestation, suggesting they are not ubiquitously present in all individuals or might be observed at lower levels (e.g., FRA2D, FRA18B, and FRA9D are expressed in <12% of individuals) (Savelyeva et al., 2006). However, many fragile sites have been categorized as “common” when they are detected by aphidicolin induction, but have not been assessed at a population level. Distinct rare and common fragile sites have also been found to cluster together, appearing either on the same or on neighboring metaphase chromosome bands; for example, the RFS FRA11B and the APH-inducible CFS FRA11G are located at 11q23.3 (Fechter et al., 2007), and the RFS FRAXA and the APH-inducible CFS FRAXD are located at Xq27.3 and Xq27.2, respectively (Hecht and Bixenman, 1990; Sutherland and Baker, 1990) (see Table 1 for complete list of known clustered fragile sites). Due to this clustering, fragile sites may often be missed or misclassified, despite being independent fragility events with their own downstream consequences. Furthermore, some very common CFSs can be induced by conditions known to induce RFSs (i.e. folate deficiency) (Kähkönen et al., 1989; Jenkins et al., 1990; Mrasek et al., 2010). This finding demonstrates that, although certain sites may be more sensitive to specific induction methods, cytogenetic expression at a given site could be achieved with other drugs, albeit at reduced levels. Our current cytological screening methods, relying on the presence of observed metaphase chromatid breaks, may not be sensitive enough to reveal less pronounced signs of fragility at many sites. In fact, all fragile loci may be inherently sensitive to any form of replicative stress, but the ability to observe cytogenetic fragile site expression at the macro level may be uniquely influenced by their specific genomic landscape–i.e., sequence, gene expression, replication timing, among other factors. As such we propose that each CFS and RFS should also be classified on the primary induction conditions used for that locus, which may more accurately reveal similarities and differences in the characteristics and mechanisms of fragility.
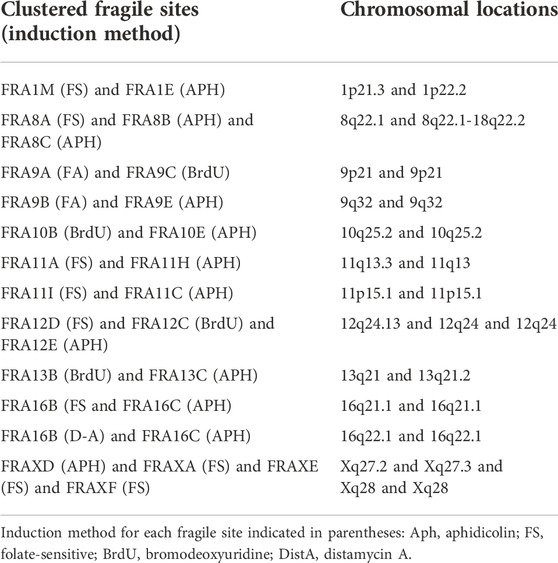
TABLE 1. Clustered fragile sites.
The most common inducer of CFSs (∼75 sites) is aphidicolin, a deoxycytidine analogue and inhibitor of DNA polymerases α, δ, and ɛ that affects replication fork progression (Glover et al., 1984; Cheng and Kuchta, 1993). There are currently 25 molecularly mapped aphidicolin-inducible CFSs, all characterized by large AT-rich regions of DNA (reviewed in (Feng and Chakraborty, 2017) and can span a region of hundreds of kilobases to megabases of a chromosome (Mishmar et al., 1999; Zlotorynski et al., 2003; Irony-Tur Sinai and Kerem, 2019). They are frequently associated with hotspots of deletions, rearrangements, and translocations in cancer. Although the exact mechanism of aphidicolin-induced fragility is unknown, it is proposed that the induced replicative stress leads to stalling and breakage at these CFS regions due to the compounded effects of late replication, origin scarcity, concurrent transcription, and structure formation (reviewed in (Glover et al., 2017; Irony-Tur Sinai and Kerem, 2019; Kaushal and Freudenreich, 2019).
Thymidylate stress, caused by folate deficiency, induces the appearance of 24 of the 30 known RFSs, hereafter identified as folate-sensitive fragile sites (FSFS). To date, 10 FSFSs have been sequence-mapped to gene-specific expanded (CGG)n repeats with the most well-known site being FRAXA which occurs at FMR1 and causes FXS. Of the remaining RFSs, three are inducible by distamycin A and three are inducible by either distamycin A or BrdU. Two of the distamycin A-inducible RFSs have been mapped to minisatellite AT-rich repeat sequences (reviewed in (Debacker and Kooy, 2007; Lukusa and Fryns, 2008). Fragility is proposed to occur when replication progression is impeded upon the binding of distamycin A (and related compounds like berenil, netropsin, Hoechst 33248, D287/170, methyl-green, and DAPI) to the minor groove of these CFS regions (Thys et al., 2015). All the currently identified RFSs, which have been cytogenetically defined and mapped and many cloned and sequenced, are highlighted in Table 2, along with known features and disease links for each. Numerous attempts to identify internal controls for diagnostic FXS by FRAXA induction revealed many sites that presented low-level (<4%) folate-sensitive fragility (reviewed in (Krawczun et al., 1991). In the proper population (disease or other) and induction systems, new rare fragile sits may be discovered. Recent discovery of new tandem repeat expansion loci could be the molecular cause of new, as yet to be observed fragile sites or chromosomal lesions (Giannuzzi et al., 2021; Altemose et al., 2022; Ebler et al., 2022; Gershman et al., 2022; Hoyt et al., 2022; Nurk et al., 2022; Talbert and Henikoff, 2022; Vollger et al., 2022; Wang et al., 2022).
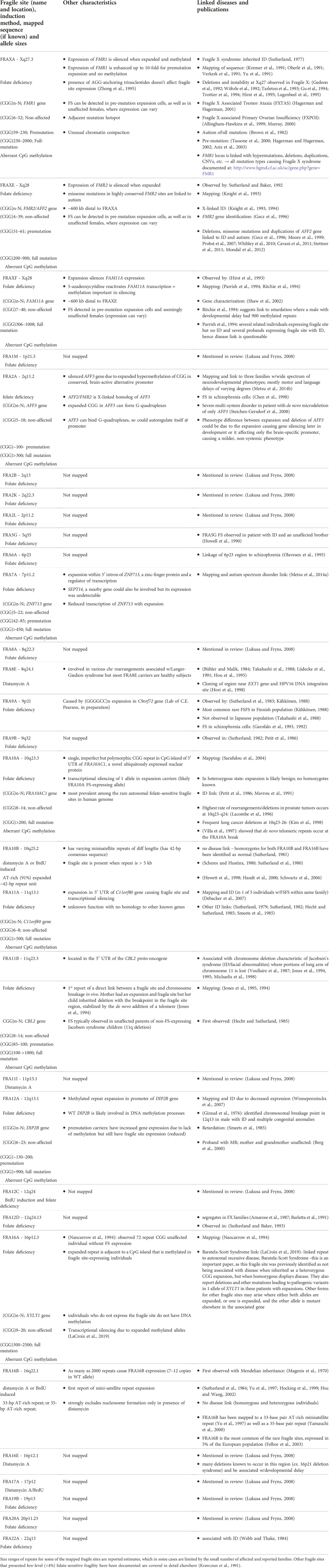
TABLE 2. Rare fragile sites (folate, distamycin A, and BrdU).
Spontaneous fragile sites occur without the need for induction at chromosomal locations distinct from either the common or rare fragile sites. These spontaneous sites can be expressed at unusually high levels, from 80 to 100% of the population, compared to the 4–30% for most fragile sites (Dar et al., 1995; Karadeniz et al., 2003; Zamani et al., 2007). The nature of the molecular cause (sequence, epigenetic, or other) of most of these spontaneous fragile sites is not known and warrants further investigation. Examples include the secondary constrictions on chromosomes 1, 9, 16, and Y, as well as FRA1R/1q41 and FRA16B/16q22. It is possible that these spontaneous sites are due to repetitive sequences, as the spontaneous FRA16B has been mapped to a 33-base pair (bp) AT-rich minisatellite repeat (Yu et al., 1997) as well as a 35 bp repeat (Yamauchi et al., 2000). FRA16B is the most common of the RFSs, expressed in 5% of the European population (Felbor et al., 2003). Other spontaneous fragile sites have been localized to intra-chromosomal telomere tracts (Musio et al., 1996), which are frequent polymorphisms of heterochromatin without known functional or phenotypic effect. The length of the chromosomal gaps or despiralized regions can vary widely between individuals, is considered to be hereditary, and due to the highly variable lengths of the satellite tracts (Craig-Holmes and Shaw, 1971; Yunis and Yasmineh, 1971; Craig-Holmes et al., 1975, 1973; McKenzie and Lubs, 1973; Podugolnikova and Korostelev, 1980). These spontaneous, heritable fragile sites often map to loci known to be prone to structural variations including microdeletions, microduplications, and copy number variations (CNVs) (Zamani et al., 2007; Szafranski et al., 2010; Gillentine and Schaaf, 2015). Viral integration can also be a driving factor for these spontaneous sites (O’Neill and Miles, 1969; Peat and Stanley, 1986) (reviewed in (Fortunato and Spector, 2003). Interestingly, chromosomal integration of tandem repeats of foreign DNA can lead to fragile site expression, further supporting the possibility that repeat tracts underlie spontaneously expressed fragile sites (Ragland et al., 2008; Jacome and Fernandez-Capetillo, 2011; Irony-Tur Sinai et al., 2019).
As mentioned, many CFSs and RFSs have been observed cytogenetically; however, only a handful have been molecularly mapped to specific genomic locations, or specific sequences. Mapping fragile sites is an investment, as the efforts from initial cytogenetic observation, to molecular mapping, to gene identification and epigenetic modifications, can be considerable and span years (Figure 2). Mapping of fragile sites dates to the 1980s, where R-banding was performed, and the general chromosomal site of the observed break was reported. This technique was utilized to determine the chromosomal location of the DAPI-inducible CFS FRA1H (Pelliccia and Rocchi, 1986), providing the basis for further, more detailed mapping. Using yeast artificial chromosomes (YACs), bacterial artificial chromosomes (BACs), and cosmid clones that span the region of the identified cytogenetic location, physical mapping and fluorescence in situ hybridization (FISH) experiments allowed for further characterization of the genomic location of these fragile sites, albeit still at a low resolution. Some examples of both CFSs and RFSs that were mapped in such a manner include FRAXA (Kremer et al., 1991; Verkerk et al., 1991), FRA11B (Jones et al., 1994), FRA3B (Boldog et al., 1994), FRA16D (Paige et al., 2000), FRAXB (Arlt et al., 2002), and FRA7B (Bosco et al., 2010). Clustered fragile sites (touched upon in section 1.1) can require finer mapping in order to be distinguished. Higher resolution mapping has been performed with the use of multi-colour FISH combined with the availability of sequence databases and programs. With this method, an initial large region spanning the cytogenetic location of the fragile site is covered with BAC probes labelled with different colors. Increasingly finer mapping is conducted with contiguous multi-colored BAC probes spanning smaller and smaller lengths across the break point until an exact breakage boundary can be determined. The specific sequence of this region along with the encompassing genes are then identified through programs such as RepeatMasker and through human genome sequence databases (Hormozian et al., 2007; Zheglo et al., 2019). The identification of these specific fragile site-associated genes can initiate further studies on the role of fragile sites in human genetic diseases and cancer.
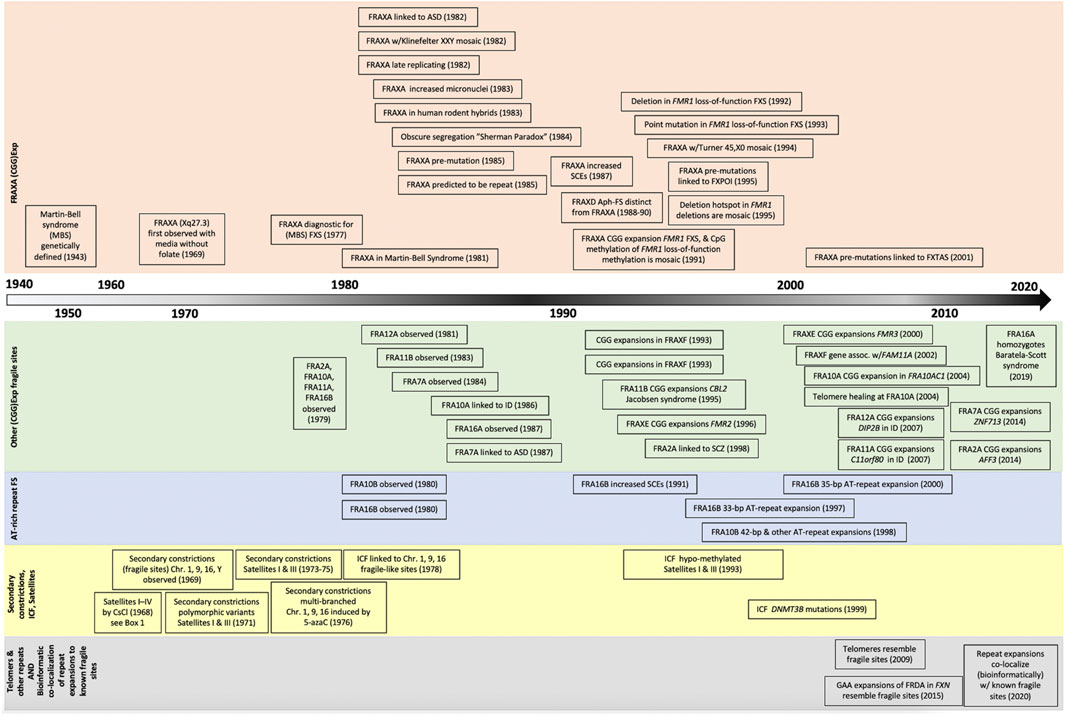
FIGURE 2. Timeline of discovery for tandem repeat expansions and chromosomal lesions. Increased awareness and improved methods of detection have fostered the identification of dozens of fragile site in the past two decades, with new bioinformatic techniques poised to launch an new era of fragile site discoveries.
As in the case of CFSs, the mechanisms and common sequence motifs that are shared between these regions are merely beginning to be elucidated, having previously been limited by early cytogenetic methods used to fine map fragile regions (i.e., physical mapping and FISH). The onset of bioinformatic methods and databases in recent years provides the potential to simultaneously identify many regions prone to fragility, making them strong candidates for further analysis. Prada and Laissue (2014) used bioinformatic methods to identify chromosomal rearrangements of the X chromosome in 13 different mammalian species (Prada and Laissue, 2014). They identified fragile sites previously associated with the human X chromosome (FRAXA, B, C, D, E, and F), and were also able to determine fragile sites that are conserved between mammalian species, implying that these regions could have functional roles. Their work characterizing the X chromosome provides exciting new avenues for expansion to the rest of the genome and in identifying novel important regions of fragility. Ji et al. (2020) provided a genome-wide mapping of CFSs by using the previous knowledge that most CFSs undergo mitotic DNA synthesis (MiDAS); by sequencing the nascent DNA in mitotic cells treated with aphidicolin, novel aphidicolin-inducible CFSs were able to be uncovered (Ji et al., 2020). The methods of mapping the molecular cause of a fragile site are outlined in Figure 3, and could include CNVs in variable number tandem repeats, identified by bioinformatic tools such as ExpansionHunter Denovo (Garg et al., 2020; Trost et al., 2020). Overall, the current improvements in methodology and technology allowing for more detailed and quicker discovery of CFSs and RFSs provides the potential to advance the understanding of these fragile regions. Further studies on common genomic features such as sequence, epigenetic landscapes, and expression profiles would allow for the development of more accurate automated programs for the discovery of novel fragile sites. Moreover, revised “gapless” reference genomes should further facilitate the suspected association of tandem repeats with fragile sites, speeding the mapping process (Figure 3).
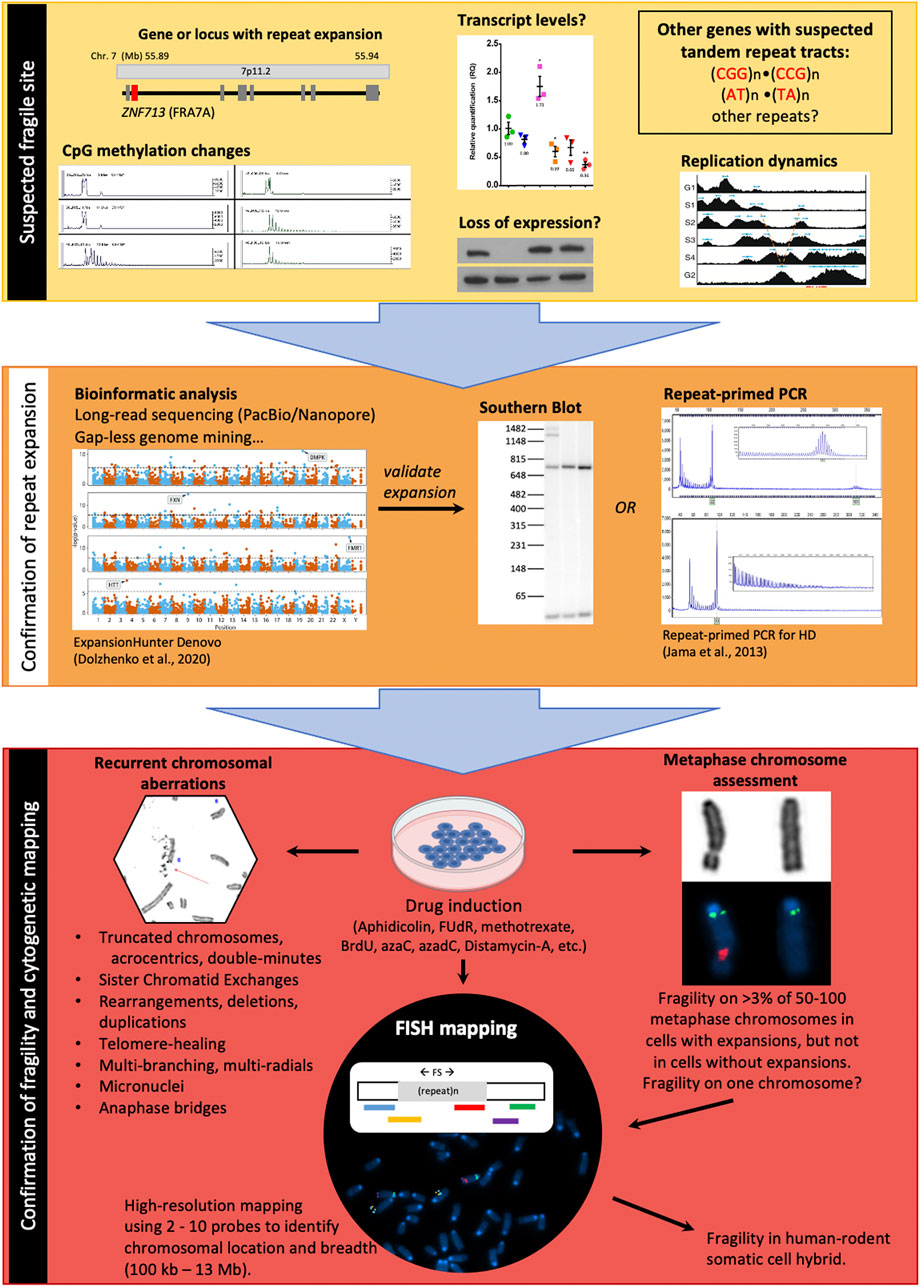
FIGURE 3. Strategies to identify and map a fragile site. Yellow segment–Repeat-associated FS breakpoints may be suspected based upon various genetic and epigenetic landmarks in normal cells. Large genes, changes in CpG methylation patterns on one chromosome, loss of expression (transcript or protein), a V-shaped replication timing pattern, and the presence of tandem repeat sequences (CGG)n, (GGGGCC)n, and (AT)n are all pre-disposing factors for FSs. Orange segment–confirmation of a repeat expansion at the suspected FS can first be carried out by bioinformatic analysis of sequenced reads, followed by validation via Southern blot (gold standard) or repeat-primed PCR. Red segment–The final step of FS localization requires drug treatment to induce expression of the FS, followed by characterization of metaphase spreads for fragile site hallmarks and FISH-based mapping of the FS using two or more coloured FISH probes.
A variety of repeat sequences–including telomeric, centromeric, classical satellite repeats I, II, and III, and various disease-related repeats–have been mapped as fragile sites, chromosomal lesions, or chromatin aberrations (Warburton et al., 1996; Sfeir et al., 2009; Bosco and de Lange, 2012; Black and Giunta, 2018) (Figure 2, see also Box 1). Unlike CFSs, which arise at genomic regions with no clear sequence motif, all mapped disease-associated RFSs arise at repeat sequence motifs, including the CGG expansion-associated sites (FRAXA, FRAXE, FRAXF, et cetera). In the past decade, there has been a steady discovery of new folate-sensitive disease-associated fragile sites, including expanded CGG tracts associated with FRA2A (Metsu et al., 2014b), and FRA7A (Metsu et al., 2014a). Most recently, the rare autosomal-recessive Baratela-Scott syndrome was reported to be associated with the FRA16A CGG expansion in the homozygous state (LaCroix et al., 2019). This site was originally reported 25 years ago as benign when heterozygous (Nancarrow et al., 1994). It should be noted that some homozygously expressed fragile sites have not been associated with disease (FRA10B, FRA16B, and FRA17A) (Berg et al., 1969; Sutherland, 1981; Voiculescu et al., 1991; Felbor et al., 2003). Thus, the phenotypic impact of a fragile site must be considered as other genetic variations.
Box 1 Satellite terminology
Acrocentric or satellited chromosomes, where the “satellited” chromosomal arm is telomeric to a secondary constriction -the centromere (Ferguson-Smith and Handmaker, 1961). The compact heterochromatic region, known as the stalk, between the centromere and the satellite arm is repetitive satellites and rDNA clusters. Satellited chromosomes were observed to form inter-chromosomal satellite associations (Ferguson-Smith and Handmaker, 1961). Similar to the multi-branched chromosomes in ICF syndrome (see section 2.3), satellite associations are genetically inherited (Ferguson-Smith and Handmaker, 1961). The term “satellite DNA” was first named where density separation (isopycnic gradients on CsCl or Ag+-Cs2S04) of genomic DNAs were found to resolve as multiple distinct bands; a major band and numerous “satellite DNA” bands (Kit, 1961). The density difference between bands was subsequently found to be due to the limited and tandem repetitive nature of the DNA sequences in the bands (Jones and Corneo, 1971; Jones et al., 1973; Jones et al., 1974; Gosden et al., 1975; Frommer et al., 1982; Prosser et al., 1986), and hence their being termed “satellite repeats” (with units of 5–171 bp), microsatellites (with motifs of 1–4 bp), minisatellites (with motifs of 5–64 bp), megasatellites/macrosatellites (motifs of up to several hundred kb), and tandem gene amplifications. Human DNA contains at least four defined isopycnic density bands: satellite I (1.687 g/ml), satellite II (1.693 g/ml), satellite III (1.696 g/ml), and satellite IV (1.700 g/ml) (Corneo et al., 1968; Corneo et al., 1970; Corneo et al., 1972). These constitute respectively ∼0.5, ∼2.0, ∼1.5, and ∼2.0% of the total genomic DNA. Interestingly, the DNA constituting the secondary constriction of some satellited chromosomes, turns out to be due to repeat expansions, as in FRAXA, ICF, etc. New sequencing and bioinformatic tools are only beginning to harness a full appreciation of these tandem repeats and their relationship to chromosome structure (Cechova, 2020; Liehr, 2021; Suzuki and Morishita, 2021; Thakur et al., 2021; Altemose, 2022; Altemose et al., 2022; Cechova and Miga, 2022; Gall-Duncan et al., 2022; Hoyt et al., 2022; Nurk et al., 2022). This nomenclature, while not comprehensive, lacks clear boundaries. It was recently suggested so as to avoid confusion, especially with the ever-increasing number of TRs with units of almost any length, to use the term “tandem repeat (TR), with a motif of X nucleotides” (Gall-Duncan et al., 2022).
Technological advances are driving the discovery of additional tandem repeats and disease-linked CGG repeat expansions (Ishiura et al., 2019, 2018; Sone et al., 2019). These repeats could be the molecular cause of novel undiscovered fragile sites and warrant further investigation. Our recent work identified over 2500 repeat motifs significantly enriched in the genomes of autistic patients (Trost et al., 2020). Many of these repeats colocalized to cytogenetically observed, but not molecularly mapped FSFSs (Trost et al., 2020). Using epigenetic-based methodologies, others have also computationally identified abnormally hypermethylated CpG-rich tandem repeat loci colocalizing to unmapped FSFSs (Garg et al., 2020). It is not clear whether these epigenetically mapped TRs actually require aberrant CpG methylation for expression (Garg et al., 2020), as other cytogenetically mapped FSFS do not require methylation with repeat expansion being sufficient for expression (Smeets et al., 1995; Perroni et al., 1996; Winnepenninckx et al., 2007). Other repeat sequences could also manifest as fragile sites under the correct inducing conditions, as the unique conditions necessary to induce fragility at different repeat sequences may not yet be understood. Additionally, there are several repeat expansion disease loci in regions not yet associated with fragile sites but may show fragility only in currently uncharacterized patient populations. Figure 4 compares the cytogenetic location of all known repeat expansions against neighboring common and rare fragile sites previously identified in the literature. In the next section we review the various types of repeats, associated disease, and fragility.
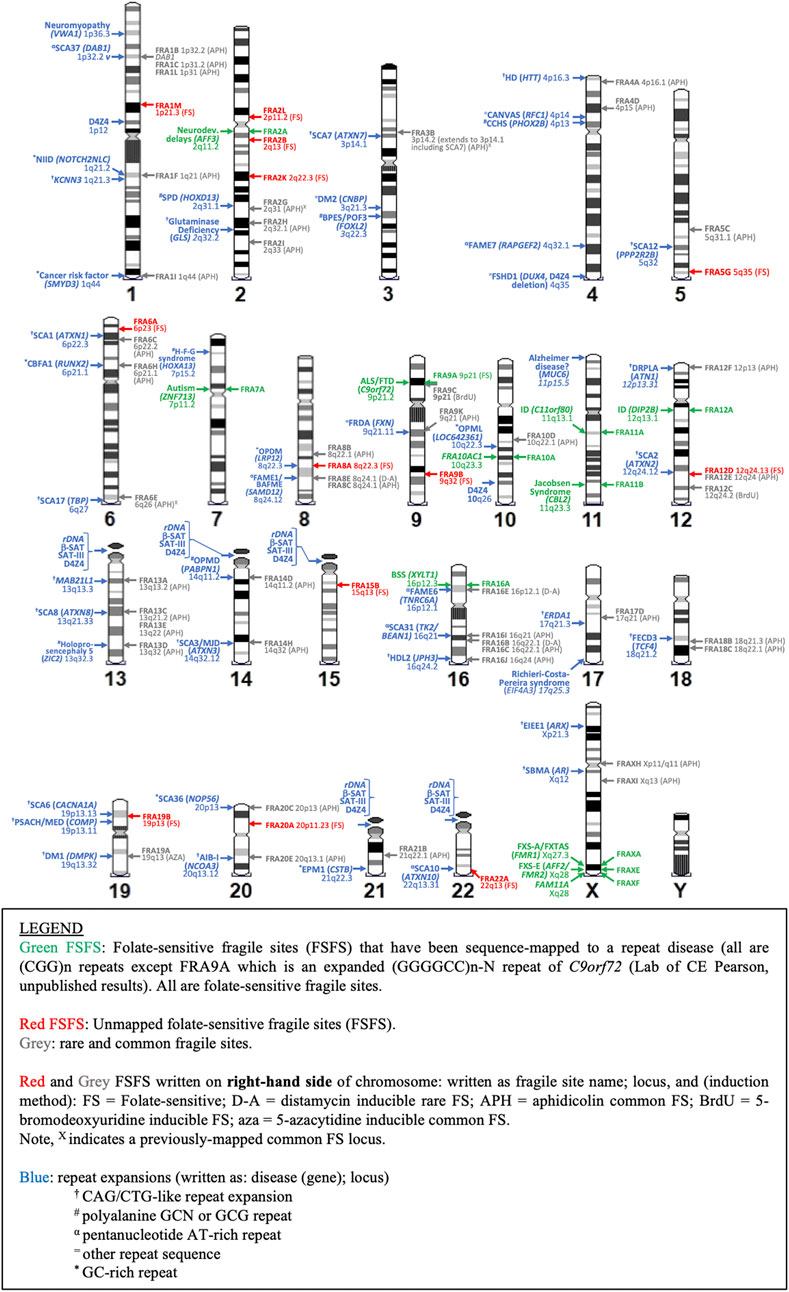
FIGURE 4. Karyotypic ideogram of repeat expansions and fragile site locations. Ideogram shows the mapping of all disease and non-disease repeat expansions (blue; on left side of chromosome) compared to all folate sensitive fragile sites (FSFS) (red and green; right side) and selected rare and common FS near disease loci (gray; right side).
Ten FSFSs have been molecularly mapped to gene-specific expanded (CGG)n repeats. These sites include FRAXA (at FMR1 - causing FXS, FRAXE, FRAXF, FRA2A, FRA7A, FRA10A, FRA11A, FRA11B, FRA12A, and FRA16A (details and relevant citations in Table 2). While it is clear that an expansion is required for fragile site expression, there is only a mild effect of larger expansions on fragility (Rousseau et al., 1994), supporting the importance of the presence of a repeat expansion over its size. Each of these sites shows aberrant CpG methylation both upstream of and at the repeat, which is associated with loss of transcription of the expanded allele. Most of these 10 characterized FSFSs have been associated with some form of neurological disease, with 16 other FSFSs remaining uncharacterized with respect to sequence and disease association. As such, it is possible that some other non-CGG repeat may be involved with the uncharacterized FSFS. Moreover, there may be additional undiscovered FSFSs for which the disease-causing mutation may be a GC-rich repeat motif.
The most extensively studied fragile site, FRAXA, provides a complex picture of the mutational and disease heterogeneity that can arise from a single fragile site. Depending on expansion size, methylation status, and sex, different diseases manifest within patients, many of whom have vastly different symptomatic features (Figure 5) (reviewed in (Nichol Edamura and Pearson, 2005; Lozano et al., 2014; Hagerman et al., 2018). Various mutation forms and epimutations at FMR1 were identified to be the cause of a broad spectrum of clinical presentations, including FXS, autism, fragile X-associated ataxia (FXTAS), premature ovarian failure/insufficiency (FXPOI), attention-deficit disorder, learning disabilities, as well as psychologic, endocrine, autoimmune, and metabolic disorders (Hagerman et al., 2018). Interestingly, this complexity in disease manifestation at the FRAXA locus has only recently become apparent, some 75 years after the initial reports of FXS as Martin-Bell syndrome (Martin and Bell, 1943). Given such a complex etiology at this particular locus, enormous unrecognized and unexplored complexity may exist at other fragile sites.
