 Evans Raballah1,2*
Evans Raballah1,2* Kristen Wilding3
Kristen Wilding3 Samuel B. Anyona1,4
Samuel B. Anyona1,4 Elly O. Munde1,5Ivy Hurwitz6Clinton O. Onyango1,7Cyrus Ayieko8Christophe G. Lambert6
Elly O. Munde1,5Ivy Hurwitz6Clinton O. Onyango1,7Cyrus Ayieko8Christophe G. Lambert6 Kristan A. Schneider9Philip D. Seidenberg10
Kristan A. Schneider9Philip D. Seidenberg10 Collins Ouma1,7
Collins Ouma1,7 Benjamin H. McMahon3Qiuying Cheng6Douglas J. Perkins1,6
Benjamin H. McMahon3Qiuying Cheng6Douglas J. Perkins1,6- 1University of New Mexico-Kenya Global Health Programs, Kisumu, Kenya
- 2Department of Medical Laboratory Sciences, School of Public Health Biomedical Sciences and Technology, Masinde Muliro University of Science and Technology, Kakamega, Kenya
- 3Theoretical Biology and Biophysics Group, Theoretical Division, Los Alamos National Laboratory, Los Alamos, NM, United States
- 4Department of Medical Biochemistry, School of Medicine, Maseno University, Maseno, Kenya
- 5Department of Clinical Medicine, School of Health Sciences, Kirinyaga University, Kerugoya, Kenya
- 6University of New Mexico, Center for Global Health, Department of Internal Medicine, Albuquerque, NM, United States
- 7Department of Biomedical Sciences and Technology, School of Public Health and Community Development, Maseno University, Maseno, Kenya
- 8Department of Zoology, Maseno University, Maseno, Kenya
- 9Department of Applied Computer and Biosciences, University of Applied Sciences Mittweida, Mittweida, Germany
- 10University of New Mexico, Department of Emergency Medicine, Albuquerque, NM, United States
Background: Severe malarial anemia (SMA; Hb < 5.0 g/dl) is a leading cause of childhood morbidity and mortality in holoendemic Plasmodium falciparum transmission regions such as western Kenya.
Methods: We investigated the relationship between two novel complement component 5 (C5) missense mutations [rs17216529:C>T, p(Val145Ile) and rs17610:C>T, p(Ser1310Asn)] and longitudinal outcomes of malaria in a cohort of Kenyan children (under 60 mos, n = 1,546). Molecular modeling was used to investigate the impact of the amino acid transitions on the C5 protein structure.
Results: Prediction of the wild-type and mutant C5 protein structures did not reveal major changes to the overall structure. However, based on the position of the variants, subtle differences could impact on the stability of C5b. The influence of the C5 genotypes/haplotypes on the number of malaria and SMA episodes over 36 months was determined by Poisson regression modeling. Genotypic analyses revealed that inheritance of the homozygous mutant (TT) for rs17216529:C>T enhanced the risk for both malaria (incidence rate ratio, IRR = 1.144, 95%CI: 1.059–1.236, p = 0.001) and SMA (IRR = 1.627, 95%CI: 1.201–2.204, p = 0.002). In the haplotypic model, carriers of TC had increased risk of malaria (IRR = 1.068, 95%CI: 1.017–1.122, p = 0.009), while carriers of both wild-type alleles (CC) were protected against SMA (IRR = 0.679, 95%CI: 0.542–0.850, p = 0.001).
Conclusion: Collectively, these findings show that the selected C5 missense mutations influence the longitudinal risk of malaria and SMA in immune-naïve children exposed to holoendemic P. falciparum transmission through a mechanism that remains to be defined.
Introduction
Malaria remains a significant global public health challenge. Recent data from the World Health Organization (WHO) show that 241 million cases resulted in 627,000 malaria-related deaths globally (WHO, 2021). The African region had 228 million cases and 602,000 deaths, accounting for approximately 95% of the cases and 96% of the deaths globally. The majority of cases in the African region are due to Plasmodium falciparum infections, with 80% of the deaths occurring in children less than 5 years of age (WHO, 2021). In holoendemic P. falciparum transmission regions such as western Kenya, falciparum malaria is a leading cause of childhood morbidity and mortality (Perkins et al., 2011). Severe malaria in holoendemic regions primarily manifests as severe malaria anemia [SMA, hemoglobin (Hb) < 5.0 g/dl] in children less than 5 years of age who are yet to develop naturally-acquired immunity to malaria (Ong’echa et al., 2006; Obonyo et al., 2007).
The pathogenesis of SMA is complex and multifactorial, resulting from overlapping sequelae that include lysis of infected and uninfected erythrocytes, splenic sequestration of erythrocytes, and inefficient erythropoietic responses in bone marrow (Awandare et al., 2011; Perkins et al., 2011). Over the last 2 decades, studies by our team in western Kenya have demonstrated that dysregulation in the innate immune genes is central to the pathogenesis of SMA, and that polymorphic variability in such genes influences susceptibility to P. falciparum infections, SMA, and mortality (Ouma et al., 2008; Munde et al., 2017; Achieng et al., 2019; Anyona et al., 2019; Kisia et al., 2019).
An integral part of the innate immune response is the complement system, comprised of three activation pathways: classical, alternative, and lectin, all of which converge at complement component 3 (C3) protein (Ricklin et al., 2010). Cleavage of C3 results in two subunits: 1) C3a, an anaphylatoxin that elicits potent pro-inflammatory mediator and chemoattractant properties that facilitate clearance of opsonized pathogens by phagocytosis (Klos et al., 2009) and 2) C3b, which binds with C2b4b to form C5 convertase, a serine protease that cleaves C5 into C5a and C5b. C5a, like C3a, is an anaphylatoxin, while C5b binds to C6, C7, C8, and C9 to initiate formation of the membrane attack complex [MAC (C5b-C9)] (Müller-Eberhard, 1985; Hadders et al., 2012). Membrane insertion of the MAC causes lysis of the target cell and the release of pro-inflammatory mediators (Kilgore et al., 1996; Morgan, 2015).
Previous investigations revealed that the complement cascade is activated in children and adults during acute malaria (Greenwood and Brueton, 1974; Helegbe et al., 2007; Nyakoe et al., 2009). In addition, relative to aparasitemic children, those with malaria have elevated C3b deposition on red blood cells, a process that can enhance phagocytosis, and consequently, promote malarial anemia (Odhiambo et al., 2008). Mechanism-based in-vitro studies show that the P. falciparum parasite can induce MAC formation through both the classical and alternative pathways (Roestenberg et al., 2007). Studies also show that protection against clinical malaria and high-density parasitemia is associated with acquired anti-malarial antibodies that inhibit P. falciparum invasion of erythrocytes, primarily through C1q fixation and activation of the classical pathway (Boyle et al., 2015). Recent longitudinal investigations by our group in Kenyan children also revealed that two missense mutations in the C3 gene [rs2230199 (2307C>G, Arg>Gly102) and rs11569534 (34420G>A, Gly>Asp1224)], altered susceptibility to both malaria and SMA during the developmental phase of naturally-acquired malarial immunity (Raballah et al., 2021).
To further characterize the role of complement in the pathogenesis of malaria and SMA, we explored two novel missense mutations in the C5 gene and its association with malaria, SMA and all-cause mortality. The human C5 gene consists of 43 exons and is located on chromosome 9q33.2 (Sherry et al., 1999). Inherited human deficiencies of C5, including [MAC (C5b-C9)] deficiency, are associated with recurrent infection by Neisseria species resulting in mild-to-moderate meningitis (Schejbel et al., 2013; Colobran et al., 2016). However, C5 deficient mice (C5−/−) are resistant to experimental cerebral malaria due to an inability to form the MAC, and as such, abrogate the immune response that drives pathogenesis (Colobran et al., 2016). Since the role of C5 mutations in human malaria pathogenesis has not been previously reported, we selected two missense mutations that had an elevated proportion of minor allele frequencies (MAF) in African populations relative to other variants that induce amino acid changes. The first variant, rs17216529:C>T, p. (Val145Ile), results in an amino acid change from valine to isoleucine at position 145, while the second variant, rs17610:C>T, p. (Ser1310Asn), causes a serine to asparagine transition at position 1,310. A previous study in a cohort of patients with symptomatic coronary artery disease, although underpowered, showed that carriers of the homozygous minor allele TT for rs17216529:C>T, p. (Ile145Ile) had a trend towards increased disease severity and bleeding events (Henes et al., 2021). However, an earlier study did not find any relationship between rs17216529:C>T and stroke in a cohort of patients with sickle cell disease (Flanagan et al., 2013). To date, there are no reports on the influence of rs17610:C>T on any human disease outcomes. Here, we present the impact of the two C5 missense mutations, and their combinations, on longitudinal susceptibility to malaria and SMA in a cohort of under- five Kenyan children residing in a holoendmic P. falciparum transmission region.
