 Sixi Hao
Sixi Hao Xiuzhen Hu
Xiuzhen Hu Zhenxing Feng1,2*
Zhenxing Feng1,2*
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 11 August 2022
Sec. Computational Genomics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.969412
This article is part of the Research Topic Deep Machine Learning and Big Data Resources for Transcriptional Regulation Analysis View all 4 articles
Proteins need to interact with different ligands to perform their functions. Among the ligands, the metal ion is a major ligand. At present, the prediction of protein metal ion ligand binding residues is a challenge. In this study, we selected Zn2+, Cu2+, Fe2+, Fe3+, Co2+, Mn2+, Ca2+ and Mg2+ metal ion ligands from the BioLip database as the research objects. Based on the amino acids, the physicochemical properties and predicted structural information, we introduced the disorder value as the feature parameter. In addition, based on the component information, position weight matrix and information entropy, we introduced the propensity factor as prediction parameters. Then, we used the deep neural network algorithm for the prediction. Furtherly, we made an optimization for the hyper-parameters of the deep learning algorithm and obtained improved results than the previous IonSeq method.
The interaction between proteins and ligands is particularly important for a variety of biological processes such as the transport of oxygen, the transfer of cellular signals, energy conversion and muscle contraction (Reif, 1992; Davis et al., 2004; Jeffrey et al., 2006). Therefore, it is valuable to accurately identify the protein-ligand binding site for understanding protein function, disease occurrence and molecular drug design (Laurie and Jackson, 2006). Among these ligands, more than one-third are metal ion ligands (Hu et al., 2020). Although the bond length, bond angle and torsion angle of each metal ion ligand binding to proteins are different, from the perspective of the spatial structure of protein binding to metal ion ligands, all metal ion ligands combine with residues on the “pocket” of the protein surface to form a complex and stable spatial structure. Therefore, we selected the eight metal ion ligands as a series of studies. Due to the small size and active chemical properties of metal ion ligands, it is a challenging work to predict the metal ion ligand binding residues with similar chemical structures by theoretical calculation methods.
In the prediction of protein-metal ion ligand binding sites, predecessors have done a lot of research work and made significant progress. At present, the feature parameters of most studies were based on the information of primary sequences, the physical and chemical and predicted structure information. For example, in 2016, Jiang et al. (2016) used the component of amino acids, the autocross covariance value, center motif and site conservative information as feature parameters for predicting the binding sites of Ca2+ ligands, and the total predict accuracy (Acc) was better than 70.0%. Then, Hu et al. (2016b) used position specific scoring matrix (PSSM), secondary structure, and the real values of phi and psi as feature parameters to predict the binding sites of Cu2+, Fe2+, Fe3+, and Zn2+, and the obtained Matthew’s correlation coefficient (MCC) was higher than 0.20, Acc was higher than 97.0%. In 2017, Cao et al. (2017) selected the sequences information, site conservative information, secondary structure information, matrix scoring values of the hydrophilic-hydrophobic and polarization charge as feature parameters to identify the binding sites of 10 metal ion ligands, the MCC value was higher than 0.502. In 2019, Wang et al. (2019) selected the component information and site conservative information of features such as amino acids, secondary structure, relative solvent accessibility, hydrophilic-hydrophobic, and polarization charge to predict binding sites of 10 metal ion ligands, the ACC was higher than 68.0%. Although the physicochemical feature of amino acid and predicted structure information were usually used as feature parameters in previous studies, the obtained prediction results by different extraction methods were also different. Therefore, the selection and extraction methods of feature parameters need to be emphasized and innovatively optimized in study.
In recent years, many traditional machine learning algorithms have been used to predict protein-metal ion ligand binding sites, such as support vector machine (SVM), random forest (RF), bayesian classifier. For example, in 2016, Hu et al. (2016a) developed a method called IonSeq based on SVM to predict 10 metal ion ligands, and the values of sensitivity (Sn), Acc and MCC were higher than 5.57%, 74.09% and 0.1516, respectively. In 2020, Liu et al. (2020) used RF algorithm to predict the binding sites of 10 metal ion ligands, the MCC and Acc were better than 0.07 and 52%, respectively. In 2021, Wang et al. (2021) applied the SVM algorithm to predict ten metal ion ligands, the Sn and MCC values were greater than 39.5% and 0.118, respectively. Although these traditional algorithms obtained good results in the prediction of protein-metal ion ligand binding residues, it is difficult for them to learn deeply and effectively from the growing amount of data in the post-genomic and big data era (Song et al., 2020). At present, deep learning is a new way to realize machine learning, and has powerful deep learning capabilities and parallel distributed processing capabilities. It has been used in the study of protein-metal ion ligand binding residues, and good prediction results have been obtained (Cui et al., 2019).