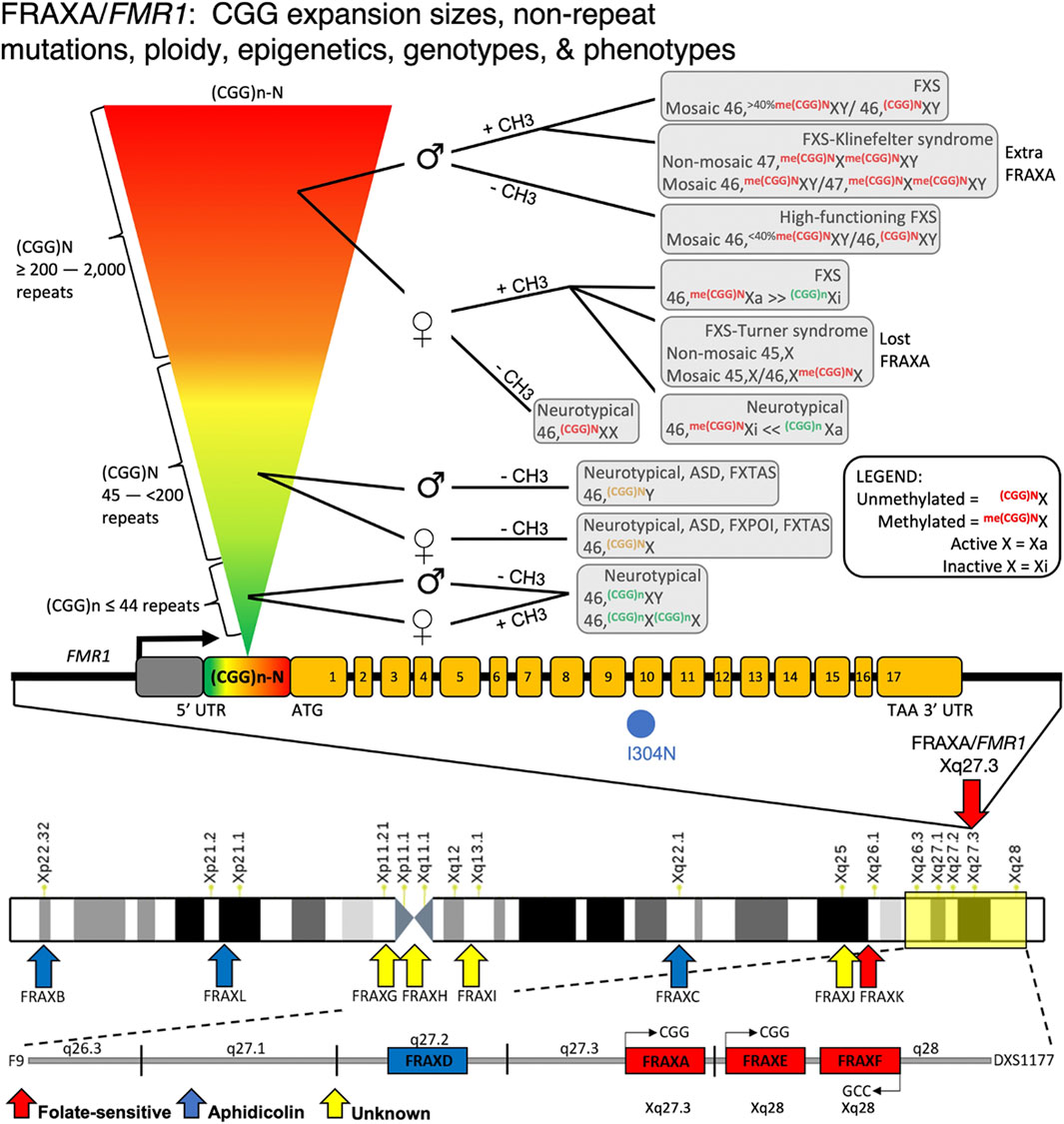
FIGURE 5. FMR1/FRAXA numerous mutations, genotypes, and phenotypes. Disease mosaicism reported at the FMR1 (CGG)n repeat locus, influenced by repeat size (rainbow inverted triangle showing increasing repeat size), DNA methylation of the repeat expansion ( ± CH3), and sex of patient (♂ = male; ♀ = female). Individuals with <44 repeats, regardless of sex, are neurotypical. Those with repeats between 45 and 200 can have Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS) or autism if male with unmethylated repeat. If female, these individuals have unmethylated alleles that are likely neurotypical or have Fragile X primary ovarian insufficiency (FXPOI). With expansions >200 repeats, the disease spectrum becomes more variable. In females, X-inactivation affects disease outcome. Due to the presence of two X alleles in females and random X-inactivation, DNA methylation effects can vary substantially between individuals depending on which allele is X-inactive (Xi) compared to active (Xa). This is also influenced by which tissues are affected and what degree of mosaicism in X-inactivation exists in the patient. If allele is methylated, females can show FXS or Turner syndrome mosaicism based on which allele is Xi vs. Xa. Males with unmethylated expanded alleles are high functioning FXS. Methylated individuals can have FXS or FXS with mosaic Klinefelter syndrome. Many deletions within this FMR1 promoter region have been reported, causing FMR1 silencing and FXS syndrome.
The sequences of all CFSs mapped thus far exhibit a strong skew towards AT-rich regions (Zlotorynski et al., 2003; Tubbs et al., 2018). Although breakage frequencies and general sequence characteristics have been described for these sites, a common causative sequence has yet to be identified. In most CFSs, several factors associated with the AT-rich sequence likely contribute to the propensity to break under replicative stress. Some mapped CFS loci, such as FRA3B and FRA6E, are coincident with repeat expansions, which could contribute to the increased frequency of fragile site expression at these specific loci under replicative stress. For RFSs, the distamycin-A/BrdU-inducible sites FRA10B/FRA16B map to uninterrupted AT-rich repeat motifs spanning several kilobases (Lukusa and Fryns, 2008). FRA16B has been mapped to a 33 bp AT-rich minisatellite repeat (Yu et al., 1997) as well as a 35 bp repeat (Yamauchi et al., 2000). Differences in repeat motif length or composition between different FRA10B families indicate multiple independent expansion events (Hewett et al., 1998). That the expanded repeats at FRA10B and FRA16B can be of various repeat motifs with various lengths seen among different individuals, supports the likelihood that various AT-rich repeat motifs - when expanded - may become fragile sites. Currently, these AT-rich expansions have not been demonstrated as requirements for fragile site expression. Interestingly, several repeat expansion diseases, such as SCA10, SCA37, FAME1/BAFME, and SCA31, are all caused by pentanucleotide AT-rich repeats that reside within known distamycin rare or aphidicolin CFS regions. At each of these loci, multiple repeat motifs can arise, but only certain motifs are associated with disease (Ishiura and Tsuji, 2020). Additionally, there are also several repeat expansion disease loci in regions not yet associated with fragile sites which could show fragility in patient populations not currently analyzed. We propose that in the large regions associated with CFS, the repeat expansions, although not necessary, could further enhance fragile site expression.
The tandem arrays of rDNA have been observed as fragile sites and other complex macro-structures (Ferraro et al., 1977; Warmerdam and Wolthuis, 2019; Zhou et al., 2021). Fragility induction at rDNA arrays by aphidicolin and actinomycin-D was recently demonstrated (Zhou et al., 2021). The multiple clusters of tandem ribosomal DNA (rDNA) repeat arrays reside in the short arms of five of the 10 human acrocentric chromosomes, 13, 14, 15, 21, and 22 (Boisvert et al., 2007; McStay, 2016). Acrocentrics have the centromere very near the end of the chromosome, have a long q-arm, a centromere (primary constriction), a stalk (secondary constriction), and a satellited arm (Figure 1G). The stalks and satellites are variably sized heterochromatin structures (Orye, 1974; Cheung et al., 1989; Heliot et al., 1997). The stalks contain the genes for 18S, 5.8S, and 28S ribosomal RNA, which occur as tandem copies, with varying lengths. It is the variation in these lengths that is thought to modulate the length of the chromosome, as this is due predominantly to length variations of the stalk (Orye, 1974; Cheung et al., 1989; Heliot et al., 1997). Each acrocentric has short satellited arms containing three bands: p11, p12, and p13. Bands p11 and p13 are composed of the heterochromatic satellite III and β-satellite repeats. Band p12 contains ∼400 copies of the 43-kb rDNA repeat unit tandemly arrayed. Each unit contains the 28S, 5.8S, and 18S rRNAs (45S rRNA) and a non-coding intergenic spacer. The size of the rDNA arrays varies between individuals and decreases with ageing and displays increased length variation in cancers (Stults et al., 2009; Xu et al., 2017; Salim and Gerton, 2019; Valori et al., 2020). The tandem arrays of rDNA are in nucleolar organizer regions (NORs) which are within nucleoli. The exact sequence of the rDNA arrays have long been elusive, but are now able to be known (Hori et al., 2021; Nurk et al., 2022). The chromatin compaction of the rDNA arrays is altered between active and inactive states. In Xenopus Laevis the transcriptionally active rDNA arrays are densely compacted in nuclease resistant chromatin (Spadafora et al., 1979; Spadafora and Crippa, 1984; Spadafora and Riccardi, 1985). NORs on metaphase chromosomes present as achromatic gaps known as secondary constrictions of undercondensed rDNA repeats within active NORs (Heliot et al., 1997). The lengths of the rDNA arrays have long been known to contract over aging, especially in the brain (Johnson and Strehler, 1972). The rDNA arrays are particularly unstable in cancers (Stults et al., 2009; Xu et al., 2017; Salim and Gerton, 2019; Valori et al., 2020), and sensitive to DNA damage (van Sluis and McStay, 2019, 2017; Salim et al., 2017). Recent advances on understanding the mechanisms of rDNA fragility have been made, revealing an involvement of transcription across the arrays and R-loop formation (Zhou et al., 2021). The D4Z4 repeat constitutes a family of subtelomeric repeats present on human chromosomes 10q26, 1p12, and the p arm of all five acrocentric chromosomes (Lyle et al., 1995; Stout et al., 1999). Telomeres avoid the nuclear periphery and tend to reside within the internal, euchromatic compartment. Exceptions to this are the telomeric q-arm 4q35 (Tam et al., 2004) and the short p-arms of the acrocentric chromosomes, 13, 14, 15, 21, and 22 (Boisvert et al., 2007; McStay, 2016). Interestingly, each of these harbors a D4Z4 repeat (Lyle et al., 1995; Stout et al., 1999). FSHD patient cells with a mutant contracted D4Z4 repeat tract (typically 11–100 repeats, down to <11 units) still colocalized to the nuclear periphery, arguing that a critical number of D4Z4 repeats is not required for localizing 4q35 (Tam et al., 2004).
Human centromeres are composed primarily of repeating ∼171 bp units known as α-satellite DNA repeats (Warburton et al., 1996). Centromeric regions are the primary constrictions of chromosomes and exhibit a high degree of heterogeneity in repeat sequence composition among individuals (Fowler et al., 1987; Altemose et al., 2014; Aldrup-MacDonald et al., 2016). Unlike many fragile sites and repeat expansion diseases, these variations are considered benign. Flanking the centromeres are pericentromeric regions, which are composed of α-satellites and other repetitive elements such as LINES, SINES, and satellites II and III (reviewed in (Plohl et al., 2014). The pericentromeric regions of chromosomes 1, 9, and 16 have large constitutive heterochromatin stretches of repetitive DNAs (see Box 1). These regions give rise to the secondary constrictions or stretched heterochromatic sites, that often appear as long over-stretched despiralized regions (Jeanpierre et al., 1993; Guttenbach and Schmid, 1994). These are constitutively seen in patients with ICF syndrome, a rare autosomal recessive disease characterized by immunodeficiency (Fryns et al., 1981; Turleau et al., 1989; Tuck-Muller et al., 2000). Like common and rare fragile sites, these chromosomal regions are prone to breakage, mis-segregation, aneuploidy, and micronuclei formation. Multi-branched inter-chromosomal associations, much like satellite chromosome associations are often observed in ICF chromosomes (see Box 1). ICF syndrome is caused predominantly by mutations in DNMT3b (the gene encoding the human de novo DNA methyltransferase) but also by mutations in the HELLS, CDCA7, and ZBTB24 genes, each involved in DNA methylation regulation (reviewed in (Wijmenga et al., 2000). ICF individuals show severe immunodeficiency, abnormal facial features, and cognitive disabilities. All ICF patients assessed to date have hypomethylation of the juxtacentromeric satellite II repeats, leading to the hypothesis that the chromosome fragility and disease symptoms are directly linked to DNA hypomethylation (Maraschio et al., 1988; Jeanpierre et al., 1993). Juxtacentromeric heterochromatin, unlike pericentromeric regions, does not include the centromeric heterochromatin. The cytogenetic observation of despiralized lesions, cytogenetically similar to fragile sites within these specific heterochromatic regions, highlights the importance of methylation in relation to fragility at various loci. This connection is supported by the observation that exposure of non-ICF cells to demethylating agents such as 5-azadeoxycytidine, leads to the induction of the same fragile sites as those endogenously expressed in ICF patient cells (Sutherland et al., 1985b). Furthermore, under replicative stress, such as in tumorigenesis, centromeric DNA rearrangements and mutations are commonly observed, just like at CFS regions. Whether the mechanisms of maintaining chromatin integrity at these various repetitive regions share common pathways has yet to be elucidated. It is notable that other inter-chromosomal associations have been reported by molecular means (Maass et al., 2019; Agelopoulos et al., 2021), however, these have not been reported to be detectable cytogenetically.
The telomeric ends of chromosomes are another site of constitutive, repetitive heterochromatin within the genome. In an attempt to identify internal controls for diagnostic FXS by FRAXA induction, telomeric fragile sites were observed (Steinbach et al., 1982). These folate-sensitive telomere fragile sites occurred more often at 4p, than on other chromosomes (Jenkins et al., 1986a). The cause of this telomeric fragility was not mapped at the sequence level, but their variable expression might be due to chromosome arm specific sub-telomeric sequences (Flint et al., 1997). Telomere repeat tracts (TTAGGG)n are typically bound and protected by the shelterin protein complex, have also been identified as aphidicolin inducible fragile sites (Sfeir et al., 2009; Bosco and de Lange, 2012). The repetitive nature of these long sequences challenges the fidelity of the replication machinery. Deletions of TRF1, a key protein of the shelterin complex, is sufficient to cause telomeric fragile sites similar in appearance to traditional fragile sites induced in replicative stress conditions (Sfeir et al., 2009). This effect can be further exacerbated in aphidicolin or ATR-knock-down replicative stress conditions (Sfeir et al., 2009). The fragile nature of this repetitive sequence is further validated by the presence of fragility at the interstitial telomeric repeat on chromosome 2q14 (Bosco and de Lange, 2012), where two stretches of TTAGGG repeats exist as remnants of telomere-telomere fusions from ancestral ape chromosomes (IJdo et al., 1991). Telomere fragility may be regulated by progerin and dNTP pools (Kychygina et al., 2021). These data argue that the telomere repeat sequence itself is prone to fragility and may share many characteristics with other fragile sites, likely due to its replicative stress response.
Viral integration into the genome has been associated in two ways with fragile sites. First, the integration of foreign DNA, including viruses and plasmids, occurs preferentially at known CFSs (Wilke et al., 1996). This selectivity has previously been harnessed to map the locations of fragile sites (Chen et al., 1976; De Ambrosis et al., 1992; Smith et al., 1992; Wilke et al., 1996; Mishmar et al., 1998). Secondly, several herpes viruses (HSV-1 and HSV-2), papilloma virus (HPV18) (Popescu and DiPaolo, 1989; Zimonjic et al., 1994), cytomegalovirus (Fortunato and Spector, 2003; Siew et al., 2009), and the oncogenic adenoviruses (Ad5 and Ad12) have been reported to induce fragile sites following integration at locations which do not normally express fragile sites (reviewed in (Fortunato and Spector, 2003). Similarly, the integration of foreign DNA into the genome can induce novel fragile sites (Matzner et al., 2003). Virally-induced fragile sites occur without chemical induction, although they can be enhanced following viral integration (Caporossi et al., 1991). The best studied of these virally-induced fragile sites are those induced by adenoviruses. Adenovirus serotype 12 induces fragile sites at four specific genomic locations where viral integration occurs at tandem repeating units. These repeats need to be actively transcribed for fragility to arise (Gargano et al., 1995; Li et al., 1998). One of the earliest studies to observe and map the location of a virally-induced fragile site was through HSV-1 and -2 induced fragile sites at the secondary constrictions of chromosomes 1, 9, and 16, each composed of satellites I-III (Fortunato and Spector, 2003). The HSV infections ultimately lead to random chromosome pulverization/fragmentation (Fortunato and Spector, 2003). It is noteworthy that many of the fragile sites that are claimed to be virally-induced often appear to express fragile sites naturally, but can be induced by exposure to demethylating agents, or in cells from an ICF-affected individual who is genetically deficient in the de novo methyltransferase, DNMT3B (see section 2.3). In this situation, it is difficult to know if cells expressing some of these fragile sites have a history of exposure to these viruses.
Fragile site expression due to thymidylate stress can be achieved through numerous induction methods that perturb the folate metabolism pathway: 1) folic acid deficient growth medium; 2) addition of methotrexate, an inhibitor of dihydrofolate reductase (DHFR); 3) addition of fluorodeoxyuridine (FUdR), an inhibitor of thymidylate synthase; 4) excess thymidine, which inhibits the ability of ribonucleotide reductase to convert cytidine diphosphate to deoxycytidine diphosphate, and inhibiting dCTP production (Jacky et al., 1991). Curiously, excess BrdU (a thymidine analog), which also decreases dCTP levels, prohibits FSFS expression (Sutherland et al., 1985b), likely due to its ability to base pair with guanosine in its enol form (Freese, 1959). This pairing allows DNA synthesis to proceed, unlike the excess thymidine treatment that leaves many guanosine molecules unpaired due to dCTP depletion (Sutherland et al., 1985a). Additionally, imbalances in dNTP pools compromise the fidelity of DNA polymerases (Das et al., 1985), increasing mutagenic products in cellulo (Mattano et al., 1990; Kunz and Kohalmi, 1991), a pre-disposing factor for fragile site expression.
The folate pathway is tightly linked with the DNA methylation pathway (Figure 6), suggesting a possible association between folate-sensitive DNA sites and the ICF-linked fragile sites covered in Section 2.3. Folic acid is a cofactor necessary in the methylation of uridine monophosphate (dUMP) to thymidine monophosphate (TMP). Through this conversion of uracil, folate prevents the toxic incorporation of uracil into genomic DNA. The folate metabolism pathway and the various folate stressors are schematized in Figure 6. Experimental evidence suggests that incorporation of uracil into the DNA gives rise to single- and double-stranded breaks, chromosomal breakage, and micronuclei formation (Blount and Ames, 1995; Blount et al., 1997; Duthie and McMillan, 1997; Duthie and Hawdon, 1998). Folic acid also has critical roles in the production of methionine and S-adenosyl methionine (SAM), a methyl donor necessary for many methylation reactions, including the maintenance of DNA methylation (Zingg and Jones, 1997). That four of the eight known human glycosylases exist specifically to remove uracil (UNG, TDG, hSMUG1, MBD4) highlights the toxicity of uracil within the genome (Lindahl and Wood, 1999).
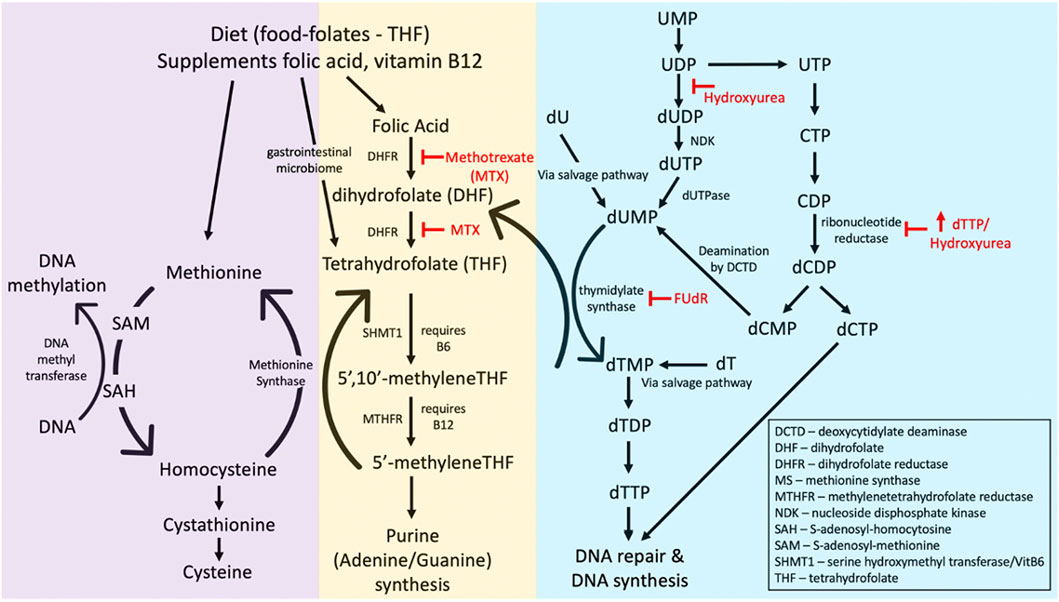
FIGURE 6. The folate metabolism pathway affecting folate-sensitive fragile site expression. Common fragile site inducers methotrexate (MTX), 5′-fluorodeoyuridine (FUdR), deoxythymidine triphosphate (dTTP), and hydroxyurea are indicated (in red) as to what enzymes they inhibit. Abbreviations of other enzymes or substrates in the pathway are given in the bottom right corner.
In vivo effects of folate upon DNA methylation have been documented in human and animal model studies, where low levels of either dietary or serum folate are significantly correlated with global DNA hypomethylation (Bekdash, 2021). Folic acid supplementation of a low folate diet over a few weeks increased genome DNA methylation (Jacob et al., 1998). Mild folate depletion caused various chromosomal rearrangements in cultured rodent prostate cells, a cell type sensitive to folate deficiency due to its high dependence on SAM for polyamine biosynthesis (Bistulfi et al., 2010). Overall, although it is difficult to observe the in vivo effects of folate deprivation, several studies on various cell types suggest genomic instability as a key feature.
CGG repeats show preferential sensitivity to fragility following folate depletion. While the nature of this sensitivity remains an enigma, one hypothesis focuses on the incorporation of uracil into DNA due to increased dUTP levels. Methotrexate treatment in culture causes a large increase in the dUTP/dTTP ratio, leading to a highly increased incorporation of uracil in DNA (Goulian et al., 1980). This uracil incorporation occurs more frequently in late than in early replicating genes in S. cerevisiae (Bryan et al., 2014) and coincidentally, most fragile sites tend to be late replicating (Webb, 1992; Hansen et al., 1993; Subramanian et al., 1996). Therefore, these FSFSs could be stuck in a recurring DNA repair cycle, attempting to excise and replace the uracil base but lacking sufficient levels of the correct dNTP (Reidy, 1987). This cycle is likely exacerbated by cytosine deamination, a naturally occurring process that increases the uracil content at CGG repeats (Feng and Chakraborty, 2017). Alternatively, certain DNA glycosylases function more efficiently at sites of DNA damage when the template contains kinks, bubbles, or gaps that are typical of secondary structures (Hedglin et al., 2015). Therefore, the higher propensity of CGG repeats to form secondary structure could allow uracil DNA glycosylase to more readily recognize misincorporated uracil, thereby setting off or exacerbating a futile DNA repair cycle (Feng and Chakraborty, 2017).
At the time of the earliest predictions of a repeat expansion (see Box 2), in 1985–86 (Sutherland et al., 1985b; Nussbaum et al., 1986; Sutherland and Baker, 1986), it was known that perturbation of one nucleotide precursor affected the levels of other nucleotides (Kunkel et al., 1982; Kunz, 1982; Meuth, 1984). Moreover, it was known that nucleotide pool perturbations can lead to altered mutation rates, another phenomenon that is better understood now (Kunz, 1988; Mathews, 2015, 2014, 2006; Mannava et al., 2013). Even damage to the nucleotide precursors themselves alters mutation rates, a phenomenon that may affect repeat instability itself (De Luca et al., 2008; Cilli et al., 2016; Mathews, 2017). However, even today, an appreciation of the precise levels of nucleotides in a cell, their effect upon each another and sub-cellular localization is poorly understood (Leeds et al., 1985; Andersson et al., 1988). This knowledge gap also extends to nucleotide activity-based localization (Mathews and Ji, 1992) and tissue- or development-specific nucleotide pool regulation (Mathews, 2019, 1975; Brachet, 1977). The role of folate in maintaining uracil levels, outlined above, likely plays a role in some of these cellular processes, a connection that will be revealed as researchers seek to better understand the connection between nucleotide levels, repeats, and fragility.
Box 2 Extended history of FXS
Based upon the knowledge that FRAXA and other folate-sensitive sites could be induced by perturbing nucleotide pools in the folate pathway (see Figure 6), Grant Sutherland’s group hypothesized in 1985 – 6 years prior to molecular proof–that the genetic cause of fragile sites would be an amplified repeat sequence (G. Sutherland, Baker, et al., 1985a; G. R. Sutherland and Baker, 1986). The repeat motif was suggested to be, but not necessarily limited to, amplified alternating polypurine/polypyrimidine sequence, (AG)n•
(CT)n at the fragile site. In 1986, Nussbaum and others further extended this amplified repeat-centric hypothesis to the genetics of FXS (Nussbaum et al., 1986). Specifically, they suggested that carrier females inheriting the amplification would have a level of clinical expression that depended upon the proportion of active X versus inactive X chromosomes harboring the repeat amplification (Nussbaum et al., 1986). This suggestion was consistent with the intermediate “premutation” state originally proposed in an effort to explain the puzzling genetic transmission of the disease. The puzzle originated as the cytogenetic fragile site was present in seemingly unaffected males, who would give rise to a definitive mutation only upon transmission to their heterozygous daughters, who themselves were rarely intellectually affected, but went on to have sons with both the fragile site expressed and the disease phenotype with near unity in incidence (Pembrey et al., 1985).
It would be 5 years before landmark back-to-back papers revealed the first evidence that genetic instability was in fact the cause of FRAXA and FXS, demonstrating the increasing size of the disease-causing DNA fragment through transmissions (Oberlé et al., 1991; S. Yu et al., 1991). Both papers suggested the involvement of an expanding repeat tract, and Oberlé specifically suggested the involvement of the CGG tract. These papers were quickly followed by those from Verkerk and others 1991) and Kremer and others 1991) showing a CGG tract was expanding (Kremer et al., 1991; Verkerk et al., 1991). Verkerk identified the novel FMR1 gene in which the repeat expansion resided. These early papers presented evidence for the mutation mechanism in FXS being an unstable DNA, with somatic instability of the DNA, and proposed the involvement of the GC-rich repeat and unusual DNA structures in the mutation process. Thus, the suspicion of an unstable repeat hypothesized by earlier papers (G. Sutherland, Baker, et al., 1985a; G. R. Sutherland et al., 1986b; Nussbaum et al., 1986) was confirmed in a flurry of papers published within months of each other, revealing that the expansion of the CGG repeat was the cause (Fu et al., 1991; Kremer et al., 1991; Verkerk et al., 1991). The unusual genetics of FXS was subsequently shown to be caused by the size of CGG expansions (Fu et al., 1991; Heitz et al., 1992) as well as the proportion of mutant chromosomes with aberrant methylation being present on the active X of females (Rousseau et al., 1991a). The mode of instability was revealed to be due to somatic repeat instability during early development (Devys et al., 1992). The observed aberrant CpG methylation of the mutant locus (Bell et al., 1991; Oberlé et al., 1991; Vincent et al., 1991) was soon after revealed to be associated with loss of FMR1 transcription (Pieretti et al., 1991). The identification of the CGG expansion had immediate implications on direct molecular diagnostic methods (Rousseau et al., 1991b; Shapiro, 1991; G. R. Sutherland et al., 1991), improving upon the cytogenetic diagnosis of the previous decade (Veenema et al., 1988; Shapiro, 1991; Shapiro et al., 1991). Thus, a strong sense of biology and genetics can lead to likely hypotheses, yet strong molecular genetics are needed to prove them. See also Figures 2, 3.
Martin-Bell syndrome, first described in 1943, was the first reported example of X-linked intellectual disability (ID) (Martin and Bell, 1943). The authors noted the unusual transmission by what appeared to be unaffected fathers and mothers. Notably, Julia Bell, a pioneer geneticist and statistician, had previously studied the unusual transmission of both myotonic dystrophy and Huntington’s disease, termed then as “antedating,” now more commonly referred to as anticipation (Bell, 1941). Later, upon examining another multi-generation family with X-linked ID, Lubs identified the first disease-linked fragile site, mapping to Xq27 (Lubs, 1969). This observation eventually led to the name “fragile X syndrome” (FXS). Sutherland revealed in 1977 that fragile site expression occurred in specific culture conditions (Sutherland, 1977), and subsequently several families of X-linked intellectually impaired families were reported to express the same fragile site (Harvey et al., 1977; Turner et al., 1980b, 1980a; Jacobs et al., 1980). The linkage between Martin-Bell syndrome and FXS was definitively made in 1981 when fragile X expression was demonstrated in the same family described by Martin and Bell (Richards et al., 1981). This rapidly lead to harnessing this cytogenetic observation as a diagnostic tool (Webb et al., 1981).
In the decade following 1981’s exciting discoveries, was the race to discover the molecular cause of the FRAXA fragile site and our understanding of FXS and its curious genetics. Based upon the biology of FRAXA induction, perturbation of nucleotide pools, led to the hypotheses that long amplified DNA repeat tracts were the cause of the FRAXA fragile site (Sutherland et al., 1985a; Nussbaum et al., 1986; Sutherland and Baker, 1986; Warren et al., 1987; Hori et al., 1988). Through exceptional and creative molecular and cellular experimentation by multiple groups, in the span of a few months in 1991, a series of papers collectively captured the involvement of an expanding tandem repeat tract with CGG sequence motif, whose expression was affected by aberrant repeat tract methylation, and in females, X-inactivation ratio. The timelines of these discoveries is expanded upon in Box 2, and are detailed further in (Depienne and Mandel, 2021; Gall-Duncan et al., 2022). See also Figure 3.
Currently, independent repeat expansion detection methods have confirmed the suspicion that most rare FSFSs are amplified CGG tracts (Garg et al., 2020; Trost et al., 2020). Recent genomic/bioinformatic and epigenetic approaches have colocalized CGG expansions to regions that have previously presented by cytogenetics as fragile sites, although none were validated by cytogenetic FISH mapping (Garg et al., 2020; Trost et al., 2020). That most appear to be CGG repeats does not exclude the possible involvement of other GC-rich motifs. Chromosomal confirmation and association of FSFS with disease phenotypes seems to have revived interest in these repeats.
Dietary compounds, environmental mutagen exposure, and chemotherapy are strongly correlated with increased FS expression within aphidicolin-treated peripheral lymphocytes (Kao-Shan et al., 1987; Sbrana and Musio, 1995; Musio and Sbrana, 1997; Richards, 2001; Stein et al., 2002; Francés et al., 2016). The list of potential environmental mutagen exposures is extensive and includes cigarette smoke, caffeine, ethanol, lysergic acid diethylamide (LSD), dilantin, pesticides, oil spills, dietary changes, and radiation (therapeutic and atomic bombs). While aphidicolin is still necessary for FS expression in this system, the current knowledge of how such exposures can lead to specific mutation signatures (Poon et al., 2014) may reveal trends related to fragility susceptibility. The increased scientific focus on environmental and dietary exposures may yield additional information on their association with chromosomal fragility.
Folate metabolism depends upon dietary folates (mostly tetrahydrofolate), folic acid as supplements, and vitamins B6 and B12. Humans are not capable of de novo production of folate, but the commensal microbiome can support production of this micronutrient. Deficiencies of vitamin B12 and/or folate, due to malnourishment or genetic defects in folate absorption/metabolism, lead to striking chromosomal aberrations in both direct marrow peripheral blood preparations, observations dating back to the 1950s (Cingam et al., 2017; Green and Datta Mitra, 2017). Chromosomal lesions include fragile sites (gaps and breaks), centromere spreading, and chromosome elongation/contraction (Heath, 1966; Jensen and Friis-Moller, 1967; Das et al., 2005, 1986). Numerical (ploidy) was unaltered. Upon proper nourishment or vitamin supplementation, the chromosomal aberrations were rescued, and hence reversible. Moreover, there is extensive knowledge of how certain drugs can lead to megaloblastic anemia where many of the drugs perturb folate, purine, or pyrimidine metabolism, with some overlap with RFSFS-inducers (Stebbins et al., 1973; Stebbins and Bertino, 1976; Hesdorffer and Longo, 2016, 2015; Ben Salem et al., 2016). Aside from the centromere, it is unknown if these chromosomal lesions arise at random or preferred chromosomal locations. The in vivo association with folate-deficiencies and the overlap of some drug inducers of chromosomal aberrations in megaloblastic anemia with FSFSs in cultured cells begs the question as to whether there may be molecular similarity to the sequences at the lesions. It is tempting to speculate that tracts of certain expanded repeats may be particularly sensitive fragile site induction upon perturbation of folate metabolism, drawing a direct parallel of disease-associated CGG expanded fragile sites and fragility in megaloblastic anemia. It is notable to some reports of localized mosaic chromosomal rearrangements, where the same rearrangement was observed in multiple metaphases [del(7q), del(3p), del(18p), del(20q)], and in malnourished individuals (Goh, 1981; Chintagumpala et al., 1996; Wollman et al., 1996; Parmentier et al., 2012; Cingam et al., 2017). And in each case the rearrangement was “reversible” upon treatment–indicating that the rearrangement was a folate-sensitive de novo event, that did not occur in the presence of folate. A similar link of low blood folate levels and a del(10)(q23), breaking at 10q23, a known folate-sensitive CGG FS FRA10A (Sarafidou et al., 2004), has been reported to be decreased upon vitamin supplementation (Maltby and Higgins, 1987; Ozisik et al., 1994; De Leon-Luis et al., 2005; Morel et al., 2005). It would be of interest to map the locations of the fragile sites in megaloblastic anemias.
Common characteristics identified amongst the various types of fragile sites provide critical clues as to why and how fragility occurs at these specific loci throughout the genome. Several of the proposed mechanisms of fragile site formation and resolution are supported by evidence provided by these common characteristics (see Figure 7).