Methods
Study site and participants
Children (aged 2–48 months, n = 1,546) were enrolled at the Siaya County Referral Hospital (SCRH) in Siaya County, Kenya between April 2004 and September 2015 who had a confirmed positive blood smear for P. falciparum malaria. In addition, children were enrolled at SCRH who were receiving routine childhood vaccinations and had a negative blood smear for P. falciparum (i.e., aparasitemic controls). Siaya County is a rural region in western Kenya with holoendemic P. falciparum transmission whose inhabitants are primarily from the Luo ethnic group (>96%), hence the participants can be considered genetically similar (Githeko et al., 1994). Upon enrollment, comprehensive evaluations were performed on the study participants to collect clinical and demographic information, and information on the history of present illness. Children were excluded from study participation if they were non- P. falciparum positive on blood films, had prior hospitalizations (for any reason), underwent recent blood transfusions, and/or had evidence of cerebral malaria.
Study participants presenting with P. falciparum malaria (any density) were stratified according to hemoglobin concentrations into uncomplicated malaria (UM, Hb ≥ 5.0 g/dl) and SMA (Hb < 5.0 g/dl). All study participants also had HIV-1/2 and bacteremia status determined since our previous studies showed that these comorbidities influence the severity of malarial anemia (Otieno et al., 2006; Were et al., 2011). Parents/legal guardians of the children were provided with pre- and post-test HIV counselling. Required treatment interventions for clinical management followed the Ministry of Health (MOH)-Kenya guidelines. The study was approved by Maseno University Ethics Review Committee (MUERC; MSU/DRPI/MUERC/00510/18) and the University of New Mexico Institutional Review Board (16–284). Written informed consent was obtained from the parent/legal guardian of the study participants in the language of choice (English, Kiswahili, or Dholuo).
Longitudinal follow-up
Parents/guardians were asked to return with their child every 3 months throughout the 36-months follow-up period (i.e. quarterly visit). If parents/guardians failed to return with their child for the scheduled quarterly visit, study personnel visited the residences to check on the health status of the children (including mortality). In addition, for proper clinical management and longitudinal documentation of childhood illnesses, parents/guardians were asked to bring their child to the hospital during any febrile episode(s) or other illnesses (i.e., acute visit). At each acute and quarterly visit, study participants underwent a complete physical and laboratory workup for proper clinical management (e.g., complete blood count, malaria parasitemia determination, evaluation of viral and bacterial infections, etc.). Throughout the follow-up period, mortality data were collected either from hospital records or by verbal autopsy in cases in which death occurred outside the hospital.
Laboratory measures
Upon informed consent from the parents/legal guardians, approximately 3.0 ml venous whole blood samples were collected into EDTA-containing vacutainer tubes for the purposes of laboratory diagnostics, care and to conduct the experiments. P. falciparum density was determined following our previously published reports (Were et al., 2006). Additionally, complete blood counts were performed using a Beckman Coulter® AcT diff2™ hematology analyzer (Beckman–Coulter Inc. Brea, CA, United States). The determination of sickle cell trait and sickle cell disease was performed using cellulose acetate electrophoresis according to the manufacturer’s standard operating procedures (Helena Bio-Sciences, Oxford, United Kingdom). It is however important to note that according to the manufacturer, this method has a limitatition in that, it may be difficult to discriminate between some abnormal hemoglobins, due to similarities in electrophoretic mobilities. Therefore, such cases must be distinguished using other available methods. Serological tests for HIV (Determine™ HIV-1/2, Abbott, Woodmead, South Africa and Uni-Gold™ HIV-1/2, Trinity Biotech Plc. Wicklow, Ireland) were used to determine HIV exposure. Proviral DNA PCR testing was performed for the participants that tested positive for HIV, with at least one the available serological tests, based on our previously published reports (Otieno et al., 2006). Bacterial comorbidity was assessed by culture and sensitivity assays following our previously published protocols (Were et al., 2011). Glucose six phosphate dehydrogenase (G6PD) deficiency (levels: normal, intermediate, deficient) was determined following the methods of Cherepnalkovski et al., (Cherepnalkovski et al., 2015). The α3.7 deletion produces an HBA2/HBA1 hybrid gene and a 3.7 kb deletion (Borg et al., 2009). The α3.7− thalassemia deletion was determined according to published methods (Dode et al., 1993).
C5 variant selection
Variants were selected by cataloging all known missense mutations reported for C5 with elevated MAFs (>10%) in African populations in the National Center for Biotechnology Information (NCBI) database (Sherry et al., 1999). This strategy was based on the rationale that low penetrance of variants in the population would likely not explain high rates of malaria in a region with long-standing holoendemic P. falciparum transmission. Allelic distributions of C5 candidate variants were determined for the following populations: global, African (AFR), Luhya (LWK), and Maasai (MWK) using data from the International HapMap and 1000 Genomes Projects. Additional criteria included selecting variants that were in linkage equilibrium, i.e., not strongly co-inherited (Machiela and Chanock, 2015). Based on the above criteria, two missense mutations were selected: 1) rs17216529:C>T - valine to isoleucine at amino acid position 145 p. (Val145Ile), and 2) rs17610:C>T - serine to asparagine at position 1,310 p. (Ser1310Asn). Analysis using the GRCh38 high coverage genome build for the 1000 Genomes Project (Luhya, Kenya, LWK) revealed that the two SNPs were not linked, i.e., in linkage equilibrium (D’ = 0.763; R2 = 0.021; χ2 = 4.226; p = 0.040] (Machiela and Chanock, 2015). Neither of the selected C5 variants, nor their combinations, have been explored in malaria.
Visualization of the C5 variants on 3-dimensional (3D) protein structures
The reference sequence for the human C5 protein was retrieved from Genbank at the NCBI with the accession number NP_001304092. The potential impact of the C5 SNPs on the overall protein structure was investigated using AlphaFold (Jumper et al., 2021; Varadi et al., 2022) with 3D protein structures visualized using UCSF Chimera (Pettersen et al., 2004). The wild-type (WT) 3D protein structure for C5 was previously determined by Fredslund et al., at 3.1 Å resolution using X-ray diffraction (Fredslund et al., 2008). The structure is deposited as code 3CU7 in the Protein Data Bank (PDB). The WT structure for the membrane attack complex, including C5b, was previously determined by Menny et al., at 5.6 Å using electron microscopy and deposited as 6h04 in the PDB (Menny et al., 2018).
Genotyping of C5 variants
Prior to performing the genotyping assays, genomic DNA was extracted from cheek cells (WBCs) collected on buccal swabs using the MasterAmp™ Buccal swab DNA Extraction kit (Epicentre Biotechnologies, Madison, WI, USA). Further, the quality of the isolated DNA was assessed using a Qubit 3.0 Fluorometer (Thermofisher Scientific, Waltham, MA, United States). High-quality DNA samples were amplified using the GenomiPhi® system (GE Healthcare, South Plainfield, NJ, United States) to obtain sufficient DNA quantities for the genotyping assays. TaqMan® 5′ allelic discrimination Assay-By-Design high-throughput method was used to genotype the C5 rs17216529:C>T (Assay ID: C_32672495_10) and rs17610:C>T (C_11720403_20) variants, following the manufacturer’s protocols (Thermofisher Scientific, Carlsbad, CA, USA), using a StepOne Plus™ Real-Time PCR System. The PCR reaction was performed in a total reaction volume of 10.0 μl with the following amplification cycles: initial denaturation (60°C for 30 s and 95°C for 10 min) followed by 40 cycles of (95°C for 15 s and 60°C for 1 min) and a final extension (60°C for 30 s) using allele-specific fluorescence. The StepOne™ Software (Version 2.3) was used for allelic discrimination (Thermofisher Scientific, Carlsbad, CA, United States).
Statistical analyses
The demographic, clinical, and laboratory characteristics collected at enrollment were analyzed using SPSS, version 23.0 (IBM SPSS Statistics Inc. Chicago, IL, United States). Across group comparisons were determined using Kruskal–Wallis tests, and when significant, pairwise comparisons were performed using the Mann-Whitney U test. Proportions across the groups were compared using Chi-square tests. Hardy-Weinberg equilibrium (HWE) was determined by the exact test (Schaid et al., 2006). We tested whether the number of malaria episodes, SMA episodes, and mortality was proportional to the genotype frequencies at the two SNPs using chi-square goodness-of-fit tests. p-values ≤ 0.05 were considered statistically significant.
Linkage disequilibrium (LD) for the cohort was determined by Multiallelic Interallelic Disequilibrium Analysis (MIDAS) software version 1.0 (Gaunt et al., 2006). The association between the C5 genotypes/haplotypes on the longitudinal episodes of malaria, SMA, and mortality were determined using R (version 3.1.3) (Team, 2019). Whenever necessary, we performed secondary analyses using the additive model, that discriminates the presence of either one or two copies of a haplotype. Phasing of the haplotypes was performed using R. The association between C5 variants and the frequency of malaria and SMA episodes was determined by a Poisson rate regression (R glm function, family = Poisson), with the (logarithm of) age at the patients’ last visits being the offset variable (rate regression). A model search based on minimizing the AIC, using a forward-backward selection was performed with C5 genotypes/haplotypes as covariates, along with additional covariates that can influence anemia: age at enrollment, sex, HIV status (unexposed uninfected, exposed-uninfected, and positive), cohort, sickle cell status (sickle trait and sickle cell disease), G6PD deficiency and α3.7− thalassemia status. In addition, Multiple-event per subject Cox model (Anderson Gill Ref) (R survival package version 2.38.2, coxph function) was performed to delineate the relationship between C5 variants and all-cause mortality (death during the follow-up period, including malaria-unrelated causes of mortality) using identical covariates as the Poisson models and a forward-backward selection. For each model, Bonferroni-Holm corrections were performed to adjust for multiple comparisons.
Results
Characteristics upon enrollment
The study included a total of 1,546 study participants who were stratified into three categories at enrollment: aparasitemic (n = 288), uncomplicated malaria (UM, Hb ≥ 5.0 g/dl, n = 971) and SMA (Hb < 5.0 g/dl, n = 287). Demographic, clinical, and laboratory characteristics of the study participants upon entry into the study are presented in Table 1. The distribution of males and females was comparable across the groups (p = 0.898). However, age differed across the three groups (p = 4.480 × 10−4), with the SMA group being the youngest.
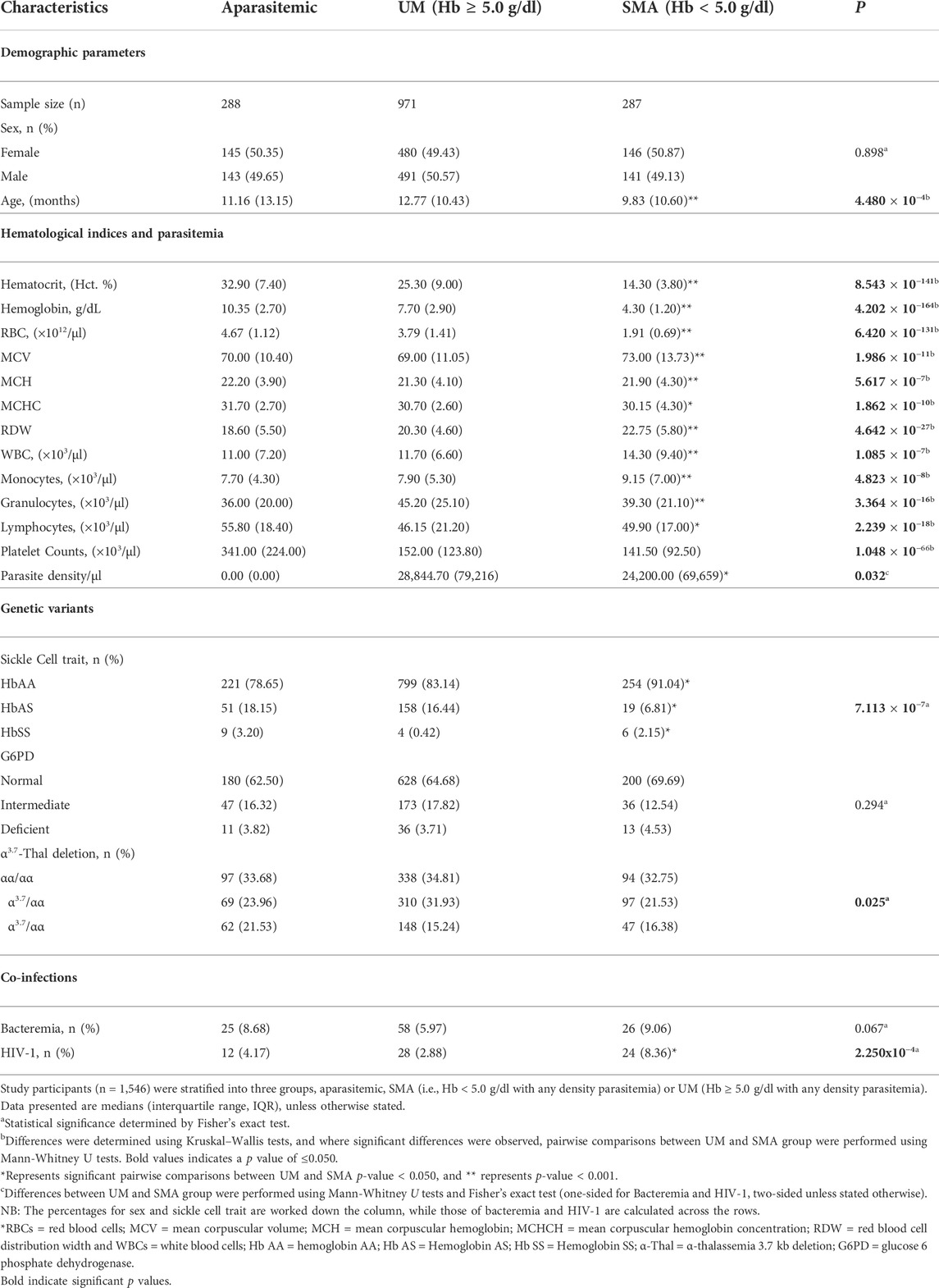
TABLE 1. Demographic, clinical, and laboratory characteristics of study participants at enrollment.
Hematocrit (Hct), Hemoglobin (Hb) concentrations, and red blood cell (RBC) counts progressively declined across the three groups (p = 8.543x10−141, p = 4.202 × 10−164, p = 6.420 × 10−131, respectively) and were lowest in children with SMA, as expected based on a priori grouping. Mean corpuscular volume (MCV) was elevated in children with SMA and differed across the groups (p = 1.986 × 10−11), while mean corpuscular hemoglobin (MCH) and mean corpuscular hemoglobin concentration (MCHC) were highest in the aparasitemic group with an overall group difference (p = 5.617 × 10−7 and p = 1.862 × 10−10, respectively). The red blood cell distribution (RDW) and counts for white blood cells (WBCs) and monocytes varied in the cohort (p = 4.642x10−27, p = 1.085 × 10−7 and p = 4.823 × 10−8, respectively) and were highest in children with SMA. Granulocyte counts also varied (3.364 × 10−16) with the UM group having the highest values. Lymphocyte counts differed in the cohort (p = 2.239 × 10−18) and were highest in aparasitemic children, while platelet counts progressively decreased across the groups (p = 1.048 × 10−66) and were lowest in children with SMA. Parasite density in peripheral blood was lower in children with SMA relative to those with milder forms of malaria (UM, p = 0.032).
Analysis of hemoglobin S (HbS) carriage revealed that the frequency of HbAS (protective alleles) was highest in aparasitemic children and lowest in those presenting with SMA (p = 7.113 × 10−7). The proportion of G6PD variants was comparable across the groups (p = 0.294), while the proportion of thalassemia variants differed across the groups (p = 0.025). In addition, results from our models show that both HbAS and HbSS reduced the risk of malaria episoded [incident risk ratio (IRR) = 0.799, 95%CI: 0.730–0.855, p = 4.666 × 10−9 and IRR = 0.528, 95%CI: 0.388–0.718, p = 4.796 × 10−5, respectuvely). Similarly, homozygous mutants (α/α) for thallassemia reduced malaria during the study period (IRR = 0.900, 95%CI: 0.837–0.967, p = 0.004). However, after acquisistion of malaria only HbAS and HbSS impacted on the development of SMA. Carriage of HbAS reduced the risk of SMA (IRR = 0.499, 95%CI: 0.334–0.746, p = 7.042 × 10−4), while inheritance of HbSS increased the risk of SMA 3-fold (IRR = 3.108, 95%CI: 1.771–5.455, p = 7.77x10−5). The proportions of these genetic variants in the study population relative to the frequencies of malaria and SMA epidosodes are shown in Supplementary Table S2. Since our previous investigations demonstrated that bacteremia and HIV exacerbate the development of SMA (Otieno et al., 2006; Were et al., 2011), the absence/presence of these co-infections was determined in the study population. The proportion of children with bacteremia did not significantly differ between the groups (p = 0.067). However, the proportion of HIV infection differed across the groups (p = 2.250x10−4) and was highest in children with SMA. As expected based on a priori grouping, the RBC indices i. e RBC counts, Hct and Hb were strongly correlated (Supplementary Figure S1). These results are consistent with our previous findings and underscore the importance of considering age, carriage of HbS, and co-infection status in the complex phenotype of SMA (Davenport et al., 2010; Novelli et al., 2010; Were et al., 2011).
Modeling of the C5 protein structure
The structure of the C5 protein is shown in Figure 1A. Analysis of the protein structure revealed that the amino acid transitions from the missense mutations were in the α−chain. rs17216529:C>T, p. (Val145Ile) lies within the macroglobulin 2 (MG2) domain, while rs17610:C>T, p. (Ser1310Asn) is in the CUB domain (for complement C1r/C1s, Uegf, Bmp1). The Val to Ile transition at position 145 does not change the amino acid charge since both Val and Ile are non-polar. Similarly, the Ser to Asn transition at position 1,310 does not impart any changes in amino acid charge since Ser and Asn are both polar residues.
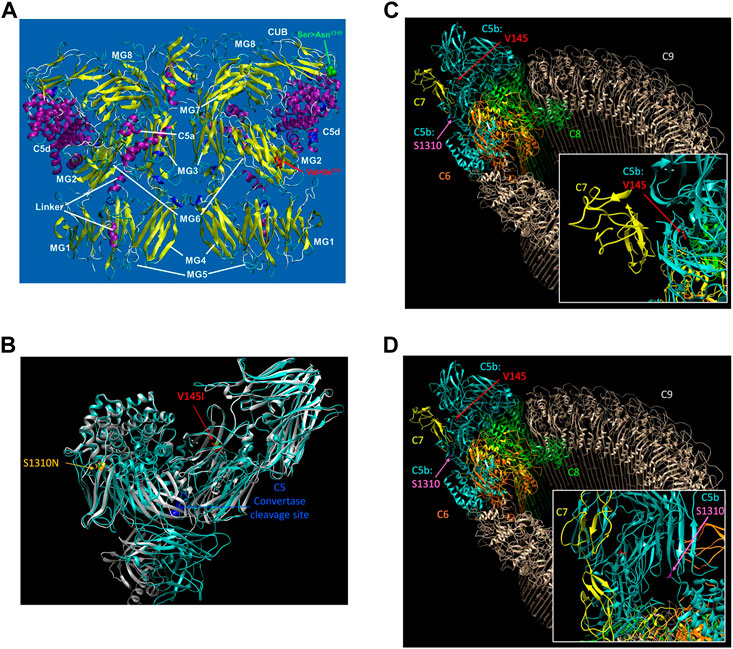
FIGURE 1. The reference sequence for the human C5 protein was retrieved from Genbank at the NCBI with the accession number NP_001304092. The potential impact of the C5 SNPs on the overall protein structure were investigated using AlphaFold (Varadi et al., 2022) with 3D protein structures visualized using UCSF Chimera (Pettersen et al., 2004) (A). The structure of C5 showing the various domains i.e., macroglobulin (MG)1-MG8. Relative to the published C5 structure (Fredslund et al., 2008), we observed that Val145le mutation is located in the macroglobulin MG2 domain, while the Ser1310Asn is located on the CUB (B). AlphaFold structure prediction of rs17216529:C>T and rs17610:C>T. Inspection of the wild-type (WT) and mutant proteins revealed no significant perturbations to the overall structure. However, subtle changes that may not be detected by AlphaFold may impact on the unstable C5b. The WT molecule for both V145I and S1310N are colored green, while the mutant for V145I is colored red and that of S1310N is colored orange (C). AlphaFold structure prediction of the two SNPs relative to the cleavage site for C5 convertase. Examination of the structure revealed that the SNPs are also distal from the C5 convertase cleavage site. Additionally, the WT structure of both C5 and C5b in the MAC, rs17216529:C>T appeared to be buried, orienting the hydrophobic side chain into the protein. On the other hand, we did not observe a clear interaction partner for rs17216529:C>T neither in the C5 nor MAC structures. However, the mutation is located near the C5b and C7 in the MAC structure (D). AlphaFold structure prediction of the location of mutation on rs17610:C>T. Examination of the C5 protein structure revealed that the mutation is exposed both on the C5 protein, and in the MAC.
Accordingly, AlphaFold structure prediction of both rs17216529:C>T, p. (Val145Ile) and rs17610:C>T, p. (Ser1310Asn) revealed no significant perturbations to the overall structure between the WT and mutant proteins (Figure 1B). However, the mutation does lie close to the interface between C5b and C7 in the MAC structure. Therefore, the mutation may still impact C7 binding through subtle perturbations e.g. through changes to the stability of the local structure (Pak et al., 2021) and/or changes in aggregation potential, as is the case with sickle cell disease (Lu et al., 2019) as current modeling techniques may not adequately capture these impacts (Pak et al., 2021). Both sites are also distal from the C5 convertase cleavage site. In the WT structure of both C5 and C5b in the MAC, rs17216529:C>T, p. (Val145Ile) is buried, with the hydrophobic side chain oriented into the interior of the protein (Figure 1C). No clear interaction partner of rs17216529:C>T, p. (Val145Ile) is evident in either the C5 or MAC structures. However, the mutation does lie close to the interface between C5b and C7 in the MAC structure (Figure 1C), so subtle structural perturbations, not anticipated by AlphaFold, could impact C7 binding. In contrast, the rs17610:C>T, p. (Ser1310Asn) mutation is located on the surface of both the C5 protein, and in the MAC (Figure 1D). The Ser in the WT structure is not located at an interface with any other components of the MAC, suggesting minimal impacts on the MAC structure.
Location and allelic distribution of the C5 SNPs
The location of the two SNPs in the C5 gene at q33.2 of chromosome nine is shown in Figure 2A. As discussed above, the selection strategy for choosing C5 variants included prioritizing those with elevated MAFs in African populations. The MAFs for rs17216529:C>T and rs17610:C>T from the International HapMap Project, 1,000 Genome Project, and genotyping of the Kenyan cohort (Figure 2B) (Sherry et al., 1999; International Hap Map, 2003). Relative to the MAF in the global population (0.147), rs17216529:C>T is enriched in AFR (0.312), and slightly higher in the cohort studied here (Luo, 0.320). The MAF for rs17610:C>T is also higher in AFR (0.120) than in the global population (0.034) which is comparable to the LWK (0.050) and MMK (0.035). Thus, the MAF for the Kenyan cohort (Luo. 0.110) is comparable to AFR (0.120) and 3-fold higher than the global population (0.034). Consistent with the linkage results in the LWK population used to guide variant selection, MIDAS revealed that rs17216529:C>T and rs17610:C>T were also in linkage equilibrium (i.e., not considered linked) in the current cohort (D’ = 0.581, LOD = 10.63, R2=0.02, Figure 2C). The distribution of genotypes and haplotypes for rs17216529:C>T, p. (Val145Ile) and rs17610:C>T, p. (Ser1310Asn), and HWE analyses are presented in Supplementary Table S1. Collectively, these results show the selected C5 variants are enriched in the Luo population and not strongly co-inherited.
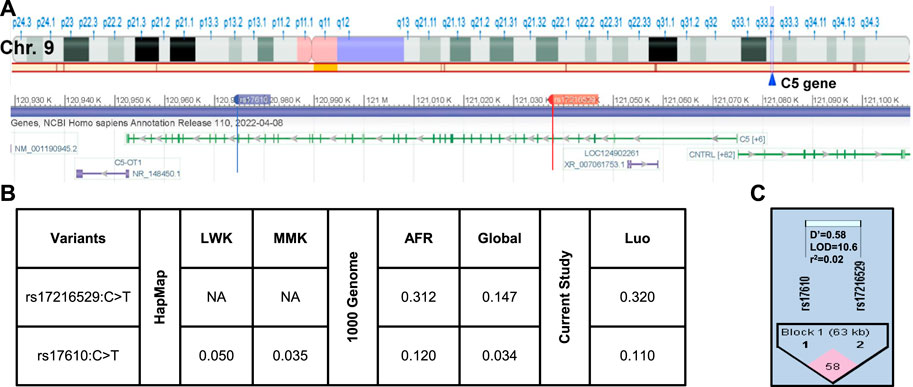
FIGURE 2. C5 chromosome location and linkage disequilibrium (LD) plot of the selected missense mutations (A). Location of C5 on Chromosome 9q33.2. C5 consists of 1,682 amino acids and consists of 43 exons. Chromosome position (build GRCh38. p14) for the investigated SNPs rs17216529:C>T and rs17610:C>T. The alignment and gene structure for C5 was retrieved from dbSNP database (B). C5 genotype minor allele frequencies (MAF) from the HapMap database for Kenyan populations [Luhya (LWK), Maasai (MKK) and calculated by MIDAS for the Luo (current study). MAFs from the 1,000 Genome project shows the African (AFR) and global allele frequencies. MAFs for the current Luo (Kenyan) population are also presented (C). Linkage disequilibrium plot for rs17216529:C>T and rs17610:C>T SNPs (D’ = 0.581, LOD = 10.63, R2 = 0.02). The two SNPs are in linkage equilibrium, that is, they are inherited independently.
Influence of C5 variants on P. falciparum infections, SMA, and all-cause mortality
Prior to the development of adaptive immunity, children in holoendemic P. falciparum transmission regions experience many episodes of malaria. As such, longitudinal studies offer robust platforms to investigate the influence of genetic variants on susceptibility to disease outcomes. The impact of the C5 variants on susceptibility to malaria, SMA, and mortality were determined throughout a 36-month period coinciding with the development of naturally-acquired malarial immunity. As shown in Table 2 (stratified according to genotype/haplotype), there was a high number of both malaria and SMA episodes in the cohort across the follow-up period, and a total of 99 deaths.
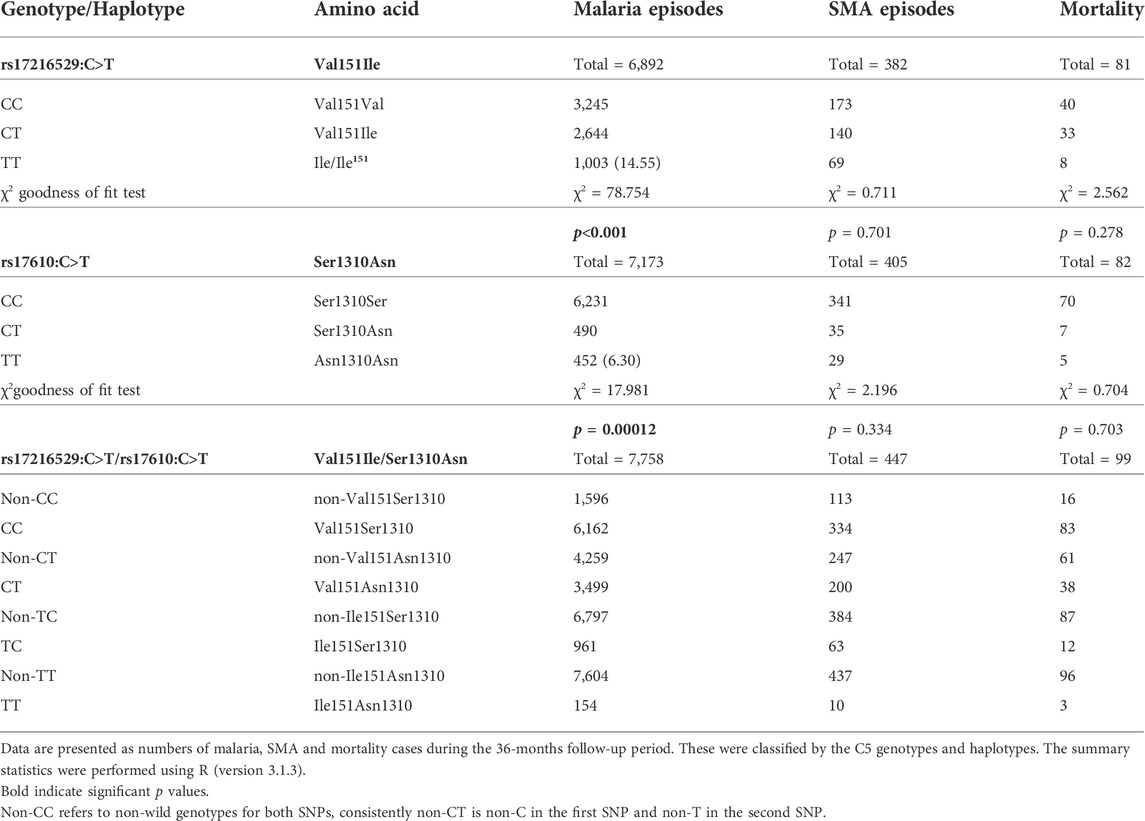
TABLE 2. Number of malaria and SMA episodes, and all-cause mortality across the 36-months follow-up period stratified by genotype/haplotype.
The number of malaria and SMA episodes, as well as mortality cases were recorded throughout the 36-months follow-up period (Table 2). A total of 6,892 malaria cases were reported for rs17216529:C>T. Children with the wild-type CC genotype had the highest number of malaria cases (3,245), followed by the heterozygous CT (2,644), and homozygous mutant TT (1,003) carriers. Cases of malaria for rs17610:C>T, 7,173 were distributed as follows: wild-type CC (6,231), heterozygous CT (490), and homozygous mutant (452). In addition, during the study period, 382 cases of SMA were observed for rs17216529:C>T. The distribution of SMA cases for rs17610:C>T were: wild-type CC (173), heterozygous CT (140) and homozygous mutant TT 69) SMA cases. Finally, mortality cases were recorded for both SNPs during the follow-up period. There were 81 deaths recorded for rs17216529:C>T, distributed into wild-type CC (40), heterozygous CT (33) and homozygous mutant TT (8). For rs17610:C>T, there were 82 cases of mortality within the 36-months study period, categorized into wild-type CC (70), heterozygous CT (7), and homozygous mutant TT (5).
The cases of malaria, SMA, and mortality were also assessed in the haplotypic combinations. There were 7,758 malaria cases for all the haplotypes. These were stratified into CC (6,162), CT (3,499), TC (961), and TT (154). The SMA cases were 447 for all the haplotypes and distributed into CC (334), CT (200), TC (63), and TT (10). Lastly, 99 mortality cases were reported across the haplotypes. The following deaths occurred in CC (83), CT (38), TC (12), and TT (3). Additionally, we performed chi-square goodness of fit test to determine if the number of malaria, SMA and death were elevated that expected in the mutant cases. These analyses revealed that the number of malaria episodes among mutant homozygotes at rs17216529:C>T were higher than expected (Table 2), but lower in the mutant homozygotes at rs17610:C>T. The distribution of SMA episodes and mortality did not differ from the genotype distribution for both SNPs. However, due to the relatively low cases of SMA and mortality there is not much discriminative power at this aggregate level.
The influence of the C5 variant genotypes and haplotypes on susceptibility to malaria and SMA is shown in Figure 3. Genotypic analyses revealed that homozygous carriage of the mutant allele (TT) for rs17216529:C>T, p. (Ile145Ile) increased the risk of both malaria [incident risk ratio (IRR) = 1.144, 95%CI: 1.059–1.236, p = 0.001) and SMA (IRR = 1.627, 95%CI: 1.201–2.204, p = 0.002], with both results remaining significant after adjusting for multiple comparisons. The genotypic model also showed that heterozygous carriers (CT) for the rs17610:C>T, p. (Ser1310Asn) variant had a reduced risk of malaria (IRR = 0.885, 95%CI: 0.794–0.987, p = 0.028], however, the results did not remain significant after correction for multiple comparisons. None of the genotypic combinations of rs17610:C>T significantly altered susceptibility to SMA.
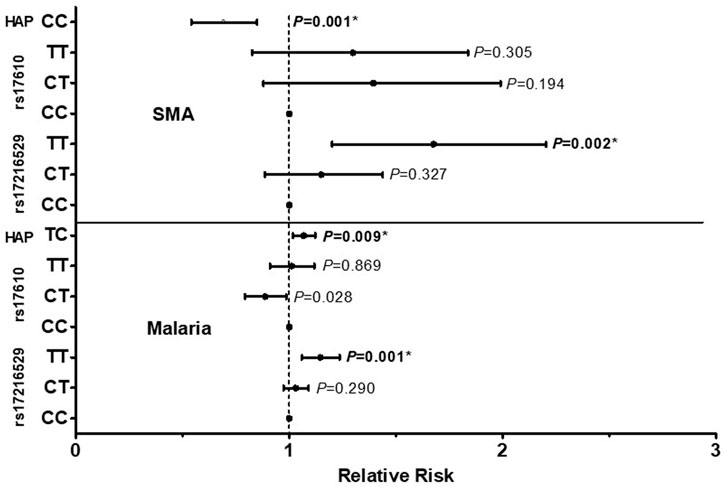
FIGURE 3. A total of 1,546 study participants were enrolled into the study. These were grouped into three categories at enrollment: aparasitemic (n = 288), uncomplicated malaria (UM, Hb ≥ 5.0 g/dl, n = 971) and SMA (Hb < 5.0 g/dl, n = 287). The influence of C5 genotypes/haplotypes on the number of malaria and SMA episodes throughout the study period was determined by a Poisson rate regression (R glm function, family = Poisson), with the (logarithm of) age at the patients last visits being the offset variable (rate regression). A forward-backward model selection that minimize the AIC was used in the Poisson regression. The first covariate inputs into the model were either C5 genotypes or haplotypes. Thereafter, additional covariates which included age at enrollment, sex, HIV, cohort, sickle cell status (sickle trait and sickle cell disease) glucose six phosphate deficiency (G6PD) and α thalassemia were entered into the model. The figure shows relative risk (midline dot) and 95% confidence intervals as determined by log-linear regression analyses, adjusting for age at enrollment, sex, HIV, cohort and sickle cell status (sickle trait and sickle cell disease). This cohort for followed quarterly for a period of 36 months *Significant after Bonferroni-Holm correction.
Further analyses exploring the impact of haplotypic combinations on susceptibility to malaria and SMA revealed that carriers of the TC p.(Ile145Asn1310) haplotype had an increased risk of malaria (IRR = 1.068, 95%CI: 1.017–1.122, p = 0.009, Bonferroni-Holm significant), while inheritance of both wild-type alleles (CC, Val145Ser1310) reduced the risk of SMA (IRR = 0.679, 95%CI: 0.542–0.850, p = 0.001, Bonferroni-Holm significant). Although only results that emerged from the forward-backward selection were reported for the haplotypic models, we performed secondary analyses by forcing the TC haplotype into a binary and additive model for SMA. Carriage of the TC haplotype non-significantly increased the risk of SMA in the binary model (IRR = 1.187, 95%CI: 0.968–1.454, p = 0.098), and significantly increased the risk of SMA in the additive model (IRR = 1.192, 95%CI: 1.038–1.368, p = 0.013). Thus, it appears that inheritance of the T allele(s) at rs17216529:C>T has a substantial impact on both P. falciparum infections and the development of SMA. None of the other haplotypic combinations emerged in the selection models for influencing susceptibility to either malaria or SMA. Moreover, neither the genotypes nor the haplotypes investigated influenced susceptibility to all-cause mortality in the Cox regression analyses.
Discussion
In holoendemic transmission regions for P. falciparum such as western Kenya, SMA has remained a leading cause of morbidity and mortality in immune naïve children. Previous studies in experimental human malaria have shown that complement activation is an early immune response marker against P. falciparum infections (Roestenberg et al., 2007). The cleavage of C5 by C5 convertase is critical for activation of the terminal complement pathway which forms the MAC (C5b-C6-C7-C8-C9) on target pathogens, resulting in cell lysis and apoptosis (Merle et al., 2015). Previous investigations demonstrated that there are elevated levels of soluble C5b-C9 in falciparum malaria (Wenisch et al., 1997). Prior studies also demonstrated C5-dependent P. falciparum sporozoite lysis, presumably by MAC formation in the presence of immune antibodies (Behet et al., 2018; Kurtovic et al., 2018). Moreover, cohort studies in malaria endemic populations have established that P. falciparum sporozoites are susceptible to antibody-mediated complement fixation and activation, with the circumsporozoite protein (CSP) being an important target of complement fixation (Kurtovic et al., 2018). Additionally, antibodies against P. falciparum merozoites can activate the classical pathway, culminating in formation of the MAC and merozoite lysis (Boyle et al., 2015). Results presented here extend previous findings by showing that missense mutations in C5 alter the longitudinal risk for both malaria and SMA.
Selection of the C5 variants for investigation was based on polymorphic variants that could impart amino acid changes, had elevated MAFs in African populations, and were in linkage equilibrium. While both mutations are distal from the C5 convertase cleavage site and are not predicted to have major structural impacts, subtle changes in protein structure may still impact protein function, e.g., by changing the propensity of a molecule to aggregate or by impacting its stability (Goldenzweig and Fleishman, 2018). Since C5b is unstable (Hadders et al., 2012), small changes in stability may result in more rapid degradation or aggregation before binding of other MAC components. Interestingly, the rs17216529:C>T, p. (Val145Ile) variant has previously been non-significantly associated with decreased risk of stroke in pediatric patients with sickle cell disease (Flanagan et al., 2013), and increased levels of the MAC in serum have been associated with incidence and poor outcome of acute ischemic stroke (Si et al., 2019). In Kenyan children, reduced complement lytic activity has been reported in SMA, despite similar levels of C5a between uncomplicated and severe cases (Nyakoe et al., 2009). Thus, although the variants selected for investigation are not predicted to substantially alter the C5 structure, they may impart subtle changes to structure and/or stability of C5b and the MAC which could result in reduced lytic activity.
The main findings that emerged when examining the influence of the C5 variants on longitudinal clinical outcomes was that inheritance of two mutant alleles TT (Ile145Ile) for rs17216529:C>T significantly increased susceptibility to both malaria and SMA. Although SMA is an important cause of childhood mortality in the region, carriage of TT alleles did not significantly alter the risk of all-cause mortality, potentially explaining the relatively high MAF (0.320) maintained in the cohort.
When investigating additional C5 variant combinations, we found that the TC haplotype significantly increased the risk of malaria, consistent with the T allele at rs17216529:C>T being associated with an increased risk of both malaria and SMA in the genotypic models. The only other haplotypic combination that significantly altered longitudinal clinical outcomes was carriage of the CC haplotype that imparted protection against SMA. This result is consistent with both wild type alleles for the two C5 variants being maintained at high proportions in a population residing in a holoendemic P. falciparum transmission region.
Consistent with our recent findings showing that C3 variants inducing non-synonymous amino acid changes failed to alter susceptibility to all-cause childhood mortality (Raballah et al., 2021), the C5 variants explored here were also not significantly associated with mortality. These results, however, should not be over interpreted since our efforts over the past 2 decades have dramatically reduced childhood deaths far below what is expected in rural holoendemic P. falciparum transmission regions. As such, the influence of the selected C5 variants on all-cause mortality may not be expected to emerge in the 99 deaths that occurred.
In conclusion, while the high frequency variants selected for investigation do not appear to confer structural changes to the protein structure, the two novel mutations in the C5 alter susceptibility to both malaria and SMA. As such, a limitation of the current findings is that the specific mechanism(s) by which the variants impart changes in genetic susceptibility remain unknown. Future investigations on the mechanistic basis of the current findings should foster a deeper understanding on how potential structural and functional changes in C5 alter susceptibility to malaria and SMA during the developmental phase of naturally-acquired malarial immunity.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving human participants were reviewed and approved by the Maseno University Ethics Review Committee. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author contributions
ER, QC, and DP, conceptualized the studies; SA, EM, IH, CA, and CO and performed data curation; CL and KS performed data analysis, KS and DP acquired funding for the studies, ER, SA and QC, performed the investigations and methodology, ER, KW, BM and DP performed data analysis, protein modeling and final edits of the manuscript, PS, BM, and CO performed project administration; provision of resources, software, and supervision, PS, BM, CO, CL, and DP performed the final reviews and edits. All authors reviewed and approved the final version of the manuscript.
Funding
These investigations were supported by grants from the National Institutes of Health (R01 AI130473-01 (DP) and D43 TW005884 (DP)). Additional support obtained from the Harvard University-Boston University-Northwestern University-University of New Mexico Consortium, Fogarty Global Health Postdoctoral Fellowship awarded to ER from the Fogarty International Center and National Institute of Mental Health, of the National Institutes of Health (D43 TW010543 (ER, DP)); the German Academic Exchange (DAAD, Project-ID 57417782 (KS); the German Federal Ministry for Research and Education (BMBF-DLR, Project-ID 01DQ20002 (KS)) and Saxony’s State Ministry of Science, Education, and Arts (SMWK-SAB, Project-ID 100257255 (KS)). The data presented in this article is the sole responsibility of the authors.
Acknowledgments
The authors gratefully acknowledge the assistance of the University of New Mexico-Kenya team: Anne A Ong’ondo for administrative support, Vincent Omanje for maintaining the databases, Vincent Odhiambo Otieno, Chrispine Wasonga Ochieng, Joan L. A. Ochieng and Duncan Njega for laboratory support, Nicholas Otieno Ondiek and Enos Osanyo for clinical support, Everlyne A. Modi, Joseph Oduor, Moses Epungure, Moses Lokorkeju, and Rodney B. Mongare for field work and study participants follow-up. We are indebted to all of the parents, guardians, and children whose participation made these studies successful.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.977810/full#supplementary-material
References
Achieng, A. O., Hengartner, N. W., Raballah, E., Cheng, Q., Anyona, S. B., Lauve, N., et al. (2019). Integrated OMICS platforms identify LAIR1 genetic variants as novel predictors of cross-sectional and longitudinal susceptibility to severe malaria and all-cause mortality in Kenyan children. EBioMedicine 45, 290–302. doi:10.1016/j.ebiom.2019.06.043
Anyona, S. B., Hengartner, N. W., Raballah, E., Ong'echa, J. M., Lauve, N., Cheng, Q., et al. (2019). Cyclooxygenase-2 haplotypes influence the longitudinal risk of malaria and severe malarial anemia in Kenyan children from a holoendemic transmission region. J. Hum. Genet. 65, 99–113. doi:10.1038/s10038-019-0692-3
Awandare, G. A., Kempaiah, P., Ochiel, D. O., Piazza, P., Keller, C. C., and Perkins, D. J. (2011). Mechanisms of erythropoiesis inhibition by malarial pigment and malaria-induced proinflammatory mediators in an in vitro model. Am. J. Hematol. 86, 155–162. doi:10.1002/ajh.21933
Behet, M. C., Kurtovic, L., Van Gemert, G. J., Haukes, C. M., Siebelink-Stoter, R., Graumans, W., et al. (2018). The complement system contributes to functional antibody-mediated responses induced by immunization with Plasmodium falciparum malaria sporozoites. Infect. Immun. 86, e00920-17. doi:10.1128/IAI.00920-17
Borg, J., Georgitsi, M., Aleporou-Marinou, V., Kollia, P., and Patrinos, G. P. (2009). Genetic recombination as a major cause of mutagenesis in the human globin gene clusters. Clin. Biochem. 42, 1839–1850. doi:10.1016/j.clinbiochem.2009.07.014
Boyle, M. J., Reiling, L., Feng, G., Langer, C., Osier, F. H., Aspeling-Jones, H., et al. (2015). Human antibodies fix complement to inhibit Plasmodium falciparum invasion of erythrocytes and are associated with protection against malaria. Immunity 42, 580–590. doi:10.1016/j.immuni.2015.02.012
Cherepnalkovski, A. P., Zemunik, T., Glamocanin, S., Piperkova, K., Gunjaca, I., Kocheva, S., et al. (2015). Molecular characterization of glucose-6-phosphate dehydrogenase deficiency in families from the republic of Macedonia and genotype-phenotype correlation. Med. Arch. 69, 284–288. doi:10.5455/medarh.2015.69.284-288
Colobran, R., Franco-Jarava, C., Martin-Nalda, A., Baena, N., Gabau, E., Padilla, N., et al. (2016). Novel mutations causing C5 deficiency in three north-african families. J. Clin. Immunol. 36, 388–396. doi:10.1007/s10875-016-0275-4
Davenport, O. C., Hittner, J. B., Were, T., Ouma, Y., Ong'echa, J. M., Perkins, D. J., et al. (2010). Hematological predictors of increased severe anemia in Kenyan children coinfected with Plasmodium falciparum and HIV-1. Am. J. Hematol. 85 (4), 227–233. doi:10.1002/ajh.21653
Dode, C., Krishnamoorthy, R., Lamb, J., and Rochette, J. (1993). Rapid analysis of -alpha 3.7 thalassaemia and alpha alpha alpha anti 3.7 triplication by enzymatic amplification analysis. Br. J. Haematol. 83, 105–111. doi:10.1111/j.1365-2141.1993.tb04639.x
Flanagan, J. M., Sheehan, V., Linder, H., Howard, T. A., Wang, Y. D., Hoppe, C. C., et al. (2013). Genetic mapping and exome sequencing identify 2 mutations associated with stroke protection in pediatric patients with sickle cell anemia. Blood 121, 3237–3245. doi:10.1182/blood-2012-10-464156
Fredslund, F., Laursen, N. S., Roversi, P., Jenner, L., Oliveira, C. L., Pedersen, J. S., et al. (2008). Structure of and influence of a tick complement inhibitor on human complement component 5. Nat. Immunol. 9, 753–760. doi:10.1038/ni.1625
Gaunt, T. R., Rodriguez, S., Zapata, C., and Day, I. N. (2006). Midas: Software for analysis and visualisation of interallelic disequilibrium between multiallelic markers. BMC Bioinforma. 7, 227. doi:10.1186/1471-2105-7-227
Githeko, A. K., Service, M. W., Mbogo, C. M., Atieli, F. K., and Juma, F. O. (1994). Origin of blood meals in indoor and outdoor resting malaria vectors in Western Kenya. Acta Trop. 58, 307–316. doi:10.1016/0001-706x(94)90024-8
Goldenzweig, A., and Fleishman, S. J. (2018). Principles of protein stability and their application in computational design. Annu. Rev. Biochem. 87, 105–129. doi:10.1146/annurev-biochem-062917-012102
Greenwood, B. M., and Brueton, M. J. (1974). Complement activation in children with acute malaria. Clin. Exp. Immunol. 18, 267–272.
Hadders, M. A., Bubeck, D., Roversi, P., Hakobyan, S., Forneris, F., Morgan, B. P., et al. (2012). Assembly and regulation of the membrane attack complex based on structures of C5b6 and sC5b9. Cell Rep. 1, 200–207. doi:10.1016/j.celrep.2012.02.003
Helegbe, G. K., Goka, B. Q., Kurtzhals, J. A., Addae, M. M., Ollaga, E., Tetteh, J. K., et al. (2007). Complement activation in Ghanaian children with severe Plasmodium falciparum malaria. Malar. J. 6, 165. doi:10.1186/1475-2875-6-165
Henes, J. K., Groga-Bada, P., Schaeffeler, E., Winter, S., Hack, L., Zdanyte, M., et al. (2021). C5 variant rs10985126 is associated with mortality in patients with symptomatic coronary artery disease. Pharmgenomics. Pers. Med. 14, 893–903. doi:10.2147/PGPM.S307827
International Hap M, C. (2003). The international HapMap project. Nature 426, 789–796. doi:10.1038/nature02168
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi:10.1038/s41586-021-03819-2
Kilgore, K. S., Flory, C. M., Miller, B. F., Evans, V. M., and Warren, J. S. (1996). The membrane attack complex of complement induces interleukin-8 and monocyte chemoattractant protein-1 secretion from human umbilical vein endothelial cells. Am. J. Pathol. 149, 953–961.
Kisia, L. E., Kempaiah, P., Anyona, S. B., Munde, E. O., Achieng, A. O., Ong’echa, J. M., et al. (2019). Genetic variation in interleukin-7 is associated with a reduced erythropoietic response in Kenyan children infected with Plasmodium falciparum. BMC Med. Genet. 20, 140. doi:10.1186/s12881-019-0866-z
Klos, A., Tenner, A. J., Johswich, K.-O., Ager, R. R., Reis, E. S., and Köhl, J. (2009). The role of the anaphylatoxins in health and disease. Mol. Immunol. 46, 2753–2766. doi:10.1016/j.molimm.2009.04.027
Kurtovic, L., Behet, M. C., Feng, G., Reiling, L., Chelimo, K., Dent, A. E., et al. (2018). Human antibodies activate complement against Plasmodium falciparum sporozoites, and are associated with protection against malaria in children. BMC Med. 16, 61. doi:10.1186/s12916-018-1054-2
Lu, L., Li, Z., Li, H., Li, X., Vekilov, P. G., and Karniadakis, G. E. (2019). Quantitative prediction of erythrocyte sickling for the development of advanced sickle cell therapies. Sci. Adv. 5, eaax3905. doi:10.1126/sciadv.aax3905
Machiela, M. J., and Chanock, S. J. (2015). LDlink: A web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31, 3555–3557. doi:10.1093/bioinformatics/btv402
Menny, A., Serna, M., Boyd, C. M., Gardner, S., Joseph, A. P., Morgan, B. P., et al. (2018). CryoEM reveals how the complement membrane attack complex ruptures lipid bilayers. Nat. Commun. 9, 5316. doi:10.1038/s41467-018-07653-5
Merle, N. S., Church, S. E., Fremeaux-Bacchi, V., and Roumenina, L. T. (2015). Complement system Part I - molecular mechanisms of activation and regulation. Front. Immunol. 6, 262. doi:10.3389/fimmu.2015.00262
Morgan, B. (2015). The membrane attack complex as an inflammatory trigger. Immunobiology 221, 747–751. doi:10.1016/j.imbio.2015.04.006
Müller-Eberhard, H. J. (1985). The killer molecule of complement. J. Invest. Dermatol. 85, 47s–52s. doi:10.1111/1523-1747.ep12275445
Munde, E. O., Raballah, E., Okeyo, W. A., Ong’echa, J. M., Perkins, D. J., and Ouma, C. (2017). Haplotype of non-synonymous mutations within IL-23R is associated with susceptibility to severe malaria anemia in a P. falciparum holoendemic transmission area of Kenya. BMC Infect. Dis. 17, 291. doi:10.1186/s12879-017-2404-y
Novelli, E. M., Hittner, J. B., Davenport, G. C., Ouma, C., Were, T., Obaro, S., et al. (2010). Clinical predictors of severe malarial anaemia in a holoendemic Plasmodium falciparum transmission area. Br. J. Haematol. 149, 711–721. doi:10.1111/j.1365-2141.2010.08147.x
Nyakoe, N. K., Taylor, R. P., Makumi, J. N., and Waitumbi, J. N. (2009). Complement consumption in children with Plasmodium falciparum malaria. Malar. J. 8, 7. doi:10.1186/1475-2875-8-7
Obonyo, C. O., Vulule, J., Akhwale, W. S., and Grobbee, D. E. (2007). In-hospital morbidity and mortality due to severe malarial anemia in Western Kenya. Am. J. Trop. Med. Hyg. 77, 23–28. doi:10.4269/ajtmh.77.6.suppl.23
Odhiambo, C. O., Otieno, W., Adhiambo, C., Odera, M. M., and Stoute, J. A. (2008). Increased deposition of C3b on red cells with low CR1 and CD55 in a malaria-endemic region of Western Kenya: Implications for the development of severe anemia. BMC Med. 6, 23. doi:10.1186/1741-7015-6-23
Ong'echa, J. M., Keller, C. C., Were, T., Ouma, C., Otieno, R. O., Landis-Lewis, Z., et al. (2006). Parasitemia, anemia, and malarial anemia in infants and young children in a rural holoendemic Plasmodium falciparum transmission area. Am. J. Trop. Med. Hyg. 74, 376–385. doi:10.4269/ajtmh.2006.74.376
Otieno, R. O., Ouma, C., Ong'echa, J. M., Keller, C. C., Were, T., Waindi, E. N., et al. (2006). Increased severe anemia in HIV-1-exposed and HIV-1-positive infants and children during acute malaria. AIDS 20, 275–280. doi:10.1097/01.aids.0000200533.56490.b7
Ouma, C., Davenport, G. C., Awandare, G. A., Keller, C. C., Were, T., Otieno, M. F., et al. (2008). Polymorphic variability in the interleukin (IL)-1beta promoter conditions susceptibility to severe malarial anemia and functional changes in IL-1beta production. J. Infect. Dis. 198, 1219–1226. doi:10.1086/592055
Pak, M. A., Markhieva, K. A., Novikova, M. S., Petrov, D. S., Vorobyev, I. S., Maksimova, E. S., et al. (2021). Using AlphaFold to predict the impact of single mutations on protein stability and function. bioRxiv, 1–7. doi:10.1101/2021.09.19.460937
Perkins, D. J., Were, T., Davenport, G. C., Kempaiah, P., Hittner, J. B., and Ong'echa, J. M. (2011). Severe malarial anemia: Innate immunity and pathogenesis. Int. J. Biol. Sci. 7, 1427–1442. doi:10.7150/ijbs.7.1427
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi:10.1002/jcc.20084
Raballah, E., Anyona, S. B., Cheng, Q., Munde, E. O., Hurwitz, I. F., Onyango, C., et al. (2021). Complement component 3 mutations alter the longitudinal risk of pediatric malaria and severe malarial anemia. Exp. Biol. Med. 247 (8), 672–682. doi:10.1177/15353702211056272
Ricklin, D., Hajishengallis, G., Yang, K., and Lambris, J. D. (2010). Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 11, 785–797. doi:10.1038/ni.1923
Roestenberg, M., Mccall, M., Mollnes, T., Deuren, M., Sprong, T., Klasen, I., et al. (2007). Complement activation in experimental human malaria infection. Trans. R. Soc. Trop. Med. Hyg. 101, 643–649. doi:10.1016/j.trstmh.2007.02.023
Schaid, D. J., Batzler, A. J., Jenkins, G. D., and Hildebrandt, M. A. (2006). Exact tests of Hardy-Weinberg equilibrium and homogeneity of disequilibrium across strata. Am. J. Hum. Genet. 79, 1071–1080. doi:10.1086/510257
Schejbel, L., Fadnes, D., Permin, H., Lappegard, K. T., Garred, P., and Mollnes, T. E. (2013). Primary complement C5 deficiencies - molecular characterization and clinical review of two families. Immunobiology 218, 1304–1310. doi:10.1016/j.imbio.2013.04.021
Sherry, S. T., Ward, M., and Sirotkin, K. (1999). dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 9, 677–679. doi:10.1101/gr.9.8.677
Si, W., He, P., Wang, Y., Fu, Y., Li, X., Lin, X., et al. (2019). Complement complex C5b-9 levels are associated with the clinical outcomes of acute ischemic stroke and carotid plaque stability. Transl. Stroke Res. 10, 279–286. doi:10.1007/s12975-018-0658-3
Team, R. C. 2019. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: Available at: https://www.R-project.org/.[Google Scholar].
Varadi, M., Anyango, S., Deshpande, M., Nair, S., Natassia, C., Yordanova, G., et al. (2022). AlphaFold protein structure database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444. doi:10.1093/nar/gkab1061
Wenisch, C., Spitzauer, S., Florris-Linau, K., Rumpold, H., Vannaphan, S., Parschalk, B., et al. (1997). Complement activation in severe Plasmodium falciparum malaria. Clin. Immunol. Immunopathol. 85, 166–171. doi:10.1006/clin.1997.4417
Were, T., Davenport, G. C., Hittner, J. B., Ouma, C., Vulule, J. M., Ong'echa, J. M., et al. (2011). Bacteremia in Kenyan children presenting with malaria. J. Clin. Microbiol. 49, 671–676. doi:10.1128/JCM.01864-10
Were, T., Hittner, J. B., Ouma, C., Otieno, R. O., Orago, A. S., Ong'echa, J. M., et al. (2006). Suppression of RANTES in children with Plasmodium falciparum malaria. Haematologica 91, 1396–1399.
Keywords: severe malarial anaemia, complement 5, missense mutations, P. facliparum, malaria
Citation: Raballah E, Wilding K, Anyona SB, Munde EO, Hurwitz I, Onyango CO, Ayieko C, Lambert CG, Schneider KA, Seidenberg PD, Ouma C, McMahon BH, Cheng Q and Perkins DJ (2022) Nonsynonymous amino acid changes in the α-chain of complement component 5 influence longitudinal susceptibility to Plasmodium falciparum infections and severe malarial anemia in kenyan children. Front. Genet. 13:977810. doi: 10.3389/fgene.2022.977810
Received: 24 June 2022; Accepted: 24 August 2022;
Published: 14 September 2022.
Edited by:
Danillo G. Augusto, University of California, San Francisco, United StatesReviewed by:
Eugenia Lo, University of North Carolina at Charlotte, United StatesEdith Christiane Bougouma, Groupe de Recherche Action en Santé (GRAS), Burkina Faso
Copyright © 2022 Raballah, Wilding, Anyona, Munde, Hurwitz, Onyango, Ayieko, Lambert, Schneider, Seidenberg, Ouma, McMahon, Cheng and Perkins. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Evans Raballah, ZXJhYmFsbGFoQGhvdG1haWwuY29t