 Ye Liu
Ye Liu William S. B. Yeung
William S. B. Yeung Philip C. N. Chiu
Philip C. N. Chiu Dandan Cao
Dandan Cao
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 29 September 2022
Sec. Computational Genomics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.981005
This article is part of the Research Topic Current State of Multi-omics Modeling and Simulations View all 5 articles
One objective of human genetics is to unveil the variants that contribute to human diseases. With the rapid development and wide use of next-generation sequencing (NGS), massive genomic sequence data have been created, making personal genetic information available. Conventional experimental evidence is critical in establishing the relationship between sequence variants and phenotype but with low efficiency. Due to the lack of comprehensive databases and resources which present clinical and experimental evidence on genotype-phenotype relationship, as well as accumulating variants found from NGS, different computational tools that can predict the impact of the variants on phenotype have been greatly developed to bridge the gap. In this review, we present a brief introduction and discussion about the computational approaches for variant impact prediction. Following an innovative manner, we mainly focus on approaches for non-synonymous variants (nsSNVs) impact prediction and categorize them into six classes. Their underlying rationale and constraints, together with the concerns and remedies raised from comparative studies are discussed. We also present how the predictive approaches employed in different research. Although diverse constraints exist, the computational predictive approaches are indispensable in exploring genotype-phenotype relationship.
One of the primary goals of human genetics is to discover the genetic variants associated with the onset and progression of human disease. The challenge is a “a needle in haystack” problem: how to pinpoint the potential causative ones from millions of individual variants (Genomes Project et al., 2015) spreading over the newly assembled, non-gap 3.055 billion–base pair human genome sequence (Nurk et al., 2022). Efforts to achieve this goal, such as linkage analysis and genome-wide association studies, were inadequately effective in identifying causative candidates and had poor clinical predictive value (Tam et al., 2019).
Over the last decade, the next generation sequencing (NGS) has been extensively utilized in biomedical research as consequences of its substantially reduced cost and generation of large volume of data. According to the fact sheets on genomic cost provided by the National Human Genome Research Institute (NHGRI) (KA., 2021), NGS technology achieved one hundred-fold cost reduction compared to Sanger sequencing, and the price is currently less than $1,000 per human genome. Nowadays, NGS platforms can finish one run within 2 days producing billions of reads for up to 48 samples (Hu et al., 2021). With the raw NGS data, standard and well-recognized variant format files can be generated using upstream analysis pipeline (Kanzi et al., 2020). Whereas the downstream disease-causing variant fishing step among ∼50,000 variants from WES, or even millions of variants from WGS is the most challenge part (Eberle et al., 2017; Koboldt, 2020).
There are plenty of data resources storing evidenced genotype-phenotype relationship information. To a certain extent, clinicians and researchers are able to utilize these records to interpret the formation, progress, diagnosis and treatment of diseases from a genetic perspective. However, even the most well-recognized databases, such as ClinVar (Landrum et al., 2020), only contain around 14,000 of highly confident variants with evidence evaluated by genetic experts, which is a small fraction compared to the huge number of variants identified from NGS. This situation dramatically reduces clinical utility from genetics. In addition, it also poses great challenges for understanding differential actions of genes between/among individuals, populations and species, as well as deciphering the genotype-phenotype relationship (Orgogozo et al., 2015). To address these issues, computational tools for predicting variant impact have emerged which can help bridge the gap between vast amount of genomic data generated and limited known genetic evidence, and finally build up the potential genotype-phenotype relationship for the newly identified variants.
Variant call format (VCF) files store identified variants providing variant genomic position, nucleotide substitution, assessed quality score, genotype and other relevant information according to alignment and variant calling information (Danecek et al., 2011). Based on the specified information, variant annotation can locate them to specific genes or transcripts, classify them into different types and conclude on their impactable consequences (Wang et al., 2010; Cingolani et al., 2012; McLaren et al., 2016). Variants causing sequence alteration are mainly categorized into four types: insertion, deletion, single nucleotide variant (SNV) and other substitution, including multiple nucleotide variant (MNV) (Eilbeck et al., 2005). Among them, SNVs are the most frequently identified (Genomes Project et al., 2015; Lek et al., 2016) and annotated (Cunningham et al., 2015). SNVs are composed of non-synonymous SNVs (nsSNVs) and synonymous SNVs (sSNVs). Comparing to sSNVs, nsSNVs, which will cause amino acid change based on the protein translation codons, are estimated at higher frequency in individuals with excess deleteriousness (Genomes Project et al., 2012). Therefore, in this review, we focus on the computational approaches which are developed to infer the impact of nsSNVs in coding regions. The database resources that are utilized by majority of the predictive methods (we name them as predictors throughout this review) are firstly introduced. Following that, we discuss the underlying motivation and constraints of those predictors with which we group them into six categories in an innovative manner. We also present their corresponding predictive performance and concerns from assessment studies. Finally, we demonstrate the application performance of the predictors in large-scale studies, as well as their ability to reveal the genotype-phenotype associations.
Models are not created out of thin air; rather, they are designed to identify hidden correlations in massive volumes of real data, allowing data to be interpreted and used to generate predictions. Since the deployment of the Human Genome Project in the 1990s, various relevant databases and knowledgebases have been established and maintained by academic institutions, organizations, consortia, and communities to collect, store, and retrieve records pertaining to genetic, clinical, and phenotypic information. They provide sufficient accessible evidences and facts to reliably demonstrate the genotype-phenotype association, which explains the functional and pathogenic importance of genetic variations (Johnston and Biesecker, 2013).
Databases can be categorized according to their scope, purpose, and scale. Several reviews (Thorisson et al., 2009; Brookes and Robinson, 2015; Zhang et al., 2019; Banck et al., 2021; Katsonis et al., 2022) provided comprehensive details of the content, usage, comparisons, and limitations for those databases. In this section, we briefly review the most frequently used databases (Table 1) containing sequence information, population-scale data, phenotype ontology, clinical and experimental evidence.
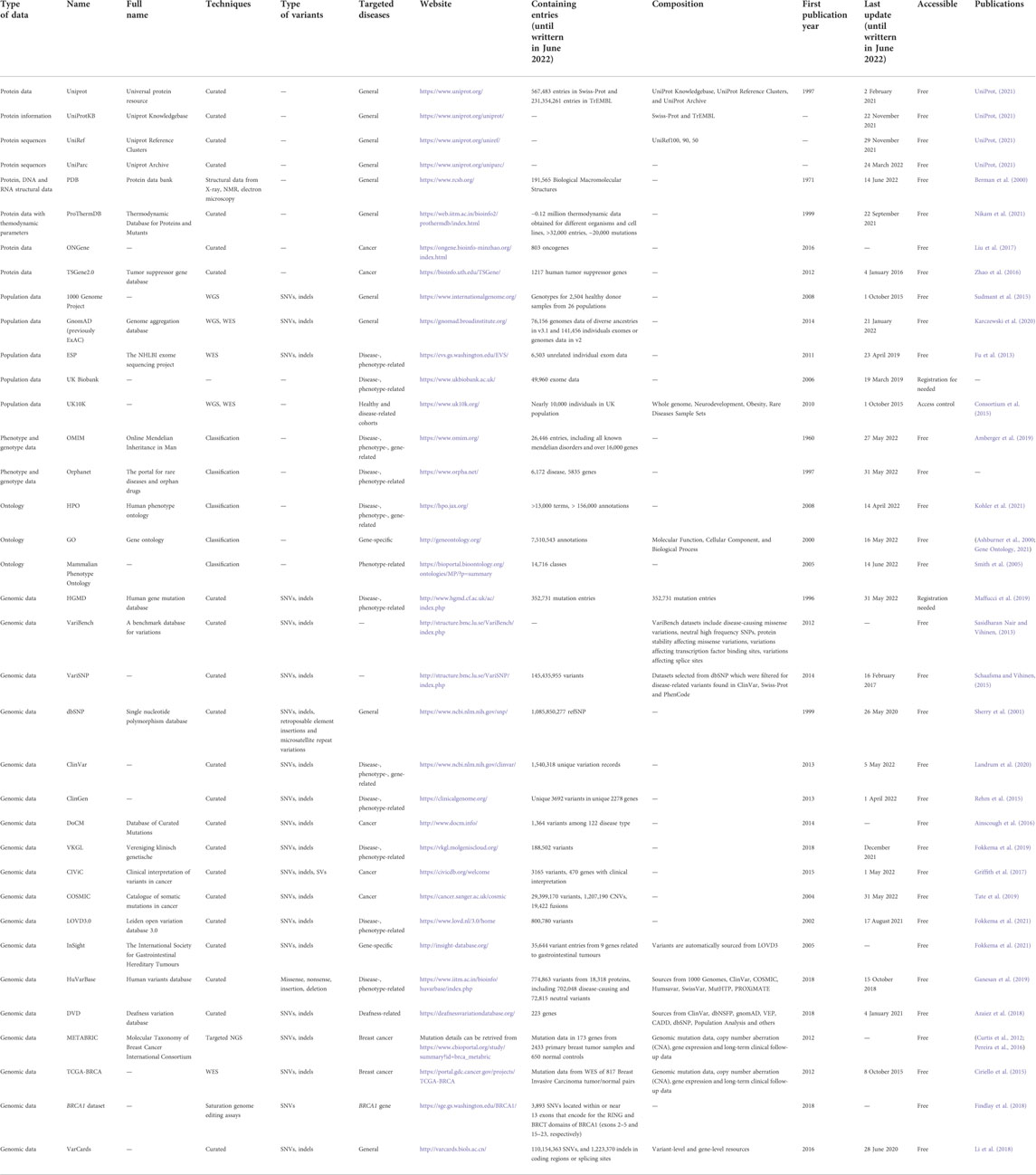
TABLE 1. Summary of resources for human genotypes and phenotypes relationships.
GenBank (Sayers et al., 2022), hosted by National Institutes of Health (NIH), European Nucleotide Archive (ENA) (Baker et al., 2000), hosted by European Molecular Biology Laboratory’s European Bioinformatics Institute (EMBL-EBI), as well as the DNA Data Bank of Japan (DDBJ) (Okido et al., 2022) are the most widely used sequence databases, storing over 2.5 billion nucleotide sequences for over 504,000 formally described species. They serve as a basis for genetic analysis since aligning clean reads to the reference genome is an indispensable step in NGS analysis. As sequences of plethora species become accessible, protein sequences with 100%, 95%, and 50% identity are assembled to create clusters that are stored in informative databases such as UniProt Reference Clusters (UniRef) (Suzek et al., 2007), a branch of Universal Protein Resource (UniProt) (UniProt, 2021). These clusters are utilized to build multiple sequence alignment (MSA) sets, which form the basis of homology sequence-based approach.
Several worldwide population projects exist, including the NCBI dbSNP (Smigielski et al., 2000), 1000 genome project (1KGP) (Sudmant et al., 2015), HapMap (International HapMap, 2003), UK10K (Consortium et al., 2015), Genome Aggregation Database (gnomAD) (Karczewski et al., 2020), and NHLBI GO Exome Sequencing Project (ESP) (Fu et al., 2013). With their progress and completion, their reports are now public and offer an exquisite view of the landscape of human genetic variants ranging from common to extremely rare ones. They also provide valuable information allowing the examination of variants between and within subpopulations with different ethnicities or disease status like heart, lung and blood disorders. Furthermore, minor allele frequency (MAF) from these databases is usually a useful indicator for prioritization or pertain as important feature for building prediction models.
Phenotype databases describe phenotypes and illnesses in conjunction with genetic information. The most widely known are OMIM (Online Mendelian Inheritance in Man) (Amberger et al., 2019) and Orphanet (Ayme et al., 1998). Their goal is to offer high-quality information on common and rare diseases or phenotypes in order to comprehensively review the genotype-phenotype association. To assist the investigation on connections between phenotypes and genes and to describe diseases in an algorithm-friendly data structure, ontology databases such as Human Phenotype Ontology (HPO) (Robinson et al., 2008), Mammalian Phenotype Ontology (Smith et al., 2005), and Gene Ontology (GO) (Ashburner et al., 2000) were developed. They are designed to annotate clinical phenotypes and genes with well-structured, computational-friendly, precise, and accurate terminology. Overall, these databases provide valuable insights for prioritization and interpretation of genetic data.
Several databases curated genetic data with clinical significance information. These databases are also known as Locus-Specific Databases (LSDB). Data and entries are often curated from literature and clinical trials. LSDBs range in scale from a single gene with roughly 4000 variants (Findlay et al., 2018) to hundreds of millions of variants (Schaafsma and Vihinen, 2015). The goal of LSDBs is to unambiguously and accurately define and categorize genotype-phenotype correlation, to understand gene functions and effects, to provide a map of genetic distribution across populations and diseases, and to assist clinicians/diagnostic laboratories in conducting further validation assays by providing detail molecule, pathogenicity, and effects of variants (Greenblatt et al., 2008). A well-curated and annotated LSDB is a valuable resource for constructing and evaluating prediction models. But note in mind that there would be overlapped variants in different LSDBs, even with contradictory classification of clinical impact due to inconsistent rules and subjective opinion of different curators. Phenotype-/disease- specific LSDBs are established, such as DVD (Deafness Variation Database) (Azaiez et al., 2018) for deafness, RAPID (Resource of Asian Primary Immunodeficiency Diseases) (Keerthikumar et al., 2009) for primary immunodeficiency disease, InSiGHT (The International Society for Gastrointestinal Hereditary Tumors) (Fokkema et al., 2021) for gastrointestinal tumors, fabry-database.org (Saito et al., 2011) for Fabry disease. Thanks to the effort of the Leiden Open source Variation Database (LOVD) (Fokkema et al., 2021) platform, a comprehensive list of public LSDBs are presented with details for researchers and clinicians to retrieve gene and mutation information from different resources.
Each predictor has a unique biochemical or biological basis. It is important to remember that the outcome of the predictor on different bases has different implications. The terms “dangerous,” “pathogenic,” “conservative,” and “damaging” do not necessarily denote causal of a specific phenotype or condition. Knowing the principles and drawbacks of each type of predictor aids in correctly interpreting the variants.
Variant impact predictors can be categorized in different ways: machine-learning (ML) and non-ML models based on the used algorithms; homology sequence-based and structural-based models regarding the features they used in prediction; supervised and unsupervised ML-models. Unlike the category of sequence-, structure- and meta-methodologies in other reviews (Hassan et al., 2019b; Yazar and Ozbek, 2021), we introduced an innovative category here based on the characteristics and included features of each type (Figure 1). We discuss these categories by outlining the rational reasoning behind the predictors and provide an overview of the constraints. Later, we discuss predictor performance evaluation and underline current concerns and remedies. Details for each tool are present in Supplementary Table S1 and Table 2.