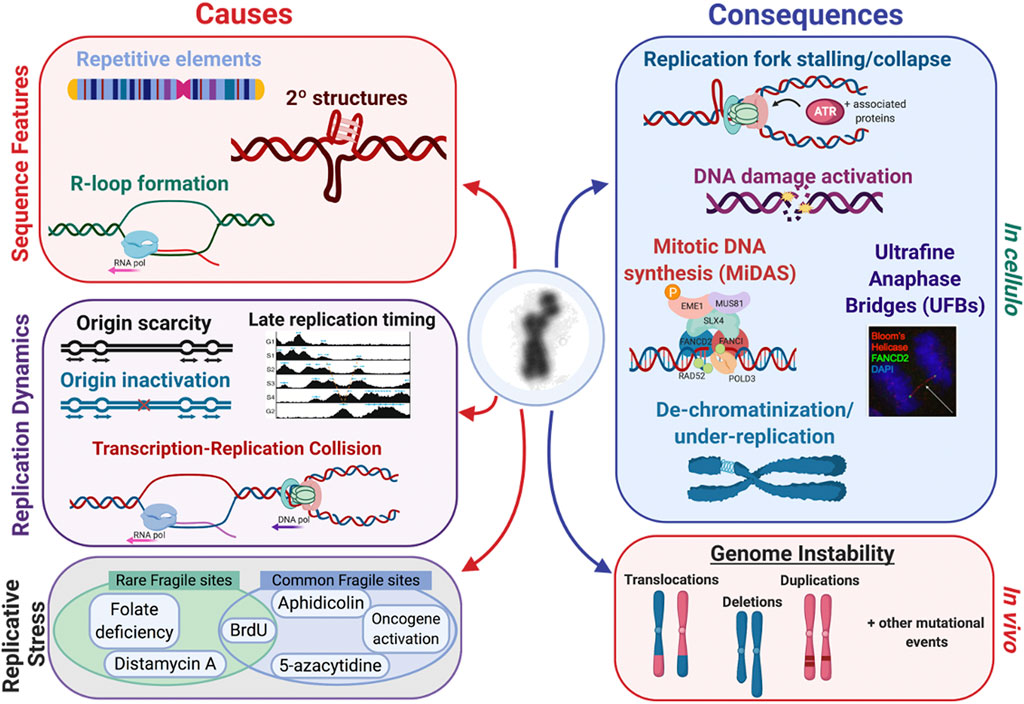
FIGURE 7. Fragile site expression is caused by several factors, such as 1) sequence features, 2) replication dynamics, and 3) replicative stress conditions. There are several inducers that act on several fragile sites, like FRA3B which can be induced by FUdR and aphidicolin (Kähkönen et al., 1989; Jenkins et al., 1990; Kuwano et al., 1990). There are many consequences both in cellulo and in vivo that are linked to fragile site expression. In cellulo, replication fork stalling and collapse leads to activation of the DNA damage response, mitotic DNA synthesis (MiDAS), ultrafine anaphase bridges (UFBs), and ultimately the appearance of de-chromatinization and under-replication if the site is not repaired in time. This can be observed in vivo through mutational events such as copy number variants, translocations, deletions, and duplications.
All fragile sites have a propensity to form higher order secondary structures more than non-fragile regions of the genome. CFSs, which are typically AT-rich, possess high DNA torsional flexibility (Chen et al., 1985), which influences formation of secondary structures and can perturb DNA replication (Zlotorynski et al., 2003; Dillon et al., 2013). In S. cerevisiae AT-rich regions cause fork stalling and breakage (Zhang and Freudenreich, 2007). Work by Burrow and others (2010) shows that only 14 copies of the 33 bp AT-rich minisatellite repeat of FRA16B is enough to cause replication fork stalling, regression, and polymerase skipping in vitro (Burrow et al., 2010). Additionally, there is a significant effect on replication due to the orientation and distance of this sequence from the replication origin with electron microscopy revealing spontaneous regression of stalled forks at these sequences (Burrow et al., 2010). While the AT-rich flexible motifs exist within or near deletion breakpoints at fragile sites (Finnis et al., 2005; Burrow et al., 2009), deletion of these motifs within FRA16D (Finnis et al., 2005) or FRA3B (Corbin et al., 2002; Durkin et al., 2008) does not block fragile site expression. This disparity suggests that higher order structures cause by AT-rich motifs at these regions cannot solely explain their fragility. FSFSs, on the other hand, are comprised of expanded CGG repeats that are capable of forming hairpins, slipped strand structures, G-quadruplexes, and i-motif structures that can hinder replication fork progression both in vitro and in vivo (Fry and Loeb, 1994; Kang et al., 1995; Usdin and Woodford, 1995; Samadashwily et al., 1997; Zamiri et al., 2018, 2015). Both rare and common fragile sites form secondary structures, albeit through very different repeat composition, with RFSs having a high predisposition to expand to greater repeat sizes (Schwartz et al., 2006).
DNA methylation status has been primarily explored in relation to RFSs, primarily FSFSs, which undergo CpG methylation at the expanded CGG repeats. Generally, folate deficiency decreases methylation levels of the DNA, because without folate, S-adenosylmethionine (the principle methyl donor) is not produced, leading to a reduction of cytosine methylation in the DNA (Giovannucci et al., 1993). All 10 mapped FSFSs are predisposed to aberrant CpG methylation which is also linked with silencing of the associated gene and development of disease phenotype. For some loci, expansions without methylation can lead to different phenotypes all together (see Section 4.4). Additionally, the FRAXA, FRAXE, and FRA12A fragile sites have been cytogenetically observed in individuals with unmethylated expanded alleles (Smeets et al., 1995; Perroni et al., 1996; Winnepenninckx et al., 2007), suggesting that methylation is not an absolute requirement for fragile site expression. However, a larger study of high-functioning males with full CGG expansions and considerably reduced aberrant CpG methylation, reveals that fragile site expression does correlate with methylation levels (Hagerman et al., 1994; Rousseau et al., 1994; Lesca et al., 2003). Thus, while DNA methylation is not required for fragile site expression, it can enhance fragility.
It is likely that CFSs are also sensitive to altered methylation status, which could give rise to DNA conformational changes or altered DNA-protein interactions that contribute to fragile site expression (Thys et al., 2015). Interestingly, cytogenetically, fragile sites appear similar to the chromosome constrictions that endogenously arise in cells of individuals with ICF, most of whom are genetically deficient in the de novo DNA methyltransferase (DNMT3B) (Figure 1). Therefore, methylation likely plays an important role in secondary structure and stability of certain DNA regions, including both FSFSs and at satellite I-III repeat sequences associated with CFS. Perturbation of methylation status at these loci likely increases the propensity for fragile site formation.
The demethylating agents, 5-azacytidine and its analog, 5-deoxyazacytidine, are able to induce CFSs. Currently, five have been found, and are predominantly at methylated heterochromatin regions (Sutherland et al., 1985b). These drugs cause widespread demethylation of DNA through both inhibition of DNMT1 and their incorporation into the genome (Christman, 2002). Additionally, since 5-azacytidine results in hypomethylation of heterochromatic satellite repeat regions, it is likely that these regions are also rich in CpG islands. Another CFS-inducing compound that can incorporate into DNA is bromodeoxyuridine (BrdU), a thymidine analog. There are currently seven CFSs and four RFSs found to be inducible by BrdU (Sutherland et al., 1985b, 1980). Neither 5-azacytidine nor BrdU CFSs have been molecularly mapped to a particular repeat motif; however, these regions are proposed to be low complexity, AT-rich repetitive sequences with a high propensity to form secondary structures (Dillon et al., 2013; Thys et al., 2015).
All fragile sites are heritable polymorphic sequence variations (Hecht, 1986), which can be inherited on one or both chromosomes (Sutherland, 1981; Izakovic, 1984; Voiculescu et al., 1991; Martínez et al., 2005) and segregate in families (Sutherland, 1982; Smeets et al., 1985; Romain et al., 1986; Sherman and Sutherland, 1986; Müller et al., 1992; Samadder et al., 1993; Hamel et al., 1994). Fragile sites display unusual patterns of segregation that depend upon the transmitting parent. In a meta-analysis, paternal transmission of the rare autosomal folate-sensitive fragile sites (2q11, 2q13, 6p23, 7p11, 8q22, 9p21, 9q31, 9q32, 10q13, 10q23, 11q13, 11q23, 12q13, 16p12, 19p13, 20p11, and 22q13) significantly deviated from the expected 50% Mendelian inheritance ratio, which is reduced by more than five-fold (Sutherland, 1982; Sherman and Sutherland, 1986; Samadder et al., 1993). However, maternal transmission of these same sites did not significantly deviate from the expected 50% ratio (Samadder et al., 1993). Maternal transmission was also observed for FRA16B (16q22), which is induced by distamycin A/berenil and maps to an expanded AT-rich repeat of approximately 33 bp (Müller et al., 1992). The unusual maternally-biased segregation of the X-linked FRAXA, FRAXE, and FRAXF sites, can in part be explained by maternal CGG expansion bias, ratios of X-inactivation, or a predisposition for CGG contractions in the male germline (Fu et al., 1991; Hamel et al., 1994; Malter et al., 1997). The reduced paternal transmissions of the autosomal fragile sites could be due to maternal genomic imprinting, selection against male gametes carrying the fragile site, or selection against paternally-derived zygotes. We note that many of these transmission reports are sparse, with limited independent confirmation. However, we include these reports here, as it is known that such rare observations can have genetic and clinical impact, as highlighted by the historical situation of FXS.
Karyotypic variations involving mosaic gains or losses of the fragile X chromosome have been observed (Figure 5). Several reports observe these mosaics at higher than expected levels and are likely under-reported owing to the absence of associated cytogenetic studies (Fryns and Van den Berghe, 1988; Santos et al., 2003; Dobkin et al., 2009). Both germline and somatic karyotypic anomalies arise in individuals with CGG-expanded FMR1 X-chromosomes. These anomalies include mosaic cells from a given individual with 46,FRAXA,Y/47,FRAXA, FRAXA,Y (male FXS-Klinefelter syndrome mosaic with an extra fragile chromosome) or 45,X/46,FRAXA,X (female FXS-Turner syndrome mosaic, where the full-mutation fragile X is lost during somatic cell division) (Banes et al., 2003; Dobkin et al., 2009; Froster-Iskenius et al., 1982, p.; Fryns et al., 1983; Milunsky et al., 1993; Seemanová et al., 1985; Shapiro et al., 1994; Tejada et al., 1994). Non-mosaic instances of such anomalies have also been reported, with cells having only 47,FRAXA, FRAXA,Y; 46,FRAXA,X (Filippi et al., 1988; Kupke et al., 1991), or 47,FRAXA,X,X (Fuster et al., 1988; Tejada et al., 1994; Dobkin et al., 2009). These cases can arise via either maternal or paternal X-chromosome non-disjunction of the CGG-expanded fragile X chromosome (Santos et al., 2003; Dobkin et al., 2009). Mosaicism occurs when the non-disjunction arises post-zygotically, whereas non-disjunction during meiosis will give rise to homogeneous cell populations. Age-dependent increases of aneuploidy involving the expanded X also occur in most FMR1 CGG expansion carriers, where the mutant X-chromosome is either lost or retained in an ongoing manner (Nielsen, 1986). FRAXA chromosome aneuploidy is observed in both young and older individuals suggesting that the fragile expanded X chromosome is prone to missegregation (loss or gains), possibly through aberrant packaging, DNA breakage, and/or arrested replication (Kerem et al., 1988; Dobkin et al., 2009; Yudkin et al., 2014). Mosaicism for the ploidy loss or gain of the FRAXA chromosome might suggest meiotic and mitotic predisposed non-disjunction of the mutant chromosome (Milunsky et al., 1993). Such cases can pose diagnostic and counselling challenges (Pandelache et al., 2021). Like mosaics, chromosome number anomalies in cells expressing fragile sites may also be underestimated and overlooked, as chromosome counting has been historically poorly appreciated (Martin, 2004).
Karyotypic variations can also arise with other FSFSs. For example, the FRA1E (1p11) and FRA1D (1p22) fragile sites have been associated with the presentation of monosomy, trisomy, and chromosome rearrangements and multiple congenital anomalies (Neu et al., 1988). In this case mosaicism was evident in multiple tissues including 45,XY,-1/46,XY/47,XY,+1 mosaicism in lymphocytic culture, a 45,XY,-1/46,XY mosaicism in skin fibroblasts, and fra(1p) sites in 2% of the metaphases from lymphocyte, fibroblast, and bone marrow cultures. Given the lack of appreciation for chromosome counting and cytogenetics in an increasingly focused “-omics” world, it is highly likely that other instances of unusual heritability, segregation, and karyotypic anomalies associated with fragile sites remain to be uncovered.
The overwhelming association of fragile sites with multiple diseases has fueled their molecular characterization. In particular, CFSs are frequently sites of CNVs and chromosomal rearrangements–deletions or translocations commonly seen in many cancers (Popescu, 1994; Mimori et al., 1999; Krummel et al., 2000; Mangelsdorf et al., 2000; Arlt et al., 2006, 2002; Burrow et al., 2009; Bignell et al., 2010). Many fragile sites also overlap with tumor suppressor genes (Iliopoulos et al., 2006), with rearrangements possibly driving oncogenesis and affect genes that are likely to further accelerate genomic instability (reviewed in (Karras et al., 2016). In addition, oncogenic activation often, due to unchecked cellular growth, causes dNTP imbalances, promoting instability at CFS regions (Bester et al., 2011). Fragile sites are also frequent integration sites of oncogenic viruses (see section 2.5), which have been used to facilitate their precise mapping (Smith et al., 1992; Wilke et al., 1996; Mishmar et al., 1998). Finally, fragile site regions are strongly correlated with chromosomal rearrangements that have contributed to the development of the vertebrate lineage, suggesting a link between fragile sites and genome reorganization through evolution (Miró et al., 1987; Ruiz-Herrera et al., 2006, 2005, 2002). These factors suggest a strong connection between fragile sites and both advantageous and deleterious chromosomal processes.
Fragile sites are associated with a number of neurological, neuropsychiatric disorders, and neurodevelopmental diseases such as autosomal recessive juvenile parkinsonism (FRA6E) (Denison et al., 2003), idiopathic autism (FRA13A) (Savelyeva et al., 2006), and schizophrenia (Demirhan et al., 2006). In particular, 28 CFSs contain genes associated with schizophrenia (reviewed in (Smith et al., 2010). There are also claims of fragility linked to bipolar disease, schizophrenia, and Rett syndrome (Archidiacono et al., 1985; Gillberg et al., 1985; Simonic et al., 1997; Fischer, 1998; Demirhan et al., 2009, 2006; Smith et al., 2010; Kharrat et al., 2017). However, despite the historical connection between fragile sites and disease, the reproducibility or genetic mapping of these types of sites has not been sufficiently followed-up.
Genomic instability at RFSs presents predominantly as expansions of the repeat motif. However, deletions of the FRAXA and FRAXE region do occur (reviewed in (Hammond et al., 1997; Nichol Edamura and Pearson, 2005; Coffee et al., 2008; Mondal et al., 2012)) and have been covered extensively for FRAXA (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=FMR1). Most of the FMR1 deletions/rearrangements are covered in Figures 8A–D (see citations therein). Breakpoints that frequently occur at RFSs, particularly under replicative stress, tend to map to regions surrounding the expanded repeat motif, such as at FRAXA (Warren et al., 1987; Oberlé et al., 1991; Dobkin et al., 2009; Verdyck et al., 2015) and FRA11B (Michaelis et al., 1998; Tunnacliffe et al., 1999). Translocations, deletions, and rearrangements at the fragile X chromosome as well as chromosome 3 were induced under replicative stress using aphidicolin or FUdR, respectively, in somatic cell hybrids (Glover and Stein, 1988). CGG expansion-associated chromosomal deletions can arise somatically and are present at barely detectable mosaic levels, suggesting that the true extent of these deletions may be underappreciated (Jiraanont et al., 2017).

FIGURE 8. (Continued). Numerous deletions and duplications around the FMR1 locus. (A) The literature reports numerous large deletions on the X chromosome in the region surrounding the FMR1 locus, and additionally (B) many small deletions that occur within the FMR1 locus itself. Large (C) and small (D) duplications (including transversions and inversions) are reported to occur around the FMR1 fragile X locus, with many of the large duplications also occurring in tandem with large deletions (see related citations).
Atypical symptoms also arise with mosaic deletions of FMR1 and contiguous genes, FMR1 duplications, and chromosome rearrangements. These rearrangements can be relatively small or large, often bridging fragile site to fragile site. Depending upon the region duplicated or deleted additional symptoms can include hemophilia, Hunter syndrome, myotubular myopathy, overgrowth, macrocephaly, seizures, and others (Figures 8A–D) (Coffee et al., 2008). While FRAXA/FMR1 is heavily studied, numerous instances of chromosomal instability with common and rare fragile sites supports this as a common attribute of FS.
Evidence that the FRAXA site is truly fragile and prone to DNA breakage arises from the many patients that have incurred loss of FMR1 function through deletions of the (CGG)n tract and part of, or all of the FMR1 gene, and often contiguous genes (Figures 8A–D). Cytogenetically, FRAXA can manifest as a truncated X chromosome with loss of the distal long arm band, Xq28 (Fitchett and Seabright, 1984; Verdyck et al., 2015). Fragile sites are mutation and epimutation hotspots. Specifically, the (CGG)n-expanded FMR1 gene incurs ongoing somatic expansions of the (CGG)-tract (Lokanga et al., 2013), variations of CpG methylation, microdeletions, duplications, and point mutations proximal to or encompassing the FMR1 (CGG)n repeat, intra- and inter-chromosomal rearrangements, as well as germline and somatic aneuploidy (gains and losses of the whole mutant X-chromosome). These various mutations and epimutations, which can arise somatically, can lead to the broad spectrum of phenotypes associated with FMR1 and its proximal genes (Figures 8A–D). Thus, FSFSs are genetically unstable loci, where the instability can have disease implications.
All of the 10 mapped CGG FSFSs have been linked in some manner to ID or autism spectrum disorders (ASDs) (previously reviewed in (Debacker and Kooy, 2007) (Table 2). Additionally many unmapped fragile sites are proposed to be associated with neurological and neuropsychiatric disorders, including schizophrenia (Debacker and Kooy, 2007). Many of these neurological disorders are complex, polygenic conditions that are heavily influenced by environmental and genetic components (reviewed in (Miles, 2011; Kerner, 2014); therefore, the effect of chromosome fragility on particular genes could cascade to other genes (Feng and Chakraborty, 2017). It is interesting that many of the mapped CGG-repeat expanded FSFSs are from genes that are highly expressed in the brain (AFF3, ZNF713, FAM10AC1, FMR1, FMR2) which are likely to have many downstream interactions that can affect global gene or protein expression contributing to disease pathogenesis (Feng and Chakraborty, 2017). Molecular mapping of additional fragile site sequences will likely unveil some of the same complexities of disease etiology for their associated diseases.
Jacobsen syndrome occurs due to deletions in the distal end of the q arm of chromosome 11, that is associated with the FRA11B fragile site (Jacobsen et al., 1973; Schinzel, 1977). The clinical presentation is highly variable and can include malformations of the heart, kidney, gastrointestinal tract, genitalia, and central nervous system; cognitive impairment, and skeletal, ocular, hearing, immunological, and hormonal problems. The varying size and locations of the deletions likely account for the variable clinical presentation (Jacobsen et al., 1973; Tootleman et al., 2019). The molecular basis of the deletions is the fragile site FRA11B (CGG)n repeat expansion, which upon transmission can result in breakpoints of the chromosome (Voullaire et al., 1987; Jones et al., 1995). These breakpoints frequently occur within the vicinity of RFS but can also occur up to 10 Mb away from the (CGG)n repeat (Michaelis et al., 1998; Tunnacliffe et al., 1999). This fragile site was the first established as causing in vivo breakage and disease manifestation, demonstrating the clinical importance of fragile sites. This connection between in vivo breakage and disease is further supported by evidence of FRAXA chromosomal breakage and rearrangement in early embryos containing the repeat expansion (Verdyck et al., 2015). Additionally, expression of the FRA10B fragile site in mothers was correlated with 10qter deletions originating from the FRA10B locus that were identified through non-invasive prenatal screening. Furthermore, the FRA18C CFS was discovered in the parent of an offspring with a chromosomal deletion truncation originating from this site (Debacker et al., 2007). Taken together these data strongly support that fragility is associated with in vivo chromosomal breakage and disease manifestation. Some evidence suggests that telomere healing can arise at broken fragile sites, leading to interstitial telomeric sequences (Bosco and de Lange, 2012; Bouffler et al., 1993; Boutouil et al., 1996; Glousker and Lingner, 2021; Gozaly-Chianea et al., 2022, p.; Musio and Mariani, 1999; Petit, 1997; Villa et al., 1997). While there is often proximity of a fragile site to these interstitial telomeric repeats, interstitial telomeric repeats do not necessarily cause fragility (IJdo et al., 1992).
Most rare FSFSs diseases show partial penetrance of clinical symptoms while still expressing the fragile site, a connection that depends upon the presence of a CGG expansion. With the exception of FRAXA/FXS, analysis of penetrance of the other CGG fragile sites has not been possible, as many sites have been observed in too few families to account for either age effects, expansion size, or degrees of aberrant CpG methylation (Debacker and Kooy, 2007). Delayed onset or incomplete penetrance is typical of diseases that display genetic anticipation (earlier manifestation or greater severity through family generations). The FRA16A (CGG)n repeat expansion, initially reported as a benign variation when heterozygous (Nancarrow et al., 1994), is the causative mutation of Baratela-Scott syndrome when found in the homozygous state (LaCroix et al., 2019). Other FSFSs may in fact be found to cause disease when in the inherited in the homozygous state but have yet to be identified due to lack of appropriate patient or patient-derived samples.
Chromosomal fragility has not been observed in other known repeat expansion disease loci outside of (CGG)n expansions. For example, the expanded (CTG)n repeats associated with myotonic dystrophy (DM1) or Huntington’s disease (HD) do not express fragile sites in a variety of induction methods (Beverstock et al., 1985; Jalal et al., 1993; Wenger et al., 1996; Barbé and Finkbeiner, 2022). The DM1 studies used multiple patient-derived cells with long DMPK CTG/CAG repeat expansions, and multiple known fragile site induction conditions, including folate-deficient media, high thymidine media, and FUdR (three folate-sensitive rare fragile site conditions), BrdU (rare and common fragile sites), aphidicolin (common fragile sites), and 5-azacytidine (common fragile sites). The HD studies also used multiple HD lines and folate-deficiency, FUdR, and BrdU. Thus, the expansion of any repeat cannot automatically be assumed to lead to fragility. While other chemicals known to induce fragile sites, like the AT-specific Hoechst 33258 and netropsin could be tried, other chemicals, not previously assessed for fragile site induction could also be tried. For example actinomycin D, which has been shown to have loose binding preference to CAG/CTG repeats (Jacky and Dill, 1983) may be considered. Altered protein regulation could be considered. For example, in the presence of an ATM protein inhibitor, the expanded GAA repeat tract at the FRDA locus associated with Friedreich’s ataxia, exhibits enhancements in a kind of fragility as assessed through rearrangements and chromosome abnormalities (Kumari et al., 2015). While the association between repeat expansions and fragility has not been a universal association, it is entirely plausible that the unique conditions or agents necessary to induce fragility across different repeat sequences have not yet been elucidated.
Fragile site expression has only been demonstrated in a limited number of tissues. Expression of fragile sites is specific to certain cell types, suggesting that epigenetic or other trans-factors could be contributing to the sensitivity of the site to replication stress. For example, although fibroblast cells of fragile X patients can be treated to induce the FRAXA site (Tommerup et al., 1981), the frequency of expression is often not comparable to that seen in the lymphocytes or lymphoblasts of the same patient (Mattei et al., 1981; Schmidt and Passarge, 1986). Differential fragile site expression has additionally been observed between chorionic villus (placental tissue), fetal blood, and amniocytes via prenatal fragile X screening (Jenkins et al., 1986b; McKinley et al., 1988). FRAXA expression between a broad range of tissues has yet to be assessed. The specificity of fragile site expression has also been documented for certain CFSs, revealing CFS loci differences across several cell types (Kuwano et al., 1990; Le Tallec et al., 2013, 2011). Letessier and others (2011) demonstrated that cell type specificity (lymphoblastoid versus fibroblasts) was linked to the density of replication origins surrounding a particular fragile site region (Letessier et al., 2011). The FRA3B site, the most common CFS site in lymphocytes, has a paucity of replication origins within the core of the region (Palakodeti et al., 2010), yet this disparity does not exist in fibroblasts, where the density of initiation events is comparable to that of the rest of the genome (Letessier et al., 2011). In line with this model, fibroblasts, but not lymphocytes, lack origin sites at the core regions of FRA1L and FRA3L, which are highly expressed in fibroblasts but not lymphocytes (Le Tallec et al., 2011). While cell type specificity has been suggested to be influenced by transcription levels of the particular locus (Helmrich et al., 2011), a later study did not find a correlation with transcription levels, instead suggesting that chromatin architecture and organization play a key role in cell type specificity (Le Tallec et al., 2013). Overall, these findings suggests that although sequence composition is a contributing factor to fragility, there are other undefined aspects influencing the propensity of these sites to experience chromosomal instability. Elucidating these contributing factors could present novel approaches to targeting genomic instability at these problematic loci.
The increased formation of micronuclei under fragile site inducing conditions has been observed for both rare and common fragile sites (Chan et al., 2009; Bjerregaard et al., 2018). The expression of micronuclei is a proxy for genomic instability, as these events form only after faulty chromosomal segregation in anaphase leads to either an entire chromosome or a chromosome fragment becoming dissociated from the remaining nuclear content (Fenech, 2007). In the absence of drug treatments or external stressors, increased micronuclei appear in cell culture for many neurodegenerative diseases, such as Huntington’s disease (Sathasivam et al., 2001), Alzheimer’s disease (Migliore et al., 1997), ataxia telangiectasia (Rosin and Ochs, 1986), and both Werner and Cockayne syndromes (Weirich-Schwaiger et al., 1994) (reviewed in (Migliore et al., 2011). Micronuclei formation, gene amplification, and chromosome damage (such as double-stranded breaks) appear in conditions of folate deficiency (Jacky et al., 1983; Duncan, 1986; Blount et al., 1997; Melnyk et al., 1999; Fenech, 2001; Fenech and Crott, 2002; Beetstra et al., 2005, p. 200). In conditions of folate deficiency, FXS cells show increased mis-segregation of the FRAXA allele, with a higher prevalence in micronuclei and at anaphase bridges (Bjerregaard et al., 2018). This finding expanded initial reports of increased levels of micronuclei in FRAXA carriers compared to controls (Jacky et al., 1983; Duncan, 1986). Taken together these in cellulo reports support a close connection between fragility and micronuclei formation.
The in cellulo effect of elevated micronuclei with folate deprivation translates to mouse models and humans. Mice treated with methotrexate, an inhibitor of dihydrofolate reductase (DHFR), exhibit increased micronuclei and chromosomal aberrations in a dose-dependent manner (Kasahara et al., 1992). Additionally, there is a significant correlation between increased micronuclei and folate deficiency in the leukocytes, reticulocytes and erythrocytes of human subjects; supplementation with folate significantly reduced the frequency of micronuclei (Everson et al., 1988; Blount and Ames, 1995; Blount et al., 1997). Recent advances in understanding the biology of micronuclei, including their involvement in DNA damage, aneuploidy, DNA repair and segregation, can be harnessed to further understand the association of micronuclei with fragile sites.
Sister chromatid exchange (SCE) is a natural phenomenon that occurs following DNA replication and causes recombination of genetic material between chromatids, typically in an error-free manner. Although SCE occurs naturally, an increase in frequency is indicative of genotoxic stress and instability. Advances in understanding the formation and resolution of SCEs, the factors involved, their involvement in genome instability, and under-replicated regions (as observed at FRAXA (Kerem et al., 1988)), all will improve our understanding of the association of SCEs with fragile sites (Baxter, 2015; Broderick and Niedzwiedz, 2015; Uchiyama and Fukui, 2015). CFSs are sites of preferential SCE, regardless of whether a visible gap exists concurrently with the SCE (Glover and Stein, 1987; Schmid et al., 1987; Feichtinger and Schmid, 1989; Hirsch, 1991; Lukusa et al., 1991; Tsuji et al., 1991).
The formation of ultrafine anaphase bridges and the presence of MiDAS (mitotic replicative-stress DNA synthesis, see section 5.4) occurring at CFSs has led to great excitement concerning the processing and resolution of SCEs at these sites (Chan et al., 2009; Naim et al., 2013; Ying et al., 2013; Minocherhomji and Hickson, 2014; Bhowmick et al., 2016). Under aphidicolin-induced replicative stress, sister chromatid bridging leads to inefficient resolution and genotoxicity (Chan et al., 2009). These stressed cells have a higher incidence of ultrafine anaphase bridges, indicative of unresolved sister chromatids during anaphase separation (Chan et al., 2007). The FRAXA locus has an increased propensity for DNA anaphase bridges and lagging chromosomes in folate stress conditions (Bjerregaard et al., 2018). These anaphase bridges associate with the FRAXA allele and differ from CFS-associated bridges in that the majority are RPA protein positive and PICH protein negative. FANCD2 also does not colocalize to these bridges, suggesting that FSFSs are processed differently than CFSs (Bjerregaard et al., 2018). This response is outlined in the mechanism of DNA repair section below (section 5.4). Increased sister telomere associations in conditions inducing telomere fragility, suggests a similar pathway at these fragile sites (Sfeir et al., 2009).
The relation between RFSs and SCEs is less clearly delineated, although most studies support an increase in SCEs at RFSs. Carriers of the distamycin A-inducible sites have elevated SCEs, with FRA16B observed in both induced and uninduced conditions (Schmid et al., 1987; Lukusa et al., 1991; Seki et al., 1992), whereas FRA16E is only observed in induced conditions (Tsuji et al., 1991). A problematic aspect in studying SCEs and rare FSFSs is that the BrdU treatment–necessary for SCE visualization–counteracts the toxic effects of folate deprivation by base-pairing with guanine (Freese, 1959). This pairing bypasses the DNA synthesis block normally observed in folate-deprived conditions, where dTTP and dCTP levels are diminished. Reports of a global increase in SCE events in folate-deficient conditions in FXS patient and control cells (Branda et al., 1984), have been countered by others arguing that SCEs are only increased locally at the Xq27 FRAXA expanded locus (Wenger et al., 1987; Tommerup, 1989, p. 1989) and global SCEs are the same between these cells. In yeast, thymidylate depletion leads to unequal SCEs and other forms of intrachromosomal rearrangements (Kunz et al., 1986). Further investigation of the relationship between RFSs and SCEs is required to better understand the connection between these two important molecular processes.
The high coincidence of SCEs and fragile sites likely occurs because fragile sites are usually late-replicating, and likely under-replicated, making them susceptible to initiation of homologous recombination to replicate the remaining DNA. If replication is not completed, there will be colocalization of a fragile site with an SCE site. Alternatively, a fragile site could still occur if replication has taken place, but it was too late for proper chromatin condensation. Considering that both rare and common fragile sites are prone to deletions, expansions and rearrangements, the process of unequal or error-prone exchange at SCEs may also contribute to instability.
Fragile sites have been observed in many non-human species, using induction methods typical of rare or common fragile sites. Aphidicolin-induced fragile sites have been observed in many animals and different cell types. These include, but are not limited to, peripheral lymphocytes from mouse (Rozier et al., 2004), cat (Stone et al., 1993; Stone and Stephens, 1993), and dog (Stone et al., 1991), fibroblasts from mouse (Sanz et al., 1986), Persian vole (Djalali et al., 1985), Chinese hamster (Coquelle et al., 1998), racoon, dogs (Wurster-Hill et al., 1988), and splenocytes from mouse (Krummel et al., 2002) and rat (Robinson and Elder, 1987). Fragile sites are induced by folate deficiency, either through FUdR induction or growth in folate deficient media. These FSFS have been observed in lymphocytes from mouse (Elder and Robinson, 1989), rat (Elder and Robinson, 1989), river buffalo (Pires et al., 1998), Indian mole rat (Tewari et al., 1987), Persian vole (Djalali et al., 1985), goats (Lopez Corrales and Arruga, 1996), cattle (Uchida et al., 1986), and domestic pig (Yang and Long, 1993). Drugs such as trimethoprim, methotrexate, 5-azacytidine, 5-aza-2′deoxycytidine, amethopterin, and BrdU have been used to induce fragile site in non-human animals as well, including in cats (Stone et al., 1993), dogs (Stone et al., 1991), Persian vole (Djalali et al., 1985), gorilla (Schmid et al., 1985), chimpanzee (Schmid et al., 1985), goats (Lopez Corrales and Arruga, 1996), river buffalo (Pires et al., 1998), Chinese hamster (Coquelle et al., 1998), and rabbit (Poulsen and Rønne, 1991). Furthermore, spontaneous (un-induced) fragile sites have been observed in cells from horse and pigs (Riggs and Rønne, 2009).
