 Teresa Arrobas Velilla1
Teresa Arrobas Velilla1 Pedro Valdivielso
Pedro Valdivielso- 1Laboratorio de Nutrición y Riesgo Cardiovascular de Bioquímica Clínica, Unidad de Lípidos, Hospital Universitario Virgen de la Macarena, Sevilla, Spain
- 2Servicio de Medicina Interna, Unidad de Lípidos, Hospital de San Pedro, Logroño, España
- 3Servicio de Medicina Interna, Unidad de Lípidos, Hospital Virgen de la Victoria, Instituto de Investigación Biomédica de Málaga (IBIMA), Universidad de Málaga, Málaga, Spain
Background: Familial hypercholesterolemia (FH) is clearly underdiagnosed and undertreated. The aim of this present study is to assess the benefits of FH screening through a joint national program implemented between clinical laboratories and lipid units.
Methods: All clinical laboratory tests from 1 January 2017 to 31 December 2018 were reviewed, and those with LDL cholesterol (LDL-C) levels >250 mg/dl were identified in subjects >18 years of age of both sexes. Once secondary causes had been ruled out, the treating physician was contacted and advised to refer the patient to an LU to perform the Dutch Lipid Clinic Network score and to request genetic testing if the score was ≥6 points. Next Generation Sequencing was used to analyse the promoter and coding DNA sequences of four genes associated with FH (LDLR, APOB, PCSK9, APOE) and two genes that have a clinical overlap with FH characteristics (LDLRAP1 and LIPA). A polygenic risk score based on 12 variants was also obtained.
Results: Of the 3,827,513 patients analyzed in 26 centers, 6,765 had LDL-C levels >250 mg/dl. Having ruled out secondary causes and known cases of FH, 3,015 subjects were included, although only 1,205 treating physicians could be contacted. 635 patients were referred to an LU and genetic testing was requested for 153 of them. This resulted in a finding of sixty-seven pathogenic variants for FH, 66 in the LDLR gene and one in APOB. The polygenic risk score was found higher in those who had no pathogenic variant compared to those with a pathogenic variant.
Conclusion: Despite its limitations, systematic collaboration between clinical laboratories and lipid units allows for the identification of large numbers of patients with a phenotypic or genetic diagnosis of FH, which will reduce their vascular risk. This activity should be part of the clinical routine.
Introduction
FH is a monogenic disease characterised by the presence of high levels of LDL cholesterol particles (LDL-C) from birth which, if not properly treated, is associated with premature coronary artery disease and mortality. The disease is caused by pathogenic variants in the LDL receptor (LDLR), apolipoprotein B (APOB) and proprotein convertase subtilisin/kexin type 9 (PCSK9) genes. A phenotypic diagnosis can be obtained using various scoring systems such as the Dutch Lipid Clinic Network (DLCN) score (Mostaza et al., 2022), which considers the LDL-C levels of patients, the levels of first-degree relatives, the presence or absence of xanthomas or corneal arcus in the affected subject, and the personal or family history of cardiovascular disease (CVD). Diagnostic confirmation requires the sequencing of candidate genes and identification of the corresponding pathogenic variant (Brown et al., 2020).
The prevalence of FH is unknown, although it was initially estimated that the disease affected one in 500 people. Two recent meta-analyses of 44 and 42 studies give a prevalence of one in 313 (Beheshti et al., 2020) and one in 311 (Hu et al., 2020), respectively. In addition, a recent study in our country suggest an even higher prevalence among patients treated in health care centres in the Catalonia area with estimates of one in every 189 people being affected (Zamora et al., 2017). It is widely believed that FH is frequently underdiagnosed and undertreated (Nordestgaard et al., 2013).
The identification of patients with FH allows the initiation of lipid-lowering treatment consisting mainly of statins in combination or not with other drugs, such as ezetimibe or PCSK9 inhibitors (PCSK9i). Prompt initiation of treatment for FH can drastically reduce the development of CVD and the risk of mortality (Versmissen et al., 2009; Luirink et al., 2019).
Universal (Groselj et al., 2018) and cascade screening programes (Leonardi-Bee et al., 2021), which allows early diagnosis especially in children (Groselj et al., 2022) now exist in some countries and around the world (Wilemon et al., 2020). Opportunistic testing is also possible for patients who have survived an acute coronary syndrome (Mortensen et al., 2016; Amor-Salamanca et al., 2017; Dyrbuś et al., 2019). Over the last decade, considerable interest has been shown in the identification of patients with FH by using analytical records from community laboratories (Bell et al., 2014; Kirke et al., 2015; Mirzaee et al., 2019). There are limited data in Spain on the use and benefits of centralised analytical data screening in testing for FH (Gutiérrez-Cortizo et al., 2021; Sabatel-Pérez et al., 2021).
The objective of the ARIAN study is to assess the benefits of FH screening through the implementation of a joint national programme between clinical laboratories and lipid units.
Materials and methods
The design of the ARIAN study has already been published (Arrobas Velilla et al., 2021). To summarise, fifty-five clinical laboratories from different Spanish hospitals, which perform biochemical analyses in their respective primary care referral areas, were invited to participate.
In 2019, a retrospective identification of patients aged ≥18 years with a direct or calculated LDL-C > 250 mg/dl (>6.7 mmol/L) and a blood triglyceride concentration <400 mg/dl during the period of 1 January 2017 to 31 December 2018 was obtained through a consultation of the laboratory information system (LIS) of each centre. After reviewing their clinical history, patients were excluded if they had secondary hyperlipidemia (pregnancy, cholestasis, non-substituted hypothyroidism, nephrotic syndrome, among others.) or if they had been previously diagnosed with FH.
Each clinical laboratory contacted the physician requesting the analysis to inform them that their patient was suspected of having FH and to recommend that they be referred to the lipid unit (LU) of the health care centre. In no case was the patient contacted directly. At the LU, the score obtained by the DLCN method was assessed and in cases where the score was ≥6, genetic testing was advised. Each centre had funding for five genetic tests.
Whole blood or saliva samples for deoxyribonucleic acid (DNA) analysis were collected from patients who were accepted into the study. The genetic study was performed using the LIPID inCode® in vitro diagnostic platform (GENinCode Plc, Oxford, United Kingdom). This platform analyses the promoter and coding DNA sequences as well as exon-intron boundaries (±25 bp) of four genes associated with FH (LDLR, APOB, PCSK9, APOE) and two genes associated with other conditions that have a partial clinical overlap with FH characteristics [autosomal recessive hypercholesterolemia (LDLRAP1) and lysosomal acid lipase deficiency (LIPA)] by Next Generation Sequencing (NGS) (Amor-Salamanca et al., 2017). The test has been optimised to detect heterozygous, homozygous, and hemizygous single nucleotide variants (SNVs) and small insertions/deletions (indels) (<15 bp) in the six analysed genes, as well as copy number variants (CNVs) in the LDLR gene. All detected variants with possible clinical relevance (pathogenic, likely pathogenic or variants of unknown significance) were confirmed by Sanger sequencing or MLPA analysis, depending on the variant type.
Variant data analysis is described in the supplementary material. Variants with a minor allele frequency <1% in the general population were considered non common variants. The potential pathogenicity of rare variants was evaluated by considering the recommendations of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen) Familial Hypercholesterolemia consensus guidelines for LDLR variant classification (Richards et al., 2015; Chora et al., 2022), by which different criteria are evaluated: type and variant frequency, functional data if available, scientific support, and computational information for predicted pathogenicity in genomic (PolyPhen2, Provean v.1.1.3, MutationTaster2 and REVEL) or intronic (MaxEntScan, NNSplice, FSPLICE, and GeneSplicer) regions, among others (Richards et al., 2015). Moreover, information on >2,200 FH-related genomic variants included in a private database was also considered to complete the evaluation of genetic variants. Variants with clinical relevance were reported as pathogenic, likely pathogenic, and variants of unknown significance (VUS).
In the case of polygenic hypercholesterolemia, the variants included in the LDL-C Score (rs7412, rs429358, rs1367117, rs4299376, rs629301, rs1564348, rs1800562, rs3757354, rs11220462, rs8017377, rs6511720, and rs2479409) were also analysed by NGS and according to Talmud et al. (Talmud et al., 2013).
Statistical analysis
Data are shown as n (%) or mean ± SD. To compare between groups, we used the χ2 test for qualitative variables and the ANOVA test for continuous variables. A p-value of <0.05 was considered significant. Data were analysed with SPSS 25.0 (IBM).
Ethical aspects
The study obtained authorisation from the ethics committee of the Virgen de la Macarena Hospital, Seville, on the 16th February 2019. The participating clinical laboratories also obtained approval from the ethics committees of their centres. The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki and all patients gave written informed consent to the genetic test and their clinical data.
Results
In the period analysed, twenty-six (45%) centres actively participated in the study, with a reference population in their health areas of 11,147,860 individuals. Serum samples from 3,887,513 adult patients (≥18 years) were analysed, of whom 6,765 had LDL-C levels above 250 mg/dl. Secondary causes that could account for hypercholesterolemia were found in 3,270 patients; 480 patients were already known to have FH (317 had been diagnosed by DLCN and a further 163 by genetic confirmation). Thus, the final sample was 3,015 patients. Of these, only 1,205 patients could be contacted and among this group, 635 (52%) visited their LU. Then, among these patients, 373 obtained a DLCN score ≥6. A genetic test was requested for 153 patients, and a pathogenic variant for FH was found in sixty-seven (45%) of them. No one patient showed a pathogenic variant at APOE gene. Figure 1 summarises these data in a flow chart.
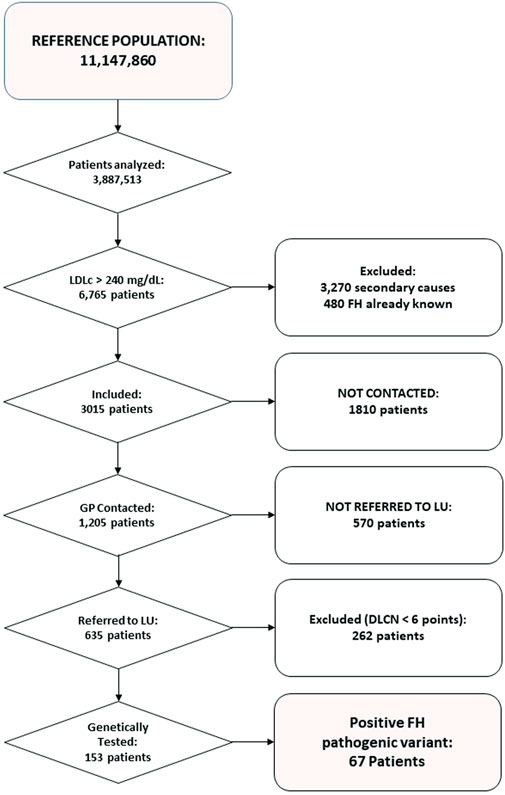
FIGURE 1. Flow-chart of the study, showing patients and analyses at each step.
Table 1 summarises the general characteristics of the sample, indicating that younger patients in whom a pathogenic variant was found exhibited higher DLCN scores and higher LDL-C than the rest of the participants. Polygenic risk score was significantly higher in subjects with genetic test negative or inconclusive compared to those with a pathogenic variant (p < 0.05, ANOVA test).
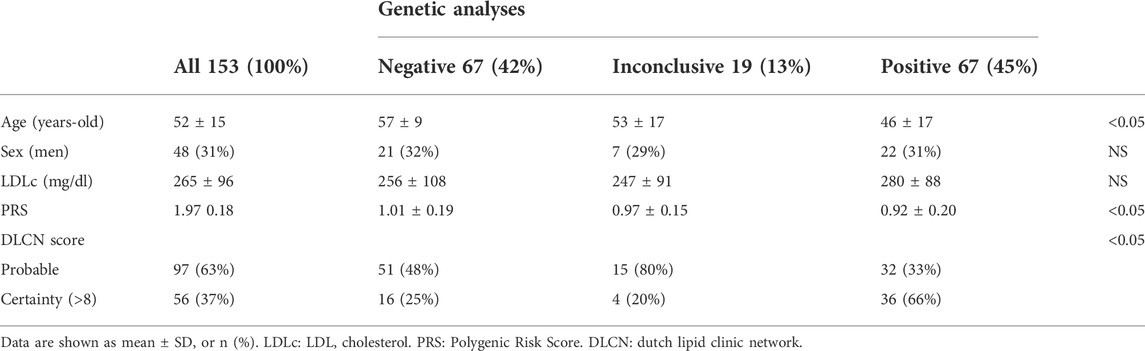
TABLE 1. Demographic and clinical data from patients genetically tested for Familial Hypercholesterolemia.
Table 2 summarizes the studies published on screening for FH using centralized laboratory data and a comparison with the present study. Rates of positive genetic testing ranges from 46 to 68%, mostly depending on the inclusion criteria.
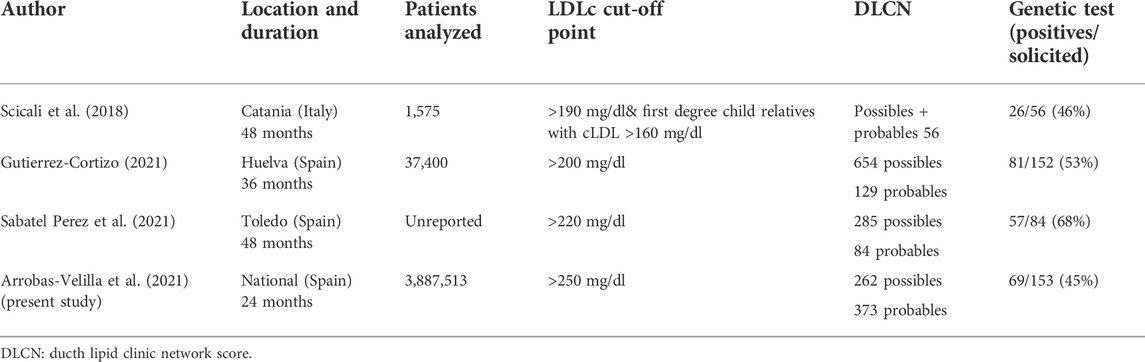
TABLE 2. Screening for Familial Hypercholesterolemia using centralized laboratory data.
Table 1 in the supplementary material presents the variants found in the patients analysed. No homozygous variants were found in the LDLRAP1 and LIPA genes.
Discussion
Our study confirms that systematic screening of LDL-C levels in the clinical laboratory and collaboration with the lipid unit allows for the identification of patients with FH phenotype and its genetic confirmation. This should lead to a better degree of lipid control and improved prevention of cardiovascular disease.
It is noteworthy that 3,270 among 6,765 patients with an LDL-C higher than 250 mg/dl were considered as secondary hypercholesterolemia. However, a recent study showed that the prevalence of secondary causes increase with LDL-C levels, reaching the 52% among those with LDL-C > 300 mg/dl (Jasani et al., 2022).
Despite widespread knowledge of genetic testing and increased accessibility, FH continues to be diagnosed at a late stage and early diagnosis is one of the main forms in reducing the clinical impact of the disease (Vallejo-Vaz et al., 2021). There is a clear need to incorporate new methods with which to identify patients with FH, such as genomic screening in populations or during blood donation, biochemical analysis at birth or during the period of childhood immunisation, and the development of algorithms derived from machine learning (Bradley et al., 2022). A recent study in Australia considered genomic screening of populations aged 18–40 years to be cost-effective from a healthcare system perspective if the cost of the test was less than 160 (Marquina et al., 2021).
As was expected in our study, the frequency of pathogenic variants gradually increased from probable FH to definite FH according to the Dutch Lipid Clinic Network criteria (Taranto et al., 2021) as 66% of the cases with a monogenic diagnosis had a DLCN score of certainty (>8) (see Table 1). In our study, only 67 out of 153 (46%) patients were found to have a pathogenic variant causing FH, a figure which is similar to that published by Scicali et al. (Scicali et al., 2018). However, in this paper, patients with LDL-C > 190 mg/dl and a DLCN score ≥4 were included. Conversely, this rate of finding pathogenic variants is slightly lower than other series of a similar design published in Spain, which range from 53 to 68% (Gutiérrez-Cortizo et al., 2021; Sabatel-Pérez et al., 2021) (see Table 2).
It is noteworthy that no pathogenic variant was found after genetic analysis in sixty-five (42%) patients in our study with LDL-C levels >250 mg/dl, and VUS variants were found in 19 (13%) patients. This highlights the importance of polygenic risk in severe primary hypercholesterolemia (Futema et al., 2018). As expected, patients with a negative or inconclusive genetic study (VUS) had a significantly higher polygenic score than in patients who were shown to have a pathogenic variant (Talmud et al., 2013) (Table 2). Although PRS is associated with increased LDL-C levels and risk of coronary heart disease, knowledge of PRS does not add value to risk calculation if incorporated into a model that includes age, sex and LDL-C levels (Tromp et al., 2021). Although severe polygenic hypercholesterolaemia does not present the same acute risk of cardiovascular disease as monogenic forms, the risk is still high (Khera and Hegele, 2020) and some authors propose family cascade screening using LDL-C as a guide (Jarauta et al., 2020).
Although the study design (Arrobas Velilla et al., 2021) allowed the identification of patients with severe hypercholesterolemia (3,015) and among them sixty-seven carriers of pathogenic FH variants, we believe the performance of the study leaves room for improvement. Having previously ruled out secondary causes and known cases of FH, the primary care physician could be contacted regarding only 1,205 (40%) of the aforementioned 3,015 individuals with LDL-C levels >250 mg/dl. Geographical dispersion among hundreds of primary care centres participants in the study and lack of personal phone number or email in the ordering test form, may have contributed to the low rate of contacting. Moreover, among those who could be contacted, only 635 (53%) were referred to the LUs. Although these findings are discouraging, they are in line with studies of a similar design to ours. Of 4,517 subjects with LDL-C levels >170 mg/dl identified by a general laboratory, only 597 (13%) agreed to be clinically evaluated for suspected FH. Of these, thirty were genetically tested and eight showed a pathogenic variant in the gene responsible for FH (Kirke et al., 2015). In another Australian study of 100 cases with LDL-C > 250 mg/dl detected in a clinical laboratory, there was a 27% referral rate to a LU when primary care physicians were informed of suspected FH cases and the high risk of CV disease and instructed to refer their patients to a LU. Furthermore, ninety-six individuals in the control group exhibited the same LDL-C levels; however, their physicians were not notified thus contributing to an even lower referral rate (4%) (Bell et al., 2014). Considering these data as a whole, there is much room for improvement in PC regarding the recognition of FH as an extremely high-risk vascular condition. At the same time, there is evidence of a certain “resistance” to refer cases to LUs, where sometimes complex diagnosis and treatment of these patients can be carried out, as many require combined treatments including PCSK9i, which is a class of prescription drugs dispensed in hospitals (Ascaso et al., 2019; Visseren et al., 2021).
Finally, it is striking that among the 6,765 patients identified in clinical laboratories with LDL-C levels >250 mg/dl, 480 already had a previous clinical or genetic diagnosis of FH (Figure 1). This finding clearly reveals undertreatment and is particularly striking considering that in Spain, FH patients benefit from a very low co-payment (reduced contribution) (Boletín Oficial del Estado, 2003), paying only 4 euros for statins and ezetimibe (regardless of type and dose), and are entitled to a full reimbursement for PCSK9i.
Among the limitations, it is worth mentioning that genetic studies were financed for only five cases per centre since many centres do not have their own means to carry out genetic studies. Furthermore, there was a low response rate to refer patients to an LU by those PC physicians who could be contacted. Other limitation of our study has been to include only adult population; that means that we have lost the opportunity to identify children with FH and, as a consequence, to delay the diagnosis in such group of patient with the highest chance to prevent atherosclerosis earlier.
Conclusion
The systematic review by a clinical laboratory of cases with extremely high LDL-C levels, communication of these to primary care physicians, and assessment of cases by lipid units allows for the identification of many patients with a phenotypic and/or genetic diagnosis of FH. This clinical activity should be part of routine clinical practice.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, Data are available from the authors upon reasonable request and with permission of Spanish Arteriosclerosis Society.
Ethics statement
The studies involving human participants were reviewed and approved by the Comite de Etica de la Investigacion Virgen de la Macarena, Sevilla, Spain. All patient gave written informed consent.
Author contributions
TA designed and coordinated the study, AB designed and coordinated the study and PV curate the database and wrote the draft. All authors analysed the data and approved the final version of the manuscript.
Funding
This ARIAN study was funded by a grant of the Spanish Arteriosclerosis Society (2018) and by SANOFI. Both funding bodies did not take part in the analyses of the data, in the decision to publish the paper and in its content.
Acknowledgments
The authors gratefully acknowledge the staff of the clinical laboratories and lipid units at each of the centers listed in the Supplementary Appendix A.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.971651/full#supplementary-material
References
Amor-Salamanca, A., Castillo, S., Gonzalez-Vioque, E., Dominguez, F., Quintana, L., Lluís-Ganella, C., et al. (2017). Genetically confirmed familial hypercholesterolemia in patients with acute coronary syndrome. J. Am. Coll. Cardiol. 70, 1732–1740. doi:10.1016/j.jacc.2017.08.009
Arrobas Velilla, T., Brea, Á., Valdivielso, P., Alguacil, B. G., Temprano, R. P., Quesada, M. M., et al. (2021). Implantación de un programa de cribado bioquímico y genético de hipercolesterolemia familiar. Colaboración entre el laboratorio clínico y las unidades de lípidos: Diseño del proyecto ARIAN. Clinica Investig. Arterioscler. 33, 289–295. doi:10.1016/J.ARTERI.2021.03.004
Ascaso, J. F., Civeira, F., Guijarro, C., López Miranda, J., Masana, L., Mostaza, J. M., et al. (2019). Indications of PCSK9 inhibitors in clinical practice. Recommendations of the Spanish Sociey of Arteriosclerosis (SEA), 2019. Clin. Investig. Arterioscler. 2019, 128–139. doi:10.1016/j.arteri.2019.04.002
Beheshti, S. O., Madsen, C. M., Varbo, A., and Nordestgaard, B. G. (2020). Worldwide prevalence of familial hypercholesterolemia: Meta-analyses of 11 million subjects. J. Am. Coll. Cardiol. 75, 2553–2566. doi:10.1016/j.jacc.2020.03.057
Bell, D. A., Hooper, A. J., Edwards, G., Southwell, L., Pang, J., van Bockxmeer, F. M., et al. (2014). Detecting familial hypercholesterolaemia in the community: Impact of a telephone call from a chemical pathologist to the requesting general practitioner. Atherosclerosis 234, 469–472. doi:10.1016/j.atherosclerosis.2014.04.002
Boletín Oficial del Estado (2003). Real decreto 1348/2003. Available at: https://www.boe.es/eli/es/rd/2003/10/31/1348.
Bradley, C. K., Khera, A., and Navar, A. M. (2022). Underdiagnosis of familial hypercholesterolaemia: Innovation is overdue. Eur. Heart J. 00, ehab869–3. doi:10.1093/EURHEARTJ/EHAB869
Brown, E. E., Sturm, A. C., Cuchel, M., Braun, L. T., Duell, P. B., Underberg, J. A., et al. (2020). Genetic testing in dyslipidemia: A scientific statement from the national lipid association. J. Clin. Lipidol. 14, 398–413. doi:10.1016/j.jacl.2020.04.011
Chora, J. R., Iacocca, M. A., Tichý, L., Wand, H., Kurtz, C. L., Zimmermann, H., et al. (2022). The clinical Genome Resource (ClinGen) familial hypercholesterolemia variant curation expert panel consensus guidelines for LDLR variant classification. Genet. Med. 24, 293–306. doi:10.1016/J.GIM.2021.09.012
Dyrbuś, K., Gąsior, M., Desperak, P., Osadnik, T., Nowak, J., and Banach, M. (2019). The prevalence and management of familial hypercholesterolemia in patients with acute coronary syndrome in the Polish tertiary centre: Results from the TERCET registry with 19, 781 individuals. Atherosclerosis 288, 33–41. doi:10.1016/J.ATHEROSCLEROSIS.2019.06.899
Futema, M., Bourbon, M., Williams, M., and Humphries, S. E. (2018). Clinical utility of the polygenic LDL-C SNP score in familial hypercholesterolemia. Atherosclerosis 277, 457–463. doi:10.1016/j.atherosclerosis.2018.06.006
Groselj, U., Kovac, J., Sustar, U., Mlinaric, M., Fras, Z., Podkrajsek, K. T., et al. (2018). Universal screening for familial hypercholesterolemia in children: The Slovenian model and literature review. Atherosclerosis 277, 383–391. doi:10.1016/j.atherosclerosis.2018.06.858
Groselj, U., Wiegman, A., and Gidding, S. S. (2022). Screening in children for familial hypercholesterolaemia: Start now. Eur. Heart J. 2022, ehac224. doi:10.1093/EURHEARTJ/EHAC224
Gutiérrez-Cortizo, E. N., Romero-Jiménez, M. J., Mansilla Rodríguez, M. E., Díaz Santos, M. A., Caballero Granado, F. J., Sánchez Ruiz-Granados, E., et al. (2021). Detection of familial hypercholesterolemia through centralized analytical data. HF HUELVA DETECTA program. Endocrinol Diabetes Nutr 2021, 1. doi:10.1016/j.endinu.2020.09.011
Hu, P., Dharmayat, K. I., Stevens, C. A. T., Sharabiani, M. T. A., Jones, R. S., Watts, G. F., et al. (2020). Prevalence of familial hypercholesterolemia among the general population and patients with atherosclerotic cardiovascular disease: A systematic review and meta-analysis. Circulation 141, 1742–1759. doi:10.1161/CIRCULATIONAHA.119.044795
Jarauta, E., Bea-Sanz, A. M., Marco-Benedi, V., and Lamiquiz-Moneo, I. (2020). Genetics of hypercholesterolemia: Comparison between familial hypercholesterolemia and hypercholesterolemia nonrelated to LDL receptor. Front. Genet. 2020, 554931. doi:10.3389/FGENE.2020.554931
Jasani, R., Ahmad, Z., Schneider, R., Tujardon, C., Basit, M., and Khera, A. (2022). Applying an LDL-C threshold-based approach to identify individuals with familial hypercholesterolemia. J. Clin. Lipidol. 2022, 1. doi:10.1016/J.JACL.2022.04.001
Khera, A. V., and Hegele, R. A. (2020). What is familial hypercholesterolemia, and why does it matter? Circulation 141, 1760–1763. doi:10.1161/CIRCULATIONAHA.120.046961
Kirke, A. B., Barbour, R. A., Burrows, S., Bell, D. A., Vickery, A. W., Emery, J., et al. (2015). Systematic detection of familial hypercholesterolaemia in primary health care: A community based prospective study of three methods. Heart Lung Circ. 24, 250–256. doi:10.1016/j.hlc.2014.09.011
Leonardi-Bee, J., Boateng, C., Faria, R., Eliman, K., Young, B., and Qureshi, N. (2021). Effectiveness of cascade testing strategies in relatives for familial hypercholesterolemia: A systematic review and meta-analysis. Atherosclerosis 338, 7–14. doi:10.1016/j.atherosclerosis.2021.09.014
Luirink, I. K., Wiegman, A., Kusters, D. M., Hof, M. H., Groothoff, J. W., de Groot, E., et al. (2019). 20-Year follow-up of statins in children with familial hypercholesterolemia. N. Engl. J. Med. 381, 1547–1556. doi:10.1056/nejmoa1816454
Marquina, C., Lacaze, P., Tiller, J., Riaz, M., Sturm, A. C., Nelson, M. R., et al. (2021). Population genomic screening of young adults for familial hypercholesterolaemia: A cost-effectiveness analysis. Eur. Heart J. 2021, ehab770. doi:10.1093/EURHEARTJ/EHAB770
Mirzaee, S., Choy, K. W., Doery, J. C. G., Zaman, S., Cameron, J. D., and Nasis, A. (2019). The tertiary hospital laboratory; a novel avenue of opportunistic screening of familial hypercholesterolemia. Int. J. Cardiol. Heart Vasc. 2019, 100354. doi:10.1016/j.ijcha.2019.100354
Mortensen, M. B., Kulenovic, I., Klausen, I. C., and Falk, E. (2016). Familial hypercholesterolemia among unselected contemporary patients presenting with first myocardial infarction: Prevalence, risk factor burden, and impact on age at presentation. J. Clin. Lipidol. 10, 1145–1152. doi:10.1016/j.jacl.2016.06.002
Mostaza, J. M., Pintó, X., Armario, P., Masana, L., Real, J. T., Valdivielso, P., et al. (2022). SEA 2022 standards for global control of cardiovascular risk. Clin. Investig. Arterioscler. 34, 130–179. doi:10.1016/J.ARTERI.2021.11.003
Nordestgaard, B. G., Chapman, M. J., Humphries, S. E., Ginsberg, H. N., Masana, L., Descamps, O. S., et al. (2013). Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease: Consensus statement of the European atherosclerosis society. Eur. Heart J. 34, 3478–390a. doi:10.1093/eurheartj/eht273
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi:10.1038/gim.2015.30
Sabatel-Pérez, F., Sánchez-Prieto, J., Becerra-Muñoz, V. M., Alonso-Briales, J. H., Mata, P., and Rodríguez-Padial, L. (2021). Improving familial hypercholesterolemia index case detection: Sequential active screening from centralized analytical data. J. Clin. Med. 10, 749. doi:10.3390/jcm10040749
Scicali, R., Di Pino, A., Platania, R., Purrazzo, G., Ferrara, V., Giannone, A., et al. (2018). Detecting familial hypercholesterolemia by serum lipid profile screening in a hospital setting: Clinical, genetic and atherosclerotic burden profile. Nutr. Metab. Cardiovasc. Dis. 28, 35–43. doi:10.1016/j.numecd.2017.07.003
Talmud, P. J., Shah, S., Whittall, R., Futema, M., Howard, P., Cooper, J. A., et al. (2013). Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: A case-control study. Lancet 381, 1293–1301. doi:10.1016/S0140-6736(12)62127-8
Taranto, M. D., Giacobbe, C., Palma, D., Iannuzzo, G., Gentile, M., Calcaterra, I., et al. (2021). Genetic spectrum of familial hypercholesterolemia and correlations with clinical expression: Implications for diagnosis improvement. Clin. Genet. 100, 529–541. doi:10.1111/CGE.14036
Tromp, T. R., Cupido, A. J., Reeskamp, L. F., Stroes, E. S. G., Hovingh, G. K., Defesche, J. C., et al. (2021). Assessment of practical applicability and clinical relevance of a commonly used LDL-C polygenic score in patients with severe hypercholesterolemia. Atherosclerosis 340, 61–67. doi:10.1016/J.ATHEROSCLEROSIS.2021.10.015
Vallejo-Vaz, A. J., Stevens, C. A. T., Lyons, A. R. M., Dharmayat, K. I., Freiberger, T., Hovingh, G. K., et al. (2021). Global perspective of familial hypercholesterolaemia: A cross-sectional study from the EAS familial hypercholesterolaemia studies collaboration (FHSC). Lancet 398, 1713–1725. doi:10.1016/s0140-6736(21)01122-3
Versmissen, J., Oosterveer, D. M., Yazdanpanah, M., Defesche, J. C., Basart, D. C. G., Liem, A. H., et al. (2009). Efficacy of statins in familial hypercholesterolaemia: A long term cohort study. BMJ 338, a2423–226. doi:10.1136/bmj.a2423
Visseren, F. L. J., Mach, F., Smulders, Y. M., Carballo, D., Koskinas, K. C., Bäck, M., et al. (2021). 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur. Heart J. 42, 3227–3337. doi:10.1093/eurheartj/ehab484
Wilemon, K. A., Patel, J., Aguilar-Salinas, C., Ahmed, C. D., Alkhnifsawi, M., Almahmeed, W., et al. (2020). Reducing the clinical and public health burden of familial hypercholesterolemia: A global call to action. JAMA Cardiol. 5, 217–229. doi:10.1001/jamacardio.2019.5173
Keywords: familial hypercholesterolemia (FH), clinical laboratory, lipid units, Dutch lipid clinic network criteria, screening
Citation: Arrobas Velilla T, Brea Á and Valdivielso P (2022) Implementation of a biochemical, clinical, and genetic screening programme for familial hypercholesterolemia in 26 centres in Spain: The ARIAN study. Front. Genet. 13:971651. doi: 10.3389/fgene.2022.971651
Received: 20 June 2022; Accepted: 03 August 2022;
Published: 29 August 2022.
Edited by:
Augusto Rojas-Martinez, Tecnológico de Monterrey, MexicoReviewed by:
Urh Groselj, University of Ljubljana, SloveniaKrzysztof Dyrbuś, Silesian Center for Heart Diseases, Poland
Copyright © 2022 Arrobas Velilla, Brea and Valdivielso. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pedro Valdivielso, dmFsZGl2aWVsc29AdW1hLmVz