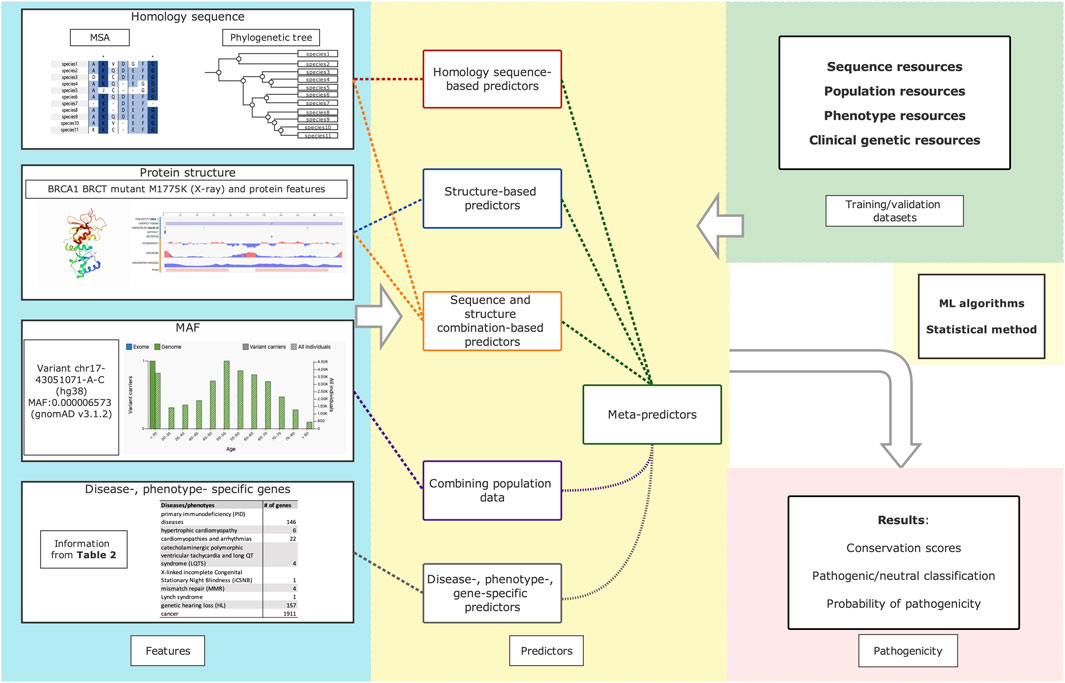
FIGURE 1. Summarized workflow of variants impact predictors. Protein structure and protein features of BRCA1 BRCT mutant M1775K are retrived from studies. (Birrane, 2006: Tischkowitz et al., 2008). The minor allele frequency (MAF) information of variant rs41293463 (chr17-43051071-A-C(GRCh38)) was retrived from gnomAD (Genome Aggregation Database).
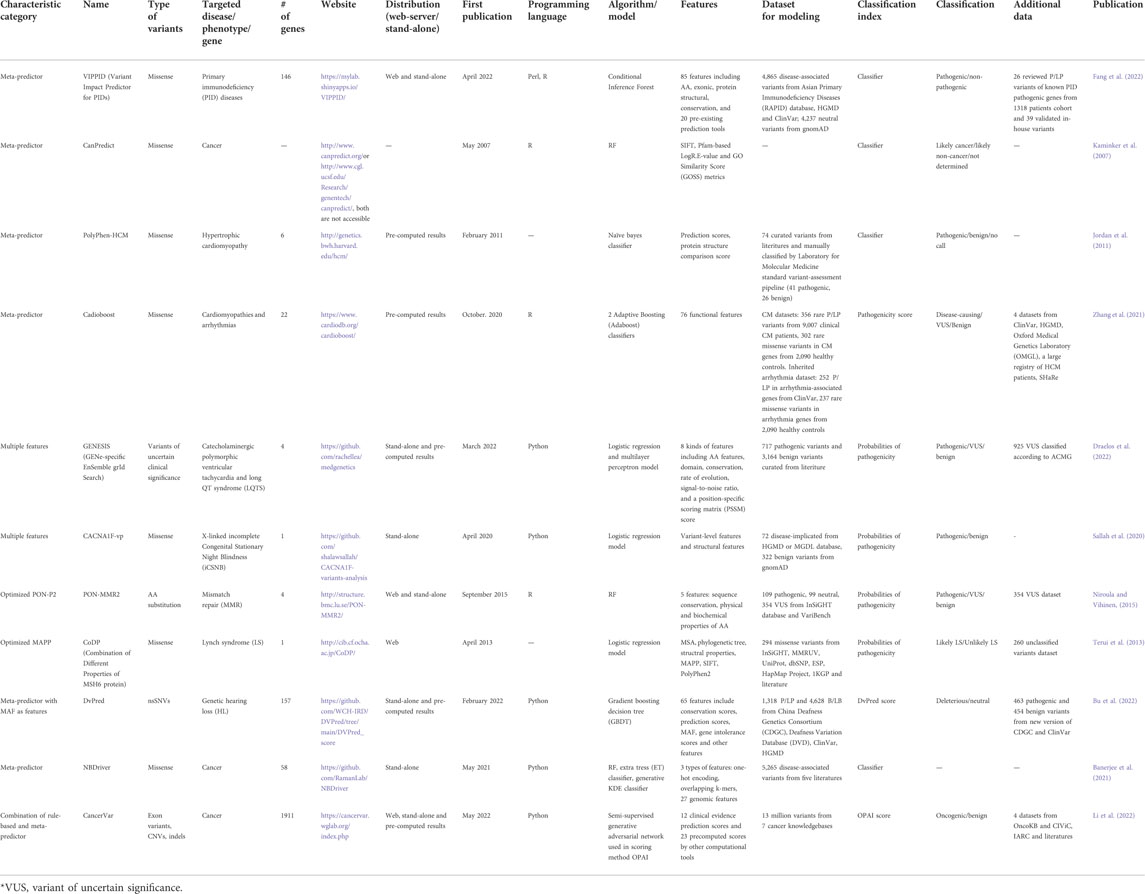
TABLE 2. Representative diseases-, phenotypes-, genes-specific variants impact predictors.
This class of predictors are derived from comparative genomics. The assumption is straightforward: under natural selection, amino acid changes in conservative sequences are more “deleterious” determined by homologous sequence searching across species, than that happened in other non-homologous positions which would be deemed as “tolerant” (Cooper and Shendure, 2011). Methodologically, these predictors firstly construct the multiple sequence alignment (MSA) either by grouping multiple protein sequences with a given similarity from BLAST alignment (Altschul et al., 1990), or just retrieval customed selective sequences from afroed-mentioned genomic databases (Section 2.1) for multiple alignment using MULTIZ (Blanchette et al., 2004), or MUSCLE (Edgar, 2004). Based on MSA, a position-specific scoring matrix (PSSM) (Gribskov et al., 1987) is computed to generate the prediction outcome with probability score (Ng and Henikoff, 2001), likelihood ratio (Chun and Fay, 2009), the average distance between targeted species and others in subfamilies (Choi et al., 2012), or the entropy difference (Reva et al., 2007; Hopf et al., 2017). The predictive outcomes are normally continuous values with the designer’s recommended threshold validated in mutation datasets.
Apart from computing scores using empirically rational equations, ML algorithms were commonly utilized as classifiers. Classical models include random forest (RF) (Capriotti et al., 2006), and hidden Markov Model (HMM) (Thomas et al., 2003; Siepel et al., 2005; Garber et al., 2009; Pollard et al., 2010; Shihab et al., 2013). Although they are both ML techniques, the attributes they employ are distinct. For example, PhD-SNP (Capriotti et al., 2006) converted MSA and mutation to a 40-feature variables in support vector machine (SVM). The 40 features are composed of two parts: the first 20 vectors explicitly define the mutation residues, with -1 for the wild-type residue, 1 for the mutation, and 0 for the others. The second set of 20 vectors represents the mutation sequence environment, which is the frequency of each 20 amino acid residue in a 20 amino acid length window centered on the targeted site. Unlike unweighted and balanced MSA, HMM is a probabilistic profile of MSA that captures position-specific information (Krogh et al., 1994). Two different configurations of HMM were observed. One assumed three hidden states: “match,” “insertion,” and “deletion” to build a profile-HMM MSA (Thomas et al., 2003; Shihab et al., 2013), while the other considered a two-hidden state as “conserved” and “non-conserved” according to the phylogenetic information from tree topologies (Siepel et al., 2005; Garber et al., 2009; Pollard et al., 2010).
More recently, a novel unsupervised ML model is utilized to discover patterns and correlations between absolute locations in the MSA, allowing direct observation of both conservation and coevolution (Riesselman et al., 2018; Frazer et al., 2021). This deep generative model captured the latent structure from MSA using Variational Autoencoders (VAEs), which was proved to be an outstanding model for separation of β-lactamase protein family, at the phyla level (Detlefsen et al., 2022). By assuming the observed data s are generated from latent variable z, the decode part of VAE consists of modeling the conditional probability. Hence, the encode part is the neural network modeling of approximate posterior distribution (Riesselman et al., 2018; Frazer et al., 2021).
ML models’ predictions were normally given as log odds ratio scores between the probabilities of “substitution” and “wild-type” or “conserved” and “non-conserved”. In other words, under wild-type or neutral model, higher scores represent higher probability of unexpected substitution, thus are more evolutionary constraint.
There are two considerations regarding homologous sequence-based predictions (Eilbeck et al., 2017). Firstly, many known disease-causing alleles reside in poorly or non-conserved regions will be false-negatively classified as neutral by predictors. Secondly, the tools are inadequate for predicting stop-gain and frameshift variations since they are not included in other organisms in the MSA (Eilbeck et al., 2017). The stop-gain and frameshift variants are rated as “HIGH” impact on biological sequence in annotation tools, e.g., VEP (McLaren et al., 2010) and SnpEff (Cingolani et al., 2012). But the impact on protein is not always concordant. The amino acid changes seem to be tolerant especially the ones located near C-terminal of protein (MacArthur et al., 2012). Some frameshift variants, even in homozygous state, were frequently observed among population suggesting limited impact on human health (Eilbeck et al., 2017). Thus, additional information such as protein structure might help improve the predictive power and efficiency of the predictors, which will be discussed in the following subsections in more detail.
Apart from the primary structure of protein, the folding and stability are also essential for protein function normally. Early findings of variants that affect protein structure leading to aberrant phenotypes can be dated back to the 1950s, when the amino acid substitution in the half molecule of hemoglobin was discovered to cause sickle cell anemia (Ingram, 1957). Since then, thousands of mutations (Giardine et al., 2014) were described to impact on the function (increase (Jones et al., 1979) or decrease (Bonaventura and Riggs, 1968) oxygen affinity), stability (Martinez et al., 1977) and conformation (Moo-Penn et al., 1988) of hemoglobin. Indeed, missense variants also affect protein expression (Haraksingh and Snyder, 2013), post-translational modification (Kim et al., 2015) or binding affinity (Pires et al., 2015; Morningstar-Kywi et al., 2021).
An estimation of ∼75% disease-causing variants directly lead to protein destabilization, making protein stability the major contributor to disease pathology (Yue et al., 2005), whereas ∼7% variants in disease dataset also have functional role (Yue et al., 2014). The location of the mutation has a preference. In comparison to polymorphisms, disease-causing mutations predominantly impact the core of the protein, whereas ∼70% are found in structural and functionally essential regions (Sunyaev et al., 2000; de Beer et al., 2013). Protein-protein interfaces are hot spots for disease-causing nsSNVs (David et al., 2012; Petukh et al., 2015). Again, disease-causing variations were 49% more likely (interface core vs interface rim odds ratio (OR) 1.49, 95% CI 1.24–1.80, p < 0.00001) to be found in the interface core than in the rim, possibly due to their differences in energy contribution to protein stability, physicochemical and evolutionary properties (David and Sternberg, 2015).
Typically, nsSNVs impact on protein stability is estimated by computing the variation of Gibbs free energy change (∆∆Gf) resulting from an amino acid substitution. Physical effective energy function, statistical potential function, and empirical defined potential function are the three types of energy computing methodologies (Guerois et al., 2002). Because the first function is computationally intensive, the latter two are more frequently utilized. Structure-based predictors of protein stability mainly attribute to empirical potentials that integrate physical and statistical structure-related energy components (Guerois et al., 2002), and ML techniques (Dehouck et al., 2009; Laimer et al., 2015).
In theory, these approaches should potentially give greater insights into the mutation effect than the homologous sequence-based predictors since they are built on the direct impact of mutation on protein structure and function. However, the truth is that protein-based predictors are still limited because of the unbalance and intrinsic variability of the thermodynamic data and their prediction performance (Sanavia et al., 2020). On one hand, despite that the Protein Data Bank (PDB) (Berman et al., 2000) contains over 50,000 human protein records, many of them are redundant, covering only 70% of reference human proteome at a sequence identity level higher than 30% (Somody et al., 2017). The development of AlphaFold2 (Tunyasuvunakool et al., 2021), to an extent increases the protein structure coverage; but its capability to predict the impact of single mutation is questionable (Pak et al., 2021; Buel and Walters, 2022). On the other hand, sequence-based techniques, under certain circumstances, outperform structure-based stability prediction tools (Hoie et al., 2022). Thus, combining sequence with structural information may aid in improving prediction capacity of variant impact.
The approaches of this category consider both the previously described homologous sequence and protein structure information. Predictions take benefit from the combination of homology sequence information (e.g., conservative scores), and the structure features, such as hydropathy, polarity, backbone angles and electrostatic interactions, supplemented with energy features and biochemical features such as solvent accessible surface area of the interface (Kulshreshtha et al., 2016). Those features are sometimes transformed or selected for model training to achieve high prediction efficiency. Sometimes hundreds of features might be incorporated into the final model (Niroula et al., 2015). Algorithmically, supervised ML approaches including SVM (Calabrese et al., 2009; Li et al., 2009), naïve bayes classifier (Adzhubei et al., 2010), neural network (NN) (Hecht et al., 2015), RF (Carter et al., 2013; Niroula et al., 2015) and boosted tree regression (Zhou et al., 2016) are commonly applied in the multiple features predictors.
Meta-predictors are tools that make predictions by integrating results of pre-existing predictors. The term “meta-” sometimes corresponds to the term “consensus” in other studies (Bendl et al., 2014). The basic idea behind meta-predictors is to leverage on potential complimentary performance of selected predictors in classifying variants.
There are mainly two improvements regarding meta-predictors comparing to aforementioned counterparts. First of all, meta-predictors give a comprehensive evaluation on the selected pre-existing tools. Each predictor has its own metric and scale making it difficult to compare across multiple predictors hindering the simultaneous usage. Meta-predictors have their own way to interpret scores from selected tools, by transforming to a comparable range as normalized scores (Bendl et al., 2014) or binary values (Gonzalez-Perez and Lopez-Bigas, 2011). In addition, meta-predictors are able to improve prediction performance by integrating prediction scores from different predictors, which allows the avoidance of bias and anti-generalization by single predictors (Kircher et al., 2014).