Evidence suggests that fragile sites and their associated genes are evolutionarily conserved, supporting a functional role, possibly in genome packaging (Berthelot et al., 2015). Many of the human disease-associated genes are evolutionarily conserved and many retain a repeat tract. For example, FMR1, for which an expanded CGG tract is the cause of FRAXA, is evolutionarily conserved. Moreover, the CGG repeat is also conserved, but is typically shorter in non-human species. An observation of a naturally-occurring CGG-expanded Fmr1 gene in a non-human species has not been reported but could theoretically exist. A repeat expansion in the dog Nhlrc1 gene, the cause of its Lafora disease (Lohi et al., 2005), does not appear to be present in humans, albeit non-repeat mutations in the same gene cause the same Lafora disease in humans (Chan et al., 2003). Human fragile sites have also been conserved within animal species, such as FRA16D, a common AT-rich repeat fragile site mapped to the gene WWOX (Lee et al., 2021). In the mouse, the Wwox gene and fragile site are highly conserved in the mouse genome, appearing as mouse fragile site Fra8E1 (Krummel et al., 2002). Similarly, a CFS induced by 5-azadeoxycytidine on human chromosome 1q42 was also induced on the homologous locus in chimpanzee and gorilla, indicating that it is also conserved (Schmid et al., 1985). The folate-sensitive FRAXA fragile site at Xq27 was observed in human-hamster and human-mouse hybrid cells, in which a human Xq24-qter from a male fragile X patient was transferred to rodent cells (Nussbaum et al., 1983, p. 983; Warren and Davidson, 1984; Warren et al., 1987). These hybrids were used to clone the FRAXA CGG repeat (Warren and Davidson, 1984; Warren et al., 1987).
The molecular mechanism of fragile site expression remains to be elucidated. Well-established link between fragile sites and cancer etiology has facilitated headway in the field of CFSs, unravelling many mechanistic aspects of their cause and the processing of DNA at these unique sites. In contrast, the field of RFSs lags behind CFS studies and could benefit from ideas gleaned from CFSs to renew progress and discovery. Several theories exist, supported by ample evidence, for the mechanisms of fragile site formation and involve 1) replication timing and origin paucity, 2) chromatin compaction, 3) replication-transcription collisions, and 4) DNA damage and repair machinery. These pathways are not mutually exclusive, and any combination could cause specific fragile sites, but not necessarily all fragile sites. Some of these causative factors and consequences are briefly summarized in Figure 7 and briefly discussed in the following sections.
Many common and rare fragile site loci are late replicating regions and often lack nearby replication origins (Le Beau et al., 1998; Wang et al., 1999; Handt et al., 2000; Hellman et al., 2000; Palakodeti et al., 2004; Pelliccia et al., 2008). However, these characteristics are not universal, as some fragile sites are in mid-replicating regions (Handt et al., 2000; El Achkar et al., 2005) and others at early replicating regions (Barlow et al., 2013). FRA3B and FRA16D, the most active CFSs in lymphoblastoid cells, are late replicating with decreased sites of replicative origin within their core regions. As such, more distant replication forks are required to traverse longer distances to eventually complete replication in these regions (Letessier et al., 2011). The same situation applies for FRA6E (Palumbo et al., 2010) and FRA7H, which have allelic asynchrony in replication (Hellman et al., 2000). These regions become particularly vulnerable when stressed with aphidicolin, since the resulting reduction of fork speed has a greater effect on longer-travelling than on shorter-travelling forks (Letessier et al., 2011). In fibroblast cell lines, the FRA3B site does not lack replication origins at this locus, hence the FRA3B fragile site is not expressed in these cells (Letessier et al., 2011). This disparity between lymphoblasts and fibroblasts provides a potential explanation as to the origin of cell-type specificity observed for most fragile sites.
The folate sensitive FRAXA (Xq27.3) and FRAXE (Xq28) regions are both in very late replicating regions in genomes that do not contain the fragile site-causing (CGG)n repeat expansion (Subramanian et al., 1996; Hansen et al., 1997). A CGG expansion at FRAXA obstructs firing from an adjacent replication origin frequently utilized by the wild-type allele. Thus, replication stress at this locus is generated from the need to rely upon more distal origins for replication (Yudkin et al., 2014). The presence of an expansion and additional thymidylate stress delays replication into G2 phase, yielding a large under-replicated region of 1 Mb for FRAXA and 300 kb for FRAXE (Subramanian et al., 1996; Hansen et al., 1997). For these FSFS, the expansion plays a critical role in influencing replication timing and related stress.
Understanding the factors at play during replication will provide clues to the association between fragile sites and replication. The origin replication complex (ORC), which is responsible for directing DNA replication throughout the genome, is assembled at specific loci through an unknown mechanism (reviewed in (Fragkos et al., 2015). Mapping of these complexes using the constituent ORC2 protein in ChIP-seq experiments demonstrated a strong association between regions of CFSs and ORC2 paucity, with 73% of all CFSs upholding this relationship (Miotto et al., 2016). Increased ORC2 correlates with regions of active chromatin, demarcated by higher levels of active transcription and histone marks (Miotto et al., 2016). What determines whether an ORC will fire is a topic of much debate but is predominantly believed to be a stochastic event influenced by factors such as chromatin architecture. A higher density of ORCs likely indicates an early replicating region (Rhind, 2006; Bechhoefer and Rhind, 2012; Gindin et al., 2014; Das et al., 2015), such that the aforementioned ORC2 paucity would be in line with general late replication of fragile sites. Determining the unique characteristics of the chromatin architecture within fragile sites could provide valuable insight into what elements and factors contribute to late replication initiation.
What does the apparent gap, constriction, or break of a fragile site represent at the chromatin level? Some data suggest an uncompacted nucleosome-free DNA (Hsu and Wang, 2002), but can also represent a true physical break within the DNA, or an as-of-yet defined epigenetic factor could be influencing these problematic regions.
Generally, CFSs are hypoacetylated compared to the rest of the genome, indicating that they exist in a compact chromatin form (Koch et al., 2007; Savelyeva and Brueckner, 2014). To probe this nuclear chromatin compaction, a widely utilized endo-exonuclease named micrococcal nuclease (MNase) is employed. MNase preferentially cleaves linker DNA between nucleosomes (Rivera and Ren, 2013; Tsompana and Buck, 2014). FRA3B is more resistant to MNase treatment when compared to non-fragile sequences at or nearby the locus, and demethylating agents trichostatin A or 5-azadeoxycytidine cause a reduction in chromosome breakage at this site (Jiang et al., 2009). Early evidence from the characterization of repetitive satellite DNA sequences from various species demonstrated that these regions are MNase resistant (Bostock et al., 1976; Bowen, 1981).
The FSFS FRAXA displays similar characteristics: in cellulo, the FRAXA locus exists as an inaccessible region, resistant to restriction enzyme digestion when compared to the unexpanded allele (Luo et al., 1993; Eberhart and Warren, 1996). This resistance is likely due to the array of repressive histone post-translational modifications that are typically associated with expanded (CGG)n repeats and the in cellulo heterochromatin-like state (Coffee et al., 2002, 1999). Fragile site expression is blocked with sodium butyrate and acetyl-carnitine, drugs which inhibits histone deacetylation, encouraging the accumulation of acetylated open chromatin (Pomponi and Neri, 1994). Curiously, in vitro, these (CGG)n expanded repeats strongly exclude nucleosome assembly, which is further exacerbated by CpG methylation (Godde et al., 1996; Wang et al., 1996; Wang and Griffith, 1996). Given the challenges in assessing fragile sites at expanded repeats, it is possible that other aberrantly bound DNA-binding proteins or changes in chromatin topology associated with these sites have yet to be elucidated and could be contributing to this inaccessibility.
The FSFS FRA2L in 2p11.2 (Lukusa and Fryns, 2008) was recently reported as the source of the unusual bending of chromosome 2 in metaphase spreads (Garribba et al., 2021). Interestingly, no fragility at 2p11.2 was identified in these experiments, performed in one FXS cell line. Chromosomal bends at this CGG-rich region were observed in the absence of any cellular treatment, together with bending of other chromosomes (chr. 1 and 3), and increased significantly under folate deficient conditions. Folate deficiency also induced chromosome 2 aneuploidy (Garribba et al., 2021). A link between this cytological phenomenon and sister chromatid missegregation is far from being identified, however the role of folate on the stability of CGG-rich regions is confirmed. It appears to be related to differential chromatin compaction and altered DNA replication (effect exacerbated by folate deficiency), which delays the condensation of mitotic chromosomes, allowing for missegregation. Such observations are not new as bending of metaphase chromosomes was first described in 1984 as a change in the direction of the longitudinal axis of the chromosome (45° fold) involving both chromatids (Flejter et al., 1984), and it was further analyzed as a possible indicator of the inactive chromosome X (Van Dyke et al., 1987, 1986). X-chromosome bending was proposed to represent a remnant of the Barr body packaging from the previous interphase or, alternatively, a structural feature that helps to provide continuity to the Barr body from one interphase to the next (Van Dyke et al., 1987, 1986; Munn et al., 1991; Walker et al., 1991; Dietzel et al., 1998). Non-random bends in autosomes have also been described, with higher incidence with increasing length of the chromosomes, and thought to be associated with chromatin compaction as residue of a folded chromosome state in the interphase nucleus (Flejter et al., 1984; Plaja et al., 2004, 2001). More observations on different cell lines are necessary to obtain robust evidence that support a biological role of chromosomal bending and its dependance upon chromatin compaction and accessibility.
Thus, unusual DNA structure formation, in addition to epigenetic factors, can affect fragile site stability. Mechanistically, these secondary structures perturb the elongation of DNA replication in vitro and in vivo (reviewed in (Kaushal and Freudenreich, 2019) and likely contribute to fragility in this manner. Additionally, proteins important for resolving secondary structures, such as helicases and topoisomerases, play a role in the stability of CFSs (Pirzio et al., 2008; Tuduri et al., 2009; Arlt and Glover, 2010; Shah et al., 2010; Murfuni et al., 2012). Aphidicolin-induced replication stress results in uncoupling of the helicase and polymerase activity, leaving up to several kilobases of separated DNA strands that may be prone to forming DNA secondary structures (Dröge et al., 1985; Lönn and Lönn, 1988). Camptothecin, a topoisomerase I inhibitor, reduces breakage at CFSs and in ATR-deficient cells, highlighting a potential role for topoisomerase I in expression of fragile sites (Arlt and Glover, 2010). Furthermore, instability at CFSs was increased upon depletion of the Rev3 subunit of polζ, polη, and possibly polκ (Bergoglio et al., 2013; Bhat et al., 2013), which are DNA polymerases specialized for synthesis through non-canonical DNA structures (Boyer et al., 2013).
The contribution of key epigenetic and DNA structural alterations to fragile site expression have yet to be fully understood. Such cis elements are known to influence the susceptibility of these DNA regions to fragility and could ultimately be harnessed to reduce instability. This possibility is particularly relevant for the rare FSFSs, which often exhibit somatic instability and expand in disease contexts. Increased knowledge of fragility leading to improved understanding and application to disease biology is a recurring theme for RFSs and highlights the importance of continued research into this often-overlooked cytogenetic phenomenon.
Typically, replication and transcriptional activities are temporally coordinated within mammalian cells to avoid problematic collisions. However, many long genes (>800 kb) initiate transcription within G2 and only complete it by late G1/early S phase increasing the chance for collision (Helmrich et al., 2011). More than 80% of human CFSs contain genes larger than 300 kb, which is in striking contrast to the median gene length of ∼20 kb (Le Tallec et al., 2013). Many CFSs harbor exceptionally long genes, which take more than a complete cell cycle to be transcribed, such as the FHIT gene (1.5 Mb) at FRA3B (Helmrich et al., 2011). This situation leads to the increased likelihood of collisions between replication and transcriptional machinery, leading to replication fork stalling or collapse and resultant instability (Prado and Aguilera, 2005; Azvolinsky et al., 2009; Merrikh et al., 2011). Supporting this connection is the observation that CFS breaks occur when the implicated genes are transcribed, but not when they are transcriptionally silent (Helmrich et al., 2011). This finding highlights the important role for transcriptional activity in chromosomal fragility.
The conflict between these two metabolic processes is further exacerbated by the propensity of nascent RNA to form RNA:DNA hybrids (R-loops) during transcription (reviewed in (Freudenreich, 2018), particularly in GC-rich regions, which all FSFSs are. (CGG)n expanded loci have been demonstrated to have increased R-loop formation (Reddy et al., 2014, 2011; Groh et al., 2014) and the link between R-loop formation and genomic instability has been a topic of intense study (reviewed in (Freudenreich, 2018; Groh et al., 2014; Santos-Pereira and Aguilera, 2015, p.). Further, knockdown or overexpression of RNase H1 (the primary enzyme responsible for resolving R-loops formed with nascent transcripts), results in increased or decreased expression of fragile sites, respectively (Helmrich et al., 2011). R-loop formation at the FRA16D locus impedes replication and causes replication fork stalling, which is a key aspect of CFS instability (Madireddy et al., 2016). R-loops can also promote the formation of repressive chromatin by altering the local epigenetic landscape (Groh et al., 2014), which also contributes to fragility. The majority of genes present in fragile site regions possess a high propensity for R-loop formation when computationally compared to the rest of the genome (Feng and Chakraborty, 2017). It is important to note that most large genes within the genome remain stable, even if they are able to form R-loops; therefore, gene size per se is not sufficient to induce fragility (Le Tallec et al., 2013). A growing awareness of R-loop formations and its connections to genomic instability may yield clues to the relationship between replication, transcription, and fragile sites.
The contribution of the DNA damage pathway to fragile sites is more extensively studied in relation to CFSs and remains largely unexplored in the field of RFSs. Most notably, the role of the DNA damage response protein kinases ATR (ataxia telangiectasia and Rad3-related), and to a lesser extent ATM (ataxia telangiectasia mutated) have been embedded at the core of the DNA lesion checkpoint response pathways in both common (Casper et al., 2002; Ozeri-Galai et al., 2008), and rare (Kumari et al., 2009) fragile sites. ATR-deficiency causes Seckle syndrome, and cells from these individuals have an increased spontaneous expression of fragile sites (Casper et al., 2004). The Fanconi anemia (FA) repair pathway, which responds to interstrand crosslink (ICL) DNA lesions amongst other functions, plays an integral role in fragile site stability. Cells from FA patients exhibit breakpoints at CFS loci at least 50% of the time (Schoder et al., 2010; Filipović et al., 2016) supporting a connection between DNA repair pathways and fragile sites.
Many other repair proteins are implicated in CFS expression, where their inhibition or knockdown enhances aphidicolin-induced fragility (refer to (Glover et al., 2017) for comprehensive review). Furthermore, proteomic studies of the FRA16D CFS locus revealed that under aphidicolin stress, several repair proteins, such as MSH3, MSH2, XRCC1, WRN, XRCC6, XPC, and CENT2 interact specifically with the locus (Beresova et al., 2016). The complex pathways and overlapping proteins involved in DNA repair of these DNA lesions suggests that other metabolic proteins may be involved in fragile site expression and remain to be identified.
Extensive work by Ian Hickson’s group and others has elucidated a key aspect of the DNA repair pathway involved in processing at CFSs during aphidicolin replicative stress (Chan et al., 2009; Naim et al., 2013; Ying et al., 2013; Minocherhomji and Hickson, 2014; Minocherhomji et al., 2015; Bhowmick et al., 2016): mitotic replicative-stress DNA synthesis, a process known as MiDAS, occurs following the onset of mitosis as a salvage pathway to complete replication of under-replicated loci (Minocherhomji et al., 2015). FANCD2, a member of the FA repair pathway and previously shown to localize to CFS loci (Chan et al., 2009), also colocalizes with >80% of newly replicated DNA foci (Minocherhomji et al., 2015). Furthermore, like fragile sites, this mitotic DNA synthesis occurs at DAPI-negative regions of chromosomes, suggesting that these fragile sites are under-replicated DNA regions, rather than distinct DNA breaks (Minocherhomji et al., 2015). Recent work demonstrated that MiDAS also occurs at the FSFS FRAXA locus (Garribba et al., 2020). While MiDAS processing at CFSs and FRAXA both involve SLX1/4 and POLD3, FRAXA differs in its requirement for RAD51 (but not RAD52 or MUS81-EME1) (Garribba et al., 2020).
Generally, DNA damage and repair at FSFSs remains understudied. ATR, ATM, and Chk1 influence fragile site expression at the FRAXA locus (Kumari et al., 2009), but no other folate-sensitive (CGG)n locus has been examined in regard to the mechanisms related to DNA repair. The depletion of ATR increases fragile site expression in fragile X patient cells, with and without FUdR treatment. ATM inhibition decreases fragile site expression upon FUdR treatment yet, without FUdR treatment, ATM inhibition can increase fragile site expression in fragile X cells (Kumari et al., 2009). Significant headway in the repair-related mechanisms of CFSs could guide studies at RFSs to reveal commonalities and differences in the mechanism of fragile site processing.
Many key questions remain to be answered in understanding fragility. The recent development of new methods to identify expanded repeats that colocalize with cytogenetically observed, but not molecularly mapped FSFS, has offered tremendous new insight into fragile sites, genome stability, and human disease (Garg et al., 2020; Trost et al., 2020). The convergence of any combination of factors described here could underlie expression of fragile sites at a particular locus, highlighting the complexity of this process. Further, what parameters are required to induce various types of fragile sites, and what commonalities and differences exist in the cellular response to each stressor have yet to be elucidated. Understanding the expression of fragility and the sensitivity of certain loci to replicative stress will be valuable to understanding the mechanisms of genomic instability and countering its effects. Many disease-causing, gene-specific repeat expansions exist at fragile site loci, hinting that mechanisms related to fragility expression could also be contributing factors to DNA expansions at these loci. Understanding the proteins and pathways that contribute to the causes and consequences of these fragile sites could provide useful targets towards therapeutic intervention to stabilize loci and prevent instability at problematic regions linked to a variety of diseases. Prior to the implementation of practical therapeutic steps aimed at improving human health and overcoming disease, it is important to lay down the foundational research to understand the fundamentals of fragile site expression and repeat expansion.
MM, NS, MS, and CP wrote the manuscript. MM, NS, and MS made the figures. MM and CP conceived of the topic and scope of this article.
Ontario Graduate Scholarship (MM, MHMS), ALS Canada (MHMS, CEP), ALS-A, Brain Canada, Weston Family Foundation (formerly Weston Brain Institute) (CEP).
We thank Carolyn J. Brown for assistance in the preparation of Figure 5. We thank Frank Kooy, Stella Lanni, and Janet Buchanan for critical review and textual suggestions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Agelopoulos, M., Foutadakis, S., and Thanos, D. (2021). The causes and consequences of spatial organization of the genome in regulation of gene expression. Front. Immunol. 12, 682397. doi:10.3389/fimmu.2021.682397
Aldrup-MacDonald, M. E., Kuo, M. E., Sullivan, L. L., Chew, K., and Sullivan, B. A. (2016). Genomic variation within alpha satellite DNA influences centromere location on human chromosomes with metastable epialleles. Genome Res. 26, 1301–1311. doi:10.1101/gr.206706.116
Allingham-Hawkins, D. J., Babul-Hirji, R., Chitayat, D., Holden, J. J., Yang, K. T., Lee, C., et al. (1999). Fragile X premutation is a significant risk factor for premature ovarian failure: The international collaborative POF in fragile X study-preliminary data. Am. J. Med. Genet. 83, 322–325. doi:10.1002/(sici)1096-8628(19990402)83:4<322::aid-ajmg17>3.0.co;2-b
Altemose, N., Miga, K. H., Maggioni, M., and Willard, H. F. (2014). Genomic characterization of large heterochromatic gaps in the human genome assembly. PLoS Comput. Biol. 10, e1003628. doi:10.1371/journal.pcbi.1003628
Altemose, N., Logsdon, G. A., Bzikadze, A. V., Sidhwani, P., Langley, S. A., Caldas, G. V., et al. (2022). Complete genomic and epigenetic maps of human centromeres. Science 376, eabl4178. doi:10.1126/science.abl4178
Altemose, N. (2022). A classical revival: Human satellite DNAs enter the genomics era. Semin. Cell Dev. Biol. 128, 2–14. doi:10.1016/j.semcdb.2022.04.012
Amarose, A. P., Huttenlocher, P. R., Sprudzs, R. M., Laitsch, T. J., and Pettenati, M. J. (1987). A heritable fragile 12q24.13 segregating in a family with the fragile X chromosome. Hum. Genet. 75, 4–6. doi:10.1007/BF00273829
Andersson, M., Lewan, L., and Stenram, U. (1988). Compartmentation of purine and pyrimidine nucleotides in animal cells. Int. J. Biochem. 20, 1039–1050. doi:10.1016/0020-711x(88)90248-0
Archidiacono, N., Rett, A., Rocchi, M., Rolando, S., Lugaresi, E., and Romeo, G. (1985). Rett syndrome and fragile site in Xp22. Lancet 2, 1242–1243. doi:10.1016/s0140-6736(85)90767-6
Arlt, M. F., and Glover, T. W. (2010). Inhibition of topoisomerase I prevents chromosome breakage at common fragile sites. DNA Repair (Amst) 9, 678–689. doi:10.1016/j.dnarep.2010.03.005
Arlt, M. F., Miller, D. E., Beer, D. G., and Glover, T. W. (2002). Molecular characterization of FRAXB and comparative common fragile site instability in cancer cells. Genes Chromosom. Cancer 33, 82–92. doi:10.1002/gcc.10000
Arlt, M. F., Durkin, S. G., Ragland, R. L., and Glover, T. W. (2006). Common fragile sites as targets for chromosome rearrangements. DNA Repair (Amst) 5, 1126–1135. doi:10.1016/j.dnarep.2006.05.010
Aziz, M., Stathopulu, E., Callias, M., Taylor, C., Turk, J., Oostra, B., et al. (2003). Clinical features of boys with fragile X premutations and intermediate alleles. Am. J. Med. Genet. B Neuropsychiatr. Genet. 121B, 119–127. doi:10.1002/ajmg.b.20030
Azvolinsky, A., Giresi, P. G., Lieb, J. D., and Zakian, V. A. (2009). Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol. Cell 34, 722–734. doi:10.1016/j.molcel.2009.05.022
Banes, S. L., Begleiter, M. L., and Butler, M. G. (2003). 45, X/46, XY mosaicism and fragile X syndrome. Am. J. Med. Genet. A 116A, 99–100. doi:10.1002/ajmg.a.10006
Barbé, L., and Finkbeiner, S. (2022). Genetic and epigenetic interplay define disease onset and severity in repeat diseases. Front. Aging Neurosci. 14, 750629. doi:10.3389/fnagi.2022.750629
Barletta, C., Ragusa, R. M., Garofalo, G., Scillato, F., and Ruggeri, M. (1991). Segregation analysis of autosomal fragile sites in three families with the fragile X chromosome. Ann. Genet. 34, 111–114.
Barlow, J. H., Faryabi, R. B., Callén, E., Wong, N., Malhowski, A., Chen, H. T., et al. (2013). Identification of early replicating fragile sites that contribute to genome instability. Cell 152, 620–632. doi:10.1016/j.cell.2013.01.006
Baxter, J. (2015). Breaking up is hard to do”: the formation and resolution of sister chromatid intertwines. J. Mol. Biol. 427, 590–607. doi:10.1016/j.jmb.2014.08.022
Bechhoefer, J., and Rhind, N. (2012). Replication timing and its emergence from stochastic processes. Trends Genet. 28, 374–381. doi:10.1016/j.tig.2012.03.011
Beetstra, S., Thomas, P., Salisbury, C., Turner, J., and Fenech, M. (2005). Folic acid deficiency increases chromosomal instability, chromosome 21 aneuploidy and sensitivity to radiation-induced micronuclei. Mutat. Res. 578, 317–326. doi:10.1016/j.mrfmmm.2005.05.012
Bekdash, R. A. (2021). Early life nutrition and mental health: The role of DNA methylation. Nutrients 13, 3111. doi:10.3390/nu13093111
Bell, M. V., Hirst, M. C., Nakahori, Y., MacKinnon, R. N., Roche, A., Flint, T. J., et al. (1991). Physical mapping across the fragile X: Hypermethylation and clinical expression of the fragile X syndrome. Cell 64, 861–866. doi:10.1016/0092-8674(91)90514-y
Bell, J. (1941). On the age of onset and age at death in hereditary muscular dystrophy with some observations bearing on the question of antedating. Ann. Eugen. 11, 272–289. doi:10.1111/j.1469-1809.1941.tb02290.x
Ben Salem, C., Sakhri, J., and Hmouda, H. (2016). Drug-induced megaloblastic anemia. N. Engl. J. Med. 374, 696–697. doi:10.1056/NEJMc1515180
Beresova, L., Vesela, E., Chamrad, I., Voller, J., Yamada, M., Furst, T., et al. (2016). Role of DNA repair factor xeroderma pigmentosum protein group C in response to replication stress as revealed by DNA fragile site affinity chromatography and quantitative proteomics. J. Proteome Res. 15, 4505–4517. doi:10.1021/acs.jproteome.6b00622
Berg, J. M., Faunch, J. A., Pendrey, M. J., Penrose, L. S., Ridler, M. A., and Shapiro, A. (1969). A homozygous chromosomal variant. Lancet 1, 531. doi:10.1016/s0140-6736(69)91627-4
Berg, J., Grace, E., Teik, K. W., Hammond, H., Tidman, M., and FitzPatrick, D. (2000). Bullous ichthyosiform erythroderma, developmental delay, aortic and pulmonary stenosis in association with a FRA12A. Clin. Dysmorphol. 9, 213–219. doi:10.1097/00019605-200009030-00012
Bergoglio, V., Boyer, A.-S., Walsh, E., Naim, V., Legube, G., Lee, M. Y. W. T., et al. (2013). DNA synthesis by Pol η promotes fragile site stability by preventing under-replicated DNA in mitosis. J. Cell Biol. 201, 395–408. doi:10.1083/jcb.201207066
Berthelot, C., Muffato, M., Abecassis, J., and Roest Crollius, H. (2015). The 3D organization of chromatin explains evolutionary fragile genomic regions. Cell Rep. 10, 1913–1924. doi:10.1016/j.celrep.2015.02.046
Bester, A. C., Roniger, M., Oren, Y. S., Im, M. M., Sarni, D., Chaoat, M., et al. (2011). Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145, 435–446. doi:10.1016/j.cell.2011.03.044
Beverstock, G. C., Mol, A., and Wienhofer, E. (1985). Absence of significant autosomal lesions in Huntington’s disease. Ann. Hum. Genet. 49, 283–290. doi:10.1111/j.1469-1809.1985.tb01704.x
Bhat, A., Andersen, P. L., Qin, Z., and Xiao, W. (2013). Rev3, the catalytic subunit of Polζ, is required for maintaining fragile site stability in human cells. Nucleic Acids Res. 41, 2328–2339. doi:10.1093/nar/gks1442
Bhowmick, R., Minocherhomji, S., and Hickson, I. D. (2016). RAD52 facilitates mitotic DNA synthesis following replication stress. Mol. Cell 64, 1117–1126. doi:10.1016/j.molcel.2016.10.037
Bignell, G. R., Greenman, C. D., Davies, H., Butler, A. P., Edkins, S., Andrews, J. M., et al. (2010). Signatures of mutation and selection in the cancer genome. Nature 463, 893–898. doi:10.1038/nature08768
Bistulfi, G., Vandette, E., Matsui, S.-I., and Smiraglia, D. J. (2010). Mild folate deficiency induces genetic and epigenetic instability and phenotype changes in prostate cancer cells. BMC Biol. 8, 6. doi:10.1186/1741-7007-8-6
Bjerregaard, V. A., Garribba, L., McMurray, C. T., Hickson, I. D., and Liu, Y. (2018). Folate deficiency drives mitotic missegregation of the human FRAXA locus. Proc. Natl. Acad. Sci. U. S. A. 115, 13003–13008. doi:10.1073/pnas.1808377115
Black, E. M., and Giunta, S. (2018). Repetitive fragile sites: Centromere satellite DNA as a source of genome instability in human diseases. Genes (Basel) 9, E615. doi:10.3390/genes9120615
Blount, B. C., and Ames, B. N. (1995). DNA damage in folate deficiency. Baillieres Clin. Haematol. 8, 461–478. doi:10.1016/s0950-3536(05)80216-1
Blount, B. C., Mack, M. M., Wehr, C. M., MacGregor, J. T., Hiatt, R. A., Wang, G., et al. (1997). Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: Implications for cancer and neuronal damage. Proc. Natl. Acad. Sci. U. S. A. 94, 3290–3295. doi:10.1073/pnas.94.7.3290
Boisvert, F.-M., van Koningsbruggen, S., Navascués, J., and Lamond, A. I. (2007). The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 8, 574–585. doi:10.1038/nrm2184
Boldog, F. L., Waggoner, B., Glover, T. W., Chumakov, I., Le Paslier, D., Cohen, D., et al. (1994). Integrated YAC contig containing the 3p14.2 hereditary renal carcinoma 3;8 translocation breakpoint and the fragile site FRA3B. Genes Chromosom. Cancer 11, 216–221. doi:10.1002/gcc.2870110403
Bosco, N., and de Lange, T. (2012). A TRF1-controlled common fragile site containing interstitial telomeric sequences. Chromosoma 121, 465–474. doi:10.1007/s00412-012-0377-6
Bosco, N., Pelliccia, F., and Rocchi, A. (2010). Characterization of FRA7B, a human common fragile site mapped at the 7p chromosome terminal region. Cancer Genet. cytogenet. 202, 47–52. doi:10.1016/j.cancergencyto.2010.06.008
Bostock, C. J., Christie, S., and Hatch, F. T. (1976). Accessibility of DNA in condensed chromatin to nuclease digestion. Nature 262, 516–519. doi:10.1038/262516a0
Bouffler, S., Silver, A., Papworth, D., Coates, J., and Cox, R. (1993). Murine radiation myeloid leukaemogenesis: Relationship between interstitial telomere-like sequences and chromosome 2 fragile sites. Genes Chromosom. Cancer 6, 98–106. doi:10.1002/gcc.2870060206
Boutouil, M., Fetni, R., Qu, J., Dallaire, L., Richer, C. L., and Lemieux, N. (1996). Fragile site and interstitial telomere repeat sequences at the fusion point of a de novo (Y;13) translocation. Hum. Genet. 98, 323–327. doi:10.1007/s004390050216
Bowen, B. C. (1981). DNA fragments associated with chromosome scaffolds. Nucleic Acids Res. 9, 5093–5108. doi:10.1093/nar/9.19.5093
Boyer, A.-S., Grgurevic, S., Cazaux, C., and Hoffmann, J.-S. (2013). The human specialized DNA polymerases and non-B DNA: Vital relationships to preserve genome integrity. J. Mol. Biol. 425, 4767–4781. doi:10.1016/j.jmb.2013.09.022
Brachet, J. (1977). Deoxyribonucleic acid synthesis during early embryogenesis. Biochem. Soc. Trans. 5, 1184–1190. doi:10.1042/bst0051184
Branda, R. F., Arthur, D. C., Woods, W. G., Danzl, T. J., and King, R. A. (1984). Folate metabolism and chromosomal stability in the fragile X syndrome. Am. J. Med. 77, 602–611. doi:10.1016/0002-9343(84)90349-8
Broderick, R., and Niedzwiedz, W. (2015). Sister chromatid decatenation: Bridging the gaps in our knowledge. Cell Cycle 14, 3040–3044. doi:10.1080/15384101.2015.1078039
Brown, W. T., Friedman, E., Jenkins, E. C., Brooks, J., Wisniewski, K., Raguthu, S., et al. (1982). Association of fragile X syndrome with autism. Lancet 1, 100. doi:10.1016/s0140-6736(82)90231-8
Bryan, D. S., Ransom, M., Adane, B., York, K., and Hesselberth, J. R. (2014). High resolution mapping of modified DNA nucleobases using excision repair enzymes. Genome Res. 24, 1534–1542. doi:10.1101/gr.174052.114
Bühler, E. M., and Malik, N. J. (1984). The tricho-rhino-phalangeal syndrome(s): Chromosome 8 long arm deletion: Is there a shortest region of overlap between reported cases? TRP I and TRP II syndromes: Are they separate entities? Am. J. Med. Genet. 19, 113–119. doi:10.1002/ajmg.1320190111
Burrow, A. A., Williams, L. E., Pierce, L. C. T., and Wang, Y.-H. (2009). Over half of breakpoints in gene pairs involved in cancer-specific recurrent translocations are mapped to human chromosomal fragile sites. BMC Genomics 10, 59. doi:10.1186/1471-2164-10-59
Burrow, A. A., Marullo, A., Holder, L. R., and Wang, Y.-H. (2010). Secondary structure formation and DNA instability at fragile site FRA16B. Nucleic Acids Res. 38, 2865–2877. doi:10.1093/nar/gkp1245
Caporossi, D., Bacchetti, S., and Nicoletti, B. (1991). Synergism between aphidicolin and adenoviruses in the induction of breaks at fragile sites on human chromosomes. Cancer Genet. cytogenet. 54, 39–53. doi:10.1016/0165-4608(91)90028-s
Casper, A. M., Nghiem, P., Arlt, M. F., and Glover, T. W. (2002). ATR regulates fragile site stability. Cell 111, 779–789. doi:10.1016/s0092-8674(02)01113-3
Casper, A. M., Durkin, S. G., Arlt, M. F., and Glover, T. W. (2004). Chromosomal instability at common fragile sites in Seckel syndrome. Am. J. Hum. Genet. 75, 654–660. doi:10.1086/422701
Cavani, S., Prontera, P., Grasso, M., Ardisia, C., Malacarne, M., Gradassi, C., et al. (2011). FMR1, FMR2, and SLITRK2 deletion inside a paracentric inversion involving bands Xq27.3-q28 in a male and his mother. Am. J. Med. Genet. A 155A, 221–224. doi:10.1002/ajmg.a.33515
Cechova, M., and Miga, K. H. (2022). Satellite DNAs and human sex chromosome variation. Semin. Cell Dev. Biol. 128, 15–25. doi:10.1016/j.semcdb.2022.04.022
Cechova, M. (2020). Probably correct: Rescuing repeats with short and long reads. Genes (Basel) 12, 48. doi:10.3390/genes12010048
Chan, E. M., Young, E. J., Ianzano, L., Munteanu, I., Zhao, X., Christopoulos, C. C., et al. (2003). Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat. Genet. 35, 125–127. doi:10.1038/ng1238
Chan, K. L., North, P. S., and Hickson, I. D. (2007). BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J. 26, 3397–3409. doi:10.1038/sj.emboj.7601777
Chan, K. L., Palmai-Pallag, T., Ying, S., and Hickson, I. D. (2009). Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 11, 753–760. doi:10.1038/ncb1882
Chen, S., McDougall, J. K., Creagan, R. P., Lewis, V., and Ruddle, F. H. (1976). Mapping of genes and adenovirus-12-induced gaps using chimpanzee-mouse somatic cell hybrids. Cytogenet. Cell Genet. 12, 412–415. doi:10.1159/000130646
Chen, H. H., Rau, D. C., and Charney, E. (1985). The flexibility of alternating dA-dT sequences. J. Biomol. Struct. Dyn. 2, 709–719. doi:10.1080/07391102.1985.10506318
Chen, C. H., Shih, H. H., Wang-Wuu, S., Tai, J. J., and Wuu, K. D. (1998). Chromosomal fragile site expression in lymphocytes from patients with schizophrenia. Hum. Genet. 103, 702–706. doi:10.1007/s004390050894
Cheng, C. H., and Kuchta, R. D. (1993). DNA polymerase epsilon: Aphidicolin inhibition and the relationship between polymerase and exonuclease activity. Biochemistry 32, 8568–8574. doi:10.1021/bi00084a025
Cheung, S. W., Sun, L., and Featherstone, T. (1989). Visualization of NORs in relation to the precise chromosomal localization of ribosomal RNA genes. Cytogenet. Cell Genet. 50, 93–97. doi:10.1159/000132731
Chintagumpala, M. M., Dreyer, Z. A., Steuber, C. P., and Cooley, L. D. (1996). Pancytopenia with chromosomal fragility: Vitamin B12 deficiency. J. Pediatr. Hematol. Oncol. 18, 166–170. doi:10.1097/00043426-199605000-00014
Christman, J. K. (2002). 5-Azacytidine and 5-aza-2’-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 21, 5483–5495. doi:10.1038/sj.onc.1205699
Cilli, P., Ventura, I., Minoprio, A., Meccia, E., Martire, A., Wilson, S. H., et al. (2016). Oxidized dNTPs and the OGG1 and MUTYH DNA glycosylases combine to induce CAG/CTG repeat instability. Nucleic Acids Res. 44, 5190–5203. doi:10.1093/nar/gkw170
Cingam, S. R., Koshy, N., Veillon, D., and Peddi, P. (2017). Reversal of isolated 20q deletion with vitamin B12 replacement in a patient with pernicious anaemia. BMJ Case Rep. 2017, bcr2016218689. doi:10.1136/bcr-2016-218689
Coffee, B., Zhang, F., Warren, S. T., and Reines, D. (1999). Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nat. Genet. 22, 98–101. doi:10.1038/8807
Coffee, B., Zhang, F., Ceman, S., Warren, S. T., and Reines, D. (2002). Histone modifications depict an aberrantly heterochromatinized FMR1 gene in fragile x syndrome. Am. J. Hum. Genet. 71, 923–932. doi:10.1086/342931
Coffee, B., Ikeda, M., Budimirovic, D. B., Hjelm, L. N., Kaufmann, W. E., and Warren, S. T. (2008). Mosaic FMR1 deletion causes fragile X syndrome and can lead to molecular misdiagnosis: A case report and review of the literature. Am. J. Med. Genet. A 146A, 1358–1367. doi:10.1002/ajmg.a.32261
Coquelle, A., Toledo, F., Stern, S., Bieth, A., Debatisse, M., and Bieth, A. (1998). A new role for hypoxia in tumor progression: Induction of fragile site triggering genomic rearrangements and Formation of complex DMs and HSRs. Mol. Cell 2, 259–265. doi:10.1016/s1097-2765(00)80137-9
Corbin, S., Neilly, M. E., Espinosa, R., Davis, E. M., McKeithan, T. W., and Le Beau, M. M. (2002). Identification of unstable sequences within the common fragile site at 3p14.2: Implications for the mechanism of deletions within fragile histidine triad gene/common fragile site at 3p14.2 in tumors. Cancer Res. 62, 3477–3484.