In this paper, the deep neural network (DNN) algorithm was used to predict the binding residues of eight metal ion ligands (Lorenzo-Trueba et al., 2018). Based on protein sequence, we selected amino acids, secondary structure, relative solvent accessibility, dihedral angles, charge and hydrophilic-hydrophobic as basic feature parameters, and added disorder values as new feature parameters. On the basis of component information, position weight matrix, information entropy and propensity factors were added as a new feature parameter. By optimizing the three hyper-parameters in the deep learning algorithm, the prediction results have been significantly improved.
The data set is the basis of prediction. To ensure the authenticity of data and the accuracy of experiment, we selected eight metal ion ligands from the BioLip database: Zn2+, Cu2+, Fe2+, Fe3+, Co2+, Mn2+, Ca2+, Mg2+ (Yang et al., 2013). In order to construct non-redundant data set, we filtered the data samples by eliminating the sequence length of less than 50 amino acids, resolution greater than 3 Å, and the sequence identity higher than 30%. The fragments were intercepted on the protein sequence by using the sliding window method. To make that every residue of the protein chain appears in the center of the fragment, we added (L−1)/2 pseudo-amino acids at both ends of the protein chain. Here the length L of the intercepted fragments was taken according to references (Hu et al., 2016a). If (L+1)/2 is a binding residue, it is defined as a positive fragment, otherwise it was a negative fragment. The non-redundant data set of eight metal ion ligands is shown in Table 1.
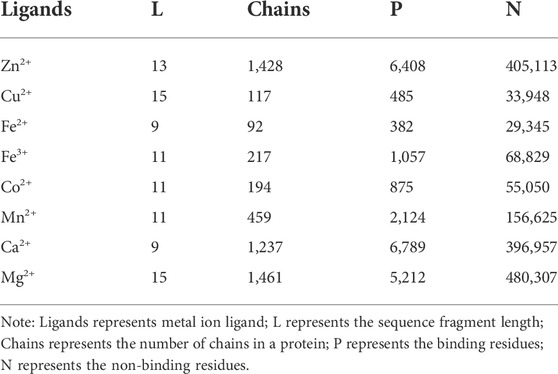
TABLE 1. The non-redundant data set for eight metal ion ligands.
In recent years, researchers have discovered a special class of amino acid fragments in protein sequences. Due to the fact that these fragments lack stable structure and are highly variable, they are called the disordered regions of proteins (Dunker et al., 2002). The instability and high variability of these disordered regions can lead to their easy interaction with ligands (Noivirt-Brik et al., 2009). In this way, it has been applied to the prediction of protein-protein interaction, and good prediction results have been obtained (Zhang et al., 2016). In this work, we used the IUPred2A software and converted the structural state of each amino acid in the protein sequence into the disorder score (Mészáros et al., 2018; Gábor and Dosztányi, 2020). The disorder score ranges from 0 to 1, and the higher the value, the more disordered the structure of amino acids. In this paper, the disorder values of positive (negative) set fragments were statistically analyzed, since the disorder value was continuous, it was divided into 10 intervals for the convenience of statistics. Taken Ca2+ and Cu2+ as examples, the distribution of disorder value of the binding residue and non-binding residue was shown in Figure 1.
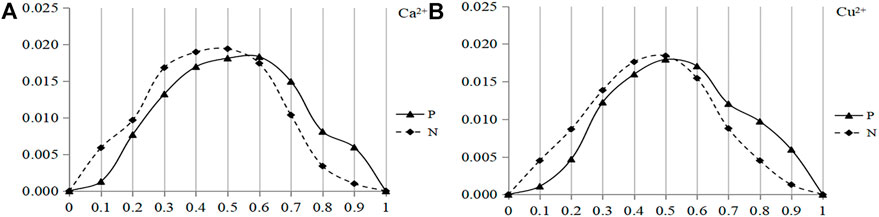
FIGURE 1. Distribution of disorder values of the binding residue and non-binding residue of Ca2+ and Cu2+ ligands. Note: The ordinate is the probability of the disorder value; P, represents the binding residues; N, represents the non-binding residues.
Note: The abscissa is the disorder value; the ordinate is the probability of the disorder value.; Solid line and dotted line are the positive and negative sets, respectively.
It can be seen from Figure 1A that the difference of the disorder values of Ca2+ ligand between the positive and negative sets was mainly concentrated in two intervals: 0–0.55 and 0.55–1, and the threshold was 0.55. In Figure 1B, the threshold for Cu2+ ligand was 0.52. Therefore, eight metal ion ligands were integrated, the disorder value was divided into two categories, the threshold value was set as 0.5, and the value greater than 0.5 tends to disorder. X represents the disorder value, and the classification threshold of the disorder value was represented by the function f(x).
Based on the sequence of amino acids, we selected amino acids, physicochemical features and predicted structural information as feature parameters. Among them, the physicochemical features of amino acids included the charge and hydrophilic-hydrophobic of amino acids. According to the charge properties of amino acids, the 20 amino acids were divided into three categories (Taylor, 1986), as shown in Figure 2A; according to the hydrophilic-hydrophobic properties of amino acids, the 20 amino acids were divided into six categories (Pánek et al., 2005), as shown in Figure 2B.
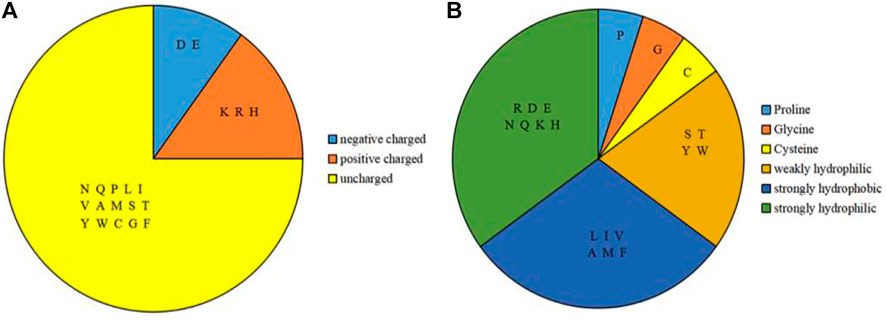
FIGURE 2. Classification of charge features and hydrophilic-hydrophobic features of amino acids. Note: (A) is 3 categories of the charge features; (B) is 6 categories of the hydrophilic-hydrophobic features.
The predicted structural information includes: secondary structure information, relative solvent accessibility and dihedral angle (phi angle and psi angle), all of which were obtained by the ANGLOR software for protein sequences (Wu and Zhang, 2008). The secondary structure information included three types: α-helix, β-sheet and coil. According to statistical analysis, the solvent accessibility was divided into four intervals (Cao et al., 2017), and its threshold was represented by r(x):
The dihedral angle information was reclassified in line with statistics (Liu et al., 2020), the threshold value of the phi angle was represented by the function g(x), and the threshold value of the psi angle was represented by the function h(x):
The previous methods of extracting feature parameters were based on sequence fragments, and the effect of binding residues and their surrounding residues on the protein-ligand binding process has been sufficiently considered. However, in the process of ligand protein binding, the specific binding residues can directly interact with the ligands. The preference for amino acids and physicochemical properties of these specific binding residues has more outstanding impact on the binding process. The propensity factors first appeared in the 1970s and was proposed by two scholars, Chou and Fasman (Chou and Fasman, 1974). It has been applied to the prediction of protein secondary structure with good prediction results. The formula of the propensity factors was expressed as follow:
where,
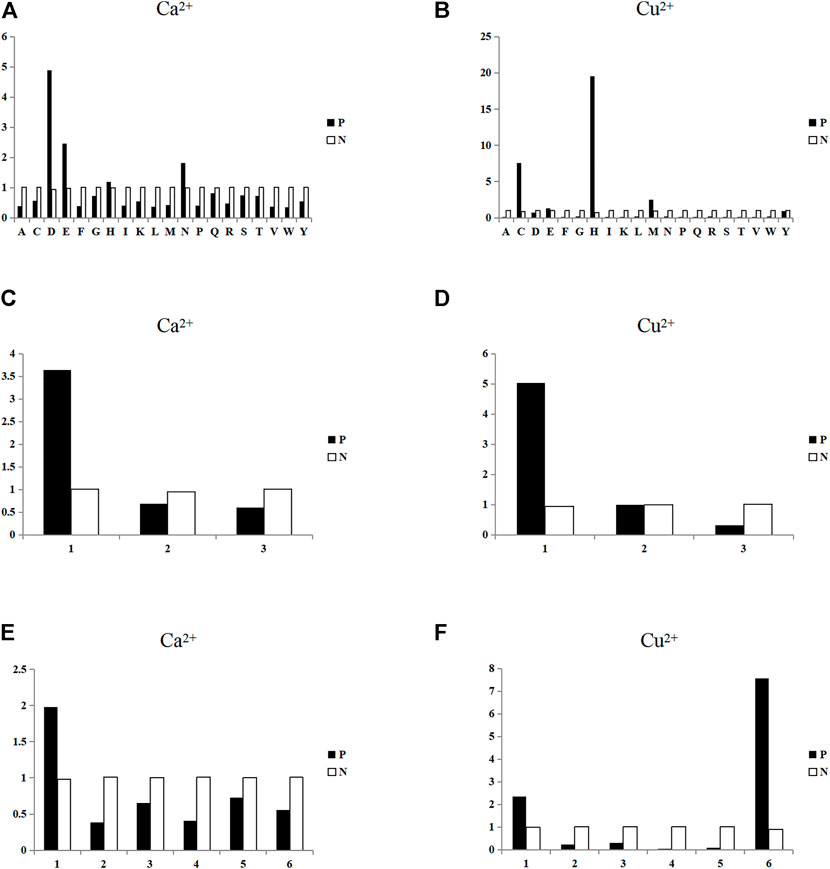
FIGURE 3. Statistical analysis of the propensity factors of binding residues and non-binding residues. Note: In Figure 3, the ordinate represents the value of propensity factors, and P and N represent binding residues and non-binding residues, respectively. Figures (A) and (B) are the statistical analysis of propensity factors of amino acids of Ca2+ and Cu2+ ligands, respectively; The abscissa represents 20 amino acids. Figures (C) and (D) are the statistical analysis of propensity factors of charge features of Ca2+ and Cu2+ ligands, respectively; and the abscissa represents the three charge classifications. Figures (E) and (F) are the statistical analysis of the propensity factors of hydrophilic-hydrophobic features of Ca2+ and Cu2+ ligands, respectively; and the abscissa represents the six hydrophilic-hydrophobic classifications.
In Figure 3A, for Ca2+ ligand, the propensity factor values of four amino acids D, E, H and N in binding residues were significantly higher than that in non-binding residues. It showed that the amino acids D, E, H, and N were more likely to be used in binding residues for Ca2+ ligand. Similarly, it can be seen from Figure 3B that the amino acids C, E, H, and M were more likely be used in binding residues for Cu2+ ligand. It can be found in Figures 3C,D that the binding residues of both Ca2+ and Cu2+ ligands tended to be positively charged. As can be seen in Figures 3E,F, the binding residues of Ca2+ ligands were more likely to strong hydrophilicity, and the binding residues of Cu2+ ligands were more likely to strong hydrophilicity and amino acid C. It can be seen from the comprehensive statistical analysis that the amino acid, charge and hydrophilic-hydrophobic had obvious preferences in binding residues and non-binding residues. Therefore, this paper used the propensity factor that can reflect the preference of binding residues as new extraction method, the above three feature parameters was extracted and used them as the predicted feature parameters. Finally, we obtain 6-dimensional propensity factor.
The position weight matrix were widely used in the prediction of protein structure and function to extract the site conservative features, and good prediction results were obtained. Here, the position weight matrix was also used to extract the site conservative features, and the matrix elements of position weight matrix were expressed as follows (Kel et al., 2003; Gao and Hu, 2014):
Where,
According to previous studies (Liu et al., 2020; Wang et al., 2021), information entropy was used to extract charge and hydrophilic-hydrophobic and better prediction performance was obtained. Here, we also use the extraction method of information entropy. The 1-dimensional information entropy was obtained from the hydropathic-hydrophobic and charge information of amino acids, respectively. Finally, we got 2-dimensional information entropy.
The information entropy formula was expressed as (Strait and Dewey, 1996):
Where,
According to previous studies (Jiang et al., 2016; Cao et al., 2017; Wang et al., 2019; Liu et al., 2020; Wang et al., 2021), it was found that good prediction results were obtained by using component information, which indicated that the component information was particularly important for predicting the binding sites of protein-metal ion ligands. Therefore, we also adopted the extraction method of component information. In the study, we extracted 21, 4, 5, 3, 4, and 3-dimensional component information for amino acids, secondary structure, relative solvent accessibility, phi angle, psi angle and disorder value, respectively. Finally, we obtained a total of 40-dimensional component information.
Deep Neural Network (DNN) is one of the common deep learning algorithms, which aims to improve the discriminative ability of the model by providing a higher level of abstraction. Its neural network layer can be divided into input layer, hidden layer and output layer. The addition of hidden layer enhances the expression ability of the model; the extension of activation functions, such as Tanx function, Softmax function, and Relu function, etc, makes that the DNN algorithm have a wider application field. Therefore, DNN algorithm is selected as the prediction tool in this paper.
This paper used the following modules of the deep learning algorithm: the DNN backpropagation algorithm was used to train samples; the sklearn-preprocessing module was used to normalize the data; the Adam module was used as optimizer; the Relu function was used as the activation function of hidden layer; using the EarlyStopping module can effectively avoid the problem of overfitting caused by continuous training; the cross entropy loss function was used to speed up the operation. These algorithm modules were implemented under the keras framework of Python deep learning, and used TensorFlow as the back-end engine to build the DNN algorithm.
In this study, the 5-fold cross-validation was generally used to predict metal ion ligand binding residues (Hu et al., 2016b; Hu et al., 2016b; Jiang et al., 2016; Cao et al., 2017; Hu et al., 2020). For the evaluation of the prediction results, we used the methods commonly used in the prediction of protein-metal ion ligand binding residues: sensitivity (Sn), specificity (Sp), accuracy (Acc), and Matthew’s correlation coefficient (MCC) (Jiao and Du, 2016; Chen et al., 2019). The expressions are:
In the formula, the number of metal ion ligand binding residues correctly predicted is TP, otherwise it is FN; the number of metal ion ligand non-binding residues correctly predicted is TN, otherwise it is FP.
The component information (37 dimensions) and site conservative information (5*2L dimensions) of amino acids, secondary structure, relative solvent accessibility and dihedral angle, and information entropy (2 dimensions) of charge and hydropathic-hydrophobic were fused as feature parameters, the DNN algorithm was used to predict, and the 5-fold cross-validation results were shown in Table 2 (DNNa). Overall, the predicted results were not ideal. The Sn value of the eight metal ion ligands was only over 11.53%, the Sp and Acc values were only better than 96.38%, and the MCC value was only better than 0.1354.
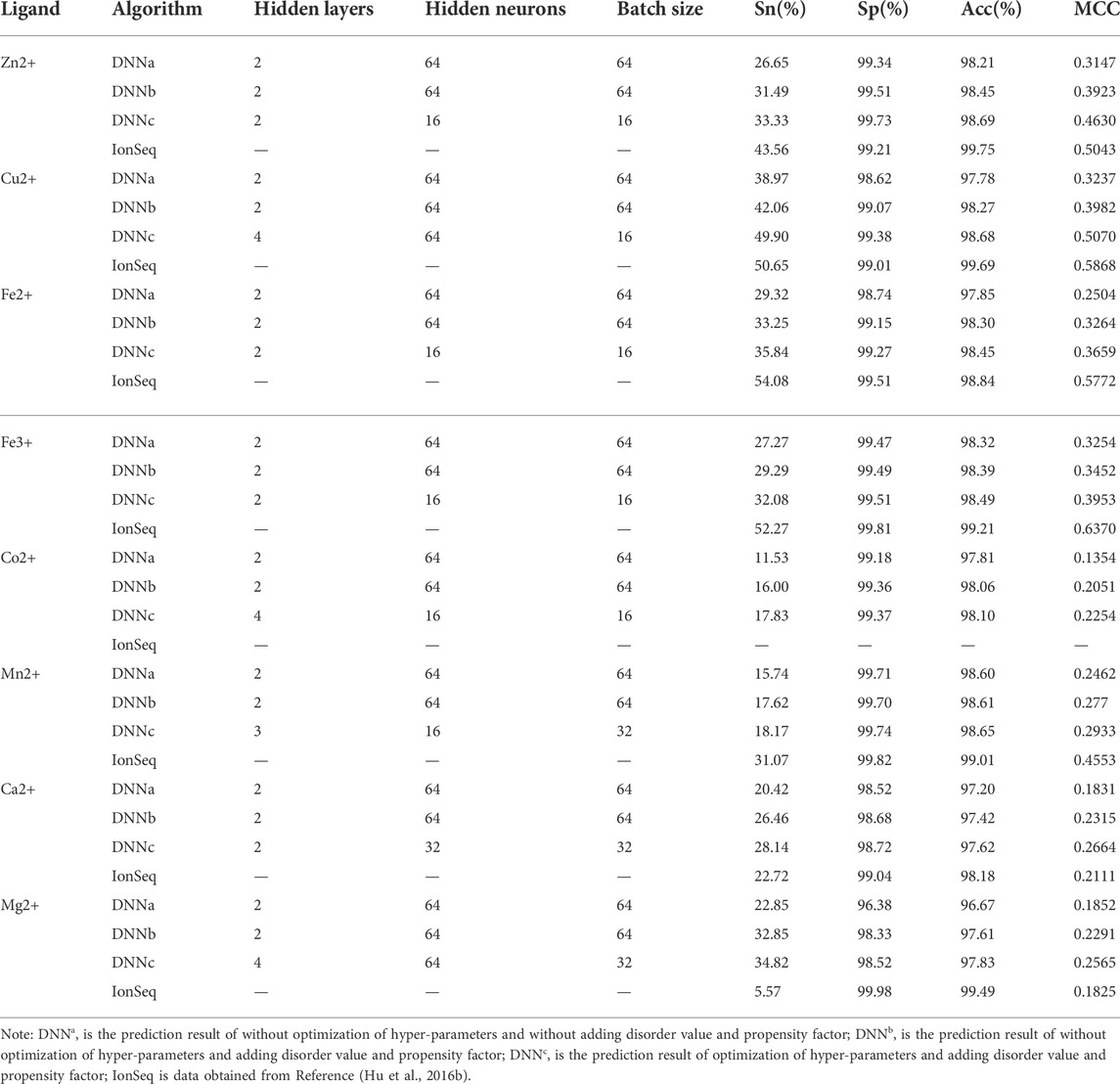
TABLE 2. Comparison of 5-fold cross-validation results.
In order to further improve the prediction performance, disorder value and propensity factor were introduced, and the DNN algorithm was used to predict the metal ion ligand binding residues. The results of 5-fold cross-validation of Ca2+ and Cu2+ ligands as examples were shown in Figure 4.
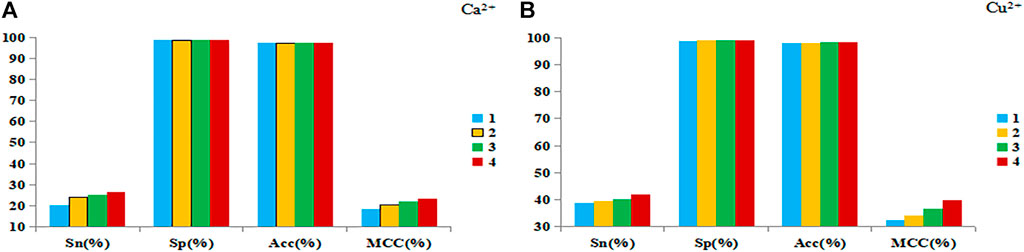
FIGURE 4. The results of 5-fold cross-validation of Ca2+ (A) and Cu2+ (B) ligands. Note: The abscissa is the four evaluation indexes, and the ordinate is the value of the evaluation index. The ordinate is the value of the evaluation index. The blue bar represents the prediction results of the basic feature parameters, the yellow bar represents the prediction results of 1+propensity factor, the green bar represents the prediction results of 1+disorder value, and the red bar represents the prediction results of 2+ disorder value.
It can be found from Figures 4A,B that when the disorder value and propensity factor were added separately, the Sn and MCC values were significantly improved, and the Sp and Acc values were almost unchanged. When the disorder values and propensity factor were used at the same time, the prediction results were the best. Therefore, we believed that both the disorder value and the propensity factor had a more positive effect on the prediction of metal ion ligand binding residues.
The prediction results of feature parameters four are listed in Table 2 (DNNb). It can be seen from Table 2 that the Sn value of the eight metal ion ligands reached 16%, the Sp and Acc values reached 97.42%, and the MCC value reached 0.2051. Compared with the prediction results of the basic parameters, it can be found that all the four evaluation indexes of the eight kinds of ions have been improved, in which the Sn and MCC values increased significantly. For example, the Sn value of Mg2+, Ca2+, Co2+ and Zn2+ ligands increased by 10%, 6.04%, 4.47% and 4.84%, respectively; the MCC value increased by 0.0439, 0.0484, 0.0697 and 0.0776, respectively. It can be seen that the adding disordered value and propensity factor can effectively improve the prediction performance.
The hyper-parameters of deep learning algorithms include: learning rate, activation function, and number of epochs, etc. The hyper-parameters had great influence on the training speed and performance of the predictor. Therefore, we optimize the hyper-parameters to improve the prediction performance. Considering the influence on model accuracy, computing resources, computing time and previous studies (Koutsoukas et al., 2017), we selected three hyper-parameters to optimize, which included the number of hidden layers, the number of hidden layer nodes (the number of hidden neurons) and the batch size. The value range of the optimized hyper-parameters was given in Table 3.

TABLE 3. Value range of hyper-parameters.
Taken Ca2+ as examples, Figure 5A is a line chart showing the MCC value and Sn value of Ca2+ ligands with the number of hidden layers. It can be seen from Figure 5A that the number of hidden layers had great influence on the performance of the predictor. When the number of hidden layers was 2, both the MCC and Sn values reached their peaks. We took the optimal layer value for Ca2+ ligand as 2. From Figure 5B, it can be known that the optimal hidden layer node value of Ca2+ ligands was 32. From Figure 5C, it could be seen that the optimal batch size of Ca2+ ligands was 32.
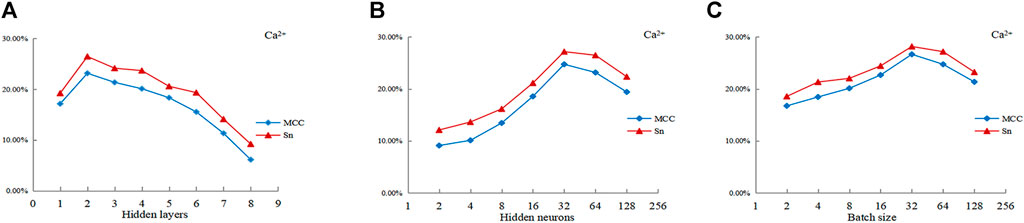
FIGURE 5. Curve of MCC value and Sn value of Ca2+ ligands with hyper-parameters. Note: The abscissas of (A-C) represent three hyperparameters, respectively. The ordinate is the value of MCC and Sn; MCC and Sn are the evaluation index.
The prediction results after optimization of hyper-parameters were shown in Table 2 (DNNc). It can be seen from the results that the Sn value of eight metal ion ligands reached 17.83%, the Sp and Acc values reached 97.62%, and the MCC value reached 0.2254. Compared with DNNb, it was found that the optimization of hyper-parameters could effectively improve the prediction performance of the DNN algorithm, and the four evaluation indexes had a certain improvement. The Sn and MCC values were significantly improved, in which the Sn value of Cu2+ and Fe3+ ligands increased by 7.84 and 2.79%, respectively. The MCC value of Fe3+, Cu2+ and Zn2+ ligands increased by 0.0501, 0.1088 and 0.0707, respectively.
In order to verify the reliability and practicability of the prediction model, the results were compared with the previous IonSeq method. For the convenience of comparison, the results of the IonSeq method were also listed in Table 2. Through analysis and comparison, it was found that the evaluation index of the prediction result of the DNN algorithm has the same characteristics as the IonSeq method. Both the methods have small Sn value and large SP value. The reason for this result was that the number of negative samples was much greater than that of positive samples in the dataset. The results of DNN algorithm for alkaline Earth metals (Mg2+ and Ca2+) were better than IonSeq method, in which the Sn and MCC values of Mg2+ ligand increased by 29.25% and 0.074, respectively. The Sn and MCC values of Ca2+ ligand increased by 5.42% and 0.0553, respectively. The prediction results of Cu2+ ligand were closest to the IonSeq method, and the Sp value was slightly higher than the IonSeq method. Co2+ ligand can’t be compared with the IonSeq method, but the prediction performance was greatly improved by comparing the prediction results. The prediction results of the other four metal ion ligands using the DNN algorithm were slightly poor. Although not all of our results were better than the IonSeq method, the DNN algorithm had a certain positive effect on the prediction of metal ion ligand residues.
In this paper, based on the information of protein sequence and sequence-derived structure, the DNN algorithm was used to predict eight types of metal ion ligands binding residues. The introduction of new feature parameters and extraction methods perfected the basic feature parameter information, which helped to identify metal ion ligand binding sites and improved the prediction performance. The hyper-parameter optimization of the model effectively improved the prediction performance of the DNN model. In comparison with IonSeq, the obtained prediction model based on sequence information, sequence-derived structure information and DNN algorithm was not very perfect. However, in view of the universality and practicability of the prediction model, DNN model can be used as a supplementary model to predict metal ion ligand residues.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
SH: Performed the experiments and wrote the paper; XH: Guided the experiments and the paper; ZF: Improved the English and assisted in the experiments; KS, XY, ZW, and CY: Gave guidance on the writing of the paper.
This work was supported by the National Natural Science Foundation of China (61961032 and 31260203) and the Natural Science Foundation of the Inner Mongolia of China (2019BS03025), and the Natural Science Foundation of Inner Mongolia University of Technology (ZY201915).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Cao, X. Y., Hu, X. Z., Zhang, X. J., Gao, S. J., Ding, C. J., Feng, Y. G., et al. (2017). Identification of metal ion binding sites based on amino acid sequences. Plos One 12, e0183756. doi:10.1371/journal.pone.0183756
Chen, Z., Zhao, P., Li, F. Y., Marquez-Lago, T. T., Leier, A., Song, J. N., et al. (2019). iLearn: an integrated platform and meta-learner for feature engineering, machine learning analysis and modeling of DNA, RNA and protein sequence data. Brief. Bioinform. 21, 1047–1057. doi:10.1093/bib/bbz041
Chou, P. Y., and Fasman, G. D. (1974). Conformational parameters for amino acids in helical, β-sheet, and random coil regions calculated from proteins. Biochemistry 13, 211–222. doi:10.1021/bi00699a001
Cui, Y., Dong, Q., Hong, D. C., and Wang, X. K. (2019). Predicting protein-ligand binding residues with deep convolutional neural networks. BMC Bioinforma. 20, 93. doi:10.1186/s12859-019-2672-1
Davis, J. P., Rall, J. A., Alionte, C., and Tikunova, S. B. (2004). Mutations of hydrophobic residues in the n-terminal domain of troponin C affect calcium binding and exchange with the troponin C-troponin I96-148 complex and muscle force production. J. Biol. Chem. 279, 17348–17360. doi:10.1074/jbc.M314095200
Dunker, A. K., Brown, C. J., Lawson, J. D., and Obradovic, Z. (2002). Intrinsic disorder and protein function. Biochemistry 41, 6573–6582. doi:10.1021/bi012159+
Gábor, E., and Dosztányi, Z. (2020). Analyzing protein disorder with IUPred2A. Curr. Protoc. Bioinforma. 70, e99. doi:10.1002/cpbi.99
Gao, S., and Hu, X. (2014). Prediction of four kinds of supersecondary structures in enzymes by using SVM based on scoring function. Biotechnol. Indian J. 10, 5986–5996.
Hu, X. Z., Dong, Q. W., Yang, J. Y., and Zhang, Y. (2016). Recognizing metal and acid radical ion binding sites by integrating ab initio modeling with template-based transferals. Bioinformatics 32, 3260–3269. doi:10.1093/bioinformatics/btw396
Hu, X. Z., Feng, Z. X., Zhang, X. J., Liu, L., and Wang, S. (2020). The identification of metal ion ligand-binding residues by adding the reclassified relative solvent accessibility. Front. Genet. 11, 214. doi:10.3389/fgene.2020.00214
Hu, X. Z., Wang, K., and Dong, Q. W. (2016). Protein ligand-specific binding residue predictions by an ensemble classifier. BMC Bioinforma. 17, 470. doi:10.1186/s12859-016-1348-3
Jeffrey, M., González, L., Espenes, A., Martin, S., Chaplin, M., Davis, L., et al. (2006). Transportation of prion protein across the intestinal mucosa of scrapie-susceptible and scrapie-resistant sheep. J. Pathol. 209, 4–14. doi:10.1002/path.1962
Jiang, Z., Hu, X. Z., Geriletu, G., Xing, H. R., and Cao, X. Y. (2016). Identification of Ca (2+)-binding residues of a protein from its primary sequence. Genet. Mol. Res. 15, 1676–1680. doi:10.4238/gmr.15027618
Jiao, Y. S., and Du, P. F. (2016). Performance measures in evaluating machine learning based bioinformatics predictors for classifications. Quant. Biol. 4, 320–330. doi:10.1007/s40484-016-0081-2
Kel, A. E., Gößling, E., Reuter, I., Cheremushkin, E., Kel-Margoulis, O., and Wingender, E. (2003). Match: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 31, 3576–3579. doi:10.1093/nar/gkg585
Koutsoukas, A., Monaghan, K. J., Li, X. L., and Huan, J. (2017). Deep-learning: Investigating deep neural networks hyper-parameters and comparison of performance to shallow methods for modeling bioactivity data. J. Cheminform. 9, 42. doi:10.1186/s13321-017-0226-y
Laurie, A. T., and Jackson, R. M. (2006). Methods for the prediction of protein-ligand binding sites for structure-based drug design and virtual ligand screening. Curr. Protein Pept. Sci. 7, 395–406. doi:10.2174/138920306778559386
Liu, L., Hu, X. Z., Feng, Z. X., Wang, S., Sun, K., Xu, S., et al. (2020). Recognizing ion ligand-binding residues by random forest algorithm based on optimized dihedral angle. Front. Bioeng. Biotechnol. 8, 493. doi:10.3389/fbioe.2020.00493
Lorenzo-Trueba, J., Henter, G. E., Takaki, S., Yamagishi, J., Morino, Y., and Ochiai, Y. (2018). Investigating different representations for modeling and controlling multiple emotions in DNN-based speech synthesis. Speech Commun. 99, 135–143. doi:10.1016/j.specom.2018.03.002
Mészáros, B., Gábor, E., and Dosztányi, Z. (2018). IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 46, W329–W337. doi:10.1093/nar/gky384
Noivirt-Brik, O., Prilusky, J., and Sussman, J. L. (2009). Assessment of disorder predictions in CASP8. Proteins 77, 210–216. doi:10.1002/prot.22586
Pánek, J., Eidhammer, I., and Aasland, R. (2005). A new method for identification of protein (sub)families in a set of proteins based on hydropathy distribution in proteins. Proteins 58, 923–934. doi:10.1002/prot.20356
Reif, D. W. (1992). Ferritin as a source of iron for oxidative damage. Free Radic. Biol. Med. 12, 417–427. doi:10.1016/0891-5849(92)90091-T
Song, J. Z., Liang, Y. C., Liu, G. X., Wang, R. Q., Sun, L. Y., Zhang, P., et al. (2020). A novel prediction method for ATP-binding sites from protein primary sequences based on fusion of deep convolutional neural network and ensemble learning. IEEE Access 8, 21485–21495. doi:10.1109/ACCESS.2020.2968847
Strait, B. J., and Dewey, T. G. (1996). The Shannon information entropy of protein sequences. Biophys. J. 71, 148–155. doi:10.1016/s0006-3495(96)79210-x
Taylor, W. R. (1986). The classification of amino acid conservation. J. Theor. Biol. 119, 205–218. doi:10.1016/s0022-5193(86)80075-3
Wang, S., Hu, X. Z., Feng, Z. X., Liu, L., Sun, K., Xu, S., et al. (2021). Recognition of ion ligand binding sites based on amino acid features with the fusion of energy. physicochemical and structural features. Curr. Pharm. Des. 27, 1093–1102. doi:10.2174/1381612826666201029100636
Wang, S., Hu, X. Z., Feng, Z. X., Zhang, X. J., Liu, L., Sun, K., et al. (2019). Recognizing ion ligand binding sites by SMO algorithm. BMC Mol. Cell Biol. 20, 53. doi:10.1186/s12860-019-0237-9
Wu, S., and Zhang, Y. (2008). Anglor: A composite machine-learning algorithm for protein backbone torsion angle prediction. Plos One 3, e3400. doi:10.1371/journal.pone.0003400
Yang, J. Y., Roy, A., and Zhang, Y. (2013). BioLiP: A semi-manually curated database for biologically relevant ligand–protein interactions. Nucleic Acids Res. 41, D1096–D1103. doi:10.1093/nar/gks966
Keywords: metal ion ligand, deep neural network algorithm, disorder value, propensity factors, binding residues
Citation: Hao S, Hu X, Feng Z, Sun K, You X, Wang Z and Yang C (2022) Prediction of metal ion ligand binding residues by adding disorder value and propensity factors based on deep learning algorithm. Front. Genet. 13:969412. doi: 10.3389/fgene.2022.969412
Received: 15 June 2022; Accepted: 04 July 2022;
Published: 11 August 2022.
Edited by:
Pu-Feng Du, Tianjin University, ChinaReviewed by:
Yongchun Zuo, Inner Mongolia University, ChinaCopyright © 2022 Hao, Hu, Feng, Sun, You, Wang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiuzhen Hu, aHh6QGltdXQuZWR1LmNu; Zhenxing Feng, enhmZW5nQGltdXQuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.