In terms of missing value, where partly pre-existing tools fail to predict, some meta-predictors impute them using deleterious/neutral threshold (Capriotti et al., 2013), average score (Kircher et al., 2014; Quang et al., 2015), fixed score (Quinodoz et al., 2022), the maximal pathogenic score (Jagadeesh et al., 2016), or a flexible imputation using average value of k-nearest neighbors (Ioannidis et al., 2016) and Bayesian principle component analysis (BPCA) (Dong et al., 2015). There is currently no gold standard for imputation. Although machine-learning imputation appears to be more accurate (Brock et al., 2008; Wei et al., 2018), meta-predictor builders revealed that missing values account for less than 10% of their training and testing datasets (Dong et al., 2015), making the imputation methods less significant difference.
While prediction performance studies suggested that meta-predictors surpassed other counterparts (Tian et al., 2019), concerns regarding circularity occurred, which will be discussed in Section 3.3.
A polymorphism is defined as an alteration in DNA sequence found in the general population at a MAF greater than 1%. According to The American College of Medical Genetics (ACMG) and the Association for Molecular Pathology (AMP) guidelines for clinical variant interpretation, a variant with >5% MAF is considered as a stand-alone support for benign interpretation for a rare Mendelian disorder (Richards et al., 2015). This is supported by the “neutral theory”, which defines neutral variants as the ones settled in the population through random genetic drift causing neither harmful nor beneficial effect to the survival of individual organisms (Kimura, 1979). When training and validating predictors, variants with higher than specified allele frequency (e.g., 5% or 1%) from population-scale databases were usually denoted as benign or neutral. However, predictors in predicting neutral variants differ greatly in capacity and specificity. For example, PON-P2 (Niroula et al., 2015) had a 95% specificity, while the poorest predictor incorrectly categorized more than one-third of polymorphisms as disease-causing (Niroula and Vihinen, 2019). Classifying the impact of variants according to their MAF were further argued by different hypothesis including “rare variant for Mendelian disease” (Pritchard and Cox, 2002), “Common disease, common variant” (CDCV) and “Common disease/rare variant” (CDRV).
Researchers now have access to an exquisitely detailed view of the landscape of common and rare human genetic variants. Another issue that predictors should be careful with when utilizing MAF is that MAF is largely dependent on the population size and varies among subpopulations leading to population stratification (Eilbeck et al., 2017). For example, rs79444516, which is common in African population (13%), exhibited its extreme rareness in European and Asian population, with MAF <0.05%. When estimated in the mixture population, the MAF is 1.2% which will cause confounding classification. Varied MAF for the same variant because of different scales of sample size could be largely mitigated with the completion of the huge population-scale projects.
To better utilize MAF in prediction, ClinPred (Alirezaie et al., 2018), a meta-predictor using ML approach, employs MAFs from diverse populations as part of their features, instead of classifying variants based on single arbitrary MAF cutoffs. Together with feature scores from 16 pre-existing tools, ClinPred trains on clinically curated pathogenic and benign datasets and outperforms other meta-predictors when applied to datasets of rare diseases and cancer (Alirezaie et al., 2018). Therefore, MAFs from population data is capable to enhance the prediction. Similarly, more and more tools (Chennen et al., 2020; do Nascimento et al., 2020; Lai et al., 2020; Li et al., 2020) integrated MAFs as predicting features and achieved competitive performance on pathogenicity prediction.
The ultimate goal of the variant prediction tools is to accelerate the development of precision medicine. Majority of the strategies discussed above aim to estimate disease occurrence based on the assumption that changes in protein function leads to a decrease in organismal fitness (Boucher et al., 2016). They are trained in a large-scale datasets in a genome-wide and pan-disease manner neglecting the complexity among different diseases and making the prediction results suboptimal (Dorfman et al., 2010). Therefore, with the necessity to precisely estimate the impact of variants on specific disease/phenotype, a class of disease-, gene-, phenotype-specific prediction has emerged.
The phenotype-targeting predictors range widely from common cardiac (Zhang et al., 2021), cancer (Kaminker et al., 2007), and neurodegenerative disease (Ahmed et al., 2015), to rare diseases, such as methylmalonic acidemia (Peng et al., 2019), X-linked incomplete Congenital Stationary Night Blindness (Sallah et al., 2020) and Pompe disease (Adhikari, 2019). More details are presented in Table 2. There are over 13,000 terms defined in HPO. While LSDBs provide benchmarked variant datasets, such as COSMIC, CIViC and OncoKB for cancer, which can be utilized for disease-specific predictors construction, limited datasets are available for majority of phenotypes. For a particular disease/phenotype, the training and validation datasets can be prepared by curation of variants and genes from literatures, or re-analysis of unpublished sequencing data of case-control cohorts, followed by manually classification using recognized guidelines such as ACMG/AMP. The scale of the curated databases for different diseases/phenotypes varies in gene (from one to hundreds) and variant number (from thousands to millions) which is largely dependent on the number of relevant publications.
With the curated databases, most of this category of predictors utilize ML methodology. They can be grouped into three classes. The first class overlaps with previous mentioned categories but has distinct characteristic. It includes sequence and structure combination-based (Sallah et al., 2020; Draelos et al., 2022), sharing the same strategy with previously mention predictors in Section 3.1.3, and meta-predictors (Jordan et al., 2011; Bu et al., 2022) similar to the ones discussed in Section 3.1.4. The distinct characteristic is the difference in training and validation datasets selection. Also, disease-related genes are known, making predictors capable of constructing sub-model for each gene, resulting in better prediction performance (Fang et al., 2022). The second class aims to optimize pre-existing predictor, usually sequence-based model, by re-constructing MSAs and phylogenetic tree of targeted gene(s) (Niroula and Vihinen, 2015; Fortuno et al., 2018). These predictors share the same strategy as their precursors, with distinct features selection. The third class predicts variants in a comprehensive and robust way, utilizing additional rule-based classification system. For example, CancerVar (Li et al., 2022), integrates rule-based categorization with ML-based meta-predictor scores to interpret the predicting clinical significance.
These well-calibrated and sculpted predictors demonstrate their capability in targeted sequencing disease-specific panels to the utmost (Peng et al., 2019). In contrast, their ability to generalize is then questioned. When utilizing these techniques, note in mind the key target phenotypes and genes.
As dozens of predictors exist, choosing the appropriate one(s) becomes challenging for end users. Several assessment criteria, such as sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV), accuracy, and Matthews correlation coefficient (MCC), are commonly used to demonstrate model performance (Vihinen, 2012). The values for sensitivity, specificity, PPV, NPV, and accuracy range from 0 to 1, with higher values indicating better performance. MCC benefits from true and false positives and negatives with values on a scale of -1 to 1, with values closer to 1 indicating perfect prediction. Furthermore, a visualization measurement, receiver operating characteristics (ROC) analysis is frequently used to intuitively compare the area under the ROC curve (AUC) of multiple predictors (Vihinen, 2012). For non-intersecting curves, the AUC value closer to 1 suggests better overall performance, while a value of 0.5 indicates random and useless classification.
Most predictors, when developed, would be assessed using respective training and validation datasets presenting supreme or acceptable performance. However, evaluation using consensus datasets would be more informative for tool selection.
There are dozens of comparison studies on the performance assessment of different selected tools using different benchmark datasets. When Performance evaluation of pathogenicity-computation methods for missense variants, meta-predictors such as REVEL, Meta-SNP, generally have better performance and stronger evidence in clinical interpretation (Accetturo et al., 2020; Cubuk et al., 2021; Anderson and Lassmann, 2022). In the assessment of 23 predictors, Li et al. (Li et al., 2018) revealed that meta-predictors achieved higher AUC than others of sequence-based and structure-based predictors using the ClinVar benchmark dataset, indicating better performance of meta-predictors. However, when regarding somatic variants and PPARG gene benchmark datasets, meta-predictors and structure-based predictors exhibited comparable performance (AUC>0.8) (Li et al., 2018), and were superior to homology sequence-based predictors (AUC>0.7). Hassan et al. (Hassan et al., 2019a) revealed that meta-predictor which integrated 4 pre-existing prediction scores, outperformed other 8 predictors achieving ∼10%, 20%, 15% improvement in specificity, sensitivity and AUC, respectively.
The performance of different categories are not always consistent, and sometimes are contradictory. Poon’s study (Poon, 2021) on BRCA1/2 datasets revealed that SIFT and PolyPhen2’s performance differed among genes. Meléndez-Aranda et al. (Melendez-Aranda et al., 2019) compared the performance of 6 in silico tools on 215 missense mutations in hemophilia B causative gene F9, and the results showed that the most popular tool, SIFT, was the most accurate. When applying to a somatic dataset containing 4319 somatic missense variants, the performance of SIFT was sub-optimal (Suybeng et al., 2020). As a result, it is critical to have pre-knowledge of your testing data and predictive goal when selecting appropriate tools.
In order to address the confounding situation and objectively determine the appropriate usage and accuracy of predictors, the Critical Assessment of Genome Interpretation (CAGI) (Andreoletti et al., 2019) community started their experiments in 2010. Until now, there are six editions with 63 challenges and over 50 articles released. Participants predict the phenotypic impact of unpublished genetic variants collected from experimental and clinical labs provided by CAGI. Later, independent assessors test the predictions against experimental characterized phenotypes, and the results will be presented at the CAGI conference and published in special journal issues. The challenges released include a wide range of topics, from nsSNVs to splicing variants, and from disease panels to databases including curated variants. However, the reality of the outcome is frequently far more complex than the challenges’ initial objective. Predictors with superior performance in one challenge, would fail to call the pathogenicity of variants in other datasets (Katsonis and Lichtarge, 2019; Savojardo et al., 2019). Complex gene datasets caused divergence predictions and confounding outcomes, raising concerns about the possibility of experimental mistakes as the basis of disagreement (Miller et al., 2019). All the above suggested the caution when interpreting the evaluation results.
Majority of predictors are trained, validated and tested using benchmarked sets of variants with explicit classification labels. When evaluated 10 predictors across major public databases, Grimm et al. (2015) raised concerns about “circularity” involving in the usage of predictors and conduction of comparative studies. The term “circularity” refers to the situation that same variants are recursively used in both training and evaluating models.
“Type 1 circularity” refers to the overlap between training and evaluation particularly for supervised ML-based predictors, resulting in poor generalization on new data (Grimm et al., 2015). Selecting predictions from unsupervised tools as features or filtering overlapping sets during training might assist to minimize the “type 1 circularity” during model construction (Alirezaie et al., 2018; Won et al., 2021). Furthermore, avoiding overuse of individual dataset (Vihinen, 2013; Weber et al., 2019) and choosing benchmark database which addressed overlapped issue (Sasidharan Nair and Vihinen, 2013; Sarkar et al., 2020) also helps when conducting comparative studies.
Grimm et al. (2015) observed that weighted FatHMM (Shihab et al., 2013) achieved outstanding performance in 2 datasets but severe drop in performance in subset from SwissVar. They found that the ratio of pathogenic and neutral variants in the same protein family was the key element for weighting scheme, leading to higher pathogenic score assigned to both neutral and pathogenic variants in the same gene with higher ratio (Grimm et al., 2015). This strategy made weighted FatHMM statistically successful in some datasets, but ultimately inappropriate. Therefore, they defined the “Type 2 circularity” as the circumstance in which all variants from the same gene are jointly labeled as pathogenic or neutral. To address this problem, it was suggested to use datasets with an appropriate pathogenic-to-neutral ratio and avoid genes with exclusive pathogenic or neutral variations when reporting performance (Bu et al., 2022; Quinodoz et al., 2022).
Another concern is about “collinearity,” which generally occurs with the regression models. ‘Collinearity’ refers to the circumstance in which significant correlation between two or more feature variables resulting in independent regression coefficients estimation problems and leading to redundancy in the set of variables (Bayman and Dexter, 2021). This situation might be mitigated via feature selection and estimator modification (Zheng et al., 2020; Chan et al., 2022). From another perspective, “collinearity” should not be a problem because more complicated machine learning algorithms including SVM, Random Forest, and Neural Network, can handle large-scale and multi-collinear datasets in a better way (Dong et al., 2015; Perez-Enciso and Zingaretti, 2019).
In-silico approaches combined mathematical strategies with expert opinion allows researchers to analyze the biological meaning of genetic data efficiently and economically (Trisilowati and Mallet, 2012). In-silico predictors on variant effect aids in genome interpretation. The prediction-based categorization provides insight into variant characterization and prioritization.
Regards to large-scale population study, in silico predictors aid in variant classification for pattern overview and comparison at subpopulation level. For example, Palmer et al. (2022) subdivided missense variants by SIFT and PolyPhen2 prediction in research on bipolar disorder (BD) and revealed an obvious enrichment in ultra-rare harmful missense variation outside of confined missense areas, particularly in bipolar II disorder (BD2). This observation contrasted with the findings in schizophrenia cases (Singh et al., 2022) of enrichment within constrained missense regions. The authors speculated this signal may capture something distinct to mood disorders relative to psychotic disorders (Palmer et al., 2022).
For large-scale population, in silico predictors also facilitate the detection of variant-level signals under natural-selection for those living in extreme environments or with a diverse geographic distribution. Deng et al. (2019) ranked variants by calculating the functional importance score (FIS) from four in silico predictors. Based on the ranking of adaptive genetic variants, they revealed a seldom studied gene, TMEM247 with a missense variant rs116983452, to be the most-differentiated functional variant identified between Tibetan and non-Tibetan populations (Deng et al., 2019). When studying non-homogeneous Taiwanese Han population, integrated selection of allele favored by evolution (iSAFE) was incorporated with the CADD functional impact score to identify 16 natural-selection signals by geographic distribution that were unambiguously localized to 5 single genes (Lo et al., 2021). Meanwhile, in the western Roma population, Font-Porterias et al. (2021) categorized missense variants based on GERP, PolyPhen2 and CADD, revealing significant difference in common deleterious variant portion between Roma and non-Roma population. Furthermore, runs of homozygozity (ROH), which are continuous homozygous regions of the DNA sequence, exhibit ancestry-specific patterns of accumulation of deleterious homozygotes.
In addition to characterization for population-level study, predictors have also been widely used for prioritization of disease-causing candidates in case-control or pedigree studies, finally leading to the identification of genotype-phenotype association. There are commonly two strategies for variant prioritization in which predictors help. Several frameworks and platforms are listed in Table 3.