Corneo, G., Ginelli, E., and Polli, E. (1968). Presence of a satellite DNA in normal and leukemic human tissues. Acta Haematol. 39, 75–84. doi:10.1159/000208946
Corneo, G., Ginelli, E., and Polli, E. (1970). Repeated sequences in human DNA. J. Mol. Biol. 48, 319–327. doi:10.1016/0022-2836(70)90163-4
Corneo, G., Zardi, L., and Polli, E. (1972). Elution of human satellite DNAs on a methylated albumin kieselguhr chromatographic column: Isolation of satellite DNA. IV. Biochim. Biophys. Acta 269, 201–204. doi:10.1016/0005-2787(72)90427-3
Craig-Holmes, A. P., and Shaw, M. W. (1971). Polymorphism of human constitutive heterochromatin. Science 174, 702–704. doi:10.1126/science.174.4010.702
Craig-Holmes, A. P., Moore, F. B., and Shaw, M. W. (1973). Polymorphism of human C-band heterochromatin. I. Frequency of variants. Am. J. Hum. Genet. 25, 181–192.
Craig-Holmes, A. P., Moore, F. B., and Shaw, M. W. (1975). Polymporphism of human C-band heterochromatin. II. Family studies with suggestive evidence for somatic crossing over. Am. J. Hum. Genet. 27, 178–189.
Crippa, L., Delozier-Blanchet, C. D., and Engel, E. (1984). [Variations in the presence of a fragile site on X-fra(X)-according to cases and methods used]. J. Genet. Hum. 32, 193–197.
Dar, H., Bar-El, H., Ziv, M., and Shapiro, I. (1995). New heritable fragile site with spontaneous expression at 1q41. Am. J. Med. Genet. 55, 145–146. doi:10.1002/ajmg.1320550202
Das, S. K., Kunkel, T. A., and Loeb, L. A. (1985). Effects of altered nucleotide concentrations on the fidelity of DNA replication. Basic Life Sci. 31, 117–126. doi:10.1007/978-1-4613-2449-2_7
Das, K. C., Mohanty, D., and Garewal, G. (1986). Cytogenetics in nutritional megaloblastic anaemia: Prolonged persistence of chromosomal abnormalities in lymphocytes after remission. Acta Haematol. 76, 146–154. doi:10.1159/000206040
Das, K. C., Das, M., Mohanty, D., Jadaon, M. M., Gupta, A., Marouf, R., et al. (2005). Megaloblastosis: From morphos to molecules. Med. Princ. Pract. 14 (1), 2–14. doi:10.1159/000086179
Das, S. P., Borrman, T., Liu, V. W. T., Yang, S. C.-H., Bechhoefer, J., and Rhind, N. (2015). Replication timing is regulated by the number of MCMs loaded at origins. Genome Res. 25, 1886–1892. doi:10.1101/gr.195305.115
De Ambrosis, A., Casciano, I., Querzola, F., Vidali, G., and Romani, M. (1992). Chromatin structure, DNA methylation, and gene expression at sites of viral integration in human fibroblasts. Implications for chromosomal fragility. Cancer Genet. cytogenet. 60, 1–7. doi:10.1016/0165-4608(92)90223-u
De Leon-Luis, J., Santolaya-Forgas, J., May, G., Tonk, V., Shelton, D., and Galan, I. (2005). Prenatal diagnosis of FRA10A: A case report and literature review. Am. J. Med. Genet. A 136, 63–65. doi:10.1002/ajmg.a.30093
De Luca, G., Russo, M. T., Degan, P., Tiveron, C., Zijno, A., Meccia, E., et al. (2008). A role for oxidized DNA precursors in Huntington’s disease-like striatal neurodegeneration. PLoS Genet. 4, e1000266. doi:10.1371/journal.pgen.1000266
Debacker, K., and Kooy, R. F. (2007). Fragile sites and human disease. Hum. Mol. Genet. 16 (2), R150–R158. doi:10.1093/hmg/ddm136
Debacker, K., Winnepenninckx, B., Longman, C., Colgan, J., Tolmie, J., Murray, R., et al. (2007). The molecular basis of the folate-sensitive fragile site FRA11A at 11q13. Cytogenet. Genome Res. 119, 9–14. doi:10.1159/000109612
Dekaban, A. (1965). Persisting clone of cells with an abnormal chromosome in a woman previously irradiated. J. Nucl. Med. 6, 740–746.
Demirhan, O., Tastemir, D., and Sertdemir, Y. (2006). Chromosomal fragile sites in schizophrenic patients. Russ. J. Genet. 42, 810–817. doi:10.1134/s1022795406070179
Demirhan, O., Tastemir, D., and Sertdemir, Y. (2009). The expression of folate sensitive fragile sites in patients with bipolar disorder. Yonsei Med. J. 50, 137–141. doi:10.3349/ymj.2009.50.1.137
Denison, S. R., Simper, R. K., and Greenbaum, I. F. (2003). How common are common fragile sites in humans: Interindividual variation in the distribution of aphidicolin-induced fragile sites. Cytogenet. Genome Res. 101, 8–16. doi:10.1159/000073411
Depienne, C., and Mandel, J.-L. (2021). 30 years of repeat expansion disorders: What have we learned and what are the remaining challenges? Am. J. Hum. Genet. 108, 764–785. doi:10.1016/j.ajhg.2021.03.011
Devys, D., Biancalana, V., Rousseau, F., Boué, J., Mandel, J. L., and Oberlé, I. (1992). Analysis of full fragile X mutations in fetal tissues and monozygotic twins indicate that abnormal methylation and somatic heterogeneity are established early in development. Am. J. Med. Genet. 43, 208–216. doi:10.1002/ajmg.1320430134
Dietzel, S., Eils, R., Sätzler, K., Bornfleth, H., Jauch, A., Cremer, C., et al. (1998). Evidence against a looped structure of the inactive human X-chromosome territory. Exp. Cell Res. 240, 187–196. doi:10.1006/excr.1998.3934
Dillon, L. W., Burrow, A. A., and Wang, Y.-H. (2010). DNA instability at chromosomal fragile sites in cancer. Curr. Genomics 11, 326–337. doi:10.2174/138920210791616699
Dillon, L. W., Pierce, L. C. T., Ng, M. C. Y., and Wang, Y.-H. (2013). Role of DNA secondary structures in fragile site breakage along human chromosome 10. Hum. Mol. Genet. 22, 1443–1456. doi:10.1093/hmg/dds561
Djalali, M., Barbi, G., and Steinbach, P. (1985). Folic acid sensitive fragile sites are not limited to the human karyotype. Demonstration of nonrandom gaps and breaks in the Persian vole Ellobius lutescens Th. inducible by methotrexate, fluorodeoxyuridine, and aphidicolin. Hum. Genet. 70, 183–185. doi:10.1007/BF00273080
Dobkin, C., Radu, G., Ding, X.-H., Brown, W. T., and Nolin, S. L. (2009). Fragile X prenatal analyses show full mutation females at high risk for mosaic Turner syndrome: fragile X leads to chromosome loss. Am. J. Med. Genet. A 149A, 2152–2157. doi:10.1002/ajmg.a.33011
Dröge, P., Sogo, J. M., and Stahl, H. (1985). Inhibition of DNA synthesis by aphidicolin induces supercoiling in simian virus 40 replicative intermediates. EMBO J. 4, 3241–3246. doi:10.1002/j.1460-2075.1985.tb04072.x
Duncan, A. M. (1986). Enhanced sensitivity of lymphoblastoid cells from individuals carrying the mutation for the fragile X syndrome to the clastogenic effects of FUdR. Mutat. Res. 173, 201–205. doi:10.1016/0165-7992(86)90036-9
Durkin, S. G., Ragland, R. L., Arlt, M. F., Mulle, J. G., Warren, S. T., and Glover, T. W. (2008). Replication stress induces tumor-like microdeletions in FHIT/FRA3B. Proc. Natl. Acad. Sci. U. S. A. 105, 246–251. doi:10.1073/pnas.0708097105
Duthie, S. J., and Hawdon, A. (1998). DNA instability (strand breakage, uracil misincorporation, and defective repair) is increased by folic acid depletion in human lymphocytes in vitro. FASEB J. 12, 1491–1497. doi:10.1096/fasebj.12.14.1491
Duthie, S. J., and McMillan, P. (1997). Uracil misincorporation in human DNA detected using single cell gel electrophoresis. Carcinogenesis 18, 1709–1714. doi:10.1093/carcin/18.9.1709
Eberhart, D. E., and Warren, S. T. (1996). Nuclease sensitivity of permeabilized cells confirms altered chromatin formation at the fragile X locus. Somat. Cell Mol. Genet. 22, 435–441. doi:10.1007/BF02369435
Ebler, J., Ebert, P., Clarke, W. E., Rausch, T., Audano, P. A., Houwaart, T., et al. (2022). Pangenome-based genome inference allows efficient and accurate genotyping across a wide spectrum of variant classes. Nat. Genet. 54, 518–525. doi:10.1038/s41588-022-01043-w
El Achkar, E., Gerbault-Seureau, M., Muleris, M., Dutrillaux, B., and Debatisse, M. (2005). Premature condensation induces breaks at the interface of early and late replicating chromosome bands bearing common fragile sites. Proc. Natl. Acad. Sci. U. S. A. 102, 18069–18074. doi:10.1073/pnas.0506497102
Elder, F. F., and Robinson, T. J. (1989). Rodent common fragile sites: Are they conserved? Evidence from mouse and rat. Chromosoma 97, 459–464. doi:10.1007/BF00295030
Everson, R. B., Wehr, C. M., Erexson, G. L., and MacGregor, J. T. (1988). Association of marginal folate depletion with increased human chromosomal damage in vivo: Demonstration by analysis of micronucleated erythrocytes. J. Natl. Cancer Inst. 80, 525–529. doi:10.1093/jnci/80.7.525
Fechter, A., Buettel, I., Kuehnel, E., Savelyeva, L., and Schwab, M. (2007). Common fragile site FRA11G and rare fragile site FRA11B at 11q23.3 encompass distinct genomic regions. Genes Chromosom. Cancer 46, 98–106. doi:10.1002/gcc.20389
Feichtinger, W., and Schmid, M. (1989). Increased frequencies of sister chromatid exchanges at common fragile sites (1)(q42) and (19)(q13). Hum. Genet. 83, 145–147. doi:10.1007/BF00286707
Felbor, U., Feichtinger, W., and Schmid, M. (2003). The rare human fragile site 16B. Cytogenet. Genome Res. 100, 85–88. doi:10.1159/000072841
Fenech, M., and Crott, J. W. (2002). Micronuclei, nucleoplasmic bridges and nuclear buds induced in folic acid deficient human lymphocytes-evidence for breakage-fusion-bridge cycles in the cytokinesis-block micronucleus assay. Mutat. Res. 504, 131–136. doi:10.1016/s0027-5107(02)00086-6
Fenech, M. (2001). The role of folic acid and Vitamin B12 in genomic stability of human cells. Mutat. Res. 475, 57–67. doi:10.1016/s0027-5107(01)00079-3
Fenech, M. (2007). Cytokinesis-block micronucleus cytome assay. Nat. Protoc. 2, 1084–1104. doi:10.1038/nprot.2007.77
Feng, W., and Chakraborty, A. (2017). Fragility extraordinaire: Unsolved mysteries of chromosome fragile sites. Adv. Exp. Med. Biol. 1042, 489–526. doi:10.1007/978-981-10-6955-0_21
Ferguson-Smith, M. A., and Handmaker, S. D. (1961). Observations on the satellited human chromosomes. Lancet 1, 638–640. doi:10.1016/s0140-6736(61)91655-5
Ferraro, M., Archidiacono, N., Pelliccia, F., Rocchi, M., Rocchi, A., and de Capoa, A. (1977). Secondary constrictions and nucleolus organizer regions in man. Exp. Cell Res. 104, 428–430. doi:10.1016/0014-4827(77)90109-4
Filipović, J., Joksić, G., Vujić, D., Joksić, I., Mrasek, K., Weise, A., et al. (2016). First molecular-cytogenetic characterization of Fanconi anemia fragile sites in primary lymphocytes of FA-D2 patients in different stages of the disease. Mol. Cytogenet. 9, 70. doi:10.1186/s13039-016-0280-6
Filippi, G., Pecile, V., Rinaldi, A., and Siniscalco, M. (1988). Fragile-X mutation and klinefelter syndrome: A reappraisal. Am. J. Med. Genet. 30, 99–107. doi:10.1002/ajmg.1320300108
Finnis, M., Dayan, S., Hobson, L., Chenevix-Trench, G., Friend, K., Ried, K., et al. (2005). Common chromosomal fragile site FRA16D mutation in cancer cells. Hum. Mol. Genet. 14, 1341–1349. doi:10.1093/hmg/ddi144
Fischer, K. M. (1998). Expanded (CAG)n, (CGG)n and (GAA)n trinucleotide repeat microsatellites, and mutant purine synthesis and pigmentation genes cause schizophrenia and autism. Med. Hypotheses 51, 223–233. doi:10.1016/s0306-9877(98)90080-9
Fitchett, M., and Seabright, M. (1984). Deleted X chromosomes in patients with the fragile X syndrome. J. Med. Genet. 21, 373. doi:10.1136/jmg.21.5.373
Flejter, W. L., Van Dyke, D. L., and Weiss, L. (1984). Bends in human mitotic metaphase chromosomes, including a bend marking the X-inactivation center. Am. J. Hum. Genet. 36, 218–226.
Flint, J., Bates, G. P., Clark, K., Dorman, A., Willingham, D., Roe, B. A., et al. (1997). Sequence comparison of human and yeast telomeres identifies structurally distinct subtelomeric domains. Hum. Mol. Genet. 6, 1305–1313. doi:10.1093/hmg/6.8.1305
Fortunato, E. A., and Spector, D. H. (2003). Viral induction of site-specific chromosome damage. Rev. Med. Virol. 13, 21–37. doi:10.1002/rmv.368
Fowler, C., Drinkwater, R., Burgoyne, L., and Skinner, J. (1987). Hypervariable lengths of human DNA associated with a human satellite III sequence found in the 3.4kb Y-specific fragment. Nucleic Acids Res. 15, 3929. doi:10.1093/nar/15.9.3929
Fragkos, M., Ganier, O., Coulombe, P., and Méchali, M. (2015). DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 16, 360–374. doi:10.1038/nrm4002
Francés, A., Hildur, K., Barberà, J. A., Rodríguez-Trigo, G., Zock, J.-P., Giraldo, J., et al. (2016). Persistence of breakage in specific chromosome bands 6 Years after acute exposure to oil. PLoS One 11, e0159404. doi:10.1371/journal.pone.0159404
Freese, E. (1959). The specific mutagenic effect of base analogues on Phage T4. J. Mol. Biol. 1, 87–105. doi:10.1016/S0022-2836(59)80038-3
Freudenreich, C. H. (2018). R-Loops: Targets for nuclease cleavage and repeat instability. Curr. Genet. 64, 789–794. doi:10.1007/s00294-018-0806-z
Frommer, M., Prosser, J., Tkachuk, D., Reisner, A. H., and Vincent, P. C. (1982). Simple repeated sequences in human satellite DNA. Nucleic Acids Res. 10, 547–563. doi:10.1093/nar/10.2.547
Froster-Iskenius, U., Schwinger, E., Weigert, M., and Fonatsch, C. (1982). Replication pattern in XXY cells with fra(X). Hum. Genet. 60, 278–280. doi:10.1007/BF00303019
Fry, M., and Loeb, L. A. (1994). The fragile X syndrome d(CGG)n nucleotide repeats form a stable tetrahelical structure. Proc. Natl. Acad. Sci. U. S. A. 91, 4950–4954. doi:10.1073/pnas.91.11.4950
Fryns, J. P., and Van den Berghe, H. (1988). The concurrence of Klinefelter syndrome and fragile X syndrome. Am. J. Med. Genet. 30, 109–113. doi:10.1002/ajmg.1320300109
Fryns, J. P., Azou, M., Jaeken, J., Eggermont, E., Pedersen, J. C., and Van den Berghe, H. (1981). Centromeric instability of chromosomes 1, 9, and 16 associated with combined immunodeficiency. Hum. Genet. 57, 108–110. doi:10.1007/BF00271181
Fryns, J. P., Kleczkowska, A., Kubień, E., Petit, P., Haspeslagh, M., Lindemans, I., et al. (1983). XY/XXY mosaicism and fragile X syndrome. Ann. Genet. 26, 251–253.
Fu, Y. H., Kuhl, D. P., Pizzuti, A., Pieretti, M., Sutcliffe, J. S., Richards, S., et al. (1991). Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the sherman paradox. Cell 67, 1047–1058. doi:10.1016/0092-8674(91)90283-5
Fuster, C., Templado, C., Miró, R., Barrios, L., and Egozcue, J. (1988). Concurrence of the triple-X syndrome and expression of the fragile site Xq27.3. Hum. Genet. 78, 293. doi:10.1007/BF00291682
Gall-Duncan, T., Sato, N., Yuen, R. K. C., and Pearson, C. E. (2022). Advancing genomic technologies and clinical awareness accelerates discovery of disease-associated tandem repeat sequences. Genome Res. 32, 1–27. doi:10.1101/gr.269530.120
Garg, P., Jadhav, B., Rodriguez, O. L., Patel, N., Martin-Trujillo, A., Jain, M., et al. (2020). A survey of rare epigenetic variation in 23, 116 human genomes identifies disease-relevant epivariations and CGG expansions. Am. J. Hum. Genet. 107, 654–669. doi:10.1016/j.ajhg.2020.08.019
Gargano, S., Wang, P., Rusanganwa, E., and Bacchetti, S. (1995). The transcriptionally competent U2 gene is necessary and sufficient for adenovirus type 12 induction of the fragile site at 17q21-22. Mol. Cell. Biol. 15, 6256–6261. doi:10.1128/MCB.15.11.6256
Garofalo, G., Ragusa, R. M., Barletta, C., and Spina, E. (1992). Schizophrenia and chromosomal fragile sites. Am. J. Psychiatry 149, 1116. doi:10.1176/ajp.149.8.1116a
Garofalo, G., Ragusa, R. M., Argiolas, A., Scavuzzo, C., Spina, E., and Barletta, C. (1993). Evidence of chromosomal fragile sites in schizophrenic patients. Ann. Genet. 36, 132–135.
Garribba, L., Bjerregaard, V. A., Gonçalves Dinis, M. M., Özer, Ö., Wu, W., Sakellariou, D., et al. (2020). Folate stress induces SLX1- and RAD51-dependent mitotic DNA synthesis at the fragile X locus in human cells. Proc. Natl. Acad. Sci. U. S. A. 117, 16527–16536. doi:10.1073/pnas.1921219117
Garribba, L., Vogel, I., Lerdrup, M., Gonçalves Dinis, M. M., Ren, L., and Liu, Y. (2021). Folate deficiency triggers the abnormal segregation of a region with large cluster of CG-rich trinucleotide repeats on human chromosome 2. Front. Genet. 12, 695124. doi:10.3389/fgene.2021.695124
Gecz, J., Gedeon, A. K., Sutherland, G. R., and Mulley, J. C. (1996). Identification of the gene FMR2, associated with FRAXE mental retardation. Nat. Genet. 13, 105–108. doi:10.1038/ng0596-105
Gedeon, A. K., Baker, E., Robinson, H., Partington, M. W., Gross, B., Manca, A., et al. (1992). Fragile X syndrome without CCG amplification has an FMR1 deletion. Nat. Genet. 1, 341–344. doi:10.1038/ng0892-341
Gershman, A., Sauria, M. E. G., Guitart, X., Vollger, M. R., Hook, P. W., Hoyt, S. J., et al. (2022). Epigenetic patterns in a complete human genome. Science 376, eabj5089. doi:10.1126/science.abj5089
Giannuzzi, G., Logsdon, G. A., Chatron, N., Miller, D. E., Reversat, J., Munson, K. M., et al. (2021). Alpha satellite insertion close to an ancestral centromeric region. Mol. Biol. Evol. 38, 5576–5587. doi:10.1093/molbev/msab244
Gillberg, C., Wahlström, J., and Hagberg, B. (1985). A “new” chromosome marker common to the Rett syndrome and infantile autism? The frequency of fragile sites at X p22 in 81 children with infantile autism, childhood psychosis and the Rett syndrome. Brain Dev. 7, 365–367. doi:10.1016/s0387-7604(85)80046-2
Gillentine, M. A., and Schaaf, C. P. (2015). The human clinical phenotypes of altered CHRNA7 copy number. Biochem. Pharmacol. 97, 352–362. doi:10.1016/j.bcp.2015.06.012
Gindin, Y., Valenzuela, M. S., Aladjem, M. I., Meltzer, P. S., and Bilke, S. (2014). A chromatin structure-based model accurately predicts DNA replication timing in human cells. Mol. Syst. Biol. 10, 722. doi:10.1002/msb.134859
Giovannucci, E., Stampfer, M. J., Colditz, G. A., Rimm, E. B., Trichopoulos, D., Rosner, B. A., et al. (1993). Folate, methionine, and alcohol intake and risk of colorectal adenoma. J. Natl. Cancer Inst. 85, 875–884. doi:10.1093/jnci/85.11.875
Giraud, F., Ayme, S., Mattei, J. F., and Mattei, M. G. (1976). Constitutional chromosomal breakage. Hum. Genet. 34, 125–136. doi:10.1007/BF00278880
Glousker, G., and Lingner, J. (2021). Challenging endings: How telomeres prevent fragility. Bioessays. 43, e2100157. doi:10.1002/bies.202100157
Glover, T. W., and Stein, C. K. (1987). Induction of sister chromatid exchanges at common fragile sites. Am. J. Hum. Genet. 41, 882–890.
Glover, T. W., and Stein, C. K. (1988). Chromosome breakage and recombination at fragile sites. Am. J. Hum. Genet. 43, 265–273.
Glover, T. W., Berger, C., Coyle, J., and Echo, B. (1984). DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum. Genet. 67, 136–142. doi:10.1007/BF00272988
Glover, T. W., Wilson, T. E., and Arlt, M. F. (2017). Fragile sites in cancer: More than meets the eye. Nat. Rev. Cancer 17, 489–501. doi:10.1038/nrc.2017.52
Godde, J. S., Kass, S. U., Hirst, M. C., and Wolffe, A. P. (1996). Nucleosome assembly on methylated CGG triplet repeats in the fragile X mental retardation gene 1 promoter. J. Biol. Chem. 271, 24325–24328. doi:10.1074/jbc.271.40.24325
Gosden, J. R., Buckland, R. A., Clayton, R. P., and Evans, H. J. (1975). Chromosomal localisation of DNA sequences in condensed and dispersed human chromatin. Exp. Cell Res. 92, 138–147. doi:10.1016/0014-4827(75)90647-3
Goulian, M., Bleile, B., and Tseng, B. Y. (1980). Methotrexate-induced misincorporation of uracil into DNA. Proc. Natl. Acad. Sci. U. S. A. 77, 1956–1960. doi:10.1073/pnas.77.4.1956
Gozaly-Chianea, Y., Roberts, T., and Slijepcevic, P. (2022). The role of BRCA2 in the fragility of interstitial telomeric sites. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 878, 503476. doi:10.1016/j.mrgentox.2022.503476
Green, R., and Datta Mitra, A. (2017). Megaloblastic anemias: Nutritional and other causes. Med. Clin. North Am. 101, 297–317. doi:10.1016/j.mcna.2016.09.013
Groh, M., Lufino, M. M., Wade-Martins, R., and Gromak, N. (2014). R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet. 10, e1004318. doi:10.1371/journal.pgen.1004318
Gu, Y., Lugenbeel, K. A., Vockley, J. G., Grody, W. W., and Nelson, D. L. (1994). A de novo deletion in FMR1 in a patient with developmental delay. Hum. Mol. Genet. 3, 1705–1706. doi:10.1093/hmg/3.9.1705
Guttenbach, M., and Schmid, M. (1994). Exclusion of specific human chromosomes into micronuclei by 5-azacytidine treatment of lymphocyte cultures. Exp. Cell Res. 211, 127–132. doi:10.1006/excr.1994.1068
Hagerman, R. J., and Hagerman, P. J. (2001). Fragile X syndrome: A model of gene-brain-behavior relationships. Mol. Genet. Metab. 74, 89–97. doi:10.1006/mgme.2001.3225
Hagerman, R. J., and Hagerman, P. J. (2002). The fragile X premutation: Into the phenotypic fold. Curr. Opin. Genet. Dev. 12, 278–283. doi:10.1016/s0959-437x(02)00299-x
Hagerman, R. J., Hull, C. E., Safanda, J. F., Carpenter, I., Staley, L. W., O’Connor, R. A., et al. (1994). High functioning fragile X males: Demonstration of an unmethylated fully expanded FMR-1 mutation associated with protein expression. Am. J. Med. Genet. 51, 298–308. doi:10.1002/ajmg.1320510404
Hagerman, R. J., Protic, D., Rajaratnam, A., Salcedo-Arellano, M. J., Aydin, E. Y., and Schneider, A. (2018). Fragile X-associated neuropsychiatric disorders (FXAND). Front. Psychiatry 9, 564. doi:10.3389/fpsyt.2018.00564
Hamel, B. C., Smits, A. P., de Graaff, E., Smeets, D. F., Schoute, F., Eussen, B. H., et al. (1994). Segregation of FRAXE in a large family: Clinical, psychometric, cytogenetic, and molecular data. Am. J. Hum. Genet. 55, 923–931.
Hammond, L. S., Macias, M. M., Tarleton, J. C., and Shashidhar Pai, G. (1997). Fragile X syndrome and deletions in FMR1: New case and review of the literature. Am. J. Med. Genet. 72, 430–434. doi:10.1002/(sici)1096-8628(19971112)72:4<430::aid-ajmg11>3.0.co;2-s
Handt, O., Baker, E., Dayan, S., Gartler, S. M., Woollatt, E., Richards, R. I., et al. (2000). Analysis of replication timing at the FRA10B and FRA16B fragile site loci. Chromosome Res. 8, 677–688. doi:10.1023/a:1026737203447
Hansen, R. S., Canfield, T. K., Lamb, M. M., Gartler, S. M., and Laird, C. D. (1993). Association of fragile X syndrome with delayed replication of the FMR1 gene. Cell 73, 1403–1409. doi:10.1016/0092-8674(93)90365-w
Hansen, R. S., Canfield, T. K., Fjeld, A. D., Mumm, S., Laird, C. D., and Gartler, S. M. (1997). A variable domain of delayed replication in FRAXA fragile X chromosomes: X inactivation-like spread of late replication. Proc. Natl. Acad. Sci. U. S. A. 94, 4587–4592. doi:10.1073/pnas.94.9.4587
Harvey, J., Judge, C., and Wiener, S. (1977). Familial X-linked mental retardation with an X chromosome abnormality. J. Med. Genet. 14, 46–50. doi:10.1136/jmg.14.1.46
Heath, C. W., and Fliegelman, L. (1966). Cytogenetic observations in vitamin B12 and folate deficiency. Blood 27, 800–815. doi:10.1182/blood.v27.6.800.800
Hecht, F., and Bixenman, H. A. (1990). Location of FRAXD in Xq27.2. Fragile sites on the X chromosome. Cancer Genet. cytogenet. 49, 137–138. doi:10.1016/0165-4608(90)90175-a
Hecht, F., and Kaiser-McCaw, B. (1979). The importance of being a fragile site. Am. J. Hum. Genet. 31, 223–225.
Hecht, F., and Sutherland, G. R. (1984). Detection of the fragile X chromosome and other fragile sites. Clin. Genet. 26, 301–303. doi:10.1111/j.1399-0004.1984.tb01063.x
Hecht, F., and Sutherland, G. R. (1985). Detection of fragile sites on human chromosomes. Clin. Genet. 28, 95–96. doi:10.1111/j.1399-0004.1985.tb01227.x
Hecht, F. (1986). Rare, polymorphic, and common fragile sites: A classification. Hum. Genet. 74, 207–208. doi:10.1007/BF00282099
Hedglin, M., Zhang, Y., and O’Brien, P. J. (2015). Probing the DNA structural requirements for facilitated diffusion. Biochemistry 54, 557–566. doi:10.1021/bi5013707
Heitz, D., Devys, D., Imbert, G., Kretz, C., and Mandel, J. L. (1992). Inheritance of the fragile X syndrome: Size of the fragile X premutation is a major determinant of the transition to full mutation. J. Med. Genet. 29, 794–801. doi:10.1136/jmg.29.11.794
Heliot, L., Kaplan, H., Lucas, L., Klein, C., Beorchia, A., Doco-Fenzy, M., et al. (1997). Electron tomography of metaphase nucleolar organizer regions: Evidence for a twisted-loop organization. Mol. Biol. Cell 8, 2199–2216. doi:10.1091/mbc.8.11.2199
Hellman, A., Rahat, A., Scherer, S. W., Darvasi, A., Tsui, L. C., and Kerem, B. (2000). Replication delay along FRA7H, a common fragile site on human chromosome 7, leads to chromosomal instability. Mol. Cell. Biol. 20, 4420–4427. doi:10.1128/MCB.20.12.4420-4427.2000
Helmrich, A., Ballarino, M., and Tora, L. (2011). Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 44, 966–977. doi:10.1016/j.molcel.2011.10.013
Hesdorffer, C. S., and Longo, D. L. (2015). Drug-induced megaloblastic anemia. N. Engl. J. Med. 373, 1649–1658. doi:10.1056/NEJMra1508861
Hesdorffer, C. S., and Longo, D. L. (2016). Drug-induced megaloblastic anemia. N. Engl. J. Med. 374, 696–697. doi:10.1056/NEJMc1515180
Hewett, D. R., Handt, O., Hobson, L., Mangelsdorf, M., Eyre, H. J., Baker, E., et al. (1998). FRA10B structure reveals common elements in repeat expansion and chromosomal fragile site Genesis. Mol. Cell 1, 773–781. doi:10.1016/s1097-2765(00)80077-5
Hirsch, B. (1991). Sister chromatid exchanges are preferentially induced at expressed and nonexpressed common fragile sites. Hum. Genet. 87, 302–306. doi:10.1007/BF00200908
Hirst, M. C., Barnicoat, A., Flynn, G., Wang, Q., Daker, M., Buckle, V. J., et al. (1993). The identification of a third fragile site, FRAXF, in Xq27-q28 distal to both FRAXA and FRAXE. Hum. Mol. Genet. 2, 197–200. doi:10.1093/hmg/2.2.197
Hirst, M., Grewal, P., Flannery, A., Slatter, R., Maher, E., Barton, D., et al. (1995). Two new cases of FMR1 deletion associated with mental impairment. Am. J. Hum. Genet. 56, 67–74.
Hocking, T., Feichtinger, W., Schmid, M., Haan, E. A., Baker, E., and Sutherland, G. R. (1999). Homozygotes for FRA16B are normal. Chromosome Res. 7, 553–556. doi:10.1023/a:1009293613064
Hori, T., Takahashi, E., Tsuji, H., Tsuji, S., and Murata, M. (1988). Fragile X expression in thymidine-prototrophic and auxotrophic human-mouse somatic cell hybrids under low and high thymidylate stress conditions. Cytogenet. Cell Genet. 47, 177–180. doi:10.1159/000132543
Hori, T., Seki, N., Ohira, M., Saito, T., Yamauchi, M., Sagara, M., et al. (1998). A distamycin A-inducible fragile site, FRA8E, located in the region of the hereditary multiple exostoses gene, is not involved in HPV16 DNA integration and amplification. Cancer Genet. cytogenet. 101, 24–34. doi:10.1016/s0165-4608(97)00222-7
Hori, Y., Shimamoto, A., and Kobayashi, T. (2021). The human ribosomal DNA array is composed of highly homogenized tandem clusters. Genome Res. 31, 1971–1982. doi:10.1101/gr.275838.121
Hormozian, F., Schmitt, J. G., Sagulenko, E., Schwab, M., and Savelyeva, L. (2007). FRA1E common fragile site breaks map within a 370kilobase pair region and disrupt the dihydropyrimidine dehydrogenase gene (DPYD). Cancer Lett. 246, 82–91. doi:10.1016/j.canlet.2006.02.004
Hou, J., Parrish, J., Lüdecke, H. J., Sapru, M., Wang, Y., Chen, W., et al. (1995). A 4-megabase YAC contig that spans the langer-giedion syndrome region on human chromosome 8q24.1: Use in refining the location of the trichorhinophalangeal syndrome and multiple exostoses genes (TRPS1 and EXT1). Genomics 29, 87–97. doi:10.1006/geno.1995.1218
Howell, R. T., McDermott, A., and Evans, J. L. (1990). A new apparently folate sensitive fragile site, 5q35. J. Med. Genet. 27, 527–528. doi:10.1136/jmg.27.8.527
Hoyt, S. J., Storer, J. M., Hartley, G. A., Grady, P. G. S., Gershman, A., de Lima, L. G., et al. (2022). From telomere to telomere: The transcriptional and epigenetic state of human repeat elements. Science 376, eabk3112. doi:10.1126/science.abk3112
Hsu, Y. Y., and Wang, Y.-H. (2002). Human fragile site FRA16B DNA excludes nucleosomes in the presence of distamycin. J. Biol. Chem. 277, 17315–17319. doi:10.1074/jbc.M200901200
Ijdo, J. W., Baldini, A., Ward, D. C., Reeders, S. T., and Wells, R. A. (1991). Origin of human chromosome 2: An ancestral telomere-telomere fusion. Proc. Natl. Acad. Sci. U. S. A. 88, 9051–9055. doi:10.1073/pnas.88.20.9051
Ijdo, J. W., Baldini, A., Wells, R. A., Ward, D. C., and Reeders, S. T. (1992). FRA2B is distinct from inverted telomere repeat arrays at 2q13. Genomics 12, 833–835. doi:10.1016/0888-7543(92)90319-n
Iliopoulos, D., Guler, G., Han, S.-Y., Druck, T., Ottey, M., McCorkell, K. A., et al. (2006). Roles of FHIT and WWOX fragile genes in cancer. Cancer Lett. 232, 27–36. doi:10.1016/j.canlet.2005.06.048
Irony-Tur Sinai, M., and Kerem, B. (2019). Genomic instability in fragile sites-still adding the pieces. Genes Chromosom. Cancer 58, 295–304. doi:10.1002/gcc.22715
Irony-Tur Sinai, M., Salamon, A., Stanleigh, N., Goldberg, T., Weiss, A., Wang, Y.-H., et al. (2019). AT-dinucleotide rich sequences drive fragile site formation. Nucleic Acids Res. 47, 9685–9695. doi:10.1093/nar/gkz689
Ishiura, H., and Tsuji, S. (2020). Advances in repeat expansion diseases and a new concept of repeat motif-phenotype correlation. Curr. Opin. Genet. Dev. 65, 176–185. doi:10.1016/j.gde.2020.05.029
Ishiura, H., Doi, K., Mitsui, J., Yoshimura, J., Matsukawa, M. K., Fujiyama, A., et al. (2018). Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat. Genet. 50, 581–590. doi:10.1038/s41588-018-0067-2
Ishiura, H., Shibata, S., Yoshimura, J., Suzuki, Y., Qu, W., Doi, K., et al. (2019). Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat. Genet. 51, 1222–1232. doi:10.1038/s41588-019-0458-z
Izakovic, V. (1984). Homozygosity for fragile site at 17p12 in a 28-year-old healthy man. Hum. Genet. 68, 340–341. doi:10.1007/BF00292597
Jacky, P. B., and Dill, F. J. (1983). Fragile X chromosome and chromosome condensation. Ann. Genet. 26, 171–173.
Jacky, P. B., Beek, B., and Sutherland, G. R. (1983). Fragile sites in chromosomes: Possible model for the study of spontaneous chromosome breakage. Science 220, 69–70. doi:10.1126/science.6828880
Jacky, P. B., Ahuja, Y. R., Anyane-Yeboa, K., Breg, W. R., Carpenter, N. J., Froster-Iskenius, U. G., et al. (1991). Guidelines for the preparation and analysis of the fragile X chromosome in lymphocytes. Am. J. Med. Genet. 38, 400–403. doi:10.1002/ajmg.1320380249
Jacob, R. A., Gretz, D. M., Taylor, P. C., James, S. J., Pogribny, I. P., Miller, B. J., et al. (1998). Moderate folate depletion increases plasma homocysteine and decreases lymphocyte DNA methylation in postmenopausal women. J. Nutr. 128, 1204–1212. doi:10.1093/jn/128.7.1204
Jacobs, P. A., Glover, T. W., Mayer, M., Fox, P., Gerrard, J. W., Dunn, H. G., et al. (1980). X-Linked mental retardation: A study of 7 families. Am. J. Med. Genet. 7, 471–489. doi:10.1002/ajmg.1320070408
Jacobsen, P., Hauge, M., Henningsen, K., Hobolth, N., Mikkelsen, M., and Philip, J. (1973). An (11;21) translocation in four generations with chromosome 11 abnormalities in the offspring. A clinical, cytogenetical, and gene marker study. Hum. Hered. 23, 568–585. doi:10.1159/000152624
Jacome, A., and Fernandez-Capetillo, O. (2011). Lac operator repeats generate a traceable fragile site in mammalian cells. EMBO Rep. 12, 1032–1038. doi:10.1038/embor.2011.158
Jalal, S. M., Lindor, N. M., Michels, V. V., Buckley, D. D., Hoppe, D. A., Sarkar, G., et al. (1993). Absence of chromosome fragility at 19q13.3 in patients with myotonic dystrophy. Am. J. Med. Genet. 46, 441–443. doi:10.1002/ajmg.1320460419
Jeanpierre, M., Turleau, C., Aurias, A., Prieur, M., Ledeist, F., Fischer, A., et al. (1993). An embryonic-like methylation pattern of classical satellite DNA is observed in ICF syndrome. Hum. Mol. Genet. 2, 731–735. doi:10.1093/hmg/2.6.731
Jenkins, E. C., Brown, W. T., Brooks, J., Duncan, C. J., Sanz, M. M., Silverman, W. P., et al. (1986a). Low frequencies of apparently fragile X chromosomes in normal control cultures: A possible explanation. Exp. Cell Biol. 54, 40–48. doi:10.1159/000163342
Jenkins, E. C., Brown, W. T., Wilson, M. G., Lin, M. S., Alfi, O. S., Wassman, E. R., et al. (1986b). The prenatal detection of the fragile X chromosome: Review of recent experience. Am. J. Med. Genet. 23, 297–311. doi:10.1002/ajmg.1320230123
Jenkins, E. C., Duncan, C. J., Sanz, M. M., Genovese, M., Gu, H., Schwartz-Richstein, C., et al. (1990). Progress toward an internal control system for fragile-X induction by 5-fluorodeoxyuridine in whole-blood cultures. Pathobiology 58, 236–240. doi:10.1159/000163591
Jensen, M. K., and Friis-Moller, A. (1967). Chromosome studies in pernicious anaemia. Acta Med. Scand. 181, 571–576. doi:10.1111/j.0954-6820.1967.tb07277.x
Ji, F., Liao, H., Pan, S., Ouyang, L., Jia, F., Fu, Z., et al. (2020). Genome-wide high-resolution mapping of mitotic DNA synthesis sites and common fragile sites by direct sequencing. Cell Res. 30, 1009–1023. doi:10.1038/s41422-020-0357-y
Jiang, Y., Lucas, I., Young, D. J., Davis, E. M., Karrison, T., Rest, J. S., et al. (2009). Common fragile sites are characterized by histone hypoacetylation. Hum. Mol. Genet. 18, 4501–4512. doi:10.1093/hmg/ddp410
Jiraanont, P., Kumar, M., Tang, H.-T., Espinal, G., Hagerman, P. J., Hagerman, R. J., et al. (2017). Size and methylation mosaicism in males with Fragile X syndrome. Expert Rev. Mol. diagn. 17, 1023–1032. doi:10.1080/14737159.2017.1377612
Johnson, R., and Strehler, B. L. (1972). Loss of genes coding for ribosomal RNA in ageing brain cells. Nature 240, 412–414. doi:10.1038/240412a0
Jones, K. W., and Corneo, G. (1971). Location of satellite and homogeneous DNA sequences on human chromosomes. Nat. New Biol. 233, 268–271. doi:10.1038/newbio233268a0
Jones, K. W., Prosser, J., Corneo, G., and Ginelli, E. (1973). The chromosomal location of human satellite DNA 3. Chromosoma 42, 445–451. doi:10.1007/BF00399411
Jones, K. W., Purdom, I. F., Prosser, J., and Corneo, G. (1974). The chromosomal localisation of human satellite DNA I. Chromosoma 49, 161–171. doi:10.1007/BF00348888
Jones, C., Slijepcevic, P., Marsh, S., Baker, E., Langdon, W. Y., Richards, R. I., et al. (1994). Physical linkage of the fragile site FRA11B and a Jacobsen syndrome chromosome deletion breakpoint in 11q23.3. Hum. Mol. Genet. 3, 2123–2130. doi:10.1093/hmg/3.12.2123
Jones, C., Penny, L., Mattina, T., Yu, S., Baker, E., Voullaire, L., et al. (1995). Association of a chromosome deletion syndrome with a fragile site within the proto-oncogene CBL2. Nature 376, 145–149. doi:10.1038/376145a0
Kähkönen, M., Tengström, C., Alitalo, T., Matilainen, R., Kaski, M., and Airaksinen, E. (1989). Population cytogenetics of folate-sensitive fragile sites. II. Autosomal rare fragile sites. Hum. Genet. 82, 3–8. doi:10.1007/BF00288261
Kähkönen, M. (1988). Population cytogenetics of folate-sensitive fragile sites. I. Common fragile sites. Hum. Genet. 80, 344–348. doi:10.1007/BF00273649
Kang, S., Ohshima, K., Shimizu, M., Amirhaeri, S., and Wells, R. D. (1995). Pausing of DNA synthesis in vitro at specific loci in CTG and CGG triplet repeats from human hereditary disease genes. J. Biol. Chem. 270, 27014–27021. doi:10.1074/jbc.270.45.27014
Kao-Shan, C. S., Fine, R. L., Whang-Peng, J., Lee, E. C., and Chabner, B. A. (1987). Increased fragile sites and sister chromatid exchanges in bone marrow and peripheral blood of young cigarette smokers. Cancer Res. 47, 6278–6282.
Karadeniz, N. N., Tunca, Y., and Imirzalioğlu, N. (2003). New heritable fragile site at 15q13 in both members of a nonconsanguineous couple. Am. J. Med. Genet. A 118A, 290–292. doi:10.1002/ajmg.a.10196
Karras, J. R., Schrock, M. S., Batar, B., and Huebner, K. (2016). Fragile genes that are frequently altered in cancer: Players not passengers. Cytogenet. Genome Res. 150, 208–216. doi:10.1159/000455753
Kasahara, Y., Nakai, Y., Miura, D., Yagi, K., Hirabayashi, K., and Makita, T. (1992). Mechanism of induction of micronuclei and chromosome aberrations in mouse bone marrow by multiple treatments of methotrexate. Mutat. Res. 280, 117–128. doi:10.1016/0165-1218(92)90007-m
Kaushal, S., and Freudenreich, C. H. (2019). The role of fork stalling and DNA structures in causing chromosome fragility. Genes Chromosom. Cancer 58, 270–283. doi:10.1002/gcc.22721
Kerem, B., Goitein, R., and Schaap, T. (1988). Cytological evidence of defective template in the fragile X chromosome. Chromosoma 97, 6–10. doi:10.1007/BF00331789
Kerner, B. (2014). Genetics of bipolar disorder. Appl. Clin. Genet. 7, 33–42. doi:10.2147/TACG.S39297
Kharrat, M., Hsairi, I., Doukali, H., Fendri-Kriaa, N., Kammoun, H., Ammar-Keskes, L., et al. (2017). Phenotypic variability in two infants sharing the same MECP2 mutation: Evidence of chromosomal rearrangements and high sister-chromatid exchange levels in Rett syndrome. Acta Neurol. belg. 117, 251–258. doi:10.1007/s13760-016-0667-5
Kim, C. H., Yoo, C. G., Han, S. K., Shim, Y. S., and Kim, Y. W. (1998). Genetic instability of microsatellite sequences in non-small cell lung cancers. Lung Cancer 21, 21–25. doi:10.1016/s0169-5002(98)00036-1
Kit, S. (1961). Equilibrium sedimentation in density gradients of DNA preparations from animal tissues. J. Mol. Biol. 3, 711–716. doi:10.1016/s0022-2836(61)80075-2
Knight, S. J., Flannery, A. V., Hirst, M. C., Campbell, L., Christodoulou, Z., Phelps, S. R., et al. (1993). Trinucleotide repeat amplification and hypermethylation of a CpG island in FRAXE mental retardation. Cell 74, 127–134. doi:10.1016/0092-8674(93)90300-f
Knight, S. J., Voelckel, M. A., Hirst, M. C., Flannery, A. V., Moncla, A., and Davies, K. E. (1994). Triplet repeat expansion at the FRAXE locus and X-linked mild mental handicap. Am. J. Hum. Genet. 55, 81–86.
Koch, C. M., Andrews, R. M., Flicek, P., Dillon, S. C., Karaöz, U., Clelland, G. K., et al. (2007). The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome Res. 17, 691–707. doi:10.1101/gr.5704207
Krawczun, M. S., Jenkins, E. C., Duncan, C. J., Stark-Houck, S. L., Kunaporn, S., Schwatz-Richstein, C., et al. (1991). Distribution of autosomal fragile sites in specimens cultured for prenatal fragile X diagnosis. Am. J. Med. Genet. 38, 456–463. doi:10.1002/ajmg.1320380264
Kremer, E. J., Pritchard, M., Lynch, M., Yu, S., Holman, K., Baker, E., et al. (1991). Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science 252, 1711–1714. doi:10.1126/science.1675488
Krummel, K. A., Roberts, L. R., Kawakami, M., Glover, T. W., and Smith, D. I. (2000). The characterization of the common fragile site FRA16D and its involvement in multiple myeloma translocations. Genomics 69, 37–46. doi:10.1006/geno.2000.6321
Krummel, K. A., Denison, S. R., Calhoun, E., Phillips, L. A., and Smith, D. I. (2002). The common fragile site FRA16D and its associated gene WWOX are highly conserved in the mouse at Fra8E1. Genes Chromosom. Cancer 34, 154–167. doi:10.1002/gcc.10047
Kumari, D., Somma, V., Nakamura, A. J., Bonner, W. M., D’Ambrosio, E., and Usdin, K. (2009). The role of DNA damage response pathways in chromosome fragility in Fragile X syndrome. Nucleic Acids Res. 37, 4385–4392. doi:10.1093/nar/gkp391
Kumari, D., Hayward, B., Nakamura, A. J., Bonner, W. M., and Usdin, K. (2015). Evidence for chromosome fragility at the frataxin locus in Friedreich ataxia. Mutat. Res. 781, 14–21. doi:10.1016/j.mrfmmm.2015.08.007
Kunkel, T. A., Silber, J. R., and Loeb, L. A. (1982). The mutagenic effect of deoxynucleotide substrate imbalances during DNA synthesis with mammalian DNA polymerases. Mutat. Res. 94, 413–419. doi:10.1016/0027-5107(82)90304-9
Kunz, B. A., and Kohalmi, S. E. (1991). Modulation of mutagenesis by deoxyribonucleotide levels. Annu. Rev. Genet. 25, 339–359. doi:10.1146/annurev.ge.25.120191.002011
Kunz, B. A., Taylor, G. R., and Haynes, R. H. (1986). Induction of intrachromosomal recombination in yeast by inhibition of thymidylate biosynthesis. Genetics 114, 375–392. doi:10.1093/genetics/114.2.375
Kunz, B. A. (1982). Genetic effects of deoxyribonucleotide pool imbalances. Environ. Mutagen. 4, 695–725. doi:10.1002/em.2860040609
Kunz, B. A. (1988). Mutagenesis and deoxyribonucleotide pool imbalance. Mutat. Res. 200, 133–147. doi:10.1016/0027-5107(88)90076-0
Kupke, K. G., Soreng, A. L., Müller, U., and Muller, U. (1991). Origin of the supernumerary X chromosome in a patient with fragile X and Klinefelter syndrome. Am. J. Med. Genet. 38, 440–444. doi:10.1002/ajmg.1320380260
Kuwano, A., Murano, I., and Kajii, T. (1990). Cell type-dependent difference in the distribution and frequency of excess thymidine-induced common fragile sites: T lymphocytes and skin fibroblasts. Hum. Genet. 84, 527–531. doi:10.1007/BF00210803
Kychygina, A., Dall’Osto, M., Allen, J. A. M., Cadoret, J.-C., Piras, V., Pickett, H. A., et al. (2021). Progerin impairs 3D genome organization and induces fragile telomeres by limiting the dNTP pools. Sci. Rep. 11, 13195. doi:10.1038/s41598-021-92631-z
Lacombe, L., Orlow, I., Reuter, V. E., Fair, W. R., Dalbagni, G., Zhang, Z. F., et al. (1996). Microsatellite instability and deletion analysis of chromosome 10 in human prostate cancer. Int. J. Cancer 69, 110–113. doi:10.1002/(SICI)1097-0215(19960422)69:2<110::AID-IJC7>3.0.CO;2-3
LaCroix, A. J., Stabley, D., Sahraoui, R., Adam, M. P., Mehaffey, M., Kernan, K., et al. (2019). GGC repeat expansion and exon 1 methylation of XYLT1 is a common pathogenic variant in baratela-scott syndrome. Am. J. Hum. Genet. 104, 35–44. doi:10.1016/j.ajhg.2018.11.005
Le Beau, M. M., Rassool, F. V., Neilly, M. E., Espinosa, R., Glover, T. W., Smith, D. I., et al. (1998). Replication of a common fragile site, FRA3B, occurs late in S phase and is delayed further upon induction: Implications for the mechanism of fragile site induction. Hum. Mol. Genet. 7, 755–761. doi:10.1093/hmg/7.4.755
Le Tallec, B., Dutrillaux, B., Lachages, A.-M., Millot, G. A., Brison, O., and Debatisse, M. (2011). Molecular profiling of common fragile sites in human fibroblasts. Nat. Struct. Mol. Biol. 18, 1421–1423. doi:10.1038/nsmb.2155
Le Tallec, B., Millot, G. A., Blin, M. E., Brison, O., Dutrillaux, B., and Debatisse, M. (2013). Common fragile site profiling in epithelial and erythroid cells reveals that most recurrent cancer deletions lie in fragile sites hosting large genes. Cell Rep. 4, 420–428. doi:10.1016/j.celrep.2013.07.003
Lee, C. S., Choo, A., Dayan, S., Richards, R. I., and O’Keefe, L. V. (2021). Molecular biology of the WWOX gene that spans chromosomal fragile site FRA16D. Cells 10, 1637. doi:10.3390/cells10071637
Leeds, J. M., Slabaugh, M. B., and Mathews, C. K. (1985). DNA precursor pools and ribonucleotide reductase activity: Distribution between the nucleus and cytoplasm of mammalian cells. Mol. Cell. Biol. 5, 3443–3450. doi:10.1128/mcb.5.12.3443
Lesca, G., Biancalana, V., Brunel, M.-J., Quack, B., Calender, A., and Lespinasse, J. (2003). Clinical, cytogenetic, and molecular description of a FRAXE French family. Psychiatr. Genet. 13, 43–46. doi:10.1097/00041444-200303000-00007
Letessier, A., Millot, G. A., Koundrioukoff, S., Lachagès, A.-M., Vogt, N., Hansen, R. S., et al. (2011). Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 470, 120–123. doi:10.1038/nature09745
Li, Z., Bailey, A. D., Buchowski, J., and Weiner, A. M. (1998). A tandem array of minimal U1 small nuclear RNA genes is sufficient to generate a new adenovirus type 12-inducible chromosome fragile site. J. Virol. 72, 4205–4211. doi:10.1128/JVI.72.5.4205-4211.1998
Liehr, T. (2021). Molecular cytogenetics in the era of chromosomics and cytogenomic approaches. Front. Genet. 12, 720507. doi:10.3389/fgene.2021.720507
Lindahl, T., and Wood, R. D. (1999). Quality control by DNA repair. Science 286, 1897–1905. doi:10.1126/science.286.5446.1897
Lohi, H., Young, E. J., Fitzmaurice, S. N., Rusbridge, C., Chan, E. M., Vervoort, M., et al. (2005). Expanded repeat in canine epilepsy. Science 307, 81. doi:10.1126/science.1102832
Lokanga, R. A., Entezam, A., Kumari, D., Yudkin, D., Qin, M., Smith, C. B., et al. (2013). Somatic expansion in mouse and human carriers of fragile X premutation alleles. Hum. Mutat. 34, 157–166. doi:10.1002/humu.22177
Lönn, U., Lönn, S., and Lonn, U. (1988). Step-wise progression of mammalian 10-kb DNA replication intermediates to mature chromatin. Eur. J. Biochem. 178, 47–51. doi:10.1111/j.1432-1033.1988.tb14427.x
Lopez Corrales, N., and Arruga, V. (1996). Induction of chromosomal fragile sites in goats: A preliminary study. Genet. Sel. Evol. 28, 129. doi:10.1186/1297-9686-28-2-129
Lozano, R., Rosero, C. A., and Hagerman, R. J. (2014). Fragile X spectrum disorders. Intractable Rare Dis. Res. 3, 134–146. doi:10.5582/irdr.2014.01022
Lüdecke, H. J., Johnson, C., Wagner, M. J., Wells, D. E., Turleau, C., Tommerup, N., et al. (1991). Molecular definition of the shortest region of deletion overlap in the Langer-Giedion syndrome. Am. J. Hum. Genet. 49, 1197–1206.
Lugenbeel, K. A., Peier, A. M., Carson, N. L., Chudley, A. E., and Nelson, D. L. (1995). Intragenic loss of function mutations demonstrate the primary role of FMR1 in fragile X syndrome. Nat. Genet. 10, 483–485. doi:10.1038/ng0895-483
Lukusa, T., and Fryns, J. P. (2008). Human chromosome fragility. Biochim. Biophys. Acta 1779, 3–16. doi:10.1016/j.bbagrm.2007.10.005
Lukusa, T., Meulepas, E., Fryns, J. P., Van den Berghe, H., and Cassiman, J. J. (1991). Spontaneous” FRA16B is a hot spot for sister chromatid exchanges. Hum. Genet. 87, 583–586. doi:10.1007/BF00209017
Luo, S., Robinson, J. C., Reiss, A. L., and Migeon, B. R. (1993). DNA methylation of the fragile X locus in somatic and germ cells during fetal development: Relevance to the fragile X syndrome and X inactivation. Somat. Cell Mol. Genet. 19, 393–404. doi:10.1007/BF01232750
Lyle, R., Wright, T. J., Clark, L. N., and Hewitt, J. E. (1995). The FSHD-associated repeat, D4Z4, is a member of a dispersed family of homeobox-containing repeats, subsets of which are clustered on the short arms of the acrocentric chromosomes. Genomics 28, 389–397. doi:10.1006/geno.1995.1166
Maass, P. G., Barutcu, A. R., and Rinn, J. L. (2019). Interchromosomal interactions: A genomic love story of kissing chromosomes. J. Cell Biol. 218, 27–38. doi:10.1083/jcb.201806052
Madireddy, A., Kosiyatrakul, S. T., Boisvert, R. A., Herrera-Moyano, E., García-Rubio, M. L., Gerhardt, J., et al. (2016). FANCD2 facilitates replication through common fragile sites. Mol. Cell 64, 388–404. doi:10.1016/j.molcel.2016.09.017
Magenis, R. E., Hecht, F., and Lovrien, E. W. (1970). Heritable fragile site on chromosome 16: Probable localization of haptoglobin locus in man. Science 170, 85–87. doi:10.1126/science.170.3953.85
Maltby, E. L., and Higgins, S. (1987). Folate sensitive site at 10q23 and its expression as a deletion. J. Med. Genet. 24, 299. doi:10.1136/jmg.24.5.299
Malter, H. E., Iber, J. C., Willemsen, R., de Graaff, E., Tarleton, J. C., Leisti, J., et al. (1997). Characterization of the full fragile X syndrome mutation in fetal gametes. Nat. Genet. 15, 165–169. doi:10.1038/ng0297-165
Mangelsdorf, M., Ried, K., Woollatt, E., Dayan, S., Eyre, H., Finnis, M., et al. (2000). Chromosomal fragile site FRA16D and DNA instability in cancer. Cancer Res. 60, 1683–1689.
Mannava, S., Moparthy, K. C., Wheeler, L. J., Natarajan, V., Zucker, S. N., Fink, E. E., et al. (2013). Depletion of deoxyribonucleotide pools is an endogenous source of DNA damage in cells undergoing oncogene-induced senescence. Am. J. Pathol. 182, 142–151. doi:10.1016/j.ajpath.2012.09.011
Maraschio, P., Zuffardi, O., Dalla Fior, T., and Tiepolo, L. (1988). Immunodeficiency, centromeric heterochromatin instability of chromosomes 1, 9, and 16, and facial anomalies: The ICF syndrome. J. Med. Genet. 25, 173–180. doi:10.1136/jmg.25.3.173
Martin, J. P., and Bell, J. (1943). A pedigree of mental defect showing sex-linkage. J. Neurol. Psychiatry 6, 154–157. doi:10.1136/jnnp.6.3-4.154
Martin, A. (2004). Can’t any body count?: Counting as an epistemic theme in the history of human chromosomes. Soc. Stud. Sci. 34, 923–948. doi:10.1177/0306312704046843
Martínez, R., Bonilla-Henao, V., Jiménez, A., Lucas, M., Vega, C., Ramos, I., et al. (2005). Skewed X inactivation of the normal allele in fully mutated female carriers determines the levels of FMRP in blood and the fragile X phenotype. Mol. Diagn. 9, 157–162. doi:10.1007/BF03260084
Mathews, C. K., and Ji, J. (1992). DNA precursor asymmetries, replication fidelity, and variable genome evolution. Bioessays 14, 295–301. doi:10.1002/bies.950140502
Mathews, C. K. (1975). Giant pools of DNA precursors in sea urchin eggs. Exp. Cell Res. 92, 47–56. doi:10.1016/0014-4827(75)90635-7
Mathews, C. K. (2006). DNA precursor metabolism and genomic stability. FASEB J. 20, 1300–1314. doi:10.1096/fj.06-5730rev
Mathews, C. K. (2014). Deoxyribonucleotides as genetic and metabolic regulators. FASEB J. 28, 3832–3840. doi:10.1096/fj.14-251249
Mathews, C. K. (2015). Deoxyribonucleotide metabolism, mutagenesis and cancer. Nat. Rev. Cancer 15, 528–539. doi:10.1038/nrc3981
Mathews, C. K. (2017). Oxidized deoxyribonucleotides, mutagenesis, and cancer. FASEB J. 31, 11–13. doi:10.1096/fj.201601100
Mathews, C. K. (2019). Deoxyribonucleotide salvage falls short in whole animals. J. Biol. Chem. 294, 15898–15899. doi:10.1074/jbc.H119.011335
Mattano, S. S., Palella, T. D., and Mitchell, B. S. (1990). Mutations induced at the hypoxanthine-guanine phosphoribosyltransferase locus of human T-lymphoblasts by perturbations of purine deoxyribonucleoside triphosphate pools. Cancer Res. 50, 4566–4571.
Mattei, M. G., Mattei, J. F., Vidal, I., and Giraud, F. (1981). Expression in lymphocyte and fibroblast culture of the fragile X chromosome: A new technical approach. Hum. Genet. 59, 166–169. doi:10.1007/BF00293069
Matzner, I., Savelyeva, L., and Schwab, M. (2003). Preferential integration of a transfected marker gene into spontaneously expressed fragile sites of a breast cancer cell line. Cancer Lett. 189, 207–219. doi:10.1016/s0304-3835(02)00504-9
Mavrou, A., Syrrou, M., Tsenghi, C., and Metaxotou, C. (1991). Autosomal folate sensitive fragile sites in normal and mentally retarded individuals in Greece. Am. J. Med. Genet. 38, 437–439. doi:10.1002/ajmg.1320380259
McKenzie, W. H., and Lubs, H. A. (1973). An analysis of the technical variables in the production of C bands. Chromosoma 41, 175–182. doi:10.1007/BF00319694
McKinley, M. J., Kearney, L. U., Nicolaides, K. H., Gosden, C. M., Webb, T. P., and Fryns, J. P. (1988). Prenatal diagnosis of fragile X syndrome by placental (chorionic villi) biopsy culture. Am. J. Med. Genet. 30, 355–368. doi:10.1002/ajmg.1320300136
McStay, B. (2016). Nucleolar organizer regions: Genomic “dark matter” requiring illumination. Genes Dev. 30, 1598–1610. doi:10.1101/gad.283838.116
Melnyk, S., Pogribna, M., Miller, B. J., Basnakian, A. G., Pogribny, I. P., and James, S. J. (1999). Uracil misincorporation, DNA strand breaks, and gene amplification are associated with tumorigenic cell transformation in folate deficient/repleted Chinese hamster ovary cells. Cancer Lett. 146, 35–44. doi:10.1016/s0304-3835(99)00213-x
Merrikh, H., Machón, C., Grainger, W. H., Grossman, A. D., and Soultanas, P. (2011). Co-directional replication-transcription conflicts lead to replication restart. Nature 470, 554–557. doi:10.1038/nature09758
Metsu, S., Rainger, J. K., Debacker, K., Bernhard, B., Rooms, L., Grafodatskaya, D., et al. (2014a). A CGG-repeat expansion mutation in ZNF713 causes FRA7A: Association with autistic spectrum disorder in two families. Hum. Mutat. 35, 1295–1300. doi:10.1002/humu.22683
Metsu, S., Rooms, L., Rainger, J., Taylor, M. S., Bengani, H., Wilson, D. I., et al. (2014b). FRA2A is a CGG repeat expansion associated with silencing of AFF3. PLoS Genet. 10, e1004242. doi:10.1371/journal.pgen.1004242
Meuth, M. (1984). The genetic consequences of nucleotide precursor pool imbalance in mammalian cells. Mutat. Res. 126, 107–112. doi:10.1016/0027-5107(84)90051-4
Michaelis, R. C., Velagaleti, G. V., Jones, C., Pivnick, E. K., Phelan, M. C., Boyd, E., et al. (1998). Most Jacobsen syndrome deletion breakpoints occur distal to FRA11B. Am. J. Med. Genet. 76, 222–228. doi:10.1002/(sici)1096-8628(19980319)76:3<222::aid-ajmg5>3.0.co;2-s
Migliore, L., Testa, A., Scarpato, R., Pavese, N., Petrozzi, L., Bonuccelli, U., et al. (1997). Spontaneous and induced aneuploidy in peripheral blood lymphocytes of patients with Alzheimer’s disease. Hum. Genet. 101, 299–305. doi:10.1007/s004390050632
Migliore, L., Coppedè, F., Fenech, M., and Thomas, P. (2011). Association of micronucleus frequency with neurodegenerative diseases. Mutagenesis 26, 85–92. doi:10.1093/mutage/geq067
Miles, J. H. (2011). Autism spectrum disorders-a genetics review. Genet. Med. 13, 278–294. doi:10.1097/GIM.0b013e3181ff67ba
Milunsky, A., Huang, X., Amos, J. A., Herskowitz, J., Farrer, L. A., and Wyandt, H. E. (1993). 46, XY/47, XYY male with the fragile X syndrome: Cytogenetic and molecular studies. Am. J. Med. Genet. 45, 589–593. doi:10.1002/ajmg.1320450514
Mimori, K., Druck, T., Inoue, H., Alder, H., Berk, L., Mori, M., et al. (1999). Cancer-specific chromosome alterations in the constitutive fragile region FRA3B. Proc. Natl. Acad. Sci. U. S. A. 96, 7456–7461. doi:10.1073/pnas.96.13.7456
Minocherhomji, S., and Hickson, I. D. (2014). Structure-specific endonucleases: Guardians of fragile site stability. Trends Cell Biol. 24, 321–327. doi:10.1016/j.tcb.2013.11.007
Minocherhomji, S., Ying, S., Bjerregaard, V. A., Bursomanno, S., Aleliunaite, A., Wu, W., et al. (2015). Replication stress activates DNA repair synthesis in mitosis. Nature 528, 286–290. doi:10.1038/nature16139
Miotto, B., Ji, Z., and Struhl, K. (2016). Selectivity of ORC binding sites and the relation to replication timing, fragile sites, and deletions in cancers. Proc. Natl. Acad. Sci. U. S. A. 113, E4810–E4819. doi:10.1073/pnas.1609060113
Miró, R., Clemente, I. C., Fuster, C., and Egozcue, J. (1987). Fragile sites, chromosome evolution, and human neoplasia. Hum. Genet. 75, 345–349. doi:10.1007/BF00284105
Mishmar, D., Rahat, A., Scherer, S. W., Nyakatura, G., Hinzmann, B., Kohwi, Y., et al. (1998). Molecular characterization of a common fragile site (FRA7H) on human chromosome 7 by the cloning of a simian virus 40 integration site. Proc. Natl. Acad. Sci. U. S. A. 95, 8141–8146. doi:10.1073/pnas.95.14.8141
Mishmar, D., Mandel-Gutfreund, Y., Margalit, H., Rahat, A., and Kerem, B. (1999). Common fragile sites: G-Band characteristics within an R-band. Am. J. Hum. Genet. 64, 908–910. doi:10.1086/302299
Mondal, K., Ramachandran, D., Patel, V. C., Hagen, K. R., Bose, P., Cutler, D. J., et al. (2012). Excess variants in AFF2 detected by massively parallel sequencing of males with autism spectrum disorder. Hum. Mol. Genet. 21, 4356–4364. doi:10.1093/hmg/dds267
Moore, S. J., Strain, L., Cole, G. F., Miedzybrodzka, Z., Kelly, K. F., and Dean, J. C. (1999). Fragile X syndrome with FMR1 and FMR2 deletion. J. Med. Genet. 36, 565–566.
Morel, C. F., Duncan, A. M. V., and Désilets, V. (2005). A fragile site at 10q23 (FRA10A) in a phenytoin-exposed fetus: A case report and review of the literature. Prenat. Diagn. 25, 318–321. doi:10.1002/pd.1134
Mrasek, K., Schoder, C., Teichmann, A.-C., Behr, K., Franze, B., Wilhelm, K., et al. (2010). Global screening and extended nomenclature for 230 aphidicolin-inducible fragile sites, including 61 yet unreported ones. Int. J. Oncol. 36, 929–940. doi:10.3892/ijo_00000572
Müller, B., Feichtinger, W., Bonaïti-Pellié, C., and Schmid, M. (1992). Fragile site (16) (q22). III. Segregation analysis. Hum. Genet. 89, 612–614. doi:10.1007/BF00221948
Munn, C. A., Wenger, S. L., and Steele, M. W. (1991). Assessment of X bends in patients with atypical X chromosome phenotypes. Ann. Genet. 34, 120–124.
Murfuni, I., De Santis, A., Federico, M., Bignami, M., Pichierri, P., and Franchitto, A. (2012). Perturbed replication induced genome wide or at common fragile sites is differently managed in the absence of WRN. Carcinogenesis 33, 1655–1663. doi:10.1093/carcin/bgs206
Murray, A. (2000). Premature ovarian failure and the FMR1 gene. Semin. Reprod. Med. 18, 59–66. doi:10.1055/s-2000-13476
Musio, A., and Mariani, T. (1999). Distribution of interstitial telomere-related sequences in the human genome and their relationship with fragile sites. J. Environ. Pathol. Toxicol. Oncol. 18, 11–15.
Musio, A., and Sbrana, I. (1997). Aphidicolin-sensitive specific common fragile sites: A biomarker of exposure to pesticides. Environ. Mol. Mutagen. 29, 250–255. doi:10.1002/(sici)1098-2280(1997)29:3<250::aid-em4>3.0.co;2-g
Musio, A., Rainaldi, G., and Sbrana, I. (1996). Spontaneous and aphidicolin-sensitive fragile site 3cen co-localizes with the (TTAGGG)n telomeric sequence in Chinese hamster cells. Cytogenet. Cell Genet. 75, 159–163. doi:10.1159/000134469
Naim, V., Wilhelm, T., Debatisse, M., and Rosselli, F. (2013). ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 15, 1008–1015. doi:10.1038/ncb2793
Nancarrow, J. K., Kremer, E., Holman, K., Eyre, H., Doggett, N. A., Le Paslier, D., et al. (1994). Implications of FRA16A structure for the mechanism of chromosomal fragile site Genesis. Science 264, 1938–1941. doi:10.1126/science.8009225
Neu, R. L., Kousseff, B. G., Madan, S., Essig, Y. P., Miller, K., and Tedesco, T. A. (1988). Monosomy, trisomy, fragile sites, and rearrangements of chromosome no. 1 in a mentally retarded male with multiple congenital anomalies. Clin. Genet. 33, 73–77. doi:10.1111/j.1399-0004.1988.tb03413.x
Nichol Edamura, K., and Pearson, C. (2005). DNA methylation and replication: Implications for the “deletion hotspot” region of FMR1. Hum. Genet. 118, 301–304. doi:10.1007/s00439-005-0037-5
Nielsen, K. B. (1986). Sex chromosome aneuploidy in fragile X carriers. Am. J. Med. Genet. 23, 537–544. doi:10.1002/ajmg.1320230146
Nurk, S., Koren, S., Rhie, A., Rautiainen, M., Bzikadze, A. V., Mikheenko, A., et al. (2022). The complete sequence of a human genome. Science 376, 44–53. doi:10.1126/science.abj6987
Nussbaum, R. L., Airhart, S. D., and Ledbetter, D. H. (1983). Expression of the fragile (X) chromosome in an interspecific somatic cell hybrid. Hum. Genet. 64, 148–150. doi:10.1007/BF00327113
Nussbaum, R. L., Airhart, S. D., and Ledbetter, D. H. (1986). Recombination and amplification of pyrimidine-rich sequences may be responsible for initiation and progression of the Xq27 fragile site: An hypothesis. Am. J. Med. Genet. 23, 715–721. doi:10.1002/ajmg.1320230162
O’Neill, F. J., and Miles, C. P. (1969). Chromosome changes in human cells induced by herpes simplex, types 1 and 2. Nature 223, 851–852. doi:10.1038/223851a0
Oberlé, I., Rousseau, F., Heitz, D., Kretz, C., Devys, D., Hanauer, A., et al. (1991). Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 252, 1097–1102. doi:10.1126/science.252.5009.1097
Olavesen, M. G., Davies, A. F., Broxholme, S. J., Wixon, J. L., Senger, G., Nizetic, D., et al. (1995). An integrated map of human chromosome 6p23. Genome Res. 5, 342–358. doi:10.1101/gr.5.4.342
Orye, E. (1974). Satellite association and variations in length of the nucleolar constriction of normal and variant human G chromosomes. Humangenetik 22, 299–309. doi:10.1007/BF00295489
Ozeri-Galai, E., Schwartz, M., Rahat, A., and Kerem, B. (2008). Interplay between ATM and ATR in the regulation of common fragile site stability. Oncogene 27, 2109–2117. doi:10.1038/sj.onc.1210849
Ozeri-Galai, E., Bester, A. C., and Kerem, B. (2012). The complex basis underlying common fragile site instability in cancer. Trends Genet. 28, 295–302. doi:10.1016/j.tig.2012.02.006
Ozisik, Y. Y., Meloni, A. M., Stone, J. F., Sandberg, A. A., and Surti, U. (1994). Spontaneous expression of the chromosome fragile site at 10q23 in leiomyoma. Cancer Genet. cytogenet. 74, 73–75. doi:10.1016/0165-4608(94)90034-5
Paige, A. J., Taylor, K. J., Stewart, A., Sgouros, J. G., Gabra, H., Sellar, G. C., et al. (2000). A 700-kb physical map of a region of 16q23.2 homozygously deleted in multiple cancers and spanning the common fragile site FRA16D. Cancer Res. 60, 1690–1697.
Palakodeti, A., Han, Y., Jiang, Y., and Le Beau, M. M. (2004). The role of late/slow replication of the FRA16D in common fragile site induction. Genes Chromosom. Cancer 39, 71–76. doi:10.1002/gcc.10290
Palakodeti, A., Lucas, I., Jiang, Y., Young, D. J., Fernald, A. A., Karrison, T., et al. (2010). Impaired replication dynamics at the FRA3B common fragile site. Hum. Mol. Genet. 19, 99–110. doi:10.1093/hmg/ddp470
Palumbo, E., Matricardi, L., Tosoni, E., Bensimon, A., and Russo, A. (2010). Replication dynamics at common fragile site FRA6E. Chromosoma 119, 575–587. doi:10.1007/s00412-010-0279-4
Pandelache, A., Francis, D., Oertel, R., Dickson, R., Sachdev, R., Ling, L., et al. (2021). Detection of cryptic fragile X full mutation alleles by southern blot in a female and her foetal DNA via chorionic villus sampling, complicated by mosaicism for 45, X0/46, XX/47, XXX. Genes (Basel) 12, 798. doi:10.3390/genes12060798
Parmentier, S., Meinel, J., Oelschlaegel, U., Mohr, B., Ehninger, G., Schaich, M., et al. (2012). Severe pernicious anemia with distinct cytogenetic and flow cytometric aberrations mimicking myelodysplastic syndrome. Ann. Hematol. 91, 1979–1981. doi:10.1007/s00277-012-1488-0
Parrish, J. E., Oostra, B. A., Verkerk, A. J., Richards, C. S., Reynolds, J., Spikes, A. S., et al. (1994). Isolation of a GCC repeat showing expansion in FRAXF, a fragile site distal to FRAXA and FRAXE. Nat. Genet. 8, 229–235. doi:10.1038/ng1194-229
Peat, D. S., and Stanley, M. A. (1986). Chromosome damage induced by herpes simplex virus type 1 in early infection. J. Gen. Virol. 67 (1), 2273–2277. doi:10.1099/0022-1317-67-10-2273
Pelliccia, F., and Rocchi, A. (1986). DAPI-inducible common fragile sites. Cytogenet. Cell Genet. 42, 174–176. doi:10.1159/000132272
Pelliccia, F., Bosco, N., Curatolo, A., and Rocchi, A. (2008). Replication timing of two human common fragile sites: FRA1H and FRA2G. Cytogenet. Genome Res. 121, 196–200. doi:10.1159/000138885
Pembrey, M. E., Winter, R. M., and Davies, K. E. (1985). A premutation that generates a defect at crossing over explains the inheritance of fragile X mental retardation. Am. J. Med. Genet. 21, 709–717. doi:10.1002/ajmg.1320210413
Perroni, L., Grasso, M., Argusti, A., Lo Nigro, C., Croci, G. F., Zelante, L., et al. (1996). Molecular and cytogenetic analysis of the fragile X syndrome in a series of 453 mentally retarded subjects: A study of 87 families. Am. J. Med. Genet. 64, 176–180. doi:10.1002/(SICI)1096-8628(19960712)64:1<176::AID-AJMG30>3.0.CO;2-I
Petit, P., Fryns, J. P., van den Berghe, H., and Hecht, F. (1986). Population cytogenetics of autosomal fragile sites. Clin. Genet. 29, 96–100. doi:10.1111/j.1399-0004.1986.tb01229.x
Petit, P. (1997). Interstitial telomere DNA sequences are not detectable at breaking sites of classical heritable fragile sites. Hum. Genet. 99, 424.
Pieretti, M., Zhang, F. P., Fu, Y. H., Warren, S. T., Oostra, B. A., Caskey, C. T., et al. (1991). Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66, 817–822. doi:10.1016/0092-8674(91)90125-i
Pires, R. M., Reichert, R. H., and Kasahara, S. (1998). Cytogenetics of three breeds of river buffalo (Bubalus bubalis L.), with evidence of a fragile site on the X chromosome. Theriogenology 49, 529–538. doi:10.1016/s0093-691x(98)00004-1
Pirzio, L. M., Pichierri, P., Bignami, M., and Franchitto, A. (2008). Werner syndrome helicase activity is essential in maintaining fragile site stability. J. Cell Biol. 180, 305–314. doi:10.1083/jcb.200705126
Plaja, A., Miró, R., Fuster, C., Perez, C., Sarret, E., Esteve, P., et al. (2001). Bends in human mitotic metaphase chromosomes revisited: 15q11-13 is the most frequent non-random autosomal bend in blood cultures. Am. J. Med. Genet. 101, 106–113. doi:10.1002/1096-8628(20010615)101:2<106::aid-ajmg1339>3.0.co;2-z
Plaja, A., Miro, R., Lloveras, E., Sarret, E., Fernandez, B., and Egozcue, J. (2004). Intranuclear arrangement of human chromosome 12 is reflected in metaphase chromosomes as non-random bending. Ann. Genet. 47, 429–432. doi:10.1016/j.anngen.2004.07.002
Plohl, M., Meštrović, N., and Mravinac, B. (2014). Centromere identity from the DNA point of view. Chromosoma 123, 313–325. doi:10.1007/s00412-014-0462-0
Podugolnikova, O. A., and Korostelev, A. P. (1980). The quantitative analysis of polymorphism on human chromosomes 1, 9, 16, and Y. IV. Heterogeneity of a normal population. Hum. Genet. 54, 163–169. doi:10.1007/BF00278966
Pomponi, M. G., and Neri, G. (1994). Butyrate and acetyl-carnitine inhibit the cytogenetic expression of the fragile X in vitro. Am. J. Med. Genet. 51, 447–450. doi:10.1002/ajmg.1320510428
Poon, S. L., McPherson, J. R., Tan, P., Teh, B. T., and Rozen, S. G. (2014). Mutation signatures of carcinogen exposure: Genome-wide detection and new opportunities for cancer prevention. Genome Med. 6, 24. doi:10.1186/gm541
Popescu, N. C., and DiPaolo, J. A. (1989). Preferential sites for viral integration on mammalian genome. Cancer Genet. cytogenet. 42, 157–171. doi:10.1016/0165-4608(89)90084-8
Popescu, N. C. (1994). Chromosome fragility and instability in human cancer. Crit. Rev. Oncog. 5, 121–140. doi:10.1615/critrevoncog.v5.i2-3.20
Poulsen, B., and Rønne, M. (1991). High-resolution R-banding and localization of fragile sites in Oryctolagus cuniculus. Genet. Sel. Evol. 23, S183. doi:10.1186/1297-9686-23-S1-S183
Prada, C. F., and Laissue, P. (2014). A high resolution map of mammalian X chromosome fragile regions assessed by large-scale comparative genomics. Mamm. Genome 25, 618–635. doi:10.1007/s00335-014-9537-8
Prado, F., and Aguilera, A. (2005). Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J. 24, 1267–1276. doi:10.1038/sj.emboj.7600602
Probst, F. J., Roeder, E. R., Enciso, V. B., Ou, Z., Cooper, M. L., Eng, P., et al. (2007). Chromosomal microarray analysis (CMA) detects a large X chromosome deletion including FMR1, FMR2, and IDS in a female patient with mental retardation. Am. J. Med. Genet. A 143A, 1358–1365. doi:10.1002/ajmg.a.31781
Prosser, J., Frommer, M., Paul, C., and Vincent, P. C. (1986). Sequence relationships of three human satellite DNAs. J. Mol. Biol. 187, 145–155. doi:10.1016/0022-2836(86)90224-x
Ragland, R. L., Glynn, M. W., Arlt, M. F., and Glover, T. W. (2008). Stably transfected common fragile site sequences exhibit instability at ectopic sites. Genes Chromosom. Cancer 47, 860–872. doi:10.1002/gcc.20591
Reddy, K., Tam, M., Bowater, R. P., Barber, M., Tomlinson, M., Nichol Edamura, K., et al. (2011). Determinants of R-loop formation at convergent bidirectionally transcribed trinucleotide repeats. Nucleic Acids Res. 39, 1749–1762. doi:10.1093/nar/gkq935
Reddy, K., Schmidt, M. H., Geist, J. M., Thakkar, N. P., Panigrahi, G. B., Wang, Y.-H. H., et al. (2014). Processing of double-R-loops in (CAG)·(CTG) and C9orf72 (GGGGCC)·(GGCCCC) repeats causes instability. Nucleic Acids Res. 42, 10473–10487. doi:10.1093/nar/gku658
Reidy, J. A. (1987). Folate- and deoxyuridine-sensitive chromatid breakage may result from DNA repair during G2. Mutat. Res. 192, 217–219. doi:10.1016/0165-7992(87)90059-5
Rhind, N. (2006). DNA replication timing: Random thoughts about origin firing. Nat. Cell Biol. 8, 1313–1316. doi:10.1038/ncb1206-1313
Richards, B. W., Sylvester, P. E., and Brooker, C. (1981). Fragile X-linked mental retardation: The martin-bell syndrome. J. Ment. Defic. Res. 25 (4), 253–256. doi:10.1111/j.1365-2788.1981.tb00115.x
Richards, R. I. (2001). Fragile and unstable chromosomes in cancer: Causes and consequences. Trends Genet. 17, 339–345. doi:10.1016/s0168-9525(01)02303-4
Riggs, P. K., and Rønne, M. (2009). Fragile sites in domestic animal chromosomes: Molecular insights and challenges. Cytogenet. Genome Res. 126, 97–109. doi:10.1159/000245910
Ritchie, R. J., Knight, S. J., Hirst, M. C., Grewal, P. K., Bobrow, M., Cross, G. S., et al. (1994). The cloning of FRAXF: Trinucleotide repeat expansion and methylation at a third fragile site in distal xqter. Hum. Mol. Genet. 3, 2115–2121. doi:10.1093/hmg/3.12.2115
Rivera, C. M., and Ren, B. (2013). Mapping human epigenomes. Cell 155, 39–55. doi:10.1016/j.cell.2013.09.011
Robinson, T. J., and Elder, F. F. (1987). Multiple common fragile sites are expressed in the genome of the laboratory rat. Chromosoma 96, 45–49. doi:10.1007/BF00285882
Romain, D. R., Columbano-Green, L. M., Smythe, R. H., Parfitt, R. G., Gebbie, O. B., and Chapman, C. J. (1986). Studies on three rare fragile sites. 2q13, 12q13, and 17p12 segregating in one family. Hum. Genet. 73, 164–170. doi:10.1007/BF00291608
Rosin, M. P., and Ochs, H. D. (1986). In vivo chromosomal instability in ataxia-telangiectasia homozygotes and heterozygotes. Hum. Genet. 74, 335–340. doi:10.1007/BF00280482
Rousseau, F., Heitz, D., Biancalana, V., Blumenfeld, S., Kretz, C., Boue, J., et al. (1991a). Direct diagnosis by DNA analysis of the fragile X syndrome of mental retardation. N. Engl. J. Med. 325, 1673–1681. doi:10.1056/NEJM199112123252401
Rousseau, F., Heitz, D., Oberlé, I., and Mandel, J. L. (1991b). Selection in blood cells from female carriers of the fragile X syndrome: Inverse correlation between age and proportion of active X chromosomes carrying the full mutation. J. Med. Genet. 28, 830–836. doi:10.1136/jmg.28.12.830
Rousseau, F., Robb, L. J., Rouillard, P., and Der Kaloustian, V. M. (1994). No mental retardation in a man with 40% abnormal methylation at the FMR-1 locus and transmission of sperm cell mutations as premutations. Hum. Mol. Genet. 3, 927–930. doi:10.1093/hmg/3.6.927
Rozier, L., El-Achkar, E., Apiou, F., and Debatisse, M. (2004). Characterization of a conserved aphidicolin-sensitive common fragile site at human 4q22 and mouse 6C1: Possible association with an inherited disease and cancer. Oncogene 23, 6872–6880. doi:10.1038/sj.onc.1207809
Ruiz-Herrera, A., Ponsà, M., García, F., Egozcue, J., and García, M. (2002). Fragile sites in human and Macaca fascicularis chromosomes are breakpoints in chromosome evolution. Chromosome Res. 10, 33–44. doi:10.1023/a:1014261909613
Ruiz-Herrera, A., García, F., Giulotto, E., Attolini, C., Egozcue, J., Ponsà, M., et al. (2005). Evolutionary breakpoints are co-localized with fragile sites and intrachromosomal telomeric sequences in primates. Cytogenet. Genome Res. 108, 234–247. doi:10.1159/000080822
Ruiz-Herrera, A., Castresana, J., and Robinson, T. J. (2006). Is mammalian chromosomal evolution driven by regions of genome fragility? Genome Biol. 7, R115. doi:10.1186/gb-2006-7-12-r115
Salim, D., and Gerton, J. L. (2019). Ribosomal DNA instability and genome adaptability. Chromosome Res. 27, 73–87. doi:10.1007/s10577-018-9599-7
Salim, D., Bradford, W. D., Freeland, A., Cady, G., Wang, J., Pruitt, S. C., et al. (2017). DNA replication stress restricts ribosomal DNA copy number. PLoS Genet. 13, e1007006. doi:10.1371/journal.pgen.1007006
Samadashwily, G. M., Raca, G., and Mirkin, S. M. (1997). Trinucleotide repeats affect DNA replication in vivo. Nat. Genet. 17, 298–304. doi:10.1038/ng1197-298
Samadder, P., Evans, J. A., and Chudley, A. E. (1993). Segregation analysis of rare autosomal folate sensitive fragile sites. Am. J. Med. Genet. 46, 165–171. doi:10.1002/ajmg.1320460213
Santos, C. B., Hjalgrim, H., Carneiro, F. R. G., Ribeiro, M., Boy, R. T., and Pimentel, M. M. G. (2003). Concurrence of fragile X and klinefelter syndromes: Report of a new case of paternal nondisjunction. Ann. Genet. 46, 53–55. doi:10.1016/s0003-3995(03)00013-3
Santos-Pereira, J. M., and Aguilera, A. (2015). R loops: New modulators of genome dynamics and function. Nat. Rev. Genet. 16, 583–597. doi:10.1038/nrg3961
Sanz, M. M., Jenkins, E. C., Brown, W. T., Davisson, M. T., Kevin, M. J., Roderick, T. H., et al. (1986). Mouse chromosome fragility. Am. J. Med. Genet. 23, 491–509. doi:10.1002/ajmg.1320230141
Sarafidou, T., Kahl, C., Martinez-Garay, I., Mangelsdorf, M., Gesk, S., Baker, E., et al. (2004). European Collaborative Consortium for the Study of ADLTEFolate-sensitive fragile site FRA10A is due to an expansion of a CGG repeat in a novel gene, FRA10AC1, encoding a nuclear protein. Genomics 84, 69–81. doi:10.1016/j.ygeno.2003.12.017
Sarni, D., and Kerem, B. (2016). The complex nature of fragile site plasticity and its importance in cancer. Curr. Opin. Cell Biol. 40, 131–136. doi:10.1016/j.ceb.2016.03.017
Sathasivam, K., Woodman, B., Mahal, A., Bertaux, F., Wanker, E. E., Shima, D. T., et al. (2001). Centrosome disorganization in fibroblast cultures derived from R6/2 Huntington’s disease (HD) transgenic mice and HD patients. Hum. Mol. Genet. 10, 2425–2435. doi:10.1093/hmg/10.21.2425
Savage, J. R., and Fitchett, M. (1988). The behaviour of fragile X and other aberrations during recovery from low folate conditions. Chromosoma 96, 391–396. doi:10.1007/BF00330707
Savelyeva, L., and Brueckner, L. M. (2014). Molecular characterization of common fragile sites as a strategy to discover cancer susceptibility genes. Cell. Mol. Life Sci. 71, 4561–4575. doi:10.1007/s00018-014-1723-z
Savelyeva, L., Sagulenko, E., Schmitt, J. G., and Schwab, M. (2006). Low-frequency common fragile sites: Link to neuropsychiatric disorders? Cancer Lett. 232, 58–69. doi:10.1016/j.canlet.2005.08.033
Sawyer, J. R., Swanson, C. M., Wheeler, G., and Cunniff, C. (1995). Chromosome instability in ICF syndrome: Formation of micronuclei from multibranched chromosomes 1 demonstrated by fluorescence in situ hybridization. Am. J. Med. Genet. 56, 203–209. doi:10.1002/ajmg.1320560218
Sbrana, I., and Musio, A. (1995). Enhanced expression of common fragile site with occupational exposure to pesticides. Cancer Genet. cytogenet. 82, 123–127. doi:10.1016/0165-4608(95)00020-p
Scheres, J. M., and Hustinx, T. W. (1980). Heritable fragile sites and lymphocyte culture medium containing BrdU. Am. J. Hum. Genet. 32, 628–629.
Schinzel, A., and Moser, H. (1977). Auf der Maur, P., Moser, HPartial deletion of long arm of chromosome 11[del(11)(q23)]: Jacobsen syndrome. Two new cases and review of the clinical findings. J. Med. Genet. 14, 438–444. doi:10.1136/jmg.14.6.438
Schmid, W., and Vischer, D. (1969). Spontaneous fragility of an abnormally wide secondary constriction region in a human chromosome no. 9. Humangenetik 7, 22–27. doi:10.1007/BF00278689
Schmid, M., Ott, G., Haaf, T., and Scheres, J. M. (1985). Evolutionary conservation of fragile sites induced by 5-azacytidine and 5-azadeoxycytidine in man, gorilla, and chimpanzee. Hum. Genet. 71, 342–350. doi:10.1007/BF00388461
Schmid, M., Feichtinger, W., Jeßberger, A., Köhler, J., Lange, R., and Jessberger, A. (1986). The fragile site (16) (q22). I. Induction by AT-specific DNA-ligands and population frequency. Hum. Genet. 74, 67–73. doi:10.1007/BF00278788
Schmid, M., Feichtinger, W., and Haaf, T. (1987). The fragile site (16)(q22). II. Sister chromatid exchanges. Hum. Genet. 76, 365–368. doi:10.1007/BF00272446
Schmidt, A., and Passarge, E. (1986). Differential expression of fragile site Xq27 in cultured fibroblasts from hemizygotes and heterozygotes and its implications for prenatal diagnosis. Am. J. Med. Genet. 23, 515–525. doi:10.1002/ajmg.1320230143
Schoder, C., Liehr, T., Velleuer, E., Wilhelm, K., Blaurock, N., Weise, A., et al. (2010). New aspects on chromosomal instability: Chromosomal break-points in fanconi anemia patients co-localize on the molecular level with fragile sites. Int. J. Oncol. 36, 307–312. doi:10.3892/ijo_00000501
Schwartz, M., Zlotorynski, E., and Kerem, B. (2006). The molecular basis of common and rare fragile sites. Cancer Lett. 232, 13–26. doi:10.1016/j.canlet.2005.07.039
Seemanová, E., Schmidt, A., Subrt, I., Passarge, E., Macek, M., and Nedomová, V. (1985). The 47, XXX syndrome in a family with the fragile X chromosome syndrome. Cas. Lek. Cesk. 124, 988–991.
Seki, N., Tsuji, H., Takahashi, E., Yamauchi, M., Saito, T., Hashimoto, T., et al. (1992). Induction of a BrdU-enhanceable fragile site-like lesion and sister chromatid exchanges at 11q23.1 in EBV-transformed lymphoblastoid cell lines. Cytogenet. Cell Genet. 61, 95–98. doi:10.1159/000133379
Sfeir, A., Kosiyatrakul, S. T., Hockemeyer, D., MacRae, S. L., Karlseder, J., Schildkraut, C. L., et al. (2009). Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138, 90–103. doi:10.1016/j.cell.2009.06.021
Shah, S. N., Opresko, P. L., Meng, X., Lee, M. Y. W. T., and Eckert, K. A. (2010). DNA structure and the Werner protein modulate human DNA polymerase delta-dependent replication dynamics within the common fragile site FRA16D. Nucleic Acids Res. 38, 1149–1162. doi:10.1093/nar/gkp1131
Shapiro, L. R., Wilmot, P. L., Shapiro, D. A., Pettersen, I. M., and Casamassima, A. C. (1991). Cytogenetic diagnosis of the fragile X syndrome: Efficiency, utilization, and trends. Am. J. Med. Genet. 38, 408–410. doi:10.1002/ajmg.1320380251
Shapiro, L. R., Simensen, R. J., Wilmot, P. L., Fisch, G. S., Vibert, B. K., Fenwick, R. G., et al. (1994). Asymmetry of methylation with FMR-1 full mutation in two 45, X/46, XX mosaic females associated with normal intellect. Am. J. Med. Genet. 51, 507–508. doi:10.1002/ajmg.1320510443
Shaw, M. A., Chiurazzi, P., Romain, D. R., Neri, G., and Gécz, J. (2002). A novel gene, FAM11A, associated with the FRAXF CpG island is transcriptionally silent in FRAXF full mutation. Eur. J. Hum. Genet. 10, 767–772. doi:10.1038/sj.ejhg.5200881
Sherman, S. L., and Sutherland, G. R. (1986). Segregation analysis of rare autosomal fragile sites. Hum. Genet. 72, 123–128. doi:10.1007/BF00283929
Siew, V.-K., Duh, C.-Y., and Wang, S.-K. (2009). Human cytomegalovirus UL76 induces chromosome aberrations. J. Biomed. Sci. 16, 107. doi:10.1186/1423-0127-16-107
Simonic, I., Gericke, G. S., Lippert, M., and Schoeman, J. F. (1997). Additional clinical and cytogenetic findings associated with Rett syndrome. Am. J. Med. Genet. 74, 331–337. doi:10.1002/(sici)1096-8628(19970531)74:3<331::aid-ajmg16>3.0.co;2-p
Smeets, D. F., Scheres, J. M., and Hustinx, T. W. (1985). Heritable fragility at 11q13 and 12q13. Clin. Genet. 28, 145–150. doi:10.1111/j.1399-0004.1985.tb00374.x
Smeets, H. J., Smits, A. P., Verheij, C. E., Theelen, J. P., Willemsen, R., van de Burgt, I., et al. (1995). Normal phenotype in two brothers with a full FMR1 mutation. Hum. Mol. Genet. 4, 2103–2108. doi:10.1093/hmg/4.11.2103
Smith, P. P., Friedman, C. L., Bryant, E. M., and McDougall, J. K. (1992). Viral integration and fragile sites in human papillomavirus-immortalized human keratinocyte cell lines. Genes Chromosom. Cancer 5, 150–157. doi:10.1002/gcc.2870050209
Smith, C. L., Bolton, A., and Nguyen, G. (2010). Genomic and epigenomic instability, fragile sites, schizophrenia and autism. Curr. Genomics 11, 447–469. doi:10.2174/138920210793176001
Sone, J., Mitsuhashi, S., Fujita, A., Mizuguchi, T., Hamanaka, K., Mori, K., et al. (2019). Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat. Genet. 51, 1215–1221. doi:10.1038/s41588-019-0459-y
Spadafora, C., and Crippa, M. (1984). Compact structure of ribosomal chromatin in Xenopus laevis. Nucleic Acids Res. 12, 2691–2704. doi:10.1093/nar/12.6.2691
Spadafora, C., and Riccardi, P. (1985). Different conformations of ribosomal DNA in active and inactive chromatin in Xenopus laevis. J. Mol. Biol. 186, 743–758. doi:10.1016/0022-2836(85)90394-8
Spadafora, C., Oudet, P., and Chambon, P. (1979). Rearrangement of chromatin structure induced by increasing ionic strength and temperature. Eur. J. Biochem. 100, 225–235. doi:10.1111/j.1432-1033.1979.tb02053.x
Stebbins, R., and Bertino, J. R. (1976). Megaloblastic anaemia produced by drugs. Clin. Haematol. 5, 619–630. doi:10.1016/s0308-2261(21)00113-2
Stebbins, R., Scott, J., and Herbert, V. (1973). Drug-induced megaloblastic anemias. Semin. Hematol. 10, 235–251.
Steichen-Gersdorf, E., Gassner, I., Superti-Furga, A., Ullmann, R., Stricker, S., Klopocki, E., et al. (2008). Triangular tibia with fibular aplasia associated with a microdeletion on 2q11.2 encompassing LAF4. Clin. Genet. 74, 560–565. doi:10.1111/j.1399-0004.2008.01050.x
Stein, C. K., Glover, T. W., Palmer, J. L., and Glisson, B. S. (2002). Direct correlation between FRA3B expression and cigarette smoking. Genes Chromosom. Cancer 34, 333–340. doi:10.1002/gcc.10061
Steinbach, P., Barbi, G., and Böller, T. (1982). On the frequency of telomeric chromosomal changes induced by culture conditions suitable for fragile X expression. Hum. Genet. 61, 160–162. doi:10.1007/BF00274209
Stettner, G. M., Shoukier, M., Höger, C., Brockmann, K., and Auber, B. (2011). Familial intellectual disability and autistic behavior caused by a small FMR2 gene deletion. Am. J. Med. Genet. A 155A, 2003–2007. doi:10.1002/ajmg.a.34122
Stone, D. M., and Stephens, K. E. (1993). Bromodeoxyuridine induces chromosomal fragile sites in the canine genome. Am. J. Med. Genet. 46, 198–202. doi:10.1002/ajmg.1320460220
Stone, D. M., Jacky, P. B., and Prieur, D. J. (1991). Chromosomal fragile site expression in dogs: II. Expression in boxer dogs with mast cell tumors. Am. J. Med. Genet. 40, 223–229. doi:10.1002/ajmg.1320400220
Stone, D. M., Stephens, K. E., and Doles, J. (1993). Folate-sensitive and aphidicolin-inducible fragile sites are expressed in the genome of the domestic cat. Cancer Genet. cytogenet. 65, 130–134. doi:10.1016/0165-4608(93)90221-7
Stout, K., van der Maarel, S., Frants, R. R., Padberg, G. W., Ropers, H. H., and Haaf, T. (1999). Somatic pairing between subtelomeric chromosome regions: Implications for human genetic disease? Chromosome Res. 7, 323–329. doi:10.1023/a:1009287111661
Stults, D. M., Killen, M. W., Williamson, E. P., Hourigan, J. S., Vargas, H. D., Arnold, S. M., et al. (2009). Human rRNA gene clusters are recombinational hotspots in cancer. Cancer Res. 69, 9096–9104. doi:10.1158/0008-5472.CAN-09-2680
Subramanian, P. S., Nelson, D. L., and Chinault, A. C. (1996). Large domains of apparent delayed replication timing associated with triplet repeat expansion at FRAXA and FRAXE. Am. J. Hum. Genet. 59, 407–416.
Sutherland, G. R., and Baker, E. (1986). Effects of nucleotides on expression of the folate sensitive fragile sites. Am. J. Med. Genet. 23, 409–417. doi:10.1002/ajmg.1320230133
Sutherland, G. R., and Baker, E. (1990). The common fragile site in band q27 of the human X chromosome is not coincident with the fragile X. Clin. Genet. 37, 167–172. doi:10.1111/j.1399-0004.1990.tb03498.x
Sutherland, G. R., and Baker, E. (1992). Characterisation of a new rare fragile site easily confused with the fragile X. Hum. Mol. Genet. 1, 111–113. doi:10.1093/hmg/1.2.111
Sutherland, G. R., and Baker, E. (1993). Unusual behaviour of a human autosome having two rare folate sensitive fragile sites. Ann. Genet. 36, 159–162.
Sutherland, G. R., Baker, E., and Seshadri, R. S. (1980). Heritable fragile sites on human chromosomes. V. A new class of fragile site requiring BrdU for expression. Am. J. Hum. Genet. 32, 542–548.
Sutherland, G. R., Jacky, P. B., Baker, E., and Manuel, A. (1983). Heritable fragile sites on human chromosomes. X. New folate-sensitive fragile sites: 6p23, 9p21, 9q32, and 11q23. Am. J. Hum. Genet. 35, 432–437.
Sutherland, G. R., Jacky, P. B., and Baker, E. G. (1984). Heritable fragile sites on human chromosomes. XI. Factors affecting expression of fragile sites at 10q25, 16q22, and 17p12. Am. J. Hum. Genet. 36, 110–122.
Sutherland, G. R., Baker, E., and Fratini, A. (1985a). Excess thymidine induces folate sensitive fragile sites. Am. J. Med. Genet. 22, 433–443. doi:10.1002/ajmg.1320220234
Sutherland, G. R., Parslow, M. I., and Baker, E. (1985b). New classes of common fragile sites induced by 5-azacytidine and bromodeoxyuridine. Hum. Genet. 69, 233–237. doi:10.1007/BF00293031
Sutherland, G. R., Haan, E. A., Kremer, E., Lynch, M., Pritchard, M., Yu, S., et al. (1991). Hereditary unstable DNA: A new explanation for some old genetic questions? Lancet 338, 289–292. doi:10.1016/0140-6736(91)90426-p
Sutherland, G. R. (1977). Fragile sites on human chromosomes: Demonstration of their dependence on the type of tissue culture medium. Science 197, 265–266. doi:10.1126/science.877551
Sutherland, G. R. (1979). Heritable fragile sites on human chromosomes II. Distribution, phenotypic effects, and cytogenetics. Am. J. Hum. Genet. 31, 136–148.
Sutherland, G. R. (1981). Heritable fragile sites on human chromosomes. VII. Children homozygous for the BrdU-requiring fra(10)(q25) are phenotypically normal. Am. J. Hum. Genet. 33, 946–949.
Sutherland, G. R. (1982). Heritable fragile sites on human chromosomes. VIII. Preliminary population cytogenetic data on the folic-acid-sensitive fragile sites. Am. J. Hum. Genet. 34, 452–458.
Sutherland, G. R. (1983). The fragile X chromosome. Int. Rev. Cytol. 81, 107–143. doi:10.1016/s0074-7696(08)62336-0
Suzuki, Y., and Morishita, S. (2021). The time is ripe to investigate human centromeres by long-read sequencing. DNA Res. 28, dsab021. doi:10.1093/dnares/dsab021
Szafranski, P., Schaaf, C. P., Person, R. E., Gibson, I. B., Xia, Z., Mahadevan, S., et al. (2010). Structures and molecular mechanisms for common 15q13.3 microduplications involving CHRNA7: Benign or pathological? Hum. Mutat. 31, 840–850. doi:10.1002/humu.21284
Takahashi, E., Hori, T., and Murata, M. (1988). A new rare heritable fragile site at 8q24.1 found in a Japanese population. Clin. Genet. 33, 91–94. doi:10.1111/j.1399-0004.1988.tb03417.x
Talbert, P. B., and Henikoff, S. (2022). The genetics and epigenetics of satellite centromeres. Genome Res. 32, 608–615. doi:10.1101/gr.275351.121
Tam, R., Smith, K. P., and Lawrence, J. B. (2004). The 4q subtelomere harboring the FSHD locus is specifically anchored with peripheral heterochromatin unlike most human telomeres. J. Cell Biol. 167, 269–279. doi:10.1083/jcb.200403128
Tarleton, J., Richie, R., Schwartz, C., Rao, K., Aylsworth, A. S., and Lachiewicz, A. (1993). An extensive de novo deletion removing FMR1 in a patient with mental retardation and the fragile X syndrome phenotype. Hum. Mol. Genet. 2, 1973–1974. doi:10.1093/hmg/2.11.1973
Tassone, F., Hagerman, R. J., Taylor, A. K., Mills, J. B., Harris, S. W., Gane, L. W., et al. (2000). Clinical involvement and protein expression in individuals with the FMR1 premutation. Am. J. Med. Genet. 91, 144–152. doi:10.1002/(sici)1096-8628(20000313)91:2<144::aid-ajmg14>3.0.co;2-v
Tejada, M. I., Mornet, E., Tizzano, E., Molina, M., Baiget, M., and Boue, A. (1994). Identification by molecular diagnosis of mosaic Turner’s syndrome in an obligate carrier female for fragile X syndrome. J. Med. Genet. 31, 76–78. doi:10.1136/jmg.31.1.76
Tewari, R., Juyal, R. C., Thelma, B. K., Das, B. C., and Rao, S. R. (1987). Folate-sensitive fragile sites on the X-chromosome heterochromatin of the Indian mole rat, Nesokia indica. Cytogenet. Cell Genet. 44, 11–17. doi:10.1159/000132334
Thakur, J., Packiaraj, J., and Henikoff, S. (2021). Sequence, chromatin and evolution of satellite DNA. Int. J. Mol. Sci. 22, 4309. doi:10.3390/ijms22094309
Thys, R. G., Lehman, C. E., Pierce, L. C. T., and Wang, Y.-H. (2015). DNA secondary structure at chromosomal fragile sites in human disease. Curr. Genomics 16, 60–70. doi:10.2174/1389202916666150114223205
Tommerup, N., Nielssen, K. B., and Mikkelsen, M. (1981). Marker X chromosome induction in fibroblasts by FUdR. Am. J. Med. Genet. 9, 263–264. doi:10.1002/ajmg.1320090313
Tommerup, N. (1989). Induction of the fragile X on BrdU-substituted chromosomes with direct visualization of sister chromatid exchanges on banded chromosomes. Hum. Genet. 81, 377–381. doi:10.1007/BF00283696
Tootleman, E., Malamut, B., Akshoomoff, N., Mattson, S. N., Hoffman, H. M., Jones, M. C., et al. (2019). Partial Jacobsen syndrome phenotype in a patient with a de novo frameshift mutation in the ETS1 transcription factor. Cold Spring Harb. Mol. Case Stud. 5, a004010. doi:10.1101/mcs.a004010
Trost, B., Engchuan, W., Nguyen, C. M., Thiruvahindrapuram, B., Dolzhenko, E., Backstrom, I., et al. (2020). Genome-wide detection of tandem DNA repeats that are expanded in autism. Nature 586, 80–86. doi:10.1038/s41586-020-2579-z
Trottier, Y., Imbert, G., Poustka, A., Fryns, J. P., and Mandel, J. L. (1994). Male with typical fragile X phenotype is deleted for part of the FMR1 gene and for about 100 kb of upstream region. Am. J. Med. Genet. 51, 454–457. doi:10.1002/ajmg.1320510431
Tsompana, M., and Buck, M. J. (2014). Chromatin accessibility: A window into the genome. Epigenetics Chromatin 7, 33. doi:10.1186/1756-8935-7-33
Tsuji, H., Hitomi, A., Takahashi, E., Murata, M., Ikeuchi, T., Yamamoto, K., et al. (1991). Induction of distamycin A-inducible rare fragile sites and increased sister chromatid exchanges at the fragile site. Hum. Genet. 87, 254–260. doi:10.1007/BF00200900
Tubbs, A., Sridharan, S., van Wietmarschen, N., Maman, Y., Callen, E., Stanlie, A., et al. (2018). Dual roles of poly(dA:dT) tracts in replication initiation and fork collapse. Cell 174, 1127–1142. e19. doi:10.1016/j.cell.2018.07.011
Tuck-Muller, C. M., Narayan, A., Tsien, F., Smeets, D. F., Sawyer, J., Fiala, E. S., et al. (2000). DNA hypomethylation and unusual chromosome instability in cell lines from ICF syndrome patients. Cytogenet. Cell Genet. 89, 121–128. doi:10.1159/000015590
Tuduri, S., Crabbé, L., Conti, C., Tourrière, H., Holtgreve-Grez, H., Jauch, A., et al. (2009). Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 11, 1315–1324. doi:10.1038/ncb1984
Tunnacliffe, A., Jones, C., Le Paslier, D., Todd, R., Cherif, D., Birdsall, M., et al. (1999). Localization of Jacobsen syndrome breakpoints on a 40-Mb physical map of distal chromosome 11q. Genome Res. 9, 44–52. doi:10.1101/gr.9.1.44
Turleau, C., Cabanis, M. O., Girault, D., Ledeist, F., Mettey, R., Puissant, H., et al. (1989). Multibranched chromosomes in the ICF syndrome: Immunodeficiency, centromeric instability, and facial anomalies. Am. J. Med. Genet. 32, 420–424. doi:10.1002/ajmg.1320320331
Turner, G., Brookwell, R., Daniel, A., Selikowitz, M., and Zilibowitz, M. (1980a). Heterozygous expression of X-linked mental retardation and X-chromosome marker fra(X)(q27). N. Engl. J. Med. 303, 662–664. doi:10.1056/NEJM198009183031202
Turner, G., Daniel, A., and Frost, M. (1980b). X-linked mental retardation, macro-orchidism, and the Xq27 fragile site. J. Pediatr. 96, 837–841. doi:10.1016/s0022-3476(80)80552-x
Uchida, I. A., Freeman, V. C., and Basrur, P. K. (1986). The fragile X in cattle. Am. J. Med. Genet. 23, 557–562. doi:10.1002/ajmg.1320230148
Uchiyama, S., and Fukui, K. (2015). Condensin in chromatid cohesion and segregation. Cytogenet. Genome Res. 147, 212–216. doi:10.1159/000444868
Usdin, K., and Woodford, K. (1995). CGG repeats associated with DNA instability and chromosome fragility form structures that block DNA synthesis in vitro. Nucleic Acids Res. 23, 4202–4209. doi:10.1093/nar/23.20.4202
Valori, V., Tus, K., Laukaitis, C., Harris, D. T., LeBeau, L., and Maggert, K. A. (2020). Human rDNA copy number is unstable in metastatic breast cancers. Epigenetics 15, 85–106. doi:10.1080/15592294.2019.1649930
van den Boogaard, M. L., Thijssen, P. E., Aytekin, C., Licciardi, F., Kıykım, A. A., Spossito, L., et al. (2017). Expanding the mutation spectrum in ICF syndrome: Evidence for a gender bias in ICF2. Clin. Genet. 92, 380–387. doi:10.1111/cge.12979
Van Dyke, D. L., Flejter, W. L., Worsham, M. J., Roberson, J. R., Higgins, J. V., Herr, H. M., et al. (1986). A practical metaphase marker of the inactive X chromosome. Am. J. Hum. Genet. 39, 88–95.
Van Dyke, D. L., Worsham, M., and Weiss, L. (1987). The human inactivated X chromosome folds in early metaphase, prometaphase, and prophase. Hum. Genet. 77, 57–59. doi:10.1007/BF00284715
van Sluis, M., and McStay, B. (2017). Nucleolar reorganization in response to rDNA damage. Curr. Opin. Cell Biol. 46, 81–86. doi:10.1016/j.ceb.2017.03.004
van Sluis, M., and McStay, B. (2019). Nucleolar DNA double-strand break responses underpinning rDNA genomic stability. Trends Genet. 35, 743–753. doi:10.1016/j.tig.2019.07.001
Verdyck, P., Berckmoes, V., De Vos, A., Verpoest, W., Liebaers, I., Bonduelle, M., et al. (2015). Chromosome fragility at FRAXA in human cleavage stage embryos at risk for fragile X syndrome. Am. J. Med. Genet. A 167A, 2306–2313. doi:10.1002/ajmg.a.37149
Verkerk, A. J., Pieretti, M., Sutcliffe, J. S., Fu, Y. H., Kuhl, D. P., Pizzuti, A., et al. (1991). Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914. doi:10.1016/0092-8674(91)90397-h
Villa, N., Dalprà, L., and Larizza, L. (1997). Expression of three rare fragile sites: Chromosomal truncation, amplification of distal segment and telomeric renewal. Chromosoma 106, 400–404. doi:10.1007/s004120050261
Vincent, A., Heitz, D., Petit, C., Kretz, C., Oberlé, I., and Mandel, J. L. (1991). Abnormal pattern detected in fragile-X patients by pulsed-field gel electrophoresis. Nature 349, 624–626. doi:10.1038/349624a0
Vogt, P. (1990). Potential genetic functions of tandem repeated DNA sequence blocks in the human genome are based on a highly conserved “chromatin folding code. Hum. Genet. 84, 301–336. doi:10.1007/BF00196228
Voiculescu, I., Back, E., and Schempp, W. (1991). Homozygous condition for a BrdU-requiring fragile site on chromosome 12. Hum. Genet. 86, 416–417. doi:10.1007/BF00201849
Vollger, M. R., Kerpedjiev, P., Phillippy, A. M., and Eichler, E. E. (2022). StainedGlass: Interactive visualization of massive tandem repeat structures with identity heatmaps. Bioinformatics 38, 2049–2051. doi:10.1093/bioinformatics/btac018
Voullaire, L. E., Webb, G. C., and Leversha, M. A. (1987). Chromosome deletion at 11q23 in an abnormal child from a family with inherited fragility at 11q23. Hum. Genet. 76, 202–204. doi:10.1007/BF00284923
Walker, C. L., Cargile, C. B., Floy, K. M., Delannoy, M., and Migeon, B. R. (1991). The Barr body is a looped X chromosome formed by telomere association. Proc. Natl. Acad. Sci. U. S. A. 88, 6191–6195. doi:10.1073/pnas.88.14.6191
Wang, Y. H., and Griffith, J. (1996). Methylation of expanded CCG triplet repeat DNA from fragile X syndrome patients enhances nucleosome exclusion. J. Biol. Chem. 271, 22937–22940. doi:10.1074/jbc.271.38.22937
Wang, Y. H., Gellibolian, R., Shimizu, M., Wells, R. D., and Griffith, J. (1996). Long CCG triplet repeat blocks exclude nucleosomes: A possible mechanism for the nature of fragile sites in chromosomes. J. Mol. Biol. 263, 511–516. doi:10.1006/jmbi.1996.0593
Wang, L., Darling, J., Zhang, J. S., Huang, H., Liu, W., and Smith, D. I. (1999). Allele-specific late replication and fragility of the most active common fragile site, FRA3B. Hum. Mol. Genet. 8, 431–437. doi:10.1093/hmg/8.3.431
Wang, T., Antonacci-Fulton, L., Howe, K., Lawson, H. A., Lucas, J. K., Phillippy, A. M., et al. (2022). Human pangenome reference ConsortiumThe human pangenome project: A global resource to map genomic diversity. Nature 604, 437–446. doi:10.1038/s41586-022-04601-8
Warburton, P. E., Haaf, T., Gosden, J., Lawson, D., and Willard, H. F. (1996). Characterization of a chromosome-specific chimpanzee alpha satellite subset: Evolutionary relationship to subsets on human chromosomes. Genomics 33, 220–228. doi:10.1006/geno.1996.0187
Warmerdam, D. O., and Wolthuis, R. M. F. (2019). Keeping ribosomal DNA intact: A repeating challenge. Chromosome Res. 27, 57–72. doi:10.1007/s10577-018-9594-z
Warren, S. T., and Davidson, R. L. (1984). Expression of fragile X chromosome in human-rodent somatic cell hybrids. Somat. Cell Mol. Genet. 10, 409–413. doi:10.1007/BF01535636
Warren, S. T., Zhang, F., Licameli, G. R., and Peters, J. F. (1987). The fragile X site in somatic cell hybrids: An approach for molecular cloning of fragile sites. Science 237, 420–423. doi:10.1126/science.3603029
Webb, T., and Thake, A. (1984). Fragile 22q13 segregating in a family. Clin. Genet. 26, 125–128. doi:10.1111/j.1399-0004.1984.tb00801.x
Webb, T., Butler, D., Insley, J., Weaver, J. B., Green, S., and Rodeck, C. (1981). Prenatal diagnosis of Martin-Bell syndrome associated with fragile site at Xq27-28. Lancet 2, 1423. doi:10.1016/s0140-6736(81)92838-5
Webb, T. (1992). Delayed replication of Xq27 in individuals with the fragile X syndrome. Am. J. Med. Genet. 43, 1057–1062. doi:10.1002/ajmg.1320430633
Weirich-Schwaiger, H., Weirich, H. G., Gruber, B., Schweiger, M., and Hirsch-Kauffmann, M. (1994). Correlation between senescence and DNA repair in cells from young and old individuals and in premature aging syndromes. Mutat. Res. 316, 37–48. doi:10.1016/0921-8734(94)90006-x
Wells, R. A., Germino, G. G., Krishna, S., Buckle, V. J., and Reeders, S. T. (1990). Telomere-related sequences at interstitial sites in the human genome. Genomics 8, 699–704. doi:10.1016/0888-7543(90)90257-u
Wenger, S. L., Hennessey, J. C., and Steele, M. W. (1987). Increased sister chromatid exchange frequency at Xq27 site in affected fragile X males. Am. J. Med. Genet. 26, 909–914. doi:10.1002/ajmg.1320260419
Wenger, S. L., Giangreco, C. A., Tarleton, J., and Wessel, H. B. (1996). Inability to induce fragile sites at CTG repeats in congenital myotonic dystrophy. Am. J. Med. Genet. 66, 60–63. doi:10.1002/(SICI)1096-8628(19961202)66:1<60::AID-AJMG13>3.0.CO;2-O
Whibley, A. C., Plagnol, V., Tarpey, P. S., Abidi, F., Fullston, T., Choma, M. K., et al. (2010). Fine-scale survey of X chromosome copy number variants and indels underlying intellectual disability. Am. J. Hum. Genet. 87, 173–188. doi:10.1016/j.ajhg.2010.06.017
Wijmenga, C., Hansen, R. S., Gimelli, G., Björck, E. J., Davies, E. G., Valentine, D., et al. (2000). Genetic variation in ICF syndrome: Evidence for genetic heterogeneity. Hum. Mutat. 16, 509–517. doi:10.1002/1098-1004(200012)16:6<509::AID-HUMU8>3.0.CO;2-V
Wilke, C. M., Hall, B. K., Hoge, A., Paradee, W., Smith, D. I., and Glover, T. W. (1996). FRA3B extends over a broad region and contains a spontaneous HPV16 integration site: Direct evidence for the coincidence of viral integration sites and fragile sites. Hum. Mol. Genet. 5, 187–195. doi:10.1093/hmg/5.2.187
Winnepenninckx, B., Debacker, K., Ramsay, J., Smeets, D., Smits, A., FitzPatrick, D. R., et al. (2007). CGG-repeat expansion in the DIP2B gene is associated with the fragile site FRA12A on chromosome 12q13.1. Am. J. Hum. Genet. 80, 221–231. doi:10.1086/510800
Wöhrle, D., Kotzot, D., Hirst, M. C., Manca, A., Korn, B., Schmidt, A., et al. (1992). A microdeletion of less than 250 kb, including the proximal part of the FMR-I gene and the fragile-X site, in a male with the clinical phenotype of fragile-X syndrome. Am. J. Hum. Genet. 51, 299–306.
Wollman, M. R., Penchansky, L., and Shekhter-Levin, S. (1996). Transient 7q- in association with megaloblastic anemia due to dietary folate and vitamin B12 deficiency. J. Pediatr. Hematol. Oncol. 18, 162–165. doi:10.1097/00043426-199605000-00013
Wurster-Hill, D. H., Ward, O. G., Davis, B. H., Park, J. P., Moyzis, R. K., and Meyne, J. (1988). Fragile sites, telomeric DNA sequences, B chromosomes, and DNA content in raccoon dogs, Nyctereutes procyonoides, with comparative notes on foxes, coyote, wolf, and raccoon. Cytogenet. Cell Genet. 49, 278–281. doi:10.1159/000132677
Xu, B., Li, H., Perry, J. M., Singh, V. P., Unruh, J., Yu, Z., et al. (2017). Ribosomal DNA copy number loss and sequence variation in cancer. PLoS Genet. 13, e1006771. doi:10.1371/journal.pgen.1006771
Yamauchi, M., Tsuji, S., Mita, K., Saito, T., and Morimyo, M. (2000). A novel minisatellite repeat expansion identified at FRA16B in a Japanese carrier. Genes Genet. Syst. 75, 149–154. doi:10.1266/ggs.75.149
Yang, M. Y., and Long, S. E. (1993). Folate sensitive common fragile sites in chromosomes of the domestic pig (Sus scrofa). Res. Vet. Sci. 55, 231–235. doi:10.1016/0034-5288(93)90086-u
Ying, S., Minocherhomji, S., Chan, K. L., Palmai-Pallag, T., Chu, W. K., Wass, T., et al. (2013). MUS81 promotes common fragile site expression. Nat. Cell Biol. 15, 1001–1007. doi:10.1038/ncb2773
Yu, S., Pritchard, M., Kremer, E., Lynch, M., Nancarrow, J., Baker, E., et al. (1991). Fragile X genotype characterized by an unstable region of DNA. Science 252, 1179–1181. doi:10.1126/science.252.5009.1179
Yu, S., Mangelsdorf, M., Hewett, D., Hobson, L., Baker, E., Eyre, H. J., et al. (1997). Human chromosomal fragile site FRA16B is an amplified AT-rich minisatellite repeat. Cell 88, 367–374. doi:10.1016/s0092-8674(00)81875-9
Yudkin, D., Hayward, B. E., Aladjem, M. I., Kumari, D., and Usdin, K. (2014). Chromosome fragility and the abnormal replication of the FMR1 locus in fragile X syndrome. Hum. Mol. Genet. 23, 2940–2952. doi:10.1093/hmg/ddu006
Yunis, J. J., and Soreng, A. L. (1984). Constitutive fragile sites and cancer. Science 226, 1199–1204. doi:10.1126/science.6239375
Yunis, J. J., and Yasmineh, W. G. (1971). Heterochromatin, satellite DNA, and cell function. Structural DNA of eucaryotes may support and protect genes and aid in speciation. Science 174, 1200–1209. doi:10.1126/science.174.4015.1200
Zamani, A. G., Durakbasi-Dursun, H. G., and Acar, A. (2007). A new heritable fragile site at 15q13 in a three-generation family. Cytogenet. Genome Res. 116, 252–255. doi:10.1159/000100408
Zamiri, B., Mirceta, M., Bomsztyk, K., Macgregor, R. B., and Pearson, C. E. (2015). Quadruplex formation by both G-rich and C-rich DNA strands of the C9orf72 (GGGGCC)8·(GGCCCC)8 repeat: Effect of CpG methylation. Nucleic Acids Res. 43, 10055–10064. doi:10.1093/nar/gkv1008
Zamiri, B., Mirceta, M., Abu-Ghazalah, R., Wold, M. S., Pearson, C. E., and Macgregor, R. B. (2018). Stress-induced acidification may contribute to formation of unusual structures in C9orf72-repeats. Biochim. Biophys. Acta. Gen. Subj. 1862, 1482–1491. doi:10.1016/j.bbagen.2018.03.001
Zhang, H., and Freudenreich, C. H. (2007). An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol. Cell 27, 367–379. doi:10.1016/j.molcel.2007.06.012
Zheglo, D., Brueckner, L. M., Sepman, O., Wecht, E. M., Kuligina, E., Suspitsin, E., et al. (2019). The FRA14B common fragile site maps to a region prone to somatic and germline rearrangements within the large GPHN gene. Genes Chromosom. Cancer 58, 284–294. doi:10.1002/gcc.22706
Zhong, N., Yang, W., Dobkin, C., and Brown, W. T. (1995). Fragile X gene instability: Anchoring AGGs and linked microsatellites. Am. J. Hum. Genet. 57, 351–361.
Zhou, H., Wang, Y., Wang, Q., Li, L., Hu, Y., Wu, Y., et al. (2021). R-loops mediate transcription-associated formation of human rDNA secondary constrictions. J. Cell. Biochem. 122, 1517–1533. doi:10.1002/jcb.30074
Zimonjic, D. B., Popescu, N. D., and DiPaolo, J. A. (1994). Chromosomal organization of viral integration sites in human papillomavirus-immortalized human keratinocyte cell lines. Cancer Genet. cytogenet. 72, 39–43. doi:10.1016/0165-4608(94)90107-4
Zingg, J. M., and Jones, P. A. (1997). Genetic and epigenetic aspects of DNA methylation on genome expression, evolution, mutation and carcinogenesis. Carcinogenesis 18, 869–882. doi:10.1093/carcin/18.5.869
Keywords: rare fragile sites, repeat expansions, folate sensitivity, chromatin structure, mechanisms of fragility
Citation: Mirceta M, Shum N, Schmidt MHM and Pearson CE (2022) Fragile sites, chromosomal lesions, tandem repeats, and disease. Front. Genet. 13:985975. doi: 10.3389/fgene.2022.985975
Received: 04 July 2022; Accepted: 02 September 2022;
Published: 17 November 2022.
Edited by:
Tao Zu, University of Florida, United StatesReviewed by:
Peng Jin, Emory University, United StatesCopyright © 2022 Mirceta, Shum, Schmidt and Pearson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher E. Pearson, Y2hyaXN0b3BoZXIucGVhcnNvbkBzaWNra2lkcy5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.