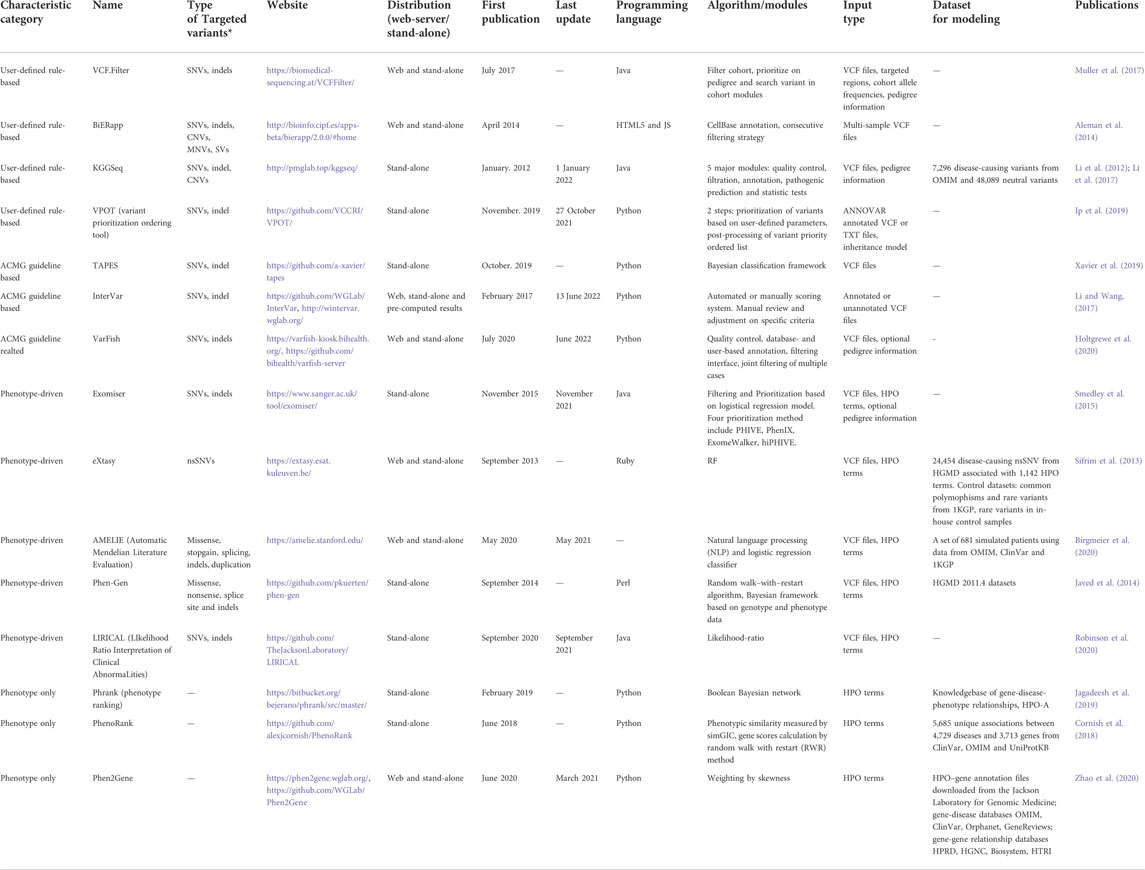
TABLE 3. Representative prioritization frameworks and tools.
First, empirical criteria are used to filter variations. With high quality variants, many studies (Ma et al., 2013; Blue et al., 2014) performed prioritization based on in silico predictions, MAF in population database and control groups, inheritance pattern, and functional effect. By this method, less than 10 variants are distilled out of hundreds of thousands obtained from WES analysis. Following the validation of orthogonal assays (e.g., Sanger sequencing), true positive causal candidates will be examined for the functional effect on protein in vitro and/or in vivo. The relationship between variants-phenotype is therefore thoroughly investigated. Several user-friendly rule-based frameworks (Coutant et al., 2012; Li et al., 2012; Aleman et al., 2014; Muller et al., 2017) have been built to make the filtering procedure easier to implement. Researchers can set their own criteria and get the findings in readable files with detailed annotation information. The prioritization can also be supplemented with adoption of consensus recommendations, such as ACMG/AMP standards and guidelines (Richards et al., 2015). The guideline includes a comprehensive set of definitions and criteria for variation interpretation, ranging from standardized nomenclature to evidence-based rating yielding a five-tier terminology system outcome. Results from in silico predictors are accounted as “supporting” evidence for benign or pathogenic classification. Some automatic tools (Li and Wang, 2017; Xavier et al., 2019) have also been developed for variant classification based on the guidelines, although the manual classification by professional geneticists would be deemed as the golden standard.
The second strategy refers to phenotype-driven frameworks, which combine phenotype and variants data for prioritization and interpretation. Clinical diagnosis would be straightforward when the disease is known. However, before the identification of candidate disease, the procedure to explain a set of clinical features is challenging due to the absence or presence of unrelated features and various degrees of specificity (Kohler et al., 2009). To extract standardized and normalized phenotypic terminologies from sparse clinical abnormalities in case studies, some tools like Phenomizer (Kohler et al., 2009) and Doc2HPO (Liu et al., 2019) are recommended to map the clinical symptoms to the list of known disorders and estimate the significance of each disease match. Prediction scores from in silico predictors are integrated in this kind of framework as “pathogenicity” or “deleteriousness” features. Most of phenotype-driven tools (Sifrim et al., 2013; Javed et al., 2014; Smedley et al., 2015; Birgmeier et al., 2020; Robinson et al., 2020) require variant files and HPO terms as input, while some tools (Cornish et al., 2018; Jagadeesh et al., 2019; Zhao et al., 2020) require only HPO terms. Yuan et al. (2022) investigated causal-gene prioritizing performance of both types on two benchmark datasets in a recent comparative study and revealed that former ones performed better overall than latter ones. Their results also indicated the complementarity of multiple phenotype-driven tools towards a viable integrated strategy may improve diagnostic efficiency (Yuan et al., 2022).
In this review, we firstly summarized the database resources frequently used during predictor development. We then discussed the rational, necessity and limitations for the newly categorized predictors: homologous sequence-based, structural-based, combination of sequence and structural, meta-predictors, population-based, and gene-, phenotype-, disease-specific predictors. Predictor performance as well as their limitations and possible remedies were then outlined. The application of the predictors in real studies was finally presented demonstrating their efficient assistance in variant characterization and prioritization, as well as the discovery of genotype-phenotype association.
When building predictors, unambiguous labeled datasets are critical. Avoiding overlapping and contradicting data, as well as balancing the positive-negative ratio in training and validation datasets, will definitely minimize the negative influence of circularity. Further examination on the collinearity between/among feature variables will facilitate the optimization of prediction models, even though some algorithms are literally not affected.
Among the predictors, meta-predictors outperform others in general; however, their prediction performance is considerably discounted in some disease-specific datasets, raising concern about their applications especially in clinical settings (Schiemann and Stowell, 2016; Mahmood et al., 2017). Employment of disease-, gene-, phenotype-specific predictors can to an extent solve the above issue. When selecting predictors for a particular study, efforts should be given on screening whether the genes and phenotype predictor calibrated perfectly matching your research, and understanding the scope and predictive performance of each predictor. On the other hand, we look forward to more specialized predictors sculpted for a variety of phenotypes covering both common and rare diseases.
According to Variation Ontology (VariO) (Vihinen, 2014), variant impacts on protein level can be annotated with effects on function, structure and property. Variants impact on protein functional or property effects can be classified as follows: abundance, which includes gene dosage, expression, degradation and mis-localization; activity, which includes enzymatic, kinetic and regulation; enzymatic specificity, and molecular affinity (Vihinen, 2021). Most of above-mentioned predictors computed the possibility of pathogenic effect on protein function and structure in a broad range, rather the effects on protein abundance, activity or affinity properties separately. This may indicate a challenging future orientation of variant predictors development.
The correlation between variants pathogenic prediction on protein function or structure and abnormal clinical outcomes are validated by experimental facts at the current stage. For certain phenotypes, an evident enrichment of deleterious variants in a set of disease-related genes, such as the increased mutational burden in essential genes in autism spectrum disorder (Ji et al., 2016), WNT signaling genes in myelomeningocele (Hebert et al., 2020), a set of 5 genes in epilepsy (Leu et al., 2015). The gap between observed higher burden genes and clinical phenotype is then bridged by functional or mechanical experimental studies. For example, meiocytes with pathogenic mutation p.S167L in HSF2BP found in premature ovarian insufficiency (POI) patients from a family, showed a reduced number of foci formed by the recombinases RAD51/DMC1, leading to crossover defect, which provided an insight into the molecular mechanism of mutation in POI and subfertility (Felipe-Medina et al., 2020). Currently, variant impact predictors are insufficient for indicating molecular mechanism of pathogenicity. However, the advancement of protein structure prediction may assist the interpretation of pathogenic variants since structural information gives useful insights in evaluating variant impact on protein or biological systems (Diwan et al., 2021).
Impacts of mutations on protein synthesis includes transcriptional and translational influences. For SNVs, the impact on transcription involves in changes in transcript sequence and influence in gene regulation (Haraksingh and Snyder, 2013). Tools for predicting impact on gene regulation have been timely and systematically reviewed by other studies (Li et al., 2015; Ohno et al., 2018; Rojano et al., 2019; Canson et al., 2020). In terms of translation, SNVs-induced amino acid substitution causes protein structure and function abnormalities, and the prediction methods have been explored in this study. The deeper association between SNVs for protein folding and post-translation modification is still being investigated.
With the development of a cutting-edge structure prediction tool, AlphaFold2, the unstructured human protein narrowed down to less than 30% (Porta-Pardo et al., 2022). However, examples showed that AlphaFold2 was not capable for predicting protein structure modification caused by pathogenic mutations, particularly those having experimentally proven destabilizing effect (Buel and Walters, 2022). The reasons for this limitation may relate to the bioinformatics and physical methodologies utilized in modeling, as well as the resources from protein sequence and PDB structure data employed, instead of the fundamental driving forces of protein folding (Jumper et al., 2021; Buel and Walters, 2022). The AlphaFold team is presently considering solutions for new mutations, which may give better prediction on unfolding to folding state, based on protein physics instead of sequence evolutionary (Callaway, 2022). We anticipate that its success will usher in a new age of human genetic research, including the acceleration of in silico functional and mechanical genotype-phenotype association investigations.
Finally, although the variant effect predictors greatly help the genomic interpretation, end-users should keep in mind that the predictor’s role is only an assistance to clinical diagnosis, and merely a starting point (Eilbeck et al., 2017). The unequal relationship between predicted damaging effect and pathogenicity warns their usage. In addition, under some circumstances, the predicted scores overstating the effect of uncommon mutations, will cause inflated estimation affecting the specificity and sensitivity (Lanktree et al., 2018). Therefore, experimental validations, the golden standard in variant impact evaluation, are still indispensable.
YL and DC designed the structure of the review. YL wrote the draft manuscript, curated data, organized tables and figure. DC supervised the work and revised the manuscript. PC and WY supervised and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Shenzhen Fundamental Research Program, China (no. JCYJ20210324114807019).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.981005/full#supplementary-material
Accetturo, M., Bartolomeo, N., and Stella, A. (2020). In-silico analysis of NF1 missense variants in ClinVar: Translating variant predictions into variant interpretation and classification. Int. J. Mol. Sci. 21 (3), E721. doi:10.3390/ijms21030721
Adhikari, A. N. (2019). Gene-specific features enhance interpretation of mutational impact on acid alpha-glucosidase enzyme activity. Hum. Mutat. 40 (9), 1507–1518. doi:10.1002/humu.23846
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7 (4), 248–249. doi:10.1038/nmeth0410-248
Ahmed, A. B., Znassi, N., Chateau, M. T., and Kajava, A. V. (2015). A structure-based approach to predict predisposition to amyloidosis. Alzheimers Dement. 11 (6), 681–690. doi:10.1016/j.jalz.2014.06.007
Ainscough, B. J., Griffith, M., Coffman, A. C., Wagner, A. H., Kunisaki, J., Choudhary, M. N., et al. (2016). DoCM: A database of curated mutations in cancer. Nat. Methods 13(10), 806–807. doi:10.1038/nmeth.4000
Aleman, A., Garcia-Garcia, F., Salavert, F., Medina, I., and Dopazo, J. (2014). A web-based interactive framework to assist in the prioritization of disease candidate genes in whole-exome sequencing studies. Nucleic Acids Res. 42, W88–W93. Web Server issue). doi:10.1093/nar/gku407
Alirezaie, N., Kernohan, K. D., Hartley, T., Majewski, J., and Hocking, T. D. (2018). ClinPred: Prediction tool to identify disease-relevant nonsynonymous single-nucleotide variants. Am. J. Hum. Genet. 103 (4), 474–483. doi:10.1016/j.ajhg.2018.08.005
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215 (3), 403–410. doi:10.1016/S0022-2836(05)80360-2
Amberger, J. S., Bocchini, C. A., Scott, A. F., and Hamosh, A. (2019). OMIM.org: Leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 47 (D1), D1038-D1043–D1043. doi:10.1093/nar/gky1151
Anderson, D., and Lassmann, T. (2022). An expanded phenotype centric benchmark of variant prioritisation tools. Hum. Mutat. 43 (5), 539–546. doi:10.1002/humu.24362
Andreoletti, G., Pal, L. R., Moult, J., and Brenner, S. E. (2019). Reports from the fifth edition of CAGI: The critical assessment of genome interpretation. Hum. Mutat. 40 (9), 1197–1201. doi:10.1002/humu.23876
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 25 (1), 25–29. doi:10.1038/75556
Ayme, S., Urbero, B., Oziel, D., Lecouturier, E., and Biscarat, A. C. (1998). Information on rare diseases: The Orphanet project. La Rev. Med. Interne 19 (3), 376S–377S. doi:10.1016/s0248-8663(98)90021-2
Azaiez, H., Booth, K. T., Ephraim, S. S., Crone, B., Black-Ziegelbein, E. A., Marini, R. J., et al. (2018). Genomic landscape and mutational Signatures of deafness-associated genes. Am. J. Hum. Genet. 103 (4), 484–497. doi:10.1016/j.ajhg.2018.08.006
Baker, W., van den Broek, A., Camon, E., Hingamp, P., Sterk, P., Stoesser, G., et al. (2000). The EMBL nucleotide sequence database. Nucleic Acids Res. 28 (1), 19–23. doi:10.1093/nar/28.1.19
Banck, H., Dugas, M., C, M. U.-T., and Sandmann, S. (2021). Comparison of open-access databases for clinical variant interpretation in cancer: A case study of MDS/AML. Cancer Genomics Proteomics 18 (2), 157–166. doi:10.21873/cgp.20250
Banerjee, S., Raman, K., and Ravindran, B. (2021). Sequence neighborhoods enable reliable prediction of pathogenic mutations in cancer genomes. Cancers (Basel) 13 (10), 2366. doi:10.3390/cancers13102366
Bayman, E. O., and Dexter, F. (2021). Multicollinearity in logistic regression models. Anesth. Analg. 133 (2), 362–365. doi:10.1213/ANE.0000000000005593
Bendl, J., Stourac, J., Salanda, O., Pavelka, A., Wieben, E. D., Zendulka, J., et al. (2014). PredictSNP: Robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput. Biol. 10 (1), e1003440. doi:10.1371/journal.pcbi.1003440
Berliner, N., Teyra, J., Colak, R., Garcia Lopez, S., and Kim, P. M. (2014). Combining structural modeling with ensemble machine learning to accurately predict protein fold stability and binding affinity effects upon mutation. PLoS One 9 (9), e107353. doi:10.1371/journal.pone.0107353
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., et al. (2000). The protein Data Bank. Nucleic Acids Res. 28 (1), 235–242. doi:10.1093/nar/28.1.235
Birgmeier, J., Haeussler, M., Deisseroth, C. A., Steinberg, E. H., Jagadeesh, K. A., Ratner, A. J., et al. (2020). AMELIE speeds Mendelian diagnosis by matching patient phenotype and genotype to primary literature. Sci. Transl. Med. 12 (544), eaau9113. doi:10.1126/scitranslmed.aau9113
Birrane, G., Soni, A., and Ladias, J. A. A. (2006). X-ray Structure of the BRCA1 BRCT mutant M1775K [Online]. Available at: https://gnomad.broadinstitute.org/variant/17-43051071-A-C?dataset=gnomad_r3 (Accessed September 13 2022) [abstract].
Blanchette, M., Kent, W. J., Riemer, C., Elnitski, L., Smit, A. F., Roskin, K. M., et al. (2004). Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 14 (4), 708–715. doi:10.1101/gr.1933104
Blue, G. M., Kirk, E. P., Giannoulatou, E., Dunwoodie, S. L., Ho, J. W., Hilton, D. C., et al. (2014). Targeted next-generation sequencing identifies pathogenic variants in familial congenital heart disease. J. Am. Coll. Cardiol. 64 (23), 2498–2506. doi:10.1016/j.jacc.2014.09.048
Bonaventura, J., and Riggs, A. (1968). Hemoglobin Kansas, a human hemoglobin with a neutral amino acid substitution and an abnormal oxygen equilibrium. J. Biol. Chem. 243 (5), 980–991. doi:10.1016/s0021-9258(18)93612-4
Boucher, J. I., Bolon, D. N., and Tawfik, D. S. (2016). Quantifying and understanding the fitness effects of protein mutations: Laboratory versus nature. Protein Sci. 25 (7), 1219–1226. doi:10.1002/pro.2928
Brock, G. N., Shaffer, J. R., Blakesley, R. E., Lotz, M. J., and Tseng, G. C. (2008). Which missing value imputation method to use in expression profiles: A comparative study and two selection schemes. BMC Bioinforma. 9, 12. doi:10.1186/1471-2105-9-12
Bromberg, Y., and Rost, B. (2007). Snap: Predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 35 (11), 3823–3835. doi:10.1093/nar/gkm238
Brookes, A. J., and Robinson, P. N. (2015). Human genotype-phenotype databases: Aims, challenges and opportunities. Nat. Rev. Genet. 16 (12), 702–715. doi:10.1038/nrg3932
Bu, F., Zhong, M., Chen, Q., Wang, Y., Zhao, X., Zhang, Q., et al. (2022). DVPred: A disease-specific prediction tool for variant pathogenicity classification for hearing loss. Hum. Genet. 141 (3-4), 401–411. doi:10.1007/s00439-022-02440-1
Buel, G. R., and Walters, K. J. (2022). Can AlphaFold2 predict the impact of missense mutations on structure? Nat. Struct. Mol. Biol. 29 (1), 1–2. doi:10.1038/s41594-021-00714-2
Calabrese, R., Capriotti, E., Fariselli, P., Martelli, P. L., and Casadio, R. (2009). Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum. Mutat. 30 (8), 1237–1244. doi:10.1002/humu.21047
Callaway, E. (2022). What's next for AlphaFold and the AI protein-folding revolution. Nature 604 (7905), 234–238. doi:10.1038/d41586-022-00997-5
Canson, D., Glubb, D., and Spurdle, A. B. (2020). Variant effect on splicing regulatory elements, branchpoint usage, and pseudoexonization: Strategies to enhance bioinformatic prediction using hereditary cancer genes as exemplars. Hum. Mutat. 41 (10), 1705–1721. doi:10.1002/humu.24074
Capriotti, E., Altman, R. B., and Bromberg, Y. (2013). Collective judgment predicts disease-associated single nucleotide variants. BMC Genomics 14 (3), S2. doi:10.1186/1471-2164-14-S3-S2
Capriotti, E., Calabrese, R., and Casadio, R. (2006). Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics 22 (22), 2729–2734. doi:10.1093/bioinformatics/btl423
Carter, H., Douville, C., Stenson, P. D., Cooper, D. N., and Karchin, R. (2013). Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genomics 14, S3. doi:10.1186/1471-2164-14-S3-S3
Chan, J. Y.-L., Leow, S. M. H., Bea, K. T., Cheng, W. K., Phoong, S. W., Hong, Z.-W., et al. (2022). Mitigating the multicollinearity problem and its machine learning approach: A review. Mathematics 10 (8), 1283. doi:10.3390/math10081283
Chennen, K., Weber, T., Lornage, X., Kress, A., Bohm, J., Thompson, J., et al. (2020). Mistic: A prediction tool to reveal disease-relevant deleterious missense variants. PLoS One 15 (7), e0236962. doi:10.1371/journal.pone.0236962
Choi, Y., and Chan, A. P. (2015). PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31 (16), 2745–2747. doi:10.1093/bioinformatics/btv195
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R., and Chan, A. P. (2012). Predicting the functional effect of amino acid substitutions and indels. PLoS One 7 (10), e46688. doi:10.1371/journal.pone.0046688
Chun, S., and Fay, J. C. (2009). Identification of deleterious mutations within three human genomes. Genome Res. 19 (9), 1553–1561. doi:10.1101/gr.092619.109
Cingolani, P., Platts, A., Wang le, L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. (Austin) 6 (2), 80–92. doi:10.4161/fly.19695
Ciriello, G., Gatza, M. L., Beck, A. H., Wilkerson, M. D., Rhie, S. K., Pastore, A., et al. (2015). Comprehensive molecular portraits of invasive lobular breast cancer. Cell. 163 (2), 506–519. doi:10.1016/j.cell.2015.09.033
Consortium, U. K., Walter, K., Min, J. L., Huang, J., Crooks, L., Memari, Y., et al. (2015). The UK10K project identifies rare variants in health and disease. Nature 526 (7571), 82–90. doi:10.1038/nature14962
Cooper, G. M., and Shendure, J. (2011). Needles in stacks of needles: Finding disease-causal variants in a wealth of genomic data. Nat. Rev. Genet. 12 (9), 628–640. doi:10.1038/nrg3046
Cooper, G. M., Stone, E. A., Asimenos, G., Program, N. C. S., Green, E. D., Batzoglou, S., et al. (2005). Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 15 (7), 901–913. doi:10.1101/gr.3577405
Cornish, A. J., David, A., and Sternberg, M. J. E. (2018). PhenoRank: Reducing study bias in gene prioritization through simulation. Bioinformatics 34 (12), 2087–2095. doi:10.1093/bioinformatics/bty028
Coutant, S., Cabot, C., Lefebvre, A., Leonard, M., Prieur-Gaston, E., Campion, D., et al. (2012). Eva: Exome Variation Analyzer, an efficient and versatile tool for filtering strategies in medical genomics. BMC Bioinforma. 13, S9. Suppl 14. doi:10.1186/1471-2105-13-S14-S9
Cubuk, C., Garrett, A., Choi, S., King, L., Loveday, C., Torr, B., et al. (2021). Clinical likelihood ratios and balanced accuracy for 44 in silico tools against multiple large-scale functional assays of cancer susceptibility genes. Genet. Med. 23 (11), 2096–2104. doi:10.1038/s41436-021-01265-z
Cunningham, F., Moore, B., Ruiz-Schultz, N., Ritchie, G. R., and Eilbeck, K. (2015). Improving the Sequence Ontology terminology for genomic variant annotation. J. Biomed. Semant. 6, 32. doi:10.1186/s13326-015-0030-4
Curtis, C., Shah, S. P., Chin, S. F., Turashvili, G., Rueda, O. M., Dunning, M. J., et al. (2012). The genomic and transcriptomic architecture of 2, 000 breast tumours reveals novel subgroups. Nature 486 (7403), 346–352. doi:10.1038/nature10983
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27 (15), 2156–2158. doi:10.1093/bioinformatics/btr330
David, A., Razali, R., Wass, M. N., and Sternberg, M. J. (2012). Protein-protein interaction sites are hot spots for disease-associated nonsynonymous SNPs. Hum. Mutat. 33 (2), 359–363. doi:10.1002/humu.21656
David, A., and Sternberg, M. J. (2015). The contribution of missense mutations in core and rim residues of protein-protein interfaces to human disease. J. Mol. Biol. 427 (17), 2886–2898. doi:10.1016/j.jmb.2015.07.004
Davydov, E. V., Goode, D. L., Sirota, M., Cooper, G. M., Sidow, A., and Batzoglou, S. (2010). Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 6 (12), e1001025. doi:10.1371/journal.pcbi.1001025
de Beer, T. A., Laskowski, R. A., Parks, S. L., Sipos, B., Goldman, N., and Thornton, J. M. (2013). Amino acid changes in disease-associated variants differ radically from variants observed in the 1000 genomes project dataset. PLoS Comput. Biol. 9 (12), e1003382. doi:10.1371/journal.pcbi.1003382
Dehouck, Y., Grosfils, A., Folch, B., Gilis, D., Bogaerts, P., and Rooman, M. (2009). Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks: PoPMuSiC-2.0. Bioinformatics 25 (19), 2537–2543. doi:10.1093/bioinformatics/btp445
Deng, L., Zhang, C., Yuan, K., Gao, Y., Pan, Y., Ge, X., et al. (2019). Prioritizing natural-selection signals from the deep-sequencing genomic data suggests multi-variant adaptation in Tibetan highlanders. Natl. Sci. Rev. 6 (6), 1201–1222. doi:10.1093/nsr/nwz108
Detlefsen, N. S., Hauberg, S., and Boomsma, W. (2022). Learning meaningful representations of protein sequences. Nat. Commun. 13 (1), 1914. doi:10.1038/s41467-022-29443-w
Diwan, G. D., Gonzalez-Sanchez, J. C., Apic, G., and Russell, R. B. (2021). Next generation protein structure predictions and genetic variant interpretation. J. Mol. Biol. 433 (20), 167180. doi:10.1016/j.jmb.2021.167180
do Nascimento, P. M., Medeiros, I. G., Falcao, R. M., Stransky, B., and de Souza, J. E. S. (2020). A decision tree to improve identification of pathogenic mutations in clinical practice. BMC Med. Inf. Decis. Mak. 20 (1), 52. doi:10.1186/s12911-020-1060-0
Dong, C., Wei, P., Jian, X., Gibbs, R., Boerwinkle, E., Wang, K., et al. (2015). Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 24 (8), 2125–2137. doi:10.1093/hmg/ddu733
Dorfman, R., Nalpathamkalam, T., Taylor, C., Gonska, T., Keenan, K., Yuan, X. W., et al. (2010). Do common in silico tools predict the clinical consequences of amino-acid substitutions in the CFTR gene? Clin. Genet. 77 (5), 464–473. doi:10.1111/j.1399-0004.2009.01351.x
Draelos, R. L., Ezekian, J. E., Zhuang, F., Moya-Mendez, M. E., Zhang, Z., Rosamilia, M. B., et al. (2022). Genesis: Gene-specific machine learning models for variants of uncertain significance found in catecholaminergic polymorphic ventricular Tachycardia and Long QT syndrome-associated genes. Circ. Arrhythm. Electrophysiol. 15 (4), e010326. doi:10.1161/CIRCEP.121.010326
Eberle, M. A., Fritzilas, E., Krusche, P., Kallberg, M., Moore, B. L., Bekritsky, M. A., et al. (2017). A reference data set of 5.4 million phased human variants validated by genetic inheritance from sequencing a three-generation 17-member pedigree. Genome Res. 27 (1), 157–164. doi:10.1101/gr.210500.116
Edgar, R. C. (2004). Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32 (5), 1792–1797. doi:10.1093/nar/gkh340
Eilbeck, K., Lewis, S. E., Mungall, C. J., Yandell, M., Stein, L., Durbin, R., et al. (2005). The sequence ontology: A tool for the unification of genome annotations. Genome Biol. 6 (5), R44. doi:10.1186/gb-2005-6-5-r44
Eilbeck, K., Quinlan, A., and Yandell, M. (2017). Settling the score: Variant prioritization and mendelian disease. Nat. Rev. Genet. 18 (10), 599–612. doi:10.1038/nrg.2017.52
Fang, M., Su, Z., Abolhassani, H., Itan, Y., Jin, X., and Hammarstrom, L. (2022). Vippid: A gene-specific single nucleotide variant pathogenicity prediction tool for primary immunodeficiency diseases. Brief. Bioinform., bbac176. doi:10.1093/bib/bbac176
Felipe-Medina, N., Caburet, S., Sanchez-Saez, F., Condezo, Y. B., de Rooij, D. G., Gomez, H. L., et al. (2020). A missense in HSF2BP causing primary ovarian insufficiency affects meiotic recombination by its novel interactor C19ORF57/BRME1. Elife 9, e56996. doi:10.7554/eLife.56996
Feng, B. J. (2017). Perch: A unified framework for disease gene prioritization. Hum. Mutat. 38 (3), 243–251. doi:10.1002/humu.23158
Findlay, G. M., Daza, R. M., Martin, B., Zhang, M. D., Leith, A. P., Gasperini, M., et al. (2018). Accurate classification of BRCA1 variants with saturation genome editing. Nature 562 (7726), 217–222. doi:10.1038/s41586-018-0461-z
Fokkema, I., Kroon, M., Lopez Hernandez, J. A., Asscheman, D., Lugtenburg, I., Hoogenboom, J., et al. (2021). The LOVD3 platform: Efficient genome-wide sharing of genetic variants. Eur. J. Hum. Genet. 29 (12), 1796–1803. doi:10.1038/s41431-021-00959-x
Fokkema, I., van der Velde, K. J., Slofstra, M. K., Ruivenkamp, C. A. L., Vogel, M. J., Pfundt, R., et al. (2019). Dutch genome diagnostic laboratories accelerated and improved variant interpretation and increased accuracy by sharing data. Hum. Mutat. 40 (12), 2230–2238. doi:10.1002/humu.23896
Font-Porterias, N., Caro-Consuegra, R., Lucas-Sanchez, M., Lopez, M., Gimenez, A., Carballo-Mesa, A., et al. (2021). The counteracting effects of demography on functional genomic variation: The roma paradigm. Mol. Biol. Evol. 38 (7), 2804–2817. doi:10.1093/molbev/msab070
Fortuno, C., James, P. A., Young, E. L., Feng, B., Olivier, M., Pesaran, T., et al. (2018). Improved, ACMG-compliant, in silico prediction of pathogenicity for missense substitutions encoded by TP53 variants. Hum. Mutat. 39 (8), 1061–1069. doi:10.1002/humu.23553
Frazer, J., Notin, P., Dias, M., Gomez, A., Min, J. K., Brock, K., et al. (2021). Disease variant prediction with deep generative models of evolutionary data. Nature 599 (7883), 91–95. doi:10.1038/s41586-021-04043-8
Frederic, M. Y., Lalande, M., Boileau, C., Hamroun, D., Claustres, M., Beroud, C., et al. (2009). UMD-Predictor, a new prediction tool for nucleotide substitution pathogenicity -- application to four genes: FBN1, FBN2, TGFBR1, and TGFBR2. Hum. Mutat. 30 (6), 952–959. doi:10.1002/humu.20970
Fu, W., O'Connor, T. D., Jun, G., Kang, H. M., Abecasis, G., Leal, S. M., et al. (2013). Analysis of 6, 515 exomes reveals the recent origin of most human protein-coding variants. Nature 493 (7431), 216–220. doi:10.1038/nature11690
Ganesan, K., Kulandaisamy, A., Binny Priya, S., and Gromiha, M. M. (2019). HuVarBase: A human variant database with comprehensive information at gene and protein levels. PLoS One 14 (1), e0210475. doi:10.1371/journal.pone.0210475
Garber, M., Guttman, M., Clamp, M., Zody, M. C., Friedman, N., and Xie, X. (2009). Identifying novel constrained elements by exploiting biased substitution patterns. Bioinformatics 25 (12), i54–62. doi:10.1093/bioinformatics/btp190
Gene Ontology, C. (2021). The gene ontology resource: Enriching a GOld mine. Nucleic Acids Res. 49 (D1), D325–D334. doi:10.1093/nar/gkaa1113
Genomes Project, C., Abecasis, G. R., Auton, A., Brooks, L. D., DePristo, M. A., Durbin, R. M., et al. (2012). An integrated map of genetic variation from 1, 092 human genomes. Nature 491 (7422), 56–65. doi:10.1038/nature11632
Genomes Project, C., Auton, A., Brooks, L. D., Durbin, R. M., Garrison, E. P., Kang, H. M., et al. (2015). A global reference for human genetic variation. Nature 526 (7571), 68–74. doi:10.1038/nature15393
Giardine, B., Borg, J., Viennas, E., Pavlidis, C., Moradkhani, K., Joly, P., et al. (2014). Updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res. 42, D1063–D1069. doi:10.1093/nar/gkt911
Gonzalez-Perez, A., and Lopez-Bigas, N. (2011). Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am. J. Hum. Genet. 88 (4), 440–449. doi:10.1016/j.ajhg.2011.03.004
Greenblatt, M. S., Brody, L. C., Foulkes, W. D., Genuardi, M., Hofstra, R. M., Olivier, M., et al. (2008). Locus-specific databases and recommendations to strengthen their contribution to the classification of variants in cancer susceptibility genes. Hum. Mutat. 29 (11), 1273–1281. doi:10.1002/humu.20889
Gribskov, M., McLachlan, A. D., and Eisenberg, D. (1987). Profile analysis: Detection of distantly related proteins. Proc. Natl. Acad. Sci. U. S. A. 84 (13), 4355–4358. doi:10.1073/pnas.84.13.4355
Griffith, M., Spies, N. C., Krysiak, K., McMichael, J. F., Coffman, A. C., Danos, A. M., et al. (2017). CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat. Genet. 49 (2), 170–174. doi:10.1038/ng.3774
Grimm, D. G., Azencott, C. A., Aicheler, F., Gieraths, U., MacArthur, D. G., Samocha, K. E., et al. (2015). The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Hum. Mutat. 36 (5), 513–523. doi:10.1002/humu.22768
Guerois, R., Nielsen, J. E., and Serrano, L. (2002). Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. J. Mol. Biol. 320 (2), 369–387. doi:10.1016/S0022-2836(02)00442-4
Haraksingh, R. R., and Snyder, M. P. (2013). Impacts of variation in the human genome on gene regulation. J. Mol. Biol. 425 (21), 3970–3977. doi:10.1016/j.jmb.2013.07.015
Hassan, M. S., Shaalan, A. A., Dessouky, M. I., Abdelnaiem, A. E., and ElHefnawi, M. (2019b). A review study: Computational techniques for expecting the impact of non-synonymous single nucleotide variants in human diseases. Gene 680, 20–33. doi:10.1016/j.gene.2018.09.028
Hassan, M. S., Shaalan, A. A., Dessouky, M. I., Abdelnaiem, A. E., and ElHefnawi, M. (2019a). Evaluation of computational techniques for predicting non-synonymous single nucleotide variants pathogenicity. Genomics 111 (4), 869–882. doi:10.1016/j.ygeno.2018.05.013
Hebert, L., Hillman, P., Baker, C., Brown, M., Ashley-Koch, A., Hixson, J. E., et al. (2020). Burden of rare deleterious variants in WNT signaling genes among 511 myelomeningocele patients. PLoS One 15 (9), e0239083. doi:10.1371/journal.pone.0239083
Hecht, M., Bromberg, Y., and Rost, B. (2015). Better prediction of functional effects for sequence variants. BMC Genomics 16 (8), S1. doi:10.1186/1471-2164-16-S8-S1
Hoie, M. H., Cagiada, M., Beck Frederiksen, A. H., Stein, A., and Lindorff-Larsen, K. (2022). Predicting and interpreting large-scale mutagenesis data using analyses of protein stability and conservation. Cell. Rep. 38 (2), 110207. doi:10.1016/j.celrep.2021.110207
Holtgrewe, M., Stolpe, O., Nieminen, M., Mundlos, S., Knaus, A., Kornak, U., et al. (2020). VarFish: Comprehensive DNA variant analysis for diagnostics and research. Nucleic Acids Res. 48 (W1), W162-W169–W169. doi:10.1093/nar/gkaa241
Hopf, T. A., Ingraham, J. B., Poelwijk, F. J., Scharfe, C. P., Springer, M., Sander, C., et al. (2017). Mutation effects predicted from sequence co-variation. Nat. Biotechnol. 35 (2), 128–135. doi:10.1038/nbt.3769
Hu, T., Chitnis, N., Monos, D., and Dinh, A. (2021). Next-generation sequencing technologies: An overview. Hum. Immunol. 82 (11), 801–811. doi:10.1016/j.humimm.2021.02.012
Ingram, V. M. (1957). Gene mutations in human haemoglobin: The chemical difference between normal and sickle cell haemoglobin. Nature 180 (4581), 326–328. doi:10.1038/180326a0
International HapMap, C. (2003). The international HapMap project. Nature 426 (6968), 789–796. doi:10.1038/nature02168
Ioannidis, N. M., Rothstein, J. H., Pejaver, V., Middha, S., McDonnell, S. K., Baheti, S., et al. (2016). Revel: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99 (4), 877–885. doi:10.1016/j.ajhg.2016.08.016
Ip, E., Chapman, G., Winlaw, D., Dunwoodie, S. L., and Giannoulatou, E. (2019). Vpot: A customizable variant prioritization ordering tool for annotated variants. Genomics Proteomics Bioinforma. 17 (5), 540–545. doi:10.1016/j.gpb.2019.11.001
Jagadeesh, K. A., Birgmeier, J., Guturu, H., Deisseroth, C. A., Wenger, A. M., Bernstein, J. A., et al. (2019). Phrank measures phenotype sets similarity to greatly improve Mendelian diagnostic disease prioritization. Genet. Med. 21 (2), 464–470. doi:10.1038/s41436-018-0072-y
Jagadeesh, K. A., Wenger, A. M., Berger, M. J., Guturu, H., Stenson, P. D., Cooper, D. N., et al. (2016). M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 48 (12), 1581–1586. doi:10.1038/ng.3703
Javed, A., Agrawal, S., and Ng, P. C. (2014). Phen-gen: Combining phenotype and genotype to analyze rare disorders. Nat. Methods 11 (9), 935–937. doi:10.1038/nmeth.3046
Ji, X., Kember, R. L., Brown, C. D., and Bucan, M. (2016). Increased burden of deleterious variants in essential genes in autism spectrum disorder. Proc. Natl. Acad. Sci. U. S. A. 113 (52), 15054–15059. doi:10.1073/pnas.1613195113
Johnston, J. J., and Biesecker, L. G. (2013). Databases of genomic variation and phenotypes: Existing resources and future needs. Hum. Mol. Genet. 22 (R1), R27–R31. doi:10.1093/hmg/ddt384
Jones, C. M., Charache, S., and Hathaway, P. J. (1979). The effect of hemoglobin F-Chesapeake (alpha 2 92 Arg. leads to Leu gamma 2) on fetal oxygen affinity and erythropoiesis. Pediatr. Res. 13 (7), 851–853. doi:10.1203/00006450-197907000-00011
Jordan, D. M., Kiezun, A., Baxter, S. M., Agarwala, V., Green, R. C., Murray, M. F., et al. (2011). Development and validation of a computational method for assessment of missense variants in hypertrophic cardiomyopathy. Am. J. Hum. Genet. 88 (2), 183–192. doi:10.1016/j.ajhg.2011.01.011
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596 (7873), 583–589. doi:10.1038/s41586-021-03819-2
Ka, W. (2021). DNA sequencing costs: Data from the NHGRI genome sequencing Program (GSP). [Online]. Available: www.genome.gov/sequencingcostsdata (Accessed June 1, 2022 2022).
Kaminker, J. S., Zhang, Y., Watanabe, C., and Zhang, Z. (2007). CanPredict: A computational tool for predicting cancer-associated missense mutations. Nucleic Acids Res. 35, W595–W598. doi:10.1093/nar/gkm405
Kanzi, A. M., San, J. E., Chimukangara, B., Wilkinson, E., Fish, M., Ramsuran, V., et al. (2020). Next generation sequencing and bioinformatics analysis of family genetic inheritance. Front. Genet. 11, 544162. doi:10.3389/fgene.2020.544162
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141, 456 humans. Nature 581 (7809), 434–443. doi:10.1038/s41586-020-2308-7
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alfoldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141, 456 humans. Nature 581 (7809), 434–443. doi:10.1038/s41586-020-2308-7
Katsonis, P., and Lichtarge, O. (2019). CAGI5: Objective performance assessments of predictions based on the Evolutionary Action equation. Hum. Mutat. 40 (9), 1436–1454. doi:10.1002/humu.23873
Katsonis, P., Wilhelm, K., Williams, A., and Lichtarge, O. (2022). Genome interpretation using in silico predictors of variant impact. Hum. Genet. doi:10.1007/s00439-022-02457-6
Keerthikumar, S., Raju, R., Kandasamy, K., Hijikata, A., Ramabadran, S., Balakrishnan, L., et al. (2009). Rapid: Resource of asian primary immunodeficiency diseases. Nucleic Acids Res. 37, D863–D867. doi:10.1093/nar/gkn682
Kim, Y., Kang, C., Min, B., and Yi, G. S. (2015). Detection and analysis of disease-associated single nucleotide polymorphism influencing post-translational modification. BMC Med. Genomics 8 (2), S7. doi:10.1186/1755-8794-8-S2-S7
Kimura, M. (1979). The neutral theory of molecular evolution. Sci. Am. 241 (5), 98–100. doi:10.1038/scientificamerican1179-98
Kircher, M., Witten, D. M., Jain, P., O'Roak, B. J., Cooper, G. M., and Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46 (3), 310–315. doi:10.1038/ng.2892
Koboldt, D. C. (2020). Best practices for variant calling in clinical sequencing. Genome Med. 12 (1), 91. doi:10.1186/s13073-020-00791-w
Kohler, S., Gargano, M., Matentzoglu, N., Carmody, L. C., Lewis-Smith, D., Vasilevsky, N. A., et al. (2021). The human phenotype ontology in 2021. Nucleic Acids Res. 49 (D1), D1207–D1217. doi:10.1093/nar/gkaa1043
Kohler, S., Schulz, M. H., Krawitz, P., Bauer, S., Dolken, S., Ott, C. E., et al. (2009). Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am. J. Hum. Genet. 85 (4), 457–464. doi:10.1016/j.ajhg.2009.09.003
Krogh, A., Brown, M., Mian, I. S., Sjolander, K., and Haussler, D. (1994). Hidden Markov models in computational biology. Applications to protein modeling. J. Mol. Biol. 235 (5), 1501–1531. doi:10.1006/jmbi.1994.1104
Kulshreshtha, S., Chaudhary, V., Goswami, G. K., and Mathur, N. (2016). Computational approaches for predicting mutant protein stability. J. Comput. Aided. Mol. Des. 30 (5), 401–412. doi:10.1007/s10822-016-9914-3
Lai, C., Zimmer, A. D., O'Connor, R., Kim, S., Chan, R., van den Akker, J., et al. (2020). Leap: Using machine learning to support variant classification in a clinical setting. Hum. Mutat. 41 (6), 1079–1090. doi:10.1002/humu.24011
Laimer, J., Hofer, H., Fritz, M., Wegenkittl, S., and Lackner, P. (2015). MAESTRO--multi agent stability prediction upon point mutations. BMC Bioinforma. 16, 116. doi:10.1186/s12859-015-0548-6
Landrum, M. J., Chitipiralla, S., Brown, G. R., Chen, C., Gu, B., Hart, J., et al. (2020). ClinVar: Improvements to accessing data. Nucleic Acids Res. 48 (D1), D835-D844–D844. doi:10.1093/nar/gkz972
Lanktree, M. B., Haghighi, A., Guiard, E., Iliuta, I. A., Song, X., Harris, P. C., et al. (2018). Prevalence estimates of polycystic kidney and liver disease by population sequencing. J. Am. Soc. Nephrol. 29 (10), 2593–2600. doi:10.1681/ASN.2018050493
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60, 706 humans. Nature 536 (7616), 285–291. doi:10.1038/nature19057
Leu, C., Balestrini, S., Maher, B., Hernandez-Hernandez, L., Gormley, P., Hamalainen, E., et al. (2015). Genome-wide polygenic burden of rare deleterious variants in sudden unexpected death in epilepsy. EBioMedicine 2 (9), 1063–1070. doi:10.1016/j.ebiom.2015.07.005
Li, B., Krishnan, V. G., Mort, M. E., Xin, F., Kamati, K. K., Cooper, D. N., et al. (2009). Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics 25 (21), 2744–2750. doi:10.1093/bioinformatics/btp528
Li, J., Shi, L., Zhang, K., Zhang, Y., Hu, S., Zhao, T., et al. (2018). VarCards: An integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res. 46 (D1), D1039-D1048–D1048. doi:10.1093/nar/gkx1039
Li, J., Zhao, T., Zhang, Y., Zhang, K., Shi, L., Chen, Y., et al. (2018). Performance evaluation of pathogenicity-computation methods for missense variants. Nucleic Acids Res. 46 (15), 7793–7804. doi:10.1093/nar/gky678
Li, M., Li, J., Li, M. J., Pan, Z., Hsu, J. S., Liu, D. J., et al. (2017). Robust and rapid algorithms facilitate large-scale whole genome sequencing downstream analysis in an integrative framework. Nucleic Acids Res. 45 (9), e75. doi:10.1093/nar/gkx019
Li, M. X., Gui, H. S., Kwan, J. S., Bao, S. Y., and Sham, P. C. (2012). A comprehensive framework for prioritizing variants in exome sequencing studies of Mendelian diseases. Nucleic Acids Res. 40 (7), e53. doi:10.1093/nar/gkr1257
Li, Q., Ren, Z., Cao, K., Li, M. M., Wang, K., and Zhou, Y. (2022). CancerVar: An artificial intelligence-empowered platform for clinical interpretation of somatic mutations in cancer. Sci. Adv. 8 (18), eabj1624. doi:10.1126/sciadv.abj1624
Li, Q., and Wang, K. (2017). InterVar: Clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am. J. Hum. Genet. 100 (2), 267–280. doi:10.1016/j.ajhg.2017.01.004
Li, S., van der Velde, K. J., de Ridder, D., van Dijk, A. D. J., Soudis, D., Zwerwer, L. R., et al. (2020). Capice: A computational method for consequence-agnostic pathogenicity interpretation of clinical exome variations. Genome Med. 12 (1), 75. doi:10.1186/s13073-020-00775-w
Li, Y., Chen, C. Y., Kaye, A. M., and Wasserman, W. W. (2015). The identification of cis-regulatory elements: A review from a machine learning perspective. Biosystems. 138, 6–17. doi:10.1016/j.biosystems.2015.10.002
Liu, C., Peres Kury, F. S., Li, Z., Ta, C., Wang, K., and Weng, C. (2019). Doc2Hpo: A web application for efficient and accurate HPO concept curation. Nucleic Acids Res. 47 (W1), W566-W570–W570. doi:10.1093/nar/gkz386
Liu, Y., Sun, J., and Zhao, M. (2017). ONGene: A literature-based database for human oncogenes. J. Genet. Genomics 44 (2), 119–121. doi:10.1016/j.jgg.2016.12.004
Lo, Y. H., Cheng, H. C., Hsiung, C. N., Yang, S. L., Wang, H. Y., Peng, C. W., et al. (2021). Detecting genetic ancestry and adaptation in the Taiwanese han people. Mol. Biol. Evol. 38 (10), 4149–4165. doi:10.1093/molbev/msaa276
Ma, L., Roman-Campos, D., Austin, E. D., Eyries, M., Sampson, K. S., Soubrier, F., et al. (2013). A novel channelopathy in pulmonary arterial hypertension. N. Engl. J. Med. 369 (4), 351–361. doi:10.1056/NEJMoa1211097
MacArthur, D. G., Balasubramanian, S., Frankish, A., Huang, N., Morris, J., Walter, K., et al. (2012). A systematic survey of loss-of-function variants in human protein-coding genes. Science 335 (6070), 823–828. doi:10.1126/science.1215040
Maffucci, P., Bigio, B., Rapaport, F., Cobat, A., Borghesi, A., Lopez, M., et al. (2019). Blacklisting variants common in private cohorts but not in public databases optimizes human exome analysis. Proc. Natl. Acad. Sci. U. S. A. 116 (3), 950–959. doi:10.1073/pnas.1808403116
Mahmood, K., Jung, C. H., Philip, G., Georgeson, P., Chung, J., Pope, B. J., et al. (2017). Variant effect prediction tools assessed using independent, functional assay-based datasets: Implications for discovery and diagnostics. Hum. Genomics 11 (1), 10. doi:10.1186/s40246-017-0104-8
Martinez, G., Lima, F., and Colombo, B. (1977). Haemoglobin J Guantanamo (alpha 2 beta 2 128 (H6) Ala replaced by Asp). A new fast unstable haemoglobin found in a Cuban family. Biochim. Biophys. Acta 491 (1), 1–6. doi:10.1016/0005-2795(77)90034-4
McLaren, W., Gil, L., Hunt, S. E., Riat, H. S., Ritchie, G. R., Thormann, A., et al. (2016). The ensembl variant effect predictor. Genome Biol. 17 (1), 122. doi:10.1186/s13059-016-0974-4
McLaren, W., Pritchard, B., Rios, D., Chen, Y., Flicek, P., and Cunningham, F. (2010). Deriving the consequences of genomic variants with the ensembl API and SNP effect predictor. Bioinformatics 26 (16), 2069–2070. doi:10.1093/bioinformatics/btq330
Melendez-Aranda, L., Jaloma-Cruz, A. R., Pastor, N., and Romero-Prado, M. M. J. (2019). In silico analysis of missense mutations in exons 1-5 of the F9 gene that cause hemophilia B. BMC Bioinforma. 20 (1), 363. doi:10.1186/s12859-019-2919-x
Mi, H., Ebert, D., Muruganujan, A., Mills, C., Albou, L. P., Mushayamaha, T., et al. (2021). PANTHER version 16: A revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 49 (D1), D394–D403. doi:10.1093/nar/gkaa1106
Miller, M., Wang, Y., and Bromberg, Y. (2019). What went wrong with variant effect predictor performance for the PCM1 challenge. Hum. Mutat. 40 (9), 1486–1494. doi:10.1002/humu.23832
Moo-Penn, W. F., Jue, D. L., Johnson, M. H., Olsen, K. W., Shih, D., Jones, R. T., et al. (1988). Hemoglobin brockton [beta 138 (H16) ala----pro]: An unstable variant near the C-terminus of the beta-subunits with normal oxygen-binding properties. Biochemistry 27 (20), 7614–7619. doi:10.1021/bi00420a007
Morningstar-Kywi, N., Haworth, I. S., and Mosley, S. A. (2021). Ligand-specific pharmacogenetic effects of nonsynonymous mutations. Pharmacogenet. Genomics 31 (4), 75–82. doi:10.1097/FPC.0000000000000424
Muller, H., Jimenez-Heredia, R., Krolo, A., Hirschmugl, T., Dmytrus, J., Boztug, K., et al. (2017). VCF.Filter: Interactive prioritization of disease-linked genetic variants from sequencing data. Nucleic Acids Res. 45 (W1), W567-W572–W572. doi:10.1093/nar/gkx425
Ng, P. C., and Henikoff, S. (2001). Predicting deleterious amino acid substitutions. Genome Res. 11 (5), 863–874. doi:10.1101/gr.176601
Ng, P. C., and Henikoff, S. (2003). Sift: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 31 (13), 3812–3814. doi:10.1093/nar/gkg509
Nikam, R., Kulandaisamy, A., Harini, K., Sharma, D., and Gromiha, M. M. (2021). ProThermDB: Thermodynamic database for proteins and mutants revisited after 15 years. Nucleic Acids Res. 49 (D1), D420–D424. doi:10.1093/nar/gkaa1035
Niroula, A., Urolagin, S., and Vihinen, M. (2015). PON-P2: Prediction method for fast and reliable identification of harmful variants. PLoS One 10 (2), e0117380. doi:10.1371/journal.pone.0117380
Niroula, A., and Vihinen, M. (2015). Classification of amino acid substitutions in Mismatch Repair proteins using PON-MMR2. Hum. Mutat. 36 (12), 1128–1134. doi:10.1002/humu.22900
Niroula, A., and Vihinen, M. (2019). How good are pathogenicity predictors in detecting benign variants? PLoS Comput. Biol. 15 (2), e1006481. doi:10.1371/journal.pcbi.1006481
Nurk, S., Koren, S., Rhie, A., Rautiainen, M., Bzikadze, A. V., Mikheenko, A., et al. (2022). The complete sequence of a human genome. Science 376 (6588), 44–53. doi:10.1126/science.abj6987
Ohno, K., Takeda, J. I., and Masuda, A. (2018). Rules and tools to predict the splicing effects of exonic and intronic mutations. WIREs RNA 9 (1). doi:10.1002/wrna.1451
Okido, T., Kodama, Y., Mashima, J., Kosuge, T., Fujisawa, T., and Ogasawara, O. (2022). DNA Data Bank of Japan (DDBJ) update report 2021. Nucleic Acids Res. 50 (D1), D102–D105. doi:10.1093/nar/gkab995
Orgogozo, V., Morizot, B., and Martin, A. (2015). The differential view of genotype-phenotype relationships. Front. Genet. 6, 179. doi:10.3389/fgene.2015.00179
Pak, M. A., Markhieva, K. A., Novikova, M. S., Petrov, D. S., Vorobyev, I. S., Maksimova, E. S., et al. (2021). Using AlphaFold to predict the impact of single mutations on protein stability and function. bioRxiv [Preprint].
Palmer, D. S., Howrigan, D. P., Chapman, S. B., Adolfsson, R., Bass, N., Blackwood, D., et al. (2022). Exome sequencing in bipolar disorder identifies AKAP11 as a risk gene shared with schizophrenia. Nat. Genet. 54 (5), 541–547. doi:10.1038/s41588-022-01034-x
Pandurangan, A. P., Ochoa-Montano, B., Ascher, D. B., and Blundell, T. L. (2017). Sdm: A server for predicting effects of mutations on protein stability. Nucleic Acids Res. 45 (W1), W229-W235–W235. doi:10.1093/nar/gkx439
Pejaver, V., Urresti, J., Lugo-Martinez, J., Pagel, K. A., Lin, G. N., Nam, H. J., et al. (2020). Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 11 (1), 5918. doi:10.1038/s41467-020-19669-x
Peng, G., Shen, P., Gandotra, N., Le, A., Fung, E., Jelliffe-Pawlowski, L., et al. (2019). Combining newborn metabolic and DNA analysis for second-tier testing of methylmalonic acidemia. Genet. Med. 21 (4), 896–903. doi:10.1038/s41436-018-0272-5
Pereira, B., Chin, S. F., Rueda, O. M., Vollan, H. K., Provenzano, E., Bardwell, H. A., et al. (2016). The somatic mutation profiles of 2, 433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 7, 11479. doi:10.1038/ncomms11479
Perez-Enciso, M., and Zingaretti, L. M. (2019). A guide for using deep learning for complex trait genomic prediction. Genes. (Basel) 10 (7), E553. doi:10.3390/genes10070553
Petukh, M., Kucukkal, T. G., and Alexov, E. (2015). On human disease-causing amino acid variants: Statistical study of sequence and structural patterns. Hum. Mutat. 36 (5), 524–534. doi:10.1002/humu.22770
Pires, D. E., Blundell, T. L., and Ascher, D. B. (2015). Platinum: A database of experimentally measured effects of mutations on structurally defined protein-ligand complexes. Nucleic Acids Res. 43, D387–D391. doi:10.1093/nar/gku966
Pollard, K. S., Hubisz, M. J., Rosenbloom, K. R., and Siepel, A. (2010). Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20 (1), 110–121. doi:10.1101/gr.097857.109
Poon, K. S. (2021). In silico analysis of BRCA1 and BRCA2 missense variants and the relevance in molecular genetic testing. Sci. Rep. 11 (1), 11114. doi:10.1038/s41598-021-88586-w
Porta-Pardo, E., Ruiz-Serra, V., Valentini, S., and Valencia, A. (2022). The structural coverage of the human proteome before and after AlphaFold. PLoS Comput. Biol. 18 (1), e1009818. doi:10.1371/journal.pcbi.1009818
Pritchard, J. K., and Cox, N. J. (2002). The allelic architecture of human disease genes: Common disease-common variant.or not? Hum. Mol. Genet. 11 (20), 2417–2423. doi:10.1093/hmg/11.20.2417
Quang, D., Chen, Y., and Xie, X. (2015). Dann: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 31 (5), 761–763. doi:10.1093/bioinformatics/btu703
Quinodoz, M., Peter, V. G., Cisarova, K., Royer-Bertrand, B., Stenson, P. D., Cooper, D. N., et al. (2022). Analysis of missense variants in the human genome reveals widespread gene-specific clustering and improves prediction of pathogenicity. Am. J. Hum. Genet. 109 (3), 457–470. doi:10.1016/j.ajhg.2022.01.006
Rehm, H. L., Berg, J. S., Brooks, L. D., Bustamante, C. D., Evans, J. P., Landrum, M. J., et al. (2015). ClinGen--the clinical genome resource. N. Engl. J. Med. 372 (23), 2235–2242. doi:10.1056/NEJMsr1406261
Reva, B., Antipin, Y., and Sander, C. (2007). Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 8 (11), R232. doi:10.1186/gb-2007-8-11-r232
Reva, B., Antipin, Y., and Sander, C. (2011). Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 39 (17), e118. doi:10.1093/nar/gkr407
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Riesselman, A. J., Ingraham, J. B., and Marks, D. S. (2018). Deep generative models of genetic variation capture the effects of mutations. Nat. Methods 15 (10), 816–822. doi:10.1038/s41592-018-0138-4
Robinson, P. N., Kohler, S., Bauer, S., Seelow, D., Horn, D., and Mundlos, S. (2008). The human phenotype ontology: A tool for annotating and analyzing human hereditary disease. Am. J. Hum. Genet. 83 (5), 610–615. doi:10.1016/j.ajhg.2008.09.017
Robinson, P. N., Ravanmehr, V., Jacobsen, J. O. B., Danis, D., Zhang, X. A., Carmody, L. C., et al. (2020). Interpretable clinical genomics with a likelihood ratio paradigm. Am. J. Hum. Genet. 107 (3), 403–417. doi:10.1016/j.ajhg.2020.06.021
Rojano, E., Seoane, P., Ranea, J. A. G., and Perkins, J. R. (2019). Regulatory variants: From detection to predicting impact. Brief. Bioinform. 20 (5), 1639–1654. doi:10.1093/bib/bby039
Saito, S., Ohno, K., and Sakuraba, H. (2011). Fabry-database.org: Database of the clinical phenotypes, genotypes and mutant alpha-galactosidase A structures in Fabry disease. J. Hum. Genet. 56 (6), 467–468. doi:10.1038/jhg.2011.31
Salgado, D., Desvignes, J. P., Rai, G., Blanchard, A., Miltgen, M., Pinard, A., et al. (2016). UMD-predictor: A high-throughput sequencing compliant system for pathogenicity prediction of any human cDNA substitution. Hum. Mutat. 37 (5), 439–446. doi:10.1002/humu.22965
Sallah, S. R., Sergouniotis, P. I., Barton, S., Ramsden, S., Taylor, R. L., Safadi, A., et al. (2020). Using an integrative machine learning approach utilising homology modelling to clinically interpret genetic variants: CACNA1F as an exemplar. Eur. J. Hum. Genet. 28 (9), 1274–1282. doi:10.1038/s41431-020-0623-y
Sanavia, T., Birolo, G., Montanucci, L., Turina, P., Capriotti, E., and Fariselli, P. (2020). Limitations and challenges in protein stability prediction upon genome variations: Towards future applications in precision medicine. Comput. Struct. Biotechnol. J. 18, 1968–1979. doi:10.1016/j.csbj.2020.07.011
Sarkar, A., Yang, Y., and Vihinen, M. (2020). Variation benchmark datasets: Update, criteria, quality and applications. Oxford: Database. doi:10.1093/database/baz117
Sasidharan Nair, P., and Vihinen, M. (2013). VariBench: A benchmark database for variations. Hum. Mutat. 34 (1), 42–49. doi:10.1002/humu.22204
Savojardo, C., Babbi, G., Bovo, S., Capriotti, E., Martelli, P. L., and Casadio, R. (2019). Are machine learning based methods suited to address complex biological problems? Lessons from CAGI-5 challenges. Hum. Mutat. 40 (9), 1455–1462. doi:10.1002/humu.23784
Sayers, E. W., Cavanaugh, M., Clark, K., Pruitt, K. D., Schoch, C. L., Sherry, S. T., et al. (2022). GenBank. GenBank. Nucleic Acids Res. 50 (D1), D161–D164. doi:10.1093/nar/gkab1135
Schaafsma, G. C., and Vihinen, M. (2015). VariSNP, a benchmark database for variations from dbSNP. Hum. Mutat. 36 (2), 161–166. doi:10.1002/humu.22727
Schiemann, A. H., and Stowell, K. M. (2016). Comparison of pathogenicity prediction tools on missense variants in RYR1 and CACNA1S associated with malignant hyperthermia. Br. J. Anaesth. 117 (1), 124–128. doi:10.1093/bja/aew065
Schwarz, J. M., Cooper, D. N., Schuelke, M., and Seelow, D. (2014). MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 11 (4), 361–362. doi:10.1038/nmeth.2890
Schwarz, J. M., Rodelsperger, C., Schuelke, M., and Seelow, D. (2010). MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7 (8), 575–576. doi:10.1038/nmeth0810-575
Sherry, S. T., Ward, M. H., Kholodov, M., Baker, J., Phan, L., Smigielski, E. M., et al. (2001). dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29 (1), 308–311. doi:10.1093/nar/29.1.308
Shihab, H. A., Gough, J., Cooper, D. N., Day, I. N., and Gaunt, T. R. (2013a). Predicting the functional consequences of cancer-associated amino acid substitutions. Bioinformatics 29 (12), 1504–1510. doi:10.1093/bioinformatics/btt182
Shihab, H. A., Gough, J., Cooper, D. N., Stenson, P. D., Barker, G. L., Edwards, K. J., et al. (2013b). Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 34 (1), 57–65. doi:10.1002/humu.22225
Shihab, H. A., Gough, J., Cooper, D. N., Stenson, P. D., Barker, G. L., Edwards, K. J., et al. (2013). Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 34 (1), 57–65. doi:10.1002/humu.22225
Shihab, H. A., Gough, J., Mort, M., Cooper, D. N., Day, I. N., and Gaunt, T. R. (2014). Ranking non-synonymous single nucleotide polymorphisms based on disease concepts. Hum. Genomics 8, 11. doi:10.1186/1479-7364-8-11
Siepel, A., Bejerano, G., Pedersen, J. S., Hinrichs, A. S., Hou, M., Rosenbloom, K., et al. (2005). Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15 (8), 1034–1050. doi:10.1101/gr.3715005
Sifrim, A., Popovic, D., Tranchevent, L. C., Ardeshirdavani, A., Sakai, R., Konings, P., et al. (2013). eXtasy: variant prioritization by genomic data fusion. Nat. Methods 10 (11), 1083–1084. doi:10.1038/nmeth.2656
Sim, N. L., Kumar, P., Hu, J., Henikoff, S., Schneider, G., and Ng, P. C. (2012). SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40, W452–W457. Web Server issue). doi:10.1093/nar/gks539
Singh, T., Poterba, T., Curtis, D., Akil, H., Al Eissa, M., Barchas, J. D., et al. (2022). Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature 604 (7906), 509–516. doi:10.1038/s41586-022-04556-w
Smedley, D., Jacobsen, J. O., Jager, M., Kohler, S., Holtgrewe, M., Schubach, M., et al. (2015). Next-generation diagnostics and disease-gene discovery with the Exomiser. Nat. Protoc. 10 (12), 2004–2015. doi:10.1038/nprot.2015.124
Smigielski, E. M., Sirotkin, K., Ward, M., and Sherry, S. T. (2000). dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Res. 28 (1), 352–355. doi:10.1093/nar/28.1.352
Smith, C. L., Goldsmith, C. A., and Eppig, J. T. (2005). The Mammalian Phenotype Ontology as a tool for annotating, analyzing and comparing phenotypic information. Genome Biol. 6 (1), R7. doi:10.1186/gb-2004-6-1-r7
Somody, J. C., MacKinnon, S. S., and Windemuth, A. (2017). Structural coverage of the proteome for pharmaceutical applications. Drug Discov. Today 22 (12), 1792–1799. doi:10.1016/j.drudis.2017.08.004
Steinhaus, R., Proft, S., Schuelke, M., Cooper, D. N., Schwarz, J. M., and Seelow, D. (2021). MutationTaster2021. Nucleic Acids Res. 49 (W1), W446–W451. doi:10.1093/nar/gkab266
Stone, E. A., and Sidow, A. (2005). Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res. 15 (7), 978–986. doi:10.1101/gr.3804205
Sudmant, P. H., Rausch, T., Gardner, E. J., Handsaker, R. E., Abyzov, A., Huddleston, J., et al. (2015). An integrated map of structural variation in 2, 504 human genomes. Nature 526 (7571), 75–81. doi:10.1038/nature15394
Sunyaev, S., Ramensky, V., and Bork, P. (2000). Towards a structural basis of human non-synonymous single nucleotide polymorphisms. Trends Genet. 16 (5), 198–200. doi:10.1016/s0168-9525(00)01988-0
Suybeng, V., Koeppel, F., Harle, A., and Rouleau, E. (2020). Comparison of pathogenicity prediction tools on somatic variants. J. Mol. Diagn. 22 (12), 1383–1392. doi:10.1016/j.jmoldx.2020.08.007
Suzek, B. E., Huang, H., McGarvey, P., Mazumder, R., and Wu, C. H. (2007). UniRef: Comprehensive and non-redundant UniProt reference clusters. Bioinformatics 23 (10), 1282–1288. doi:10.1093/bioinformatics/btm098
Tam, V., Patel, N., Turcotte, M., Bosse, Y., Pare, G., and Meyre, D. (2019). Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 20 (8), 467–484. doi:10.1038/s41576-019-0127-1
Tate, J. G., Bamford, S., Jubb, H. C., Sondka, Z., Beare, D. M., Bindal, N., et al. (2019). Cosmic: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 47 (D1), D941-D947–D947. doi:10.1093/nar/gky1015
Terui, H., Akagi, K., Kawame, H., and Yura, K. (2013). CoDP: Predicting the impact of unclassified genetic variants in MSH6 by the combination of different properties of the protein. J. Biomed. Sci. 20, 25. doi:10.1186/1423-0127-20-25
Thomas, P. D., Campbell, M. J., Kejariwal, A., Mi, H., Karlak, B., Daverman, R., et al. (2003). Panther: A library of protein families and subfamilies indexed by function. Genome Res. 13 (9), 2129–2141. doi:10.1101/gr.772403
Thomas, P. D., and Kejariwal, A. (2004). Coding single-nucleotide polymorphisms associated with complex vs. Mendelian disease: Evolutionary evidence for differences in molecular effects. Proc. Natl. Acad. Sci. U. S. A. 101 (43), 15398–15403. doi:10.1073/pnas.0404380101
Thomas, P. D., Kejariwal, A., Guo, N., Mi, H., Campbell, M. J., Muruganujan, A., et al. (2006). Applications for protein sequence-function evolution data: mRNA/protein expression analysis and coding SNP scoring tools. Nucleic Acids Res. 34, W645–W650. Web Server issue). doi:10.1093/nar/gkl229
Thorisson, G. A., Muilu, J., and Brookes, A. J. (2009). Genotype-phenotype databases: Challenges and solutions for the post-genomic era. Nat. Rev. Genet. 10 (1), 9–18. doi:10.1038/nrg2483
Tian, Y., Pesaran, T., Chamberlin, A., Fenwick, R. B., Li, S., Gau, C. L., et al. (2019). REVEL and BayesDel outperform other in silico meta-predictors for clinical variant classification. Sci. Rep. 9 (1), 12752. doi:10.1038/s41598-019-49224-8
Tischkowitz, M., Hamel, M. A, Carvalho, M., Birrane, G., Soni, A., and van Beers, E. H. (2008). Pathogenicity of the BRCA1 missense variant M1775K is determined by the disruption of the BRCT phosphopeptide-binding pocket: a multi-modal approach. Eur J Hum Genet 16 (7), 820–832. doi:10.1038/ejhg.2008.13
Trisilowati, , , and Mallet, D. G. (2012). In silico experimental modeling of cancer treatment. ISRN Oncol. 2012, 828701. doi:10.5402/2012/828701
Tunyasuvunakool, K., Adler, J., Wu, Z., Green, T., Zielinski, M., Zidek, A., et al. (2021). Highly accurate protein structure prediction for the human proteome. Nature 596 (7873), 590–596. doi:10.1038/s41586-021-03828-1
UniProt, C. (2021). UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 49 (D1), D480–D489. doi:10.1093/nar/gkaa1100
Vaser, R., Adusumalli, S., Leng, S. N., Sikic, M., and Ng, P. C. (2016). SIFT missense predictions for genomes. Nat. Protoc. 11 (1), 1–9. doi:10.1038/nprot.2015.123
Vihinen, M. (2021). Functional effects of protein variants. Biochimie 180, 104–120. doi:10.1016/j.biochi.2020.10.009
Vihinen, M. (2013). Guidelines for reporting and using prediction tools for genetic variation analysis. Hum. Mutat. 34 (2), 275–282. doi:10.1002/humu.22253
Vihinen, M. (2012). How to evaluate performance of prediction methods? Measures and their interpretation in variation effect analysis. BMC Genomics 13 (4), S2. doi:10.1186/1471-2164-13-S4-S2
Vihinen, M. (2014). Variation Ontology for annotation of variation effects and mechanisms. Genome Res. 24 (2), 356–364. doi:10.1101/gr.157495.113
Wang, K., Li, M., and Hakonarson, H. (2010). Annovar: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38 (16), e164. doi:10.1093/nar/gkq603
Weber, L. M., Saelens, W., Cannoodt, R., Soneson, C., Hapfelmeier, A., Gardner, P. P., et al. (2019). Essential guidelines for computational method benchmarking. Genome Biol. 20 (1), 125. doi:10.1186/s13059-019-1738-8
Wei, R., Wang, J., Su, M., Jia, E., Chen, S., Chen, T., et al. (2018). Missing value imputation approach for mass spectrometry-based metabolomics data. Sci. Rep. 8 (1), 663. doi:10.1038/s41598-017-19120-0
Witvliet, D. K., Strokach, A., Giraldo-Forero, A. F., Teyra, J., Colak, R., and Kim, P. M. (2016). ELASPIC web-server: Proteome-wide structure-based prediction of mutation effects on protein stability and binding affinity. Bioinformatics 32 (10), 1589–1591. doi:10.1093/bioinformatics/btw031
Won, D. G., Kim, D. W., Woo, J., and Lee, K. (2021). 3Cnet: Pathogenicity prediction of human variants using multitask learning with evolutionary constraints. Bioinformatics 37, 4626–4634. doi:10.1093/bioinformatics/btab529
Worth, C. L., Preissner, R., and Blundell, T. L. (2011). SDM--a server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 39, W215–W222. Web Server issue). doi:10.1093/nar/gkr363
Xavier, A., Scott, R. J., and Talseth-Palmer, B. A. (2019). TAPES: A tool for assessment and prioritisation in exome studies. PLoS Comput. Biol. 15 (10), e1007453. doi:10.1371/journal.pcbi.1007453
Yazar, M., and Ozbek, P. (2021). In silico tools and approaches for the prediction of functional and structural effects of single-nucleotide polymorphisms on proteins: An expert review. OMICS 25 (1), 23–37. doi:10.1089/omi.2020.0141
Yuan, X., Wang, J., Dai, B., Sun, Y., Zhang, K., Chen, F., et al. (2022). Evaluation of phenotype-driven gene prioritization methods for Mendelian diseases. Brief. Bioinform. 23 (2), bbac019. doi:10.1093/bib/bbac019
Yue, P., Li, Z., and Moult, J. (2005). Loss of protein structure stability as a major causative factor in monogenic disease. J. Mol. Biol. 353 (2), 459–473. doi:10.1016/j.jmb.2005.08.020
Yue, P., Melamud, E., and Moult, J. (2006). SNPs3D: Candidate gene and SNP selection for association studies. BMC Bioinforma. 7, 166. doi:10.1186/1471-2105-7-166
Yue, W. W., Froese, D. S., and Brennan, P. E. (2014). The role of protein structural analysis in the next generation sequencing era. Top. Curr. Chem. 336, 67–98. doi:10.1007/128_2012_326
Zhang, W., Zhang, H., Yang, H., Li, M., Xie, Z., and Li, W. (2019). Computational resources associating diseases with genotypes, phenotypes and exposures. Brief. Bioinform. 20 (6), 2098–2115. doi:10.1093/bib/bby071
Zhang, X., Walsh, R., Whiffin, N., Buchan, R., Midwinter, W., Wilk, A., et al. (2021). Disease-specific variant pathogenicity prediction significantly improves variant interpretation in inherited cardiac conditions. Genet. Med. 23 (1), 69–79. doi:10.1038/s41436-020-00972-3
Zhao, M., Havrilla, J. M., Fang, L., Chen, Y., Peng, J., Liu, C., et al. (2020). Phen2Gene: Rapid phenotype-driven gene prioritization for rare diseases. Nar. Genom. Bioinform. 2, lqaa032. doi:10.1093/nargab/lqaa032
Zhao, M., Kim, P., Mitra, R., Zhao, J., and Zhao, Z. (2016). TSGene 2.0: An updated literature-based knowledgebase for tumor suppressor genes. Nucleic Acids Res. 44 (D1), D1023–D1031. doi:10.1093/nar/gkv1268
Zheng, X., Amos, C. I., and Frost, H. R. (2020). Cancer prognosis prediction using somatic point mutation and copy number variation data: A comparison of gene-level and pathway-based models. BMC Bioinforma. 21 (1), 467. doi:10.1186/s12859-020-03791-0
Keywords: in silico prediction, human genetics, genotype-phenotype relationship, nonsynonymous variants, variant impact
Citation: Liu Y, Yeung WSB, Chiu PCN and Cao D (2022) Computational approaches for predicting variant impact: An overview from resources, principles to applications. Front. Genet. 13:981005. doi: 10.3389/fgene.2022.981005
Received: 29 June 2022; Accepted: 08 August 2022;
Published: 29 September 2022.
Edited by:
Shoba Ranganathan, Macquarie University, AustraliaReviewed by:
Simone Furini, University of Siena, ItalyCopyright © 2022 Liu, Yeung, Chiu and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Philip C. N. Chiu, cGNoaXVjbkBoa3UuaGs=; Dandan Cao, Y2FvZGRAaGt1LXN6aC